

Abstract
Antiviral innate immunity is initiated in response to RNA molecules that are produced in virus-infected cells1. These RNAs activate signalling cascades that activate the genes that encode α- and β-interferon (IFN). Signalling occurs through the interaction of the RNAs with either of two pathogen recognition receptors, retinoic acid-inducible gene-I (RIG-I, also known as DDX58) and melanoma differentiation associated gene-5 (MDA5, also known as IFIH1), which contain amino-terminal caspase activation and recruitment domains (CARD) and carboxy-terminal DExD/H Box RNA helicase motifs2-5. RIG-I and MDA5 interact with another CARD protein, interferon-β promotor stimulator protein-1 (IPS-1, also known as MAVS, VISA and Cardif), in the mitochondrial membrane, which relays the signal through the transcription factors interferon regulatory factor 3 (IRF-3) and nuclear factor (NF)-κB to the IFN-β gene6-10. Although the signalling pathway is well understood, the origin of the RNA molecules that initiate these processes is not. Here we show that activation of the antiviral endoribonuclease, RNase L11, by 2′,5′-linked oligoadenylate (2-5A)12 produces small RNA cleavage products from self-RNA that initiate IFN production. Accordingly, mouse embryonic fibroblasts lacking RNase L were resistant to the induction of IFN-β expression in response to 2-5A, dsRNA or viral infection. Single-stranded regions of RNA are cleaved 3′ of UpUp and UpAp sequences by RNase L during viral infections, resulting in small, often duplex, RNAs13,14. We show that small self-RNAs produced by the action of RNase L on cellular RNA induce IFN-β expression and that the signalling involves RIG-I, MDA5 and IPS-1. Mice lacking RNase L produce significantly less IFN-β during viral infections than infected wild-type mice. Furthermore, activation of RNase L with 2-5A in vivo induced the expression of IFN-β in wild-type but not RNase L-deficient mice. Our results indicate that RNase L has an essential role in the innate antiviral immune response that relieves the requirement for direct sensing of non-self RNA.
To determine the effect of RNase L on the induction of IFN-β, we transfected wild-type and Rnasel−/− (RNase L-deficient)15 mouse embryonic fibroblasts (MEFs) with either dephosphorylated (inactive) or triphosphorylated (active) trimer 2-5A16. Transfection of dephosphorylated 2-5A ((2′-5′)A3) did not induce the expression of IFN-β, as determined by a specific enzyme-linked immunosorbent assay (ELISA; Fig. 1a). However, triphosphorylated 2-5A ((2′-5′)p3A3), the naturally occurring activator of RNase L, potently induced IFN-β production in a dose-dependent manner in wild-type but not in RNase L-deficient cells. The RNase L-deficient cells were also relatively resistant to the induction of IFN-β expression in response to the synthetic dsRNA poly(I):poly(C) in comparison to the wild-type cells (Fig. 1b). In addition, RNase L-deficient cells were partially resistant to the induction of IFN-β expression by the paramyxovirus, Sendai virus (SeV), a negative RNA strand virus (infection of wild-type cells produced 7.5-fold more IFN-β than did infection of RNase L-deficient cells; Fig. 1c). Ectopic expression of RNase L in RNase L-deficient MEFs restored the induction of IFN-β expression by SeV, poly(I):poly(C) or 2-5A (Supplementary Fig. 1). Showing that the nuclease function of RNase L is essential for signalling, the nuclease-dead mutants of RNase L (R667A or H672A) failed to restore the induction of IFN-β expression by 2-5A (Supplementary Fig. 1c, d).
Figure 1. Involvement of RNase L in induction of IFN-β expression by 2-5A, dsRNA or viral infection.

a–c, IFN-β concentration from wild-type (WT) or RNase L-deficient MEFs that were mock transfected (Control) or transfected with (2′-5′)A3 or (2′-5′) p3A3 for 16 h (a); mock transfected or transfected with poly(I):poly(C) for 18 h (b); or mock infected or infected with SeV for 18 h (c). d–f, IFN-β concentration from wild-type MEFs and RIG-I-deficient MEFs (d), MDA5-deficient MEFs (e) or IPS-1-deficient MEFs that were either mock transfected or transfected with unfractionated 2-5A for 16 h (f). Values are means from triplicate assays with standard deviations (s.d.).
