

Abstract
Toxicity and chemoresistance are two major issues to hamper the success of current standard tumor chemotherapy. Combined therapy of agents with different mechanisms of action is a feasible and effective means to minimize the side effects and avoid the resistance to chemotherapeutic drugs while improving the antitumor effects. As the most essential tumor suppressor, p53 or its pathway has been an attractive target to develop a new type of molecule-targeting anticancer therapy. Recently, we identified a small molecule, Inauhzin (INZ), which can specifically activate p53 by inducing its deacetylation. In this study, we tested if combination with INZ could sensitize tumor cells to the current chemotherapeutic drugs, cisplatin (CIS) and doxorubicin (DOX). We found that compared with any single treatment, combination of lower doses of INZ and CIS or DOX significantly promoted apoptosis and cell growth inhibition in human non-small lung cancer and colon cancer cell lines in a p53-dependent fashion. This cooperative effect between INZ and CIS on tumor suppression was also confirmed in a xenograft tumor model. Therefore, this study suggests that specifically targeting the p53 pathway could enhance the sensitivity of cancer cells to chemotherapeutic agents and markedly reduce the doses of the chemotherapy, possibly decreasing its adverse side effects.
Introduction
The modern chemotherapy of cancers, which mainly refers to genotoxic/cytotoxic drugs [1], started around 1940s and has developed into several types, including alkylating agents, anthracyclines, plant alkaloids, topoisomerase inhibitors, and antimetabolites. Most of the above chemotherapeutic drugs inhibit tumor growth by causing DNA damages, arresting DNA replication, and cell division. As the representatives of alkylating agents and anthracyclines, respectively, cisplatin (CIS) and doxorubicin (DOX) are among the most potent drugs to fight against many kinds of hematologic malignancies and solid tumors [2,3]. Unfortunately, the dosage and treatment duration of these drugs, which are essential for maximizing their antitumor effects, are often limited due to severe toxicities to normal tissues, such as CIS caused nephrotoxicity and DOX caused cardiomyopathy [3–8]. These detrimental effects are usually cumulative, dose-dependent, and irreversible [5,8]. Besides toxicity, chemoresistance is another major obstacle for effective cancer chemotherapy [9]. This can result from spontaneous mutations occurring at the rate of 1 of 105 cells and be obtained inevitably with cell proliferation. However, resistance to two drugs occurs much less frequently (fewer than 1 in 1010 cells) [1]. Therefore, combined therapy using agents with different mechanisms of action and resistance has become an intriguing and promising strategy to overcome side effects and drug resistance as well as to obtain synergistic efficiency [1].
New strategies targeting aberrant pathways, dysregulated signaling molecules in tumors, and tumor-specific antigens have been developed during the past decade [1]. The tumor suppressor p53 is essentially important for preventing mammalian cells from undergoing neoplasia and tumorigenesis, primarily due to its ability to activate the transcription of numerous genes and some miRNA responsible for executing p53-dependent apoptosis, autophagy, senescence, and DNA repair as well as suppression of cell proliferation, growth, migration, and angiogenesis [10–12]. About half of human tumors contain a mutation or deletion of theTP53 gene [13–15], and the tumors retaining wild-type p53 usually have other aberrations in their p53 pathway, such as amplified expression of MDM2 and/or MDMX [16]. MDM2 and MDMX are two physiological repressors of p53, which inactivate the latter by directly inhibiting its transcriptional activity and mediating its ubiquitination in a feedback fashion, as they are also the transcriptional targets of p53 [17–24]. On account of the importance of the p53-MDM2/MDMX pathway in the initiation and development of wild-type p53-containing tumors, intensive studies over the past decade have been aiming to identify small molecules that could specifically target individual protein molecules of this pathway for developing a better molecule-targeting anticancer therapy [25]. Several small molecules or peptides have been reported to activate p53 by either blocking its binding to MDM2 [26–28], inhibiting MDM2 E3 ubiquitin ligase activity [29], or inhibiting MDMX-p53 binding [30]. Activating p53 by targeting its deacetylase(s) is another new strategy. p53 acetylation by p300/CREB-binding protein (CBP) and ubiquitination by MDM2 are mutually exclusive [31–35], so increased acetylation could attenuate, yet increased deacetylation could facilitate, MDM2-mediated p53 ubiquitination and degradation [32,34,35]. On the basis of the high expression level of SIRT1, an enzyme to catalyze deacetylation of p53, in a number of human cancers [36–38], several inhibitors of SIRT1 [39], including Inauhzin (INZ) by our group [16], were identified to stop MDM2-mediated p53 degradation and induce p53 activation. Therefore, indirectly interrupting the MDM2-p53 negative feedback loop by inhibiting SIRT1 activity to enhance p53 acetylation could serve as an alternative strategy for the development of anticancer therapy.
