

Abstract
Ca2+ may selectively activate various processes in part by the cell's ability to localize changes in the concentration of the ion to specific subcellular sites. Interestingly, these Ca2+ signals begin most often at the plasma membrane space so that understanding subplasma membrane signals is central to an appreciation of local signaling. Several experimental procedures have been developed to study Ca2+ signals near the plasma membrane, but probably the most prevalent involve the use of fluorescent Ca2+ indicators and fall into two general approaches. In the first, the Ca2+ indicators themselves are specifically targeted to the subplasma membrane space to measure Ca2+ only there. Alternatively, the indicators are allowed to be dispersed throughout the cytoplasm, but the fluorescence emanating from the Ca2+ signals at the subplasma membrane space is selectively measured using high resolution imaging procedures. Although the targeted indicators offer an immediate appeal because of selectivity and ease of use, their limited dynamic range and slow response to changes in Ca2+ are a shortcoming. Use of targeted indicators is also largely restricted to cultured cells. High resolution imaging applied with rapidly responding small molecule Ca2+ indicators can be used in all cells and offers significant improvements in dynamic range and speed of response of the indicator. The approach is technically difficult, however, and realistic calibration of signals is not possible. In this review, a brief overview of local subplasma membrane Ca2+ signals and methods for their measurement is provided. © 2012 IUBMB IUBMB Life, 64(7): 573–585, 2012
Keywords: Ca2+ signaling, calcium mediated signaling, signal transduction
Increases in the cytoplasmic Ca2+ concentration (Ca2+ signals) participate in virtually every physiological process (1). At rest, the cytoplasmic free Ca2+ concentration ([Ca2+]c) is maintained at a low (100 nM) value against a constant inward leak from the higher [Ca2+] in the extracellular medium (∼2 mM) and in internal stores (several hundred micromolar). A fundamental element of signaling is the controlled entry of the ion into the cytoplasm via channels that are often specific for Ca2+ and which respond to extracellular stimulation. This channel activity generates [Ca2+] increases of various time courses and locations that are translated to physiological responses. For example, exocytosis is triggered within microseconds by locally high [Ca2+] near vesicle release sites (2), contraction occurs over the timescale of hundreds of milliseconds to seconds by a global rise in [Ca2+] throughout the cell (a “global” signal), gene expression is regulated in minutes (3) and proliferation over the timescale of days. An understanding of how various Ca2+ signals arise and are controlled is, therefore, central to much of biology.
Ca2+ signals begin with the opening of one or a few channels. Influx to the cytoplasm occurs at rates of ∼0.6 million Ca2+ ions per second per channel (0.2 pA current). The influx generates a significant local concentration gradient near the channel in which [Ca2+] declines from ∼10 μM to ∼100 nM over a few hundred nanometers from the plasma membrane (4–10) (Fig. 1). Channel open time is brief (∼1 ms) so that the gradient dissipates rapidly with rates of change in the subplasma membrane space of the order of ∼5000 μM s−1, when compared with a maximum of ∼0.5 μM s−1 in the bulk cytoplasm (Fig. 1) after a global [Ca2+] rise. High local [Ca2+] and the rapid rates of change near channels may target processes with rapid Ca2+ binding kinetics to selectively activate particular functions (11–15). The high local [Ca2+] signals, in turn, may activate neighboring IP3 or ryanodine receptor channels (IP3R or RyR, respectively) to amplify the local signals or propagate through the cell as global signals with slower but more widespread effects throughout the cell (16–23). The transition of signals from those involving single to multiple channels and from local to global [Ca2+] increases creates a multitude of signals with various locations, magnitudes, and time courses (24–27) so that various cellular functions may be selectively activated.
Figure 1.
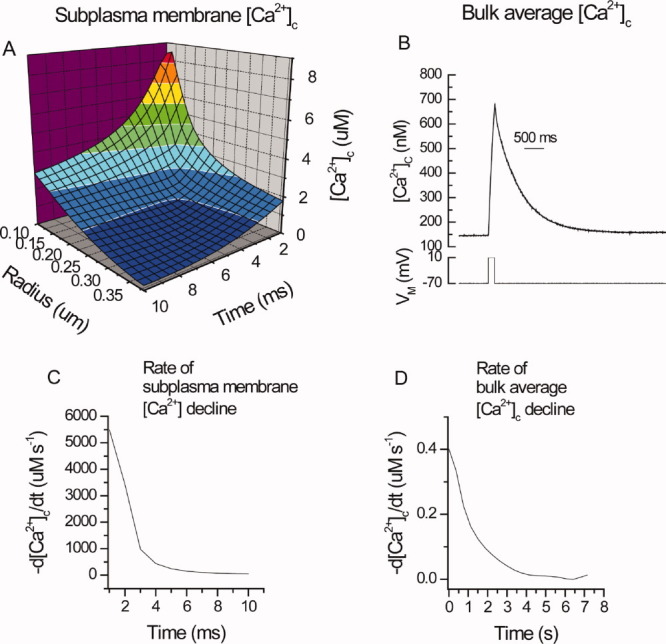
Simulated subplasma membrane [Ca2+] changes following the opening of a single Ca2+ channel in smooth muscle. A: Microdomains of [Ca2+] were produced by the opening of single Ca2+ channels. A Ca2+ channel opening for on average 2.9 ms with a current of 0.18 pA [39] will increase local [Ca2+] near the cytoplasmic aspect of the channel. The magnitude of Ca2+ entry (moles) was calculated from the time integrated current: 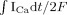 where
where 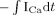 is the total charge entry and F the Faraday constant. The ion will diffuse from the channel randomly creating a hemisphere of increased [Ca2+]. The outer radius of the hemisphere was described ((6Dt)1/2) to express the three dimensional root mean square displacement of an ion through a medium with a diffusion coefficient (D) equal to 2.2 × 10−6 cm2 s−1 and t, time. A hemisphere volume (not shown) was calculated from the radius at various times and the [Ca2+] within determined from the Ca2+ moles influx via the single channel divided by the buffer power (100). High [Ca2+] (tens of micromolar) within the hemisphere volume exist in the first few milliseconds after channel opening. [Ca2+] is a function of both distance from the influx channel and time after channel opening. Note, the [Ca2+] was estimated by assuming a uniform distribution of Ca2+ in the microdomain (which may not be valid) and Ca2+ being in equilibrium with the buffers. The latter may only occur when the association rate constant exceeds 108 M−1 s−1. (B) The [Ca2+]c increase in the bulk cytoplasm in response to entry via voltage-dependent Ca2+ channels was substantially less than that of the subplasma membrane; maximum values were in the hundred of nanomolar range. Thus, depolarization of the plasma membrane (−70 mV to +10 mV B lower panel) increased [Ca2+]c uniformly throughout the bulk of the cytoplasm. [Ca2+]c was restored toward resting levels when the depolarization ended by removal of Ca2+ from the cytoplasm by Ca2+ pump activity. (C, D) Rate of [Ca2+] decline in the subplasma membrane microdomain and bulk cytoplasm following voltage-dependent Ca2+ entry. (C) In the subplasma membrane space, the maximum rates of decline were ∼ 5500 μM s−1. The data for this figure was derived from Fig 1A. (D) Decline (−d[Ca2+]c/dt μM s−1) in the bulk cytoplasm following depolarization-evoked Ca2+ entry from the data in Fig 1B. Maximum rates of decline in the bulk cytoplasm approached 0.4 μM s−1. Note the scale difference in the abscissae in C, D. (Modified from ref. 4, with permission from Elsevier).
is the total charge entry and F the Faraday constant. The ion will diffuse from the channel randomly creating a hemisphere of increased [Ca2+]. The outer radius of the hemisphere was described ((6Dt)1/2) to express the three dimensional root mean square displacement of an ion through a medium with a diffusion coefficient (D) equal to 2.2 × 10−6 cm2 s−1 and t, time. A hemisphere volume (not shown) was calculated from the radius at various times and the [Ca2+] within determined from the Ca2+ moles influx via the single channel divided by the buffer power (100). High [Ca2+] (tens of micromolar) within the hemisphere volume exist in the first few milliseconds after channel opening. [Ca2+] is a function of both distance from the influx channel and time after channel opening. Note, the [Ca2+] was estimated by assuming a uniform distribution of Ca2+ in the microdomain (which may not be valid) and Ca2+ being in equilibrium with the buffers. The latter may only occur when the association rate constant exceeds 108 M−1 s−1. (B) The [Ca2+]c increase in the bulk cytoplasm in response to entry via voltage-dependent Ca2+ channels was substantially less than that of the subplasma membrane; maximum values were in the hundred of nanomolar range. Thus, depolarization of the plasma membrane (−70 mV to +10 mV B lower panel) increased [Ca2+]c uniformly throughout the bulk of the cytoplasm. [Ca2+]c was restored toward resting levels when the depolarization ended by removal of Ca2+ from the cytoplasm by Ca2+ pump activity. (C, D) Rate of [Ca2+] decline in the subplasma membrane microdomain and bulk cytoplasm following voltage-dependent Ca2+ entry. (C) In the subplasma membrane space, the maximum rates of decline were ∼ 5500 μM s−1. The data for this figure was derived from Fig 1A. (D) Decline (−d[Ca2+]c/dt μM s−1) in the bulk cytoplasm following depolarization-evoked Ca2+ entry from the data in Fig 1B. Maximum rates of decline in the bulk cytoplasm approached 0.4 μM s−1. Note the scale difference in the abscissae in C, D. (Modified from ref. 4, with permission from Elsevier).