To test our hypothesis that RNA cleavage products generated by RNase L were responsible for the induction of IFN-β expression, we performed experiments with primary MEFs lacking the RNA helicases RIG-I or MDA5, or their adaptor, IPS-1. The induction of IFN-β in response to treatment with 2-5A was greatly reduced (by > 14-fold) in Rig-i−/− (RIG- I-deficient)3 MEFs as compared to wild-type MEFs (Fig. 1d). In addition, Mda5−/− (MDA5-deficient) MEFs4 were about fourfold less responsive to the induction of IFN-β by 2-5A than were wild-type MEFs (Fig. 1e). Both RIG-I and MDA5 relay signals through IPS-16-9. As a result, Ips1−/− (IPS-1-deficient) MEFs17 were unresponsive to the induction of IFN-β by 2-5A (Fig. 1f).
To extend these findings to human cells, we used small inhibitory (si)RNA oligonucleotides to suppress individually the levels of RIG-I, MDA5 and IPS-1 in the human prostate cancer cell line DU145 (Fig. 2a). A decrease in the level of each of these signalling proteins inhibited the induction of a human IFN-β promoter by 2-5A (by more than sevenfold compared to the untreated control; Fig. 2b). By contrast, non-specific short interfering (si)RNA failed to inhibit the induction of the IFN-β promoter by 2-5A. Simultaneous knock-down of RIG-I and MDA5 nearly prevented the induction of the IFN-β promoter by 2-5A. By contrast, a reduction in the level of the RNA helicase laboratory of genetics and physiology 2 (LGP2)18 slightly increased signalling to the IFN-β promoter by 2-5A (Supplementary Fig. 2). In addition, 2-5A-induction of the IFN-β promoter in the human hepatoma cell line Huh7.5, which has a mutation in RIG-I (T55I)19, was 28% that of 2-5A-induced Huh7 cells, which contain wild-type RIG-I (Fig. 2c). The impact of the RIG-I mutation on autocrine IFN signalling was apparent from the lower levels of phosphorylation of signal transducer and activator of transcription-1 (STAT-1) after 2-5A treatment in the Huh7.5 cells compared with identically treated Huh7 cells (Fig. 2d, compare lanes 3 and 6).
Figure 2. 2-5A induces transcriptional activation of the IFN-β promoter.

a, RIG-I, MDA5, IPS-1 and β-actin in DU145 cells treated for 48 h with siRNAs (shown above) in immunoblots. b, Activation of the IFN-β promoter in DU145 cells treated with siRNAs (5 nM) and 1.0 μM of (2′-5′)A3 or (2′-5′)p3A3. c, Activation of the IFN-β promoter in Huh7 or Huh7.5 cells transfected with 1.0 μM of (2′-5′)A3 or (2′-5′) p3A3 for 18 h. Relative luciferase (LUC) activity, firefly LUC/Renilla LUC. d, Stat1 phosphorylation in Huh7 or Huh7.5 cells treated with (2′-5′)p3A3 or (2′-5′)A3 for 18 h. Stat1-P, phosphorylated Stat1; Stat1-T, total Stat1. Error bars, s.d.
To provide direct evidence that RNA cleavage products generated by RNase L are responsible for signalling, we isolated total cellular RNA from RNase L-deficient MEFs and incubated it with recombinant, purified RNase L in the presence and absence of 2-5A. The reactions were allowed to proceed until no further RNA cleavage could be detected, as monitored by fluorescence resonance energy transfer (FRET) assays using an internal RNA FRET probe20 and by RNA size analysis using RNA chips (Fig. 3a and Supplementary Fig. 3). The production of IFN-β by MEFs increased (to sixfold higher levels) after they were transfected with RNA pre-incubated with RNase L plus 2-5A, as compared to the production of IFN-β after transfection with RNA incubated in the absence of RNase L and 2-5A (Fig. 3b). Incubation of RNA with RNase L without 2-5A provided a much smaller (twofold) increase in IFN-β induction when the RNA was subsequently transfected into MEFs, probably owing to a low level of RNA cleavage under these conditions (Supplementary Fig. 3). The RNA cleavage products that were <200 nucleotides long were isolated by a solid-phase fractionation method (Fig. 3a, lanes 5–7; see Methods). Using equivalent amounts of total RNA as substrate, IFN-β was induced to > 10-fold higher levels in response to transfection of MEFs with small RNAs produced by incubation with RNase L plus 2-5A compared to transfection with small RNAs isolated from an equivalent amount of total RNA incubated with or without RNase L alone (Fig. 3c). Small RNA at ≤50 ng ml−1 produced by incubation with 2-5A-activated RNase L induced IFN-β and its promoter in MEFs and in human HT1080 cells, respectively (Supplementary Fig. 4). By contrast, up to 1 μg ml−1 of the small RNA fraction obtained in the absence of RNase L digestion had little or no effect.