Activation of the p53 signaling pathway is one of the central mechanisms for most of the genotoxic drugs to suppress tumor growth, while silence of p53 can lead to chemoresistance [40–43]. Restoring and maximizing p53 activity in tumor cells through combination of various means could be a favorable strategy to enhance the sensitivity and reduce the toxicity of chemotherapy. INZ, as a specific activator of the p53 pathway, bypassing DNA damage with retaining antitumor activity and exerting a minimal effect on the cell viability of normal human cells [16], was hypothesized to present an advantage to increase p53 activity and enhance tumor suppression, meanwhile lowering the toxicity to normal tissues and drug resistance by combination with lower dose of chemotherapeutic drugs. In our previous study [44], we demonstrated the synergistic effects of combining INZ and Nutlin-3, an inhibitor targeting the MDM2-p53 interaction [26], on p53 activation and tumor suppression. Here, we further tested the combined antitumor effects of INZ and current first-line chemotherapeutic drugs, CIS and DOX, using cell-based assays and a xenograft tumor model system. As a result, we found that the combination of two compounds at much lower doses synergistically activates p53 and induces the proapoptotic activity in human lung and colon cancer cell lines. We also observed the enhanced growth suppression of xenografted lung cancer with combination of INZ and CIS at lower doses. Therefore, this study suggests that INZ, as a new-type non-genotoxic antitumor drug candidate, could serve as a potent component of the combined therapy to improve the antitumor effects, combat the drug resistance, and reduce the side effects of chemotherapeutic drugs.
Materials and Methods
Chemicals and Antibodies
INZ was purchased from ChemDiv (San Diego, CA) or ChemBridge (San Diego, CA) and dissolved with DMSO to a concentration of 50 mM and stored at -20°C. CIS and DOX were purchased from Sigma (St Louis, MO) and dissolved with ddH2O to a concentration of 10 mM and stored at -20°C. Cell Counting Kit was purchased from Dojindo Molecular Technologies, Inc (Gaithersburg, MD). FluoresceinIn Situ Apoptosis Detection Kit was from Roche (Indianapolis, IN). Antibodies for Western blot (WB) included rabbit polyclonal anti-p53 (FL-393; Santa Cruz Biotechnology, Dallas, TX), rabbit polyclonal anti-p21 (M19; Santa Cruz Biotechnology), mouse monoclonal anti-p21 (CP74; NeoMarkers, Fremont, CA), mouse monoclonal anti-p21 (F-5; Santa Cruz Biotechnology), rabbit polyclonal anticleaved poly (ADP-ribose) polymerase (PARP) (9542 Cell Signaling Technology, Danvers, MA), and mouse anti-β-actin (Santa Cruz Biotechnology). Mouse monoclonal anti-5-bromo-2′-deoxyuridine (BrdU) antibodies (IIB5; Santa Cruz Biotechnology) were used for immunofluorescence staining.
Cell Culture
Human non-small cell lung carcinoma wild-type p53-containing H460 and A549, human non-small cell lung carcinoma p53-null H1299, human colon cancer HCT116 (p53+/+ and p53-/-), and normal human fibroblast cell line NHF-1 were maintained in Dulbecco's modified Eagle's medium (Gibco BRL, Carlsbad, CA) supplemented with 10% FBS and Penicillin-Streptomycin antibiotic mixture (10 ml/l; Gibco BRL) at 37°C in an incubator containing humidified air with 5% CO2.