An understanding of Ca2+ signaling requires an appreciation of how Ca2+ is selectively localized to certain regions of the cell. Interestingly, in this respect, the global Ca2+ signals may arise not from the combined activity of channels throughout the cell but from only a small fraction of the overall channel complement, and significantly, often those active channels are largely located close to (IP3R and RyR) or on (voltage-dependent Ca2+ channels) the plasma membrane (28–30). For example, [Ca2+] increases in response to IP3-generating agonists may involve <1% of the overall IP3R complement with 75% of the active channels being exceedingly close (∼100 nm) to the plasma membrane in SH-SY5Y cells (30). Active RyR are also mainly at the plasma membrane to regulate ion channel activity there (31–37). The positioning of active sarcoplasmic reticulum (SR) Ca2+ release channels near the plasma membrane provides a mechanism which may enable agonist activation to target specific types of cellular response, that is, by generating [Ca2+] rises in specific regions of the cell. To facilitate this process, some plasma membrane located receptors or signaling cascades co-localize with IP3R (38–40) that are positioned near the plasma membrane. For example, while muscarinic and bradykinin receptors each stimulate phopholipase C, only bradykinin receptors coimmunoprecipitate with, and activate, IP3R (38) in neurons. The clustering of receptors for neurotransmitters or hormones in certain regions on the plasma membrane (41) may also contribute to the selective targeting of particular responses by providing areas with increased sensitivity to extracellular stimuli. Agonists which operate by releasing Ca2+ via IP3R may also evoke specific cellular responses by linking to additional signaling pathways to increase flexibility and complexity of the response. Parathyroid hormone stimulates adenylate cyclase to deliver cyclic adenosine monophosphate (cAMP) via “cAMP junctions” to sensitize IP3R2 to IP3 (39) to enable generation of Ca2+ events in specific parts of the cell. Thus, there are direct, specific associations between signaling proteins located at the plasma membrane and those on that part of the SR membrane which is close to the plasma membrane (42). Such associations enable messengers which have the potential to evoke activity throughout the cell (e.g. Ca2+), to selectively target particular physiological responses, hence the interest in understanding subplasma membrane Ca2+ signaling.
Although voltage-dependent Ca2+ channels appear uniformly distributed across the membrane (43), as with IP3R, signals arise from a small fraction of the overall channel complement (44) and active channels cluster (45) to facilitate localized specific activation of nearby effectors [Figs. 2 and 3; (see also 44, 46, 47)]. The clustering enables uniform cell wide activation via depolarization to locally activate selected specific cell processes. In vascular smooth muscle, voltage-dependent Ca2+ channels were suggested to be clustered and constitutively active (47). Although our studies show voltage-dependent Ca2+ channels are clustered, the channels are controlled by membrane potential (i.e., they lack constitutive activity), a feature which will enable the channels to remain under physiological control (45). Interestingly, during prolonged depolarization, some small sites near the plasma membrane display repetitive Ca2+ rises (Fig. 3) (45, 47), which suggests some channels are more readily activated than others and which presumably target specific cellular responses.
Figure 2.
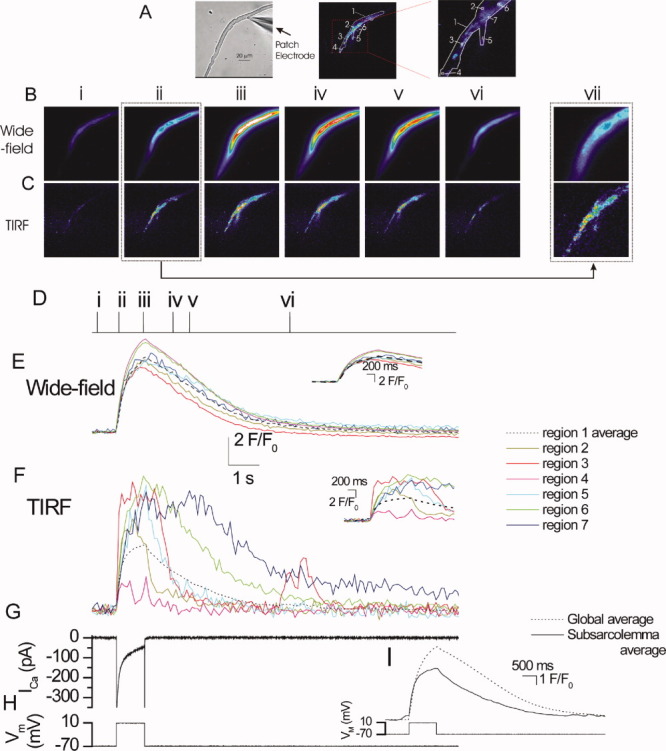
Simultaneous wide-field epi-fluorescence and TIRF [Ca2+] measurements in a voltage-clamped single smooth muscle cell. Depolarization (−70 mV to +10 mV; H), activated a voltage-dependent Ca2+ current (ICa; G) to evoke a rise in [Ca2+] (B,C,E,F). The rise in [Ca2+] which occurred in the subplasma membrane space (measured by TIRF) (C,F) was more rapid in onset than that seen in the bulk cytoplasm (measured by wide-field epi-fluorescence) (B,E). The [Ca2+] images (B, C) are derived from the time points indicated by the corresponding numerals in D. [Ca2+] changes in B and C are represented by color; blue low and red/white high [Ca2+]. The images (B and C) were taken before (i), during (ii-iii), and after (iv-vi) depolarization and show the resulting [Ca2+] changes. Changes in the fluorescence ratio with time (E and F) are derived from 2 × 2 pixel boxes (regions 1-6 in A, middle and right (expanded) panel; drawn at a 3 × 3 pixel size to facilitate visualization) and from a larger region encompassing the entire TIRF region (region 7). The latter was used to obtain an average subplasma membrane and bulk average [Ca2+]c increase (I). Significantly, although the [Ca2+]c increase which occurred in the bulk cytoplasm (B and E) was approximately uniform and simultaneous throughout the cell, those in subplasma membrane space (C and F) had a wide range of amplitudes and various time courses. Note the spark like events in region 3 toward the end of the recording. (A) left panel show a bright field image of the cell; see also whole cell electrode (right side). Insets in E and F show the rising phase of the transients on an expanded time base. For comparison, I shows the average subplasma membrane and bulk average [Ca2+]c rise as measured in region 7 (A, right hand panel). B and C (vii) shows an enlargement of ii to illustrate the localized nature of the rise in [Ca2+] in the subplasma membrane space (Reproduced from ref. 45, with permission from Rockefeller University Press).
Figure 3.