Figure 3. Cleavage of cellular RNA by RNase L produces small RNAs that activate the IFN-β gene.

a, RNA separated in RNA chip. b, c, IFN-β from MEFs transfected (for 18 h) with total (10 μg) RNA (b) or <200-nucleotide (nt) RNA (from 20 μg total RNA) (c). d, RIG-I (upper) and IRF-3 (lower) in RNase L-deficient MEFs and HT1080 cells, respectively, with <200-nt RNA (lanes 1–4) or wild-type MEFs (upper) or HT1080 cells (lower) infected with SeV (for 18 h) (lane 5). e–g, Levels of IFN-β from MEFs (wild-type, RIG-I-deficient, MDA5-deficient or IPS-1-deficient) incubated for 18 h with or without <200-nt RNA. Error bars, s.d.
It has been reported that the presence of free 5′ triphosphorylated or duplex structures in RNA serve to discriminate self (cellular) from non-self (viral) RNA substrates and is used by RIG-I to recognise viral RNA and to initiate innate immune signalling21,22. However, the small RNAs produced by RNase L contain 3′-, and not 5′-, monophosphoryl groups13. To determine how the 3′-phosphoryls might contribute to the induction of IFN expression, we treated the small RNA cleavage products with calf intestinal phosphatase (CIP) under conditions that completely dephosphorylated an internal control radiolabelled RNA substrate (data not shown). Dephosphorylation with CIP reduced the production of IFN-β by 50% (Fig. 3c). These results indicate that RIG-I and/or MDA5 are stimulated by 3′-phosphorylated RNAs. However, the small RNAs that lacked terminal phosphates were still capable of signalling, albeit at a reduced level, perhaps owing to duplex structures.
RIG-I forms multimeric complexes and IRF-3 dimerizes during signalling initiated by SeV infections, as determined by native polyacrylamide gel electrophoresis (PAGE) and immunoblotting23 (see also Fig. 3d, lane 5). Similarly, RIG-I formed multimers and IRF-3 dimerized in response to the small RNAs (produced by RNase L plus 2-5A) in RNase L-deficient MEFs and HT1080 cells, respectively (Fig. 3d, lanes 2). However, RNAs less than 200 nucleotides long that were isolated after incubation of total RNA with RNase L in the absence of 2-5A or without RNase L failed to induce either RIG-I multimers or IRF-3 dimers (Fig. 3d, lanes 3 and 4). Furthermore, IFN-β was potently induced in wild-type MEFs in response to treatment with the small RNAs (Fig. 3e-g). By contrast, IFN-β production by RIG-I-deficient and MDA5-deficient cells transfected with the small RNAs was 17.5% and 40% of the levels obtained in identically treated wild-type cells, whereas there was no detectable IFN-β produced in IPS-1-deficient cells that had been treated with the small RNAs. These results indicate that the small RNAs signal through RIG-I and MDA5 to IPS-1, thereby activating IRF-3.
To establish how RNase L contributes to IFN-β production in vivo, we infected wild-type and RNase L-deficient mice with the picornavirus, encephalomyocarditis virus (EMCV), a positive RNA strand virus or SeV. Infection of wild-type mice with EMCV produced, on average, 2.2- and 3.5-fold greater levels of IFN-β when measured 6 and 24 h after infection, respectively, when compared with similarly infected RNase L-deficient mice (Fig. 4a). The effect of RNase L was even more pronounced after SeV infection: wild-type mice produced, on average, sixfold greater levels of IFN-β when measured 6 h after infection than did RNase L-deficient mice (Fig. 4b). The increased IFN production in the wild-type mice, compared with the RNase L-deficient mice, was highly significant with both types of virus.
Figure 4. RNase L contributes to induction of IFN-β by virus or 2-5A in vivo.