WB Analysis
H460, A549, H1299, and HCT116 cells cultured in six-well culture plates were grown to 80% confluence and then treated with various concentrations of INZ together with or without CIS or DOX for indicated time periods. After being washed with ice-cold phosphate-buffered saline (PBS) three times, cells were lysed with 70 µl of ice-cold lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 8.0), 5 mM EDTA, 0.5% NP-40, 1 mM DTT, 0.2 mM PMSF, 0.3 µM aprotinin, 130 µM bestatin, 1 µM leupeptin, and 1 µg/ml pepstatin A. Tumor tissues from the control and treatment groups saved in liquid nitrogen were rinsed in precooled PBS, minced, and homogenized on ice with a Dounce homogenizer in RIPA lysis buffer [150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris (pH 7.5), 1 mM DTT, 0.2 mM PMSF, 0.3 µM aprotinin, 130 µM bestatin, 1 µM leupeptin, and 1 µg/ml pepstatin A]. After incubation at 4°C for 30 minutes, cell or tumor tissue lysates were obtained by centrifugation at 18,000g at 4°C for 15 minutes, and the concentration of total protein was determined by the Bradford method. Equal amounts (50 µg per lane) of proteins obtained as described above were loaded to each lane and ran in a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA) using a semidry transfer apparatus (Bio-Rad). After blocking in 5% nonfat dry milk in 1x TBS containing 0.1% Tween 20 (TBST) at room temperature for 1 hour, membranes were incubated with primary antibodies in the blocking buffer at 4°C overnight, followed by three washes in TBST and incubation with secondary HRP-conjugated antibody (Bio-Rad) at room temperature for 1 hour. After three washes in TBST, blots were detected with SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology Inc, Rockford, IL) and developed using Kodak Biomax film (PerkinElmer, Norwalk, CT). All blots were normalized against β-actin as a loading control.
Cell Viability Assay and Assessment of Combined Drug Effect
To assess cell growth, the Cell Counting Kit (Dojindo Molecular Technologies, Inc) was used according to the manufacturer's instructions. Briefly, cells (5000/100 µl per well) were seeded in 96-well flat-bottom plates, incubated overnight at 37°C, then treated with each compound individually at serial dilutions or both compounds simultaneously at serial dilutions with fixed molar drug ratios (INZ/CIS = 1:2 for H460 cells, INZ/CIS = 1:7 for HCT116p53+/+, and INZ/DOX = 20:1 for H460 and HCT116p53+/+). After treatment for 72 hours, WST-8 was added to each well at a final concentration of 10%, and the absorbance of the samples was recorded at 450 nm on a microplate reader (Molecular Devices, Sunnyvale, CA; SpectraMax M5e) after 3 hours of incubation at 37°C. The dose-effect curves were generated for each treatment to estimate the individual fractional survival (f).
The combination index (CI) values were calculated to determine the combined effects of INZ and CIS or DOX, based on the dose-effect curves obtained above, according to the following formula as previously developed [45]:
In this formula, (D)1 and (D)2 are the concentrations of each compound of combination required to produce the fraction (f), (Df)1 and (Df)2 are the concentrations of individuals required to producef, andα = 1 or 0 depending on whether the drugs are assumed to be mutually nonexclusive or mutually exclusive, respectively. In this method, CI < 1, =1, or >1 indicate synergism, additivity, or antagonism, respectively [45].
Cell Apoptosis Analysis by Flow Cytometry
Cells (1.5 x 105) were plated into six-well plates and incubated at 37°C overnight. After treatment of INZ and CIS or DOX at the indicated concentrations for 48 hours, cells were harvested, fixed in 70% ice-cold ethanol overnight at -20°C, resuspended in propidium iodide (PI) solution (50 µg/ml PI, 0.1 mg/ml RNase A, 0.05% Triton X-100 in PBS) for 40 minutes at 37°C, then analyzed for DNA content using a flow cytometer (FACSCalibur; Becton Dickinson, Franklin Lakes, NJ) and proprietary software (ModFit LT; Verity Software House, Topsham, ME).