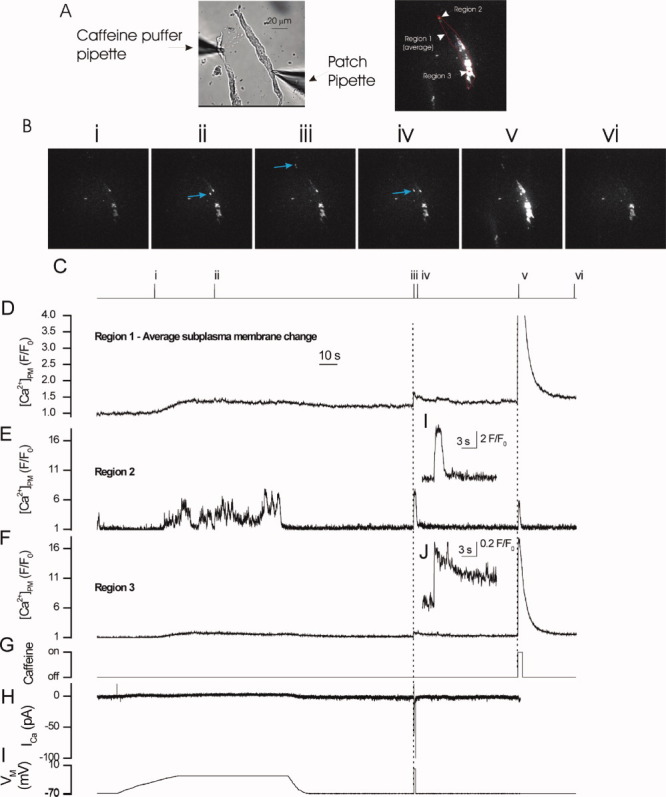
Local subplasma membrane Ca2+ transients in response to plasma membrane depolarization and caffeine in a voltage-clamped single colonic myocyte. Gradual depolarization from −70 mV to −20 mV (I) elevated [Ca2+] in the subplasma membrane space as measured in TIRF (D-F). The [Ca2+] increase was more localized and substantially larger in some regions than others (compare E and F). Two example regions (regions 2 and 3) are shown (panels E and F); localized rises occurred in region 2 but not region 3. Blue arrows in the frames in B (ii-iv) show examples of the localized rises in [Ca2+]. The [Ca2+] increase which occurred in region 3 (F) was slow and of smaller magnitude than that of region 2 (E). When the membrane potential was restored to −70 mV (I) [Ca2+] was returned towards resting levels. A subsequent transient depolarization to 0 mV (H) activated voltage-dependent Ca2+ current (ICa; H) and increased [Ca2+] (D-F). The increase was larger and declined more rapidly in region 2 (D and inset I) than region 3 (E and inset J). Those regions (region 2, E) which showed large responses to depolarization had small responses to RyR activation by caffeine (G). Conversely, a large response (region 3, F) to caffeine (G) occurred in regions with small responses to depolarization. Region 1 (D) is a subplasma membrane average change. Numerals in images (B) correspond to those in C. Changes in the fluorescence ratio with time (D,E,F) are derived from 1 pixel boxes (regions 2 and 3; shown at 3 × 3 pixels to facilitate visualization) and from a larger region the entire TIRF image (region 1) to obtain an average subplasma membrane [Ca2+] change (D). The position of the regions from which the transients (D,E,F) were obtained are shown in A (right-hand panel). A bright field image of the cell is shown in (A; left panel); see also whole cell electrode (right side) and puffer pipette which contained caffeine (10 mM; left side) (Reproduced from ref. 45, with permission from Rockefeller University Press).
The growing appreciation of the significance of local signals near the plasma membrane in controlling cell function has led to the development of various methodologies to study subplasma membrane [Ca2+]. Some approaches have been based on the sensitivities and activities of Ca2+- activated ion channels, such as the Ca2+-activated K+ channel (KCa) to estimate subplasma membrane Ca2+ concentrations. Using this approach, concentrations of the order of ∼10 μM have been estimated to occur in the subplasma membrane space (48, 49). In our studies on smooth muscle cells, depolarization-evoked [Ca2+]c increases were calculated to raise subplasma membrane [Ca2+] to ∼50 μM in the space between the plasma membrane and SR (50). In other experiments, more direct measurements were made using Ca2+ indicators. In smooth muscle cells, a fluorescent Ca2+ indicator was targetted to the plasma membrane using a lipophilic tail (FFP18) (51). The [Ca2+] transients measured in response to plasma membrane depolarization were of the order of micromolar concentrations, though the bulk average [Ca2+]c (measured using a cytoplasmic fluorescent Ca2+ indicator fura-2) rose by only a few hundred nanomolar (51). In squid giant axons, a microinjected low-affinity aequorin mutant measured “hot spots” of [Ca2+] in the range of 200–300μM in the synapse during action potentials (5). Later studies in the large synapses on the calices of Held indicated that, under physiological conditions, fast vesicle release could be explained by smaller (20 μM) (7) increases in subplasma membrane [Ca2+]. Together, these early studies were instrumental in highlighting different [Ca2+] in the subplasma membrane and bulk cytoplasm and prompted more advanced methodological development to study subplasma membrane [Ca2+]. Recently, the two main experimental approaches for visualizing Ca2+ signals have been (1) to specifically localize Ca2+ indicators to the subplasma membrane space to measure [Ca2+] only there or (2) to allow indicators to be dispersed throughout the cytoplasm but to measure the fluorescence emanating from the Ca2+ signals only at the subplasma membrane space using high resolution imaging procedures. These procedures will now be briefly reviewed.
TARGETED Ca2+ INDICATORS
In cultured cells, the most prevalent approach to measure Ca2+ signals in specific regions of the cell is to use genetically encoded Ca2+ indicator proteins (fluorescent or luminescent), which can be targeted to organelles or subcellular locations, for example, the plasma membrane. Probes localized to the inner surface of the plasma membranes have detected Ca2+ concentrations in the subplasma membrane space, which significantly exceed those of the bulk cytosol. A chimera of the luminescent protein aequorin measured the mean [Ca2+] on the inner surfaceof the plasma membrane of the A7r5 smooth muscle cell line to be ∼1–2 μM at rest (6). The high concentration may reflect the presence of “hot spots” in the vicinity of sporadically opening channels, rather than a homogeneous high [Ca2+] in the subplasma membrane space. During store-operated Ca2+ influx, in the same cells, subplasma membrane [Ca2+]increased to 50 μM (6). Although targeting to the plasma membrane was successful, aequorin has some experimental limitations. The low levels of luminescence and destruction of aequorin by Ca2+ complicate analysis of results and, significantly, prevent single cell imaging and high-time resolution comparisons with electrophysiology. To overcome this issue, targeted fluorescent protein based sensors of Ca2+ such as the “cameleons”(52) offered some advantages particularly in photostability. However, cameleons are large (650 amino acids), often bigger than the host proteins to which they are fused, which limits their use. In addition, the dynamic response of the cameleons, and indeed other Ca2+ sensitive photoproteins, are considerably slower than the physiological response they are used to report. This property may result in the indicator not accurately following the rapid changes in subplasma membrane [Ca2+]. In another parallel approach, a calmodulin-based fluorescent protein-based indicator (GCaMP2) was tethered to Na+ pumps to report subplasma membrane Ca2+ signals. Although the indicator reported changes over timescales of minutes (53), it was again too slow for the milliseconds transients close to the plasma membrane. In an attempt to overcome this speed of response issue, a troponin-based indicator which possesses a relatively fast off-rate was developed (54). Even this indicator is still more than two orders of magnitude slower than typical small molecule Ca2+ indictors such as fluo-3 (kon of 28.2 μM−1 s−1 and koff of 7.04 s−1 (54) compared to 7100 μM−1 s−1 and 369 s−1 for fluo-3 (55). The mean times for TN-XL and fluo-3 to capture (and so detect) a Ca2+ change is 35.5 ms (TN-XL) and 0.14 ms (fluo-3)). The mean capture times were calculated from 1/KonB (56, 57), where B is the free Ca2+ binding sites on the indicator and was taken to be 1 μM for each indicator. As shown in Fig. 1, the highly localized subplasma-membrane [Ca2+] arising from the opening of a single Ca2+ channel declines within 10 ms. Since the mean capture time of the “fast” troponin-based Ca2+ indicator (TN-XL) is 35.5 ms, this probe would fail to detect the Ca2+ change. Fluo-3 is on the other hand fast enough to detect the Ca2+ signal.
Thus, despite continuous improvements, the Ca2+ indicator proteins typically have a fairly restricted dynamic range (ratio of signals from Ca2+ bound and Ca2+ free forms, ∼1–4-fold (54) compared to >100-fold for fluo-3 (manufacturer's literature)) and can be physically large. However, probably most significantly the photoprotein indicators are slow when compared with the rate of change in physiological Ca2+ signals beneath the plasma membrane. Together, these features present significant drawbacks in measuring subplasma membrane [Ca2+].