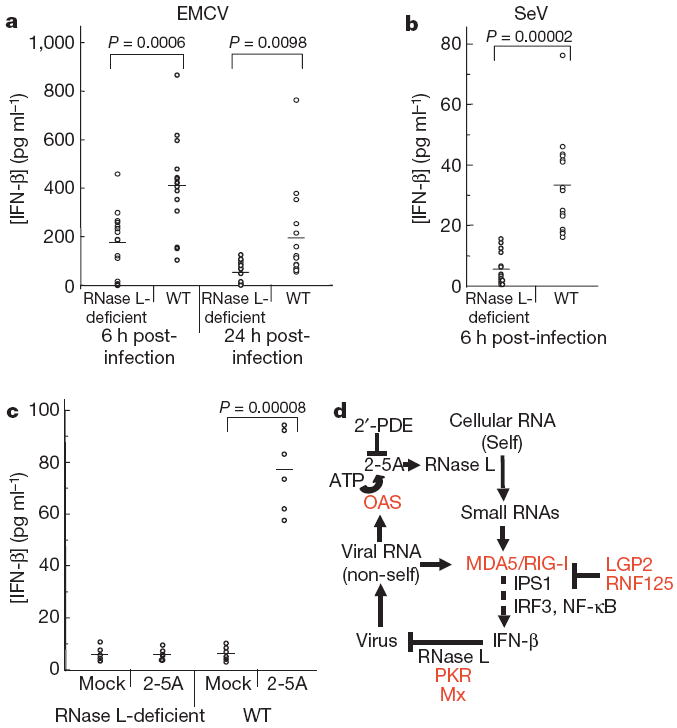
Serum levels of IFN-β from RNase L-deficient or wild-type mice infected with EMCV (a) or SeV for the indicated times (b), or injected intraperitoneally with 2-5A or mock-treated for 5 h (c). Horizontal lines show mean levels of IFN-β. Two-tailed, paired Student’s t-tests were performed. d, Model for transcriptional activation of the IFN-β gene by small RNAs produced by cleavage of cellular (self) RNA with RNase L. Red, IFN-inducible proteins; 2′-PDE, 2′-phosphodiesterase; OAS, 2′-5′-oligoadenylate synthetase.
To establish that self-RNA cleavage products could signal to the IFN-β gene in vivo, we injected 2-5A into uninfected mice by the intraperitoneal route. Mock-treated wild-type and RNase L-deficient mice showed no induction of IFN-β. There was also no induction of IFN-β by 2-5A in the RNase L-deficient mice. In contrast, 2-5A induced circulating IFN-β in all wild-type mice (n = 6; Fig. 4c).
These findings show that RNase L is crucial for enhancing IFN-β production through the RIG-I–MDA5–IPS-1 cascade. Previously, we showed that activation of RNase L by 2-5A induced the expression of twice as many messenger RNA species, including mRNAs for many IFN-β-stimulated genes, as it suppressed24. The current study provides evidence that transcriptional signalling is initiated by RNase L-generated RNA cleavage products of cellular (self)-RNA (Fig. 4d). Viruses produce ‘non-self’ RNAs consisting of double-stranded (ds)RNA and 5′-phosphorylated RNAs that signal through RIG-I and/or MDA5 to IFN-β genes21-23. In addition, viral dsRNA directly activates 2′-5′-oligoadenylate synthetase (OAS), resulting in the production of 2-5A from ATP12. 2-5A stimulates RNase L and leads to the production of small RNA cleavage products that terminate in 3′-phosphoryls from cellular self-RNA, as well as from some viral RNAs14. The products of RNase L cleavage of cellular and viral RNAs probably perform similar functions. Because RNase L cleaves exclusively single-stranded regions in RNA, the cleavage products are often duplex structures. These RNAs activate signalling by RIG-I and MDA5 to IRF-3 through the IPS-1 adaptor protein and its signalling partners, which results in the production of IFN. OAS, RIG-I and MDA5 are all induced by IFN-β at a transcriptional level, thereby further amplifying the production of IFN-β (Fig. 4d; IFN-β-induced proteins are shown in red). Negative regulators that prevent the continuous auto-amplification of IFN-β include 2′-phosphodiesterase, which degrades 2-5A25, and the IFN-β-inducible inhibitors LGP2 (ref. 18) and ubiquitin ligase RNF125 (ref. 26). Although the idea is not explored here, it is also possible that small RNAs generated from RNase L activity could activate toll-like receptor (TLR)3-dependent signalling to drive further IFN-β production1.