In Vivo Studies
Five-week-old female SCID mice were purchased fromIn vivo Therapeutics Core, Indiana University Simon Cancer Center (Indianapolis, IN). Mice were subcutaneously inoculated with 2 x 106 H460 cells in the right flank, and tumor growth was monitored with calipers. After the tumors became palpable, tumor-bearing mice were randomly divided into four groups and intraperitoneally (i.p.) administered INZ (dissolved in 4% DMSO, 20 mg/kg, i.p.) once per day for 21 days as described previously [16,44], CIS (dissolved in 1% DMSO, 3 mg/kg, i.p.) once per week for 3 weeks based on previous studies [46,47], or vehicles (4% DMSO once per day and 1% DMSO once per week). Tumor volume was measured every other day, and fractional inhibition of tumor growth was calculated on the basis of the tumor volume. Mice were sacrificed by euthanasia and tumors were harvested on the last day of treatment. Tumor weight was measured and presented in histograms.
To determine the induction of p53 and apoptotic signalsin vivo, tumor homogenates were analyzed by WB as described above.
Cell proliferation in tumors was assessed by BrdU labeling and immunofluorescence; 200 mg/kg body weight of BrdU was administrated to mice through i.p. injection 2 hours before the mice were sacrificed.In situ p53 expression was detected by immunofluorescence. Apoptosis was also determined byin situ terminal-deoxynucleoitidyl transferase-mediated nick end labelling (TUNEL) staining, using the FluoresceinIn Situ Cell Death Detection Kit (Roche) according to the manufacturer's instructions. Briefly, tumors were fixed in 4% paraformaldehyde overnight at 4°C, embedded in paraffin, and cut into 6-µm-thick sections. Slides were boiled in fresh 10 mM sodium citrate (pH 6.0) in a steamer for 30 minutes for antigen retrieval, cooled for 30 minutes at room temperature, and washed with PBS (PBS, 0.1% Tween 20). After blocked for 1 hour in blocking buffer (PBS containing 5% goat serum and 0.3% Triton X-100), sections were incubated overnight at 4°C in a humidity chamber with a mouse anti-BrdU monoclonal antibody or a rabbit anti-p53 polyclonal antibody (FL393) diluted 1:100 in the blocking buffer, followed by incubation with an Alexa Fluor 594-conjugated or an Alexa Fluor 488-conjugated secondary antibody for 30 minutes at room temperature. Images were obtained under a fluorescence microscope (Zeiss 200), and quantitative analysis was performed using ImageJ software, by counting the positive cells in six randomly chosen fields of view, with a minimum number of 1000 cells scored for each condition.
All animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine.
Statistics
Data were reported as means ± SEM with N being the sample size. Comparisons among groups were analyzed by using one-way analysis of variance. Probability values ofP < .05 were considered statistically significant.