Although small synthetic molecule Ca2+ indicators, for example, fluo-3 and fura-2 are considerably faster in detecting Ca2+ and have wider dynamic ranges than the fluorescent protein Ca2+ indicators, targeting of these indicators to specific subcellular sites has been problematic, limiting the indicators' use in reporting subcellular [Ca2+]. In some cases, the intrinsic properties of particular indicators reportedly help localize them to certain regions of the cell (e.g., mitochondria) but results have not been universally positive. For example, rhod-2 has been used in some studies to successfully measure mitochondrial matrix [Ca2+]. Rhod-2 in its membrane permeant acetoxymethyl (AM) ester form bears a net positive charge which, it is suggested, results in mitochondrial accumulation of the dye because of the mitochondria's negative membrane potential. However, rhod-2 bears one net positive charge only in the AM form. When fully de-esterified, rhod-2 bears three net negative charges which is likely to preclude significant mitochondrial accumulation. The balance of esterase activity in the cytoplasm and mitochondria will determine the major source of the signal from rhod-2 and is likely to contribute to variations in results among studies.
If the small molecule indicators could be selectively targeted to subcellular regions, their speed and wide dynamic range could offer significant advantages over fluorescent protein Ca2+ indicators in measuring [Ca2+]. Several attempts have been made to target these indicators. In one proof of concept study, peptides which carry organelle-specific import or retention sequences (58) were added to a small synthetic molecule fluorophore (BODIPY). The peptides resulted in the fluorophore being selectively localized to the SR, Golgi, peroxisomes, or nucleus within minutes (58). Although not yet used to target Ca2+ indicators, it seems likely that this procedure will prove useful. In an alternative approach, a biarsenical derivative of the small synthetic Ca2+ indicator Calcium Green [calcium green FlAsH (CaGF)] has been synthesized that specifically labels fusion proteins that possess four cysteines (tetracysteine tag) (44). This indicator has been genetically targeted to plasma membrane proteins (connexion 43) to measure subplasma membrane [Ca2+]. The results showed an uneven distribution and wide variations in the open probability of voltage-dependent Ca2+ channels across the plasma membrane and validate the procedure as a way of localizing small synthetic molecule Ca2+ indicators to measure fast Ca2+ signals (44). In yet another approach which enabled fast small molecule indicators to be targeted, a membrane permeant form of indo-1 which possessed an O6-benzylguanine side group was synthesized. O6-benzylguanine specifically interacts with SNAP-tag, an engineered human alkyguanine-DNA alkytransferase. The indicator (indo-1) was localized in cells engineered to express SNAP-tag fusion proteins in the nucleus (59). A fura-2FF Ca2+ indicator (a derivative of fura-2 with lower affinity for Ca2+ and faster kinetics) also with a O6-benzylguanine side group has been synthesized to permit subcellular localization again via links to SNAP-tag fusion proteins (60). These procedures enable the spatial specificity of the protein biosensors approach with the fast kinetics (kon in the 1–10 × 108 M−1 s−1 range) and high dynamic range (20–100 fold increase in fluorescence from Ca2+-free to Ca2+-bound) of the small synthetic indicators and await full experimental exploitation.
HIGH RESOLUTION IMAGING FOR MEASURING SUBPLASMA MEMBRANE [Ca2+]
Rather than targeting the Ca2+ indicator to specific regions of the cell, the other major approach to selectively measure Ca2+ signals from only certain regions has been to use high resolution imaging methods. Indeed, the desire to image Ca2+ at subcellular resolution has been a driving force, which led to significant advances in optics and technology to overcome the constraints imposed on optical microscopy. The main physical limitation imposed by the use of light for high resolution imaging is that of diffraction, which was first fully explained by Ernst Abbe in 1873 (61). Overcoming the “diffraction limit” had been a significant challenge but has become a reality in the last 15 years. Several methods now permit subcellular imaging at resolutions well beyond the diffraction limit. The application of these methods to real biological challenges requires an understanding of the basic physical processes behind the various methods so that the most suitable can be selected.
Diffraction occurs when a wave meets a physical stop, such as the edges of an aperture. If apertures, including the transmissive part of a lens, were infinite then diffraction would not be the limiting factor (62). At the front of a wave, each point acts as a small emitting source for a new wave, (Fig. 4A) and normally, these small sources add together to produce a uniform wavefront. However, at the edge of an aperture, some of these secondary sources are blocked and some let through and thus light appears to come through the block in certain places, with the positions of these points of light being determined by interference of the light waves (Fig. 4B). A full explanation of this effect can be found in any basic optical text book (63). In an optical microscope, the ability to resolve two closely spaced features in the lateral and axial direction is given by
Figure 4.
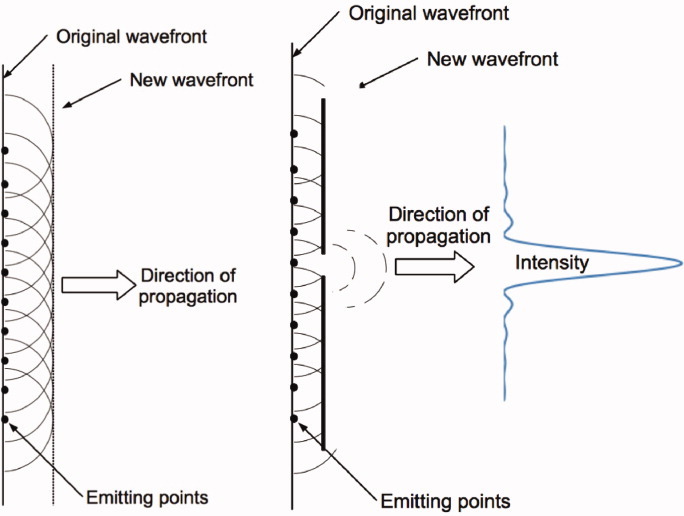
Propagating waves. (A) Shows wave propagating in free space which acts as a series of individual wavelets adding to form the overall light wave (Huygens Wavelets). (B) Shows the effect of an aperture on wave propagation as the wavelets interfere with each other leaving the diffraction intensity pattern shown for a circular aperture. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
However, the theoretical limit of resolution is difficult to achieve in biological samples. Most samples, even small cells, have a thickness of at least a few microns. As a result of the sample thickness, there will be light from outside the focal plane which will contribute to the image in the focal plane. The light from outside the focal plane will limit the resolution and quality of the image.
Confocal microscopy removes this out of focus light but can only return the image to, at best, the diffraction limit set above. One way to increase the resolution and to overcome the diffraction limit is through the use of fluorescence methods in which either only a single fluorescent molecule is excited (STochastic Optical Reconstruction Microscopy, STORM, or Photo-Activated Localization Microscopy, PALM) (64, 65) or a diffraction limited volume is excited and then most of the fluorescence removed using a second, stimulating light beam, leaving a subdiffraction limited volume to continue fluorescing, before the detector is activated (STimulated Emission Depletion microscopy, STED) (66). More recently, in another approach, a structured illumination (SIM) method has been developed. In this case, a pattern of light is directed onto the sample, and multiple images taken, which, after mathematical treatment, enables improvement of the diffraction limit (67). SIM currently has lead to imaging with a resolution of around 10 nm, that is, ∼30 times less than the diffraction limit. However, each of the above methods is comparatively slow and requires repeated exposures (STORM, PALM, and SIM) or sample scanning (STED). Thus, these methods are not suitable for observing rapid transient events such as the [Ca2+] changes in the subplasma membrane space. An alternative method, which provides sub-diffraction limited axial resolution, is total internal reflection fluorescence microscopy (TIRF) (68). In TIRF, the speed limitation is not determined by the excitation beam but only by the speed of the imaging camera and kinetics and brightness of the indicators to enable imaging rates of ∼300 frames per second with adequate signal intensity.
PRINCIPLES OF TIRF MICROSCOPY
Total internal reflection occurs when light traveling in a material of high refractive index meets a material of lower refractive index (refractive index being a measure of the speed of light in the material). If the light hits the interface at normal incidence (zero degrees), it will pass through undeviated (Fig. 5A). As the angle of incidence increases so the light will be refracted according to Snell's law (Fig. 5B) until a critical angle is reached when the light is totally reflected, that is, the surface acts like a perfect mirror (Fig. 5C). This effect is used in the prisms of binoculars for example and is also the basis of how light can be sent down optical fibers.