The effect of RNase L on viral infections in vivo has been established in studies with RNase L-deficient mice. The ability of mice to survive various viral infections, including EMCV, West Nile virus and Coxsackievirus B4, is compromised by the absence of RNase L15,27,28. In addition, an antiviral role for RNase L in humans is evident from the increased rate of prostate infections by the retrovirus, XMRV, in men that are homozygous for a reduced activity variant of RNase L29. RNase L contributes to the IFN antiviral response by directly cleaving viral and cellular RNA. However, it is now apparent that by generating small RNAs that induce IFN, the effects of the OAS–RNase L pathway extend beyond initially infected cells to support a broader antiviral state in the organism. Furthermore, RNase L circumvents the need for non-self viral RNAs in perpetuating and amplifying innate immunity, as by cleaving cellular RNA it produces RNA activators of RIG-I and MDA5. Because many pathogenic viruses have mechanisms to block RIG-I signalling9,10 or OAS function30 during infection, strategies to regulate RNase L activation could have direct immunoenhancing and antiviral therapeutic applications.
METHODS
Plasmids, reagents, cells and viruses
Plasmids were hIFNβ-luciferase reporter (hIFN-β-luc)19, Renilla luciferase vector (pRL-TK) (Promega), pcDNA3-hRNaseL (human RNase L) and the same construct with R667A or H672A mutations in RNase L11,24. Recombinant, purified human RNase L16, RNA FRET probe20, and 2′-5′-oligoadenylates24 were as described. Poly(I):poly(C) was from Amersham Biosciences (GE Healthcare). Antibodies were rabbit polyclonal antibodies against human proteins, IRF3 (a gift from M. David), RIG-I and LGP2 (M. Gale, Seattle, Washington, USA), MAVS (IPS-1) (Bethyl Labs), MDA5 (Alexis Biochemicals), STAT1 (Santa Cruz), phosphoSTAT1 (Cell Signalling Technologies) and anti-β-actin (Sigma). IFN-β was measured using mouse IFN-β ELISA kits (PBL Laboratories). Wild-type and RNaseL-deficient primary MEFs15 on C57/bl6 background were transformed with SV40 T antigen and cultured in complete RPMI medium. DU145 cells, HT1080 cells, Huh7 cells, Huh7.5 cells, RIG-I-deficient MEFs (a gift from S. Akira)3, MDA5-deficient MEFs (a gift from M. Diamond and M. Colonna)4 and IPS-1-deficient MEFs (a gift from S. Akira)17, and culture conditions, have been described19,24. SeV (Cantell strain) from Charles River Laboratories was a gift from G. Sen and EMCV was a gift from I. M. Kerr.
Isolation and purification of RNA
Total RNA from RNase L-deficient MEFs (2.0 mg isolated using Trizol Reagent (Invitrogen)) was cleaved with RNase L (20 μg) activated by unfractionated 2-5A (10 μM) in the presence of a FRET RNA probe (1.0 μg) in 500 μl containing 25 mM Tris HCl pH 7.4, 100 mM KCl, 10 mM MgCl2, 50 μM ATP and 7 mM β-mercaptoethanol for 1 h at 22 °C. Control reactions lacked RNase L and/or 2-5A. RNA cleavage at 22 °C was monitored in 5-μl samples withdrawn at different times by fluorescence with a Wallac 1420 Victor2 multilabel counter (Perkin-Elmer Life Sciences) (excitation 485 nm; emission 535 nm; 0.1 s) and in RNA chips using an Agilent Bioanalyser 2100 (Agilent Technologies). RNA size markers were from Ambion (AM7152 RNA 6000 Ladder). Small (<200-nucleotide) RNAs were isolated using mirVana miRNA Isolation Kit (Ambion). Small RNAs (150 μg per reaction) were incubated without or with 30 U CIP (NEB) for 2 h at 37 °C and 10 min for 75 °C and recovered using mirVana miRNA Isolation Kit.