Results
INZ Enhances the Potential of CIS or DOX to Activate p53
As canonic DNA-damaging anticancer drugs, CIS and DOX have been proven to kill cancer cells through multiple pathways [48], and activation of the tumor suppressor p53 is one of their major mechanisms to induce cell apoptosis and growth suppression in the cancers harboring wild-type p53 [48,49]. We recently found that INZ induces p53 by preventing the SIRT1-mediated p53 deacetylation, further inhibiting MDM2-mediated p53 degradation [16]. To test the hypothesis that INZ may potentiate the ability of CIS and DOX to activate p53, we treated p53-positive or null human non-small cell lung carcinoma cell lines H460 and H1299 with various doses of INZ, CIS or DOX alone, or of their mixtures for 18 hours, followed by WB analyses with the cell lysates as shown inFigures 1A,2A, andW2A. Consistent with previous studies [16,44], INZ, CIS, or DOX by itself induced p53 level and activity (as indicated by the level of p21 and cleaved PARP) in a dose-dependent fashion in H460 cells (Figure 1A). Interestingly, although either of INZ at 0.5 µM, CIS at 1, 3, and 5 µM, or DOX at 0.2 µM slightly induced p53 and its target expression, combined treatment of INZ and CIS or DOX at the same lower doses significantly induced the level of p53, p21, and cleaved PARP (Figures 1A and2A). This cooperative effect on p53 activation was also evident and reproducible in p53-positive human colon cancer cell line HCT116 (Figures 1B and2B) and p53-positive human non-small cell lung cancer cell A549 (Figure W1). By contrast, co-treatment of p53-null cell H1299 (Figure W2A) or HCT116p53-/- (Figure W2B) with the same combination of the compounds did not show any significant enhancement on the level of p21 and cleaved PARP, compared to either of the single treatment, suggesting a p53-dependent cooperation of the combination. Taken together, these results indicate that INZ can cooperate with DNA-damaging reagents to activate p53 in human cancer cells.
Figure 1.
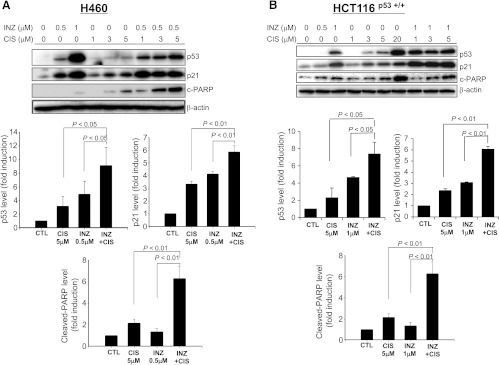
INZ significantly enhances CIS-induced p53 level and activity in H460 and HCT116 cells. H460 (A) or HCT116p53+/+ (B) cells were treated with INZ or/and CIS at the indicated concentrations for 18 hours and harvested for WB analysis. Fifty micrograms of proteins was loaded to each lane, and an anti-β-actin antibody was used as a loading control. Densitometric analyses of immunoreactive bands of p53, p21, and cleaved PARP were expressed as fold change relative to the control group from at least three independent experiments (n = 3).
Figure 2.
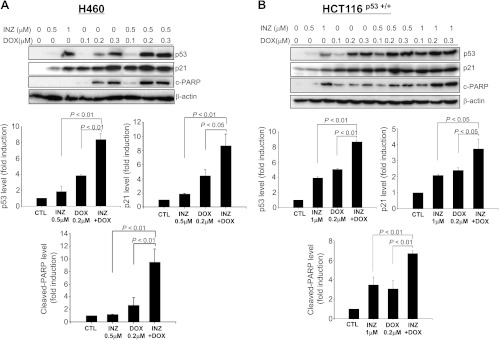
INZ significantly enhances DOX-induced p53 level and activity in H460 and HCT116 cells. H460 (A) or HCT116p53+/+ (B) cells were treated with INZ or/and DOX at the indicated concentrations for 18 hours and harvested for WB analysis. Fifty micrograms of proteins was loaded to each lane, and an anti-β-actin antibody was used as a loading control. Densitometric analyses of immunoreactive bands of p53, p21, and cleaved PARP were expressed as fold change relative to the control group from three independent experiments.
INZ Cooperates with CIS or DOX to Inhibit p53-Dependent Cell Growth
To translate the synergy of INZ and CIS or DOX on the p53 pathway into cytotoxic effects and to demonstrate that the co-activation of the p53 pathway is indeed required for their cooperative inhibition of cancer cell growth, we carried out a set of cell survival assays by treating either H460 or HCT116p53+/+ cells with each compound individually or two compounds simultaneously at serial dilutions in fixed molar ratios for 72 hours, as described in Materials and Methods section as well as inFigures 3 and4. The molar concentration of a compound required for 50% inhibition of cellular proliferation (IC50) was determined for each compound to establish equipotency. The IC50 of INZ and CIS in H460 cells was 3 and 6 µM, respectively, indicating that the molar ratio for INZ and CIS to produce one-fold equipotency is 1:2, which was then maintained for the serial dilutions of two compounds for co-treatment (Figure 3A). The survival fractions of cells after single treatment or co-treatment with INZ and CIS were shown by the curves inFigure 3A.