Figure 5.
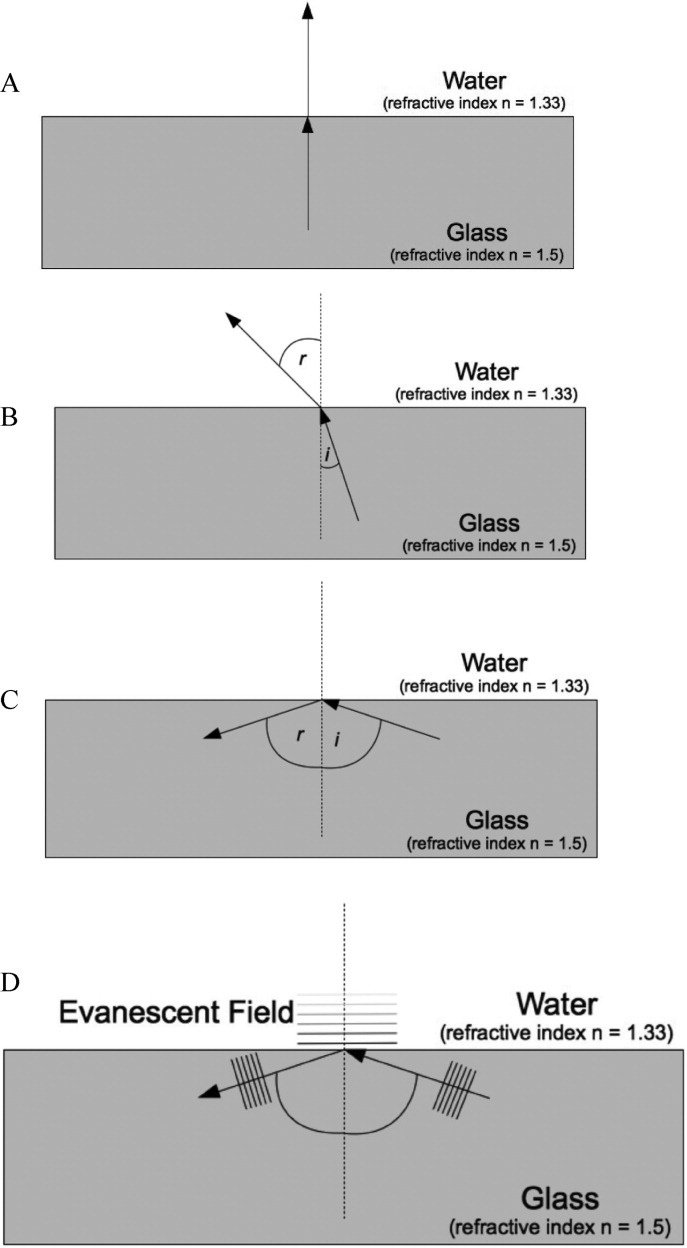
Light, refraction and total internal reflection. (A) Light is undeviated as it passes from high (glass) to low (water) refractive index when at normal incidence. (B) Light bent by refraction (Snell's law) as it passes from high to low refractive indices. (C) Total internal reflection as the angle of incidence exceeds the critical angle. (D) This figure shows the evanescent field propagating from a glass/water interface when angle of incidence exceeds the critical angle. The evanescent field intensity declines exponentially with distance from the interface.
Although the light is reflected according to Maxwell's equations (which govern the propagation of light) (63), an electric field is present on the other side of the interface which is known as an evanescent field and has the ability to excite fluorophores (Fig. 5D). In Fig. 5D, a light wave is shown hitting an interface with the light being reflected and the so-called evanescent field propagated across the interface. This field decays rapidly with distance from the interface following an exponential fall off. If a fluorescent molecule is close to the interface, in the evanescent field, then it will be excited. In this way, it is possible to obtain fluorescent images of samples very close to the surface, typically closer than 200 nm, and in certain cases, using a low excitation power, less than 100 nm—significantly better than the diffraction limit. Multiple fluorescent labeling is possible through the use of two excitation fields and different indicators, though care has to be exercised here as the evanescent field created by different wavelengths of light will fall off in slightly different ways. Even though TIRF imaging exceeds the diffraction limit in the axial direction, the limit of resolution in the lateral direction remains at the diffraction limit given above.
TIRF, thus, provides a method of selectively exciting molecules close to an interface, and two standard methods have been developed to enable this form of imaging. In the first, a prism is used to direct the light into a glass substrate (Fig. 6A). A collimated beam of light (typically a laser) is directed into the prism and initially the light will pass out of the glass slide. However, as the angle is increased, the light will eventually hit the outer surface of the microscope slide, and undergo total internal reflection and be guided along the slide, or coverslip. An imaging objective observing a short way down field from the prism, can then be used to record fluorescence from samples on the surface being excited by the evanescent field propagating with the light as it makes its way down the slide. Optically, this method works well and enables excellent control of the evanescent field. However, from a practical perspective in a biology laboratory, the exposed optics and laser beams raise laser safety issues, and the prism system requires careful adjustment and repeated alignment to ensure optimal performance which limits usefulness.
Figure 6.
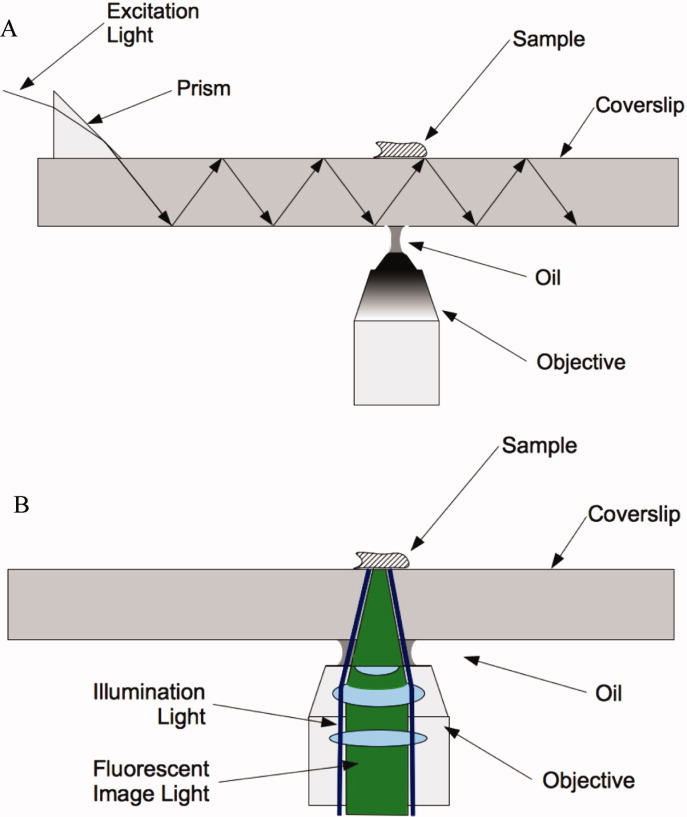
Methods to generate total internal reflection. (A) Prism coupling for TIRF microscopy and (B) TIRF via a very high numerical aperture objective. Excitation light is shown as a dark blue line traveling along the over edge of the objective while the emitted light from the sample fluorophore is shown traveling down through the objective in green.
An alternative method, and that used by nearly all commercial TIRF microscopes, is the use of a high numerical aperture objective in an epi-illumination configuration (Fig. 6B). In basic terms (specific systems can vary slightly), an annular ring of light is directed into the back aperture of a very high numerical aperture lens (Fig. 6B). The numerical aperture is such that this light is totally internally reflected when it tries to exit the sample slide to create the required evanescent field. The same objective lens then observes fluorescence emitted from the sample in a conventional epi-configuration using the central portion of the lens. The advantage of this method is that the light annulus can be controlled at the rear of the microscope enabling a simple adjustment of the angle of the light to be made to set the TIRF angle through the objective. Thus, no optical coupling (except the normal oil for a high NA objective) is required near the sample, and all the illumination optics can be kept away from the wet (physiological solutions), and less than ideally clean, biological area. In addition, the laser beams can be fully enclosed, reducing the laser safety concerns that exist with exposed beams. Thus, it is clear why this method is preferred by commercial microscope manufacturers. Our system, described below, is a variation on this basic set up and is designed to enable high-speed TIRF imaging and near simultaneous wide-field epi-fluorescence. The method enables measurement of subplasma membrane Ca2+ signals and comparison of bulk average [Ca2+]c(global signals) so that an understanding to the initiation and propagation of the signals is possible.