Transfections, luciferase assays, and infections of cells
Transfections of 2-5A, RNA or plasmids were done with lipofectamine 2000 (Invitrogen) whereas poly(I):poly(C) was transfected with Fugene 6 (Roche Applied Science). Cells (2 × 105 per well in six-well plates) were transiently transfected with hIFN-β-luc (1 μg) and pRL-TK (0.1 μg) and subjected to dual luciferase assays (Promega) in triplicate. siRNA oligonucleotides (5 nM) targeting human RIG-I, MDA5, IPS1 or LGP2 (see below), or non-specific oligonucleotides (Dharmacon), were transfected into DU145 cells with DharmaFECT 1. After 48 h, cells were transfected with luciferase-reporter plasmids and after another 24 h with 2-5A. Luciferase activity was measured 18 h later. Alternately, 24 h after transfecting with the luciferase plasmids, cells were transfected with 2-5A or RNA for 18 h. SeV in serum-free media was added to cells in six-well plates. After 1 h, the medium was replaced with growth medium for an additional 18 h. Error bars shown are standard deviations (s.d.). Two-tailed paired Student’s t-tests were used to calculate P values.
Immunoblots
Proteins in cell extracts were separated in 10% polyacrylamide/SDS gels or under native conditions through pre-cast 7.5% or 10% Tris.HCl gels (BioRad) (for Rig-I multimerization and IRF-3 dimerization assays) and transferred to nitrocellulose membrane (BioRad). Blots were incubated with various primary antibodies. Secondary goat anti-rabbit antibody tagged with horseradish peroxidase was from Cell Signalling. Immunoreactive bands were detected by enhanced chemiluminescence (ECL) (Amersham Biosciences) and subsequent exposure to X-ray film (Eastman Kodak).
Infections and 2-5A treatment of mice
Wild-type and Rnasel−/− mice (n = 15 per group, 5–6 weeks old) on a C57BL/6 background were inoculated intraperitoneally with 1 × 103 pfu of EMCV or intranasally with 320 HAU of SeV. Sera were collected (at the times indicated) after infection and IFN-β levels determined by ELISA (PBL Biomedical Labs). Control mice received only PBS. For 2-5A treatment, we injected wild-type and Rnasel−/− mice (n = 6 per group, 5–6 weeks old) intraperitoneally with 500 μl of 0.6 mM unfractionated 2-5A with 80 μl of fugene 6 for 5 h. Mock treatments were with fugene 6 alone.
Sequences of siRNA oligonucleotides
The following oligonucleotides were used (for each target mRNA, the sense sequence is given first followed by the antisense version). Human LGP2: GCAAUGUGGUGGUGCGUUAUU/5′-p-UAACGCACCACCACAUUGCUU; GCCAGUACCUAGAACUUAAUU/5′-p-UUAAGUUCUAGGUACUGGCUU; ACAGGGAGCACGUCACUAAUU/5′-p-UUAGUGACGUGCUCCCUGUUU; CAACUUCUCGAACUACUAUUU/5′-p-AUAGUAGUUCGAGAAGUUGUU; Human RIG-I: GCACAGAAGUGUAUAUUGGUU/5′-p-CCAAUAUACACUUCUGUGCUU; CCACAACACUAGUAAACAAUU/5′-p-UUGUUUACUAGUGUUGUGGUU; CGGAUUAGCGACAAAUUUAUU/5′-p-UAAAUUUGUCGCUAAUCCGUU; UCGAUGAGAUUGAGCAAGAUU/5′-p-UCUUGCUCAAUCUCAUCGAUU; Human MDA5: GAAUAACCCAUCACUAAUAUU/5′-p-UAUUAGUGAUGGGUUAUUCUU; GCACGAGGAAUAAUCUUUAUU/5′-p-UAAAGAUUAUUCCUCGUGCUU; UGACACAAUUCGAAUGAUAUU/5′-p-UAUCAUUCGAAUUGUGUCAUU; CAAUGAGGCCCUACAAAUUUU/5′-p-AAUUUGUAGGGCCUCAUUGUU; Human IPS-1: AAGUAUAUCUGCCGCAAUUUU/5′-p-AAUUGCGGCAGAUAUACUUUU; CAUCCAAAGUGCCUACUAGUU/5′-p-CUAGUAGGCACUUUGGAUGUU; GCAAUGUGGAUGUUGUAGAUU/5′-p-UCUACAACAUCCACAUUGCUU; CAUCCAAAUUGCCCAUCAAUU/5′-p-UUGAUGGGCAAUUUGGAUGUU.
Supplementary Material
Acknowledgments
We thank M. Diamond and M. Colonna (St Louis, Missouri, USA) for the Mda5−/− MEFs, S. Akira (Osaka, Japan) for Ips1−/− and Rig-i−/− cells, M. David (San Diego, California, USA) for IRF3 antibodies, C.M. Rice (New York, New York, USA) for Huh7 and Huh7.5 cells, I.M. Kerr (London, UK) for EMCV, P.J. Sims (Rochester, New York, USA) for discussions, and (all from Cleveland, Ohio, USA) G. Sen for Sendai virus, J. Paranjape for cell line preparations and RNA chip analysis, B.K. Jha for RNase L, B.K. Jha, C. Thakur and Z. Novince for preparing 2-5A, and S. Shelby for technical assistance with mice. These studies were supported by grants from the NIH to R.H.S. and M.G. and by a grant from the Burroughs Wellcome Fund to M.G.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The authors declare no competing financial interests.
References
- 1.Saito T, Gale M., Jr Principles of intracellular viral recognition. Curr Opin Immunol. 2007;19:17–23. doi: 10.1016/j.coi.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 3.Kato H, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Gitlin L, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 6.Kawai T, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nature Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 7.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Xu LG, et al. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Meylan E, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 10.Loo YM, et al. Viral and therapeutic control of IFN-β promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci USA. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou A, Hassel BA, Silverman RH. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell. 1993;72:753–765. doi: 10.1016/0092-8674(93)90403-d. [DOI] [PubMed] [Google Scholar]
- 12.Kerr IM, Brown RE. pppA2′p5′A2′p5′A: an inhibitor of protein synthesis synthesized with an enzyme fraction from interferon-treated cells. Proc Natl Acad Sci USA. 1978;75:256–260. doi: 10.1073/pnas.75.1.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wreschner DH, McCauley JW, Skehel JJ, Kerr IM. Interferon action–sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature. 1981;289:414–417. doi: 10.1038/289414a0. [DOI] [PubMed] [Google Scholar]
- 14.Han JQ, Wroblewski G, Xu Z, Silverman RH, Barton DJ. Sensitivity of hepatitis C virus RNA to the antiviral enzyme ribonuclease L is determined by a subset of efficient cleavage sites. J Interferon Cytokine Res. 2004;24:664–676. doi: 10.1089/jir.2004.24.664. [DOI] [PubMed] [Google Scholar]
- 15.Zhou A, et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong B, et al. Intrinsic molecular activities of the interferon-induced 2–5A-dependent RNase. J Biol Chem. 1994;269:14153–14158. [PubMed] [Google Scholar]
- 17.Kumar H, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. 2006;203:1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoneyama M, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 19.Sumpter R, Jr, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thakur CS, Xu Z, Wang Z, Novince Z, Silverman RH. A convenient and sensitive fluorescence resonance energy transfer assay for RNase L and 2′,5′ oligoadenylates. Methods Mol Med. 2005;116:103–113. doi: 10.1385/1-59259-939-7:103. [DOI] [PubMed] [Google Scholar]
- 21.Hornung V, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 22.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 23.Saito T, et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci USA. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malathi K, et al. A transcriptional signaling pathway in the IFN system mediated by 2′-5′-oligoadenylate activation of RNase L. Proc Natl Acad Sci USA. 2005;102:14533–14538. doi: 10.1073/pnas.0507551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubota K, et al. Identification of 2′-phosphodiesterase, which plays a role in the 2–5A system regulated by interferon. J Biol Chem. 2004;279:37832–37841. doi: 10.1074/jbc.M400089200. [DOI] [PubMed] [Google Scholar]
- 26.Arimoto K, et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA. 2007;104:7500–7505. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flodstrom-Tullberg M, et al. RNase L and double-stranded RNA-dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. J Immunol. 2005;174:1171–1177. doi: 10.4049/jimmunol.174.3.1171. [DOI] [PubMed] [Google Scholar]
- 28.Samuel MA, et al. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urisman A, et al. Identification of a novel gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006;2:e25. doi: 10.1371/journal.ppat.0020025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Beattie E, et al. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J Virol. 1995;69:499–505. doi: 10.1128/jvi.69.1.499-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.