OPTICAL INSTRUMENTATION
The complete optical system is shown in Fig. 7. This system is based upon a conventional Nikon TE2000 inverted microscope and TIRF lens (×60, 1.49NA Nikon Plan Apo). The microscope has two filter turrets for dichroic mirrors on a dual epi-illuminator before the objective lens, enabling both conventional fluorescence excitation and TIRF excitation without moving any components on the microscope. The TIRF light is provided by a frequency-doubled diode-pumped laser operating at 473 nm with a noise of less than 0.01% rms from 1 Hz to 10 MHz. The TEM00 output (single spot beam with no mode patterns) was coupled using a home built system into a single-mode optical fiber with a numerical aperture (N.A.) of 0.1 and a core diameter of 3 μm. The laser excitation had ∼1.5 mW of light emerging from the end of the optical fiber. The fiber was connected into the Nikon TIRF attachment at the rear of the TE2000, and the laser excitation was directed to the objective by a custom designed dichroic mirror present in the top turret (Fig. 7A). The objective lens allowed the laser beam to be introduced at the outer edge of the objective aperture to achieve a high angle of incidence of the beam on the coverslip surface. The angle of illumination was adjusted to exceed the critical angle to generate TIRF and an evanescent excitation field giving a penetration ∼100 nm. To determine optimal imaging conditions, a test sample which consisted of subresolution fluorescent beads (170 nm diameter) bathed in fluorescein was imaged. This sample allowed imaging parameters (illumination angle and intensity) which produced maximum image contrast to be determined and an illumination field that was generally even to be generated.
Figure 7.
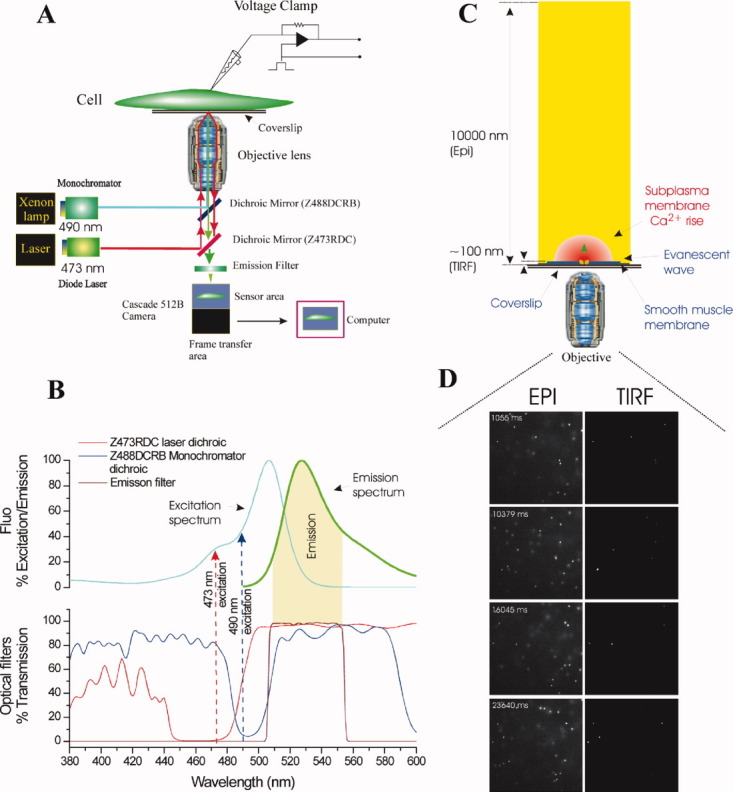
Schematic of the TIRF and wide-field epi-fluorescence system. (A) The experimental system was constructed around a Nikon TE2000U microscope; excitation and emission light paths are shown. Single cells were voltage clamped in whole cell configuration. Cells were loaded with fluo-5F and illuminated by two separate excitation sources (at 490 nm and 473 nm) via a dual epi-illuminator. The first wavelength (490 nm, bandpass 5 nm; blue), was provided by a monochromator and guided via a fiber optic guide through a field stop diaphragm, a neutral density filter and HQ480/40 excitation filter (not shown) before being reflected off a custom-made long-pass dichroic mirror (Z488DCRB). The latter was transmissive in the ranges 380–480 nm and 506–589 nm and reflective from 485 to 499 nm (B). The second excitation wavelength (473 nm; red), for TIRF illumination, was provided by a blue diode pumped laser and guided via a fiber optic coupling through an iris and 10× beam expander. The 473 nm light was then reflected off a dichroic mirror (Z473RDC) and transmitted through the upper dichroic (Z488DCRB) (A and B) and focused to a spot on the back focal plane of the objective lens (Nikon 60×, oil immersion, NA 1.49). The laser focusing lens was mounted on a micrometer driven translation system so that the laser beam could be adjusted to enter the periphery of the objective aperture to achieve total internal reflection at the interface between the cover glass and the aqueous bathing medium. Fluorescence excited in the specimen by the evanescent wave or wide-field epi-illumination was collected through the objective lens, passed through the dichroic mirrors and a barrier filter and imaged by a Phometrics Cascade 512B or evolve 128 cameras (Roper Scientific). The cameras uses a back illuminated frame transfer CCD with on-chip electron multiplication. (C) Enlarged view illustrating the imaging of near membrane Ca2+ from the microdomain (red) around a single open channel by evanescent wave (blue) formed by the TIRF objective lens and the simultaneously measured wide-field epi-fluorescence depth of field. The depth of the TIRF field was ∼ 100 nm and that of the wide-field epi-fluorescent field ∼ 10000 nm. (D) Single image frames of sub-resolution (100 nm) fluorescent latex beads floating in solution obtained in TIRF (right) and wide-field epi-fluorescence (left). Under wide-field epi-illumination (EPI, D, left) subresolution fluorescent latex beads floating in solution are illuminated through the field. Individual beads drift through Brownian motion. In TIRF illumination (D,TIRF, right) beads are illuminated only when they are within the evanescent field, that is, within ∼ 100 nm of the coverslip. Beads diffusing in and out of the evanescent field appear and disappear suddenly (Reproduced from ref. 45, with permission from Rockefeller University Press).
Wide-field epi-fluorescence excitation illumination was provided by an arc lamp with the wavelength controlled via a monochomator to give 490 nm (bandpass 5 nm) coupled via a liquid light guide. The monochromator light was directed to the objective by a custom dichroic on the lower epi-illuminator (Fig. 7A). This light passed through the dichroic in the top epi-illminator and onto the sample.
Rapid (2 ms) switching between TIRF and wide-field illumination was achieved by triggering the laser shutter open using a TTL pulse which simultaneously drove the monochomator to 250 nm (a wavelength that was optically blocked in the system). Terminating the TTL pulse shuttered the laser closed and moved the monochromator to 490 nm.
One unexpected issue arose from the movement of the monochomator between 250 nm and 470 nm. Although the transition only took 2 ms when compared to the CCD cameras frame switch (∼2 μs) in frame transfer mode, it was relatively slow. As a result, a small amount of light initially was recorded during the 2 ms monochromator switch before the final desired wavelength was reached. The light recorded during the monochromator wavelength switch created a slightly increased background fluorescence which limited the ability to resolve of small signals. After exploring several possibilities, the simplest solution was to add a 490 nm narrow bandpass excitation filter to the monochromator excitation light path. Imaging was achieved using EMCCD camera's (a Photometrics Evolve 128 (∼300 frames s−1) or Cascade 512B (∼30 frames s−1)) (Fig. 7A).
COMPLICATIONS WITH TIRF
One difficulty which arises when using freshly isolated smooth muscle cells comes from their crenulated structure, which may result in the cells not lying completely flat on the coverslip. The evanescent wave extends only ∼100 nm from the coverslip (Fig. 7C) and if the cells are not flat there is only a small footprint of cell that can be viewed. A major benefit of TIRF, the ability to view channel activity over a large surface of the cell, is restricted by the small imaging footprint. Another issue which may limit full analysis of results is that the Ca2+ signals measured in the evanescent field are the sum of signals ranging from saturated fluorophore (near to the channel) to infrequent binding events (a small distance from the channel). Fluorophore saturation will mean the gradient cannot easily be deconvolved so that the fluorescent signal is likely to be an especially nonlinear representation of [Ca2+] changes. Even if the dye did not saturate, converting the fluorescent signal to a Ca2+ concentration relies on calibration carried out in steady-state conditions. Such conditions are unlikely to be valid in the rapidly changing microdomains under in the subplasma membrane space during Ca2+ influx. Indeed it is likely that the assumption that the dye equilibrates instantly with Ca2+ will not be correct during influx near a Ca2+ channel which is rapid (∼0.6 million ions per second), that is, there is local nonequilibrium within the system (69). Even the rapid small molecule Ca2+ indicators are too slow to follow these fast [Ca2+] increases. Notwithstanding these difficulties, TIRF is a powerful method to study subplasma membrane events and has generated significant new insights to channel behavior and Ca2+ signaling.
CONCLUSIONS
Subplasma membrane Ca2+ signals may be highly localized and extremely rapid events that have a timecourse approximately four orders of magnitude faster than changes in bulk average [Ca2+]c (Fig. 1). These subplasma membrane signals target particular responses not only by location but by matching the kinetics of the [Ca2+] changes with the speed of the effectors Ca2+ binding. Local subplasma membrane Ca2+ signals have been studied using targeted Ca2+ indicators but the limited dynamic range and slow reaction to changes in [Ca2+] may not fully describe the response. Rapid small molecule Ca2+ indicators used with high resolution imaging have significantly improved dynamic range and speed but the approach is technically difficult and realistic calibration of signals is not possible. Synthetic indicators that are targeted to particular subcellular sites would combine specificity of the protein biosensors approach with the fast kinetics and high dynamic range of small molecular indicators; these are under development and await full experimental exploitation.
Acknowledgments
This work was funded by the Wellcome Trust (092292/Z/10/Z) and British Heart Foundation PG/11/70/29086; their support is gratefully acknowledged.
REFERENCES
- 1.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 2.Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr. Opin. Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 4.McCarron JG, Chalmers S, Bradley KN, Macmillan D, Muir TC. Ca2+ microdomains in smooth muscle. Cell Calcium. 2006;40:461–493. doi: 10.1016/j.ceca.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Llinas R, Sugimori M, Silver RB. Microdomains of high calcium concentration in a presynaptic terminal. Science. 1992;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- 6.Marsault R, Murgia M, Pozzan T, Rizzuto R. Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J. 1997;16:1575–1581. doi: 10.1093/emboj/16.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- 8.Aharon S, Bercovier M, Parnas H. Parallel computation enables precise description of Ca2+ distribution in nerve terminals. Bull. Math. Biol. 1996;58:1075–1097. doi: 10.1007/BF02458384. [DOI] [PubMed] [Google Scholar]
- 9.Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J. Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chalmers S, Olson ML, MacMillan D, Rainbow RD, McCarron JG. Ion channels in smooth muscle: regulation by the sarcoplasmic reticulum and mitochondria. Cell Calcium. 2007;42:447–466. doi: 10.1016/j.ceca.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Zhuge R, Bao R, Fogarty KE, Lifshitz LM. Ca2+ sparks act as potent regulators of excitation-contraction coupling in airway smooth muscle. J. Biol. Chem. 2009;285:2203–2210. doi: 10.1074/jbc.M109.067546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kargacin GJ. Responses of Ca2+ -binding proteins to localized, transient changes in intracellular [Ca2+] J. Theor. Biol. 2003;221:245–258. doi: 10.1006/jtbi.2003.3187. [DOI] [PubMed] [Google Scholar]
- 13.Bao R, Lifshitz LM, Tuft RA, Bellve K, Fogarty KE, et al. A close association of RyRs with highly dense clusters of Ca2+ activated Cl− channels underlies the activation of STICs by Ca2+ sparks in mouse airway smooth muscle. J. Gen. Physiol. 2008;132:145–160. doi: 10.1085/jgp.200709933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macrez N, Mironneau J. Local Ca2+ signals in cellular signalling. Curr. Mol. Med. 2004;4:263–275. doi: 10.2174/1566524043360762. [DOI] [PubMed] [Google Scholar]
- 15.Bazzazi H, Kargacin ME, Kargacin GJ. Ca2+ regulation in the near-membrane microenvironment in smooth muscle cells. Biophys. J. 2003;85:1754–1765. doi: 10.1016/S0006-3495(03)74605-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordienko DV, Harhun MI, Kustov MV, Pucovsky V, Bolton TB. Sub-plasmalemmal [Ca2+]i upstroke in myocytes of the guinea-pig small intestine evoked by muscarinic stimulation: IP3R- mediated Ca2+ release induced by voltage-gated Ca2+ entry. Cell Calcium. 2008;43:122–141. doi: 10.1016/j.ceca.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boittin FX, Macrez N, Halet G, Mironneau J. Norepinephrine-induced Ca2+ waves depend on InsP3 and ryanodine receptor activation in vascular myocytes. Am. J. Physiol. 1999;277:C139–C151. doi: 10.1152/ajpcell.1999.277.1.C139. [DOI] [PubMed] [Google Scholar]
- 18.Jaggar JH, Nelson MT. Differential regulation of Ca2+ sparks and Ca2+ waves by UTP in rat cerebral artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2000;279:C1528–C1539. doi: 10.1152/ajpcell.2000.279.5.C1528. [DOI] [PubMed] [Google Scholar]
- 19.McCarron JG, MacMillan D, Bradley KN, Chalmers S, Muir TC. Origin and mechanisms of Ca2+ waves in smooth muscle as revealed by localized photolysis of caged inositol 1,4,5-trisphospate. J. Biol. Chem. 2004;279:8417–8427. doi: 10.1074/jbc.M311797200. [DOI] [PubMed] [Google Scholar]
- 20.McCarron JG, Chalmers S, MacMillan D, Olson ML. Agonist-evoked Ca2+ wave progression requires Ca2+ and IP3. J. Cell Physiol. 2010;244:334–344. doi: 10.1002/jcp.22103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai Y, Edelmann M, Sanderson MJ. The contribution of inositol 1,4,5-trisphosphate and ryanodine receptors to agonist-induced Ca2+ signaling of airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;297:L347–L361. doi: 10.1152/ajplung.90559.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balemba OB, Heppner TJ, Bonev AD, Nelson MT, Mawe GM. Calcium waves in intact guinea pig gallbladder smooth muscle cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;291:G717–G727. doi: 10.1152/ajpgi.00035.2006. [DOI] [PubMed] [Google Scholar]
- 23.McCarron JG, Bradley KN, MacMillan D, Muir TC. Sarcolemma agonist-induced interactions between InsP3 and ryanodine receptors in Ca2+ oscillations and waves in smooth muscle. Biochem. Soc. Trans. 2003;31:920–924. doi: 10.1042/bst0310920. [DOI] [PubMed] [Google Scholar]
- 24.Bai Y, Sanderson MJ. Airway smooth muscle relaxation results from a reduction in the frequency of Ca2+ oscillations induced by a cAMP-mediated inhibition of the 1P3 receptor. Respir. Res. 2006;7:34. doi: 10.1186/1465-9921-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 26.Marchant JS, Parker I. Role of elementary Ca2+ puffs in generating repetitive Ca2+ oscillations. EMBO J. 2001;20:65–76. doi: 10.1093/emboj/20.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 28.Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J. Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demuro A, Parker I. Imaging the activity and localization of single voltage-gated Ca2+ channels by total internal reflection fluorescence microscopy. Biophys. J. 2004;86:3250–3259. doi: 10.1016/S0006-3495(04)74373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith IF, Wiltgen SM, Parker I. Localization of puff sites adjacent to the plasma membrane: functional and spatial characterization of Ca2+ signaling in SH-SY5Y cells utilizing membrane-permeant caged IP3. Cell Calcium. 2009;45:65–76. doi: 10.1016/j.ceca.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore ED, Voigt T, Kobayashi YM, Isenberg G, Fay FS, et al. Organization of Ca2+ release units in excitable smooth muscle of the guinea-pig urinary bladder. Biophys. J. 2004;87:1836–1847. doi: 10.1529/biophysj.104.044123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarron JG, Bradley KN, Muir TC. Ca2+ signalling and Ca2+ activated K+ channels in smooth muscle. Novartis Found. Symp. 246, 52 – 64; discussion 64 –. 2002;70:221–227. [PubMed] [Google Scholar]
- 33.McCarron JG, Craig JW, Bradley KN, Muir TC. Agonist-induced phasic and tonic responses in smooth muscle are mediated by InsP3. J. Cell Sci. 2002;115:2207–2218. doi: 10.1242/jcs.115.10.2207. [DOI] [PubMed] [Google Scholar]
- 34.Gordienko DV, Bolton TB, Cannell MB. Variability in spontaneous subcellular calcium release in guinea-pig ileum smooth muscle cells. J. Physiol. 1998;507(Pt 3):707–720. doi: 10.1111/j.1469-7793.1998.707bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, et al. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 36.Zhuge R, Fogarty KE, Baker SP, McCarron JG, Tuft RA, et al. Ca2+ spark sites in smooth muscle cells are numerous and differ in number of ryanodine receptors, large-conductance K+ channels and coupling ratio between them. Am. J. Physiol. Cell Physiol. 2004;287:C1577–C1588. doi: 10.1152/ajpcell.00153.2004. [DOI] [PubMed] [Google Scholar]
- 37.ZhuGe R, Sims SM, Tuft RA, Fogarty KE, Walsh JV., Jr Ca2+ sparks activate K+ and Cl− channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J. Physiol. 1998;513(Pt 3):711–718. doi: 10.1111/j.1469-7793.1998.711ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signalling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron. 2002;34:209–220. doi: 10.1016/s0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 39.Tovey SC, Dedos SG, Taylor EJ, Church JE, Taylor CW. Selective coupling of type 6 adenylyl cyclase with type 2 IP3 receptors mediates direct sensitization of IP3 receptors by cAMP. J. Cell Biol. 2008;183:297–311. doi: 10.1083/jcb.200803172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pucovsky V, Bolton TB. Localisation, function and composition of primary Ca2+ spark discharge region in isolated smooth muscle cells from guinea-pig mesenteric arteries. Cell Calcium. 2006;39:113–129. doi: 10.1016/j.ceca.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Thomason PA, Wolanin PM, Stock JB. Signal transduction: receptor clusters as information processing arrays. Curr. Biol. 2002;12:R399–R401. doi: 10.1016/s0960-9822(02)00885-0. [DOI] [PubMed] [Google Scholar]
- 42.Yan HD, Okamoto H, Unno T, Tsytsyura YD, Prestwich SA, et al. Effects of G-protein-specific antibodies and G beta gamma subunits on the muscarinic receptor-operated cation current in guinea-pig ileal smooth muscle cells. Br. J. Pharmacol. 2003;139:605–615. doi: 10.1038/sj.bjp.0705289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohi Y, Yamamura H, Nagano N, Ohya S, Muraki K, et al. Local Ca2+ transients and distribution of BK channels and ryanodine receptors in smooth muscle cells of guinea-pig vas deferens and urinay bladder. J. Physiol. 2001;534:313–326. doi: 10.1111/j.1469-7793.2001.t01-3-00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tour O, Adams SR, Kerr RA, Meijer RM, Sejnowski TJ, et al. Calcium Green FLASH as a genetically targeted small-molecule calcium indicator. Nat. Chem. Biol. 2007;3:423–431. doi: 10.1038/nchembio.2007.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarron JG, Olson ML, Currie S, Wright AJ, Anderson KI, et al. Elevations of intracellular calcium reflect normal voltage-dependent behavior, and not constitutive activity, of voltage-dependent calcium channels in gastrointestinal and vascular smooth muscle. J. Gen. Physiol. 2009;133:439–457. doi: 10.1085/jgp.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zenisek D, Davila V, Wan L, Almers W. Imaging calcium entry sites and ribbon strucutres in two presynaptic cells. J. Neurosci. 2003;23:2538–2548. doi: 10.1523/JNEUROSCI.23-07-02538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Navedo MF, Amberg GC, Votaw VS, Santana LF. Constitutively active L-type Ca2+ channels. Proc. Natl. Acad. Sci. USA. 2005;102:11112–11117. doi: 10.1073/pnas.0500360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhuge R, Fogarty KE, Tuft RA, Walsh JV., Jr Spontaneous transient outward currents arise from microdomains where BK channels are exposed to a mean Ca2+ concentration on the order of 10 microM during a Ca2+ spark. J. Gen. Physiol. 2002;120:15–27. doi: 10.1085/jgp.20028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou H, Lifshitz LM, Tuft RA, Fogarty KE, Singer JJ. Imaging Ca2+ entering the cytoplasm through a single opening of a plasma membrane cation channel. J. Gen. Physiol. 1999;114:575–588. doi: 10.1085/jgp.114.4.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bradley KN, Craig JW, Muir TC, McCarron JG. The sarcoplasmic reticulum and sarcolemma together form a passive Ca2+ trap in colonic smooth muscle. Cell Calcium. 2004;36:29–41. doi: 10.1016/j.ceca.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 51.Etter EF, Minta A, Poenie M, Fay FS. Near-membrane [Ca2+] transients resolved using the Ca2+ indicator FFP18. Proc. Natl. Acad. Sci. USA. 1996;93:5368–5373. doi: 10.1073/pnas.93.11.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 53.Lee MY, Song H, Nakai J, Ohkura M, Kotlikoff MI, et al. Local subplasma membrane Ca2+ signals detected by a tethered Ca2+ sensor. Proc. Natl. Acad. Sci. USA. 2006;103:13232–13237. doi: 10.1073/pnas.0605757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mank M, Reiff DF, Heim N, Friedrich MW, Borst A, et al. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophys. J. 2006;90:1790–1796. doi: 10.1529/biophysj.105.073536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- 56.Dargan SL, Parker I. Buffer kinetics shape the spatiotemporal patterns of IP3-evoked Ca2+ signals. J. Physiol. 2003;553:775–788. doi: 10.1113/jphysiol.2003.054247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stern MD. Buffering of calcium in the vicnity of a channel pore. Cell Calcium. 1992;13:183–192. doi: 10.1016/0143-4160(92)90046-u. [DOI] [PubMed] [Google Scholar]
- 58.Pap EH, Dansen TB, van Summeren R, Wirtz KW. Peptide-based targeting of fluorophores to organelles in living cells. Exp. Cell Res. 2001;265:288–293. doi: 10.1006/excr.2001.5190. [DOI] [PubMed] [Google Scholar]
- 59.Bannwarth M, Correa IR, Sztretye M, Pouvreau S, Fellay C, et al. Indo-1 derivatives for local calcium sensing. ACS Chem. Biol. 2009;4:179–190. doi: 10.1021/cb800258g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruggiu AA, Bannwarth M, Johnsson K. Fura-2FF-based calcium indicator for protein labeling. Org. Biomol. Chem. 8:3398–3401. doi: 10.1039/c000158a. [DOI] [PubMed] [Google Scholar]
- 61.Abbe E. Beitrage zur Theorie des Mikroskopd und der mikroskopischen Wahrnehmung. Archi. Mikroskopische. Anat. 1873;9:413. [Google Scholar]
- 62.Padgett M. On the focussing of light, as limited by the uncertainty principle. J. Mod. Opt. 2008;55:3083–3089. [Google Scholar]
- 63.Hecht E. Optics. Fourth Edition. USA: Addison-Wesley, MA; 2002. [Google Scholar]
- 64.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 65.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 67.Heintzmann R,, Gustafsson MGL. Subdiffraction resolution in continuous samples. Nat. Photonics. 2009;3:362–364. [Google Scholar]
- 68.Axelrod D, Burghardt TP, Thompson NL. Total internal reflection fluorescence. Annu. Rev. Biophys. Bioeng. 1984;13:247–268. doi: 10.1146/annurev.bb.13.060184.001335. [DOI] [PubMed] [Google Scholar]
- 69.Bortolozzi M, Lelli A, Mammano F. Calcium microdomains at presynaptic active zones of vertebrate hair cells unmasked by stochastic deconvolution. Cell Calcium. 2008;44:158–168. doi: 10.1016/j.ceca.2007.11.007. [DOI] [PubMed] [Google Scholar]