Figure 3.
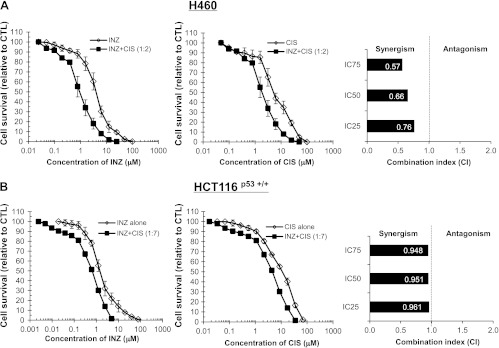
INZ and CIS demonstrate synergistic cytotoxicity in H460 and HCT116 cells. H460 (A) and HCT116p53+/+ (B) cells were plated in 96-well plates and treated with INZ or CIS individually at serial dilutions or both simultaneously at fixed molar drug ratios (INZ/CIS = 1:2 for H460, INZ/CIS = 1:7 for HCT116p53+/+) for 72 hours. Cell viability was determined using a WST cell growth assay. The data here represent the mean of three independent experiments ± SEM. CI values presented indicate the interaction of INZ with CIS when the combined treatment inhibits cell growth by 25% (IC25), 50% (IC50), or 75% (IC75), compared to the control for their individual treatment alone.
Figure 4.
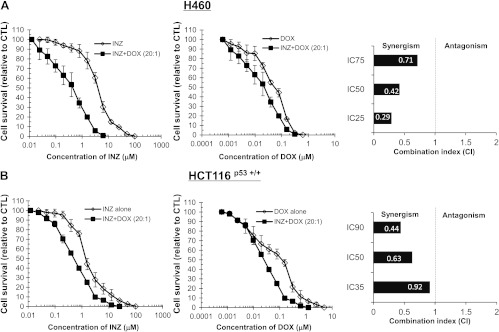
INZ and DOX demonstrate synergistic cytotoxicity in H460 and HCT116p53+/+ cells. H460 (A) and HCT116p53+/+ (B) cells were plated in 96-well plates and treated with INZ or DOX individually at serial dilutions or both simultaneously at fixed molar drug ratios (INZ/DOX = 20:1 for H460 and HCT116p53+/+) for 72 hours. Cell viability was determined using a WST cell growth assay. The data here represent the mean of three independent experiments ± SEM. CI values presented indicate the synergy of INZ with DOX assessed as that for INZ and CIS inFigure 3.
The values for CI, which indicate either synergism (less than 1) or antagonism (greater than 1), were calculated as described in Materials and Methods section when the co-treatment of H460 cells with INZ and CIS reduced cellular survival by 25% (IC25), 50% (IC50), or 75% (IC75) and plotted in the right panels ofFigure 3A. In line with the results shown inFigure 1A, combination of INZ and CIS displayed obvious synergism in suppressing the growth of H460, as the CI values at a range of effective doses were clearly less than 1 (Figure 3A). The interplay between INZ and CIS in HCT116p53+/+ cells (Figure 3B), or between INZ and DOX in H460 and HCT116p53+/+ cells (Figure 4,A andB), was also assessed by employing the same method. As shown inFigure 4,A andB, in H460 and HCT116p53+/+ cells, apparent synergy was evident in the co-treatment with INZ and DOX, as their CI values were obviously less than 1. The CI values for combination of INZ and CIS in HCT116p53+/+ cells were close to but still lower than 1 (Figure 3A), suggesting the possible additive effect between these two compounds in this cancer cell line. However, the synergistic cell growth inhibition of INZ and CIS or DOX was not observed in p53-null cells and normal human fibroblast cell NHF-1 (Figures W2 andW3), suggesting that the synergistic suppression of cell growth by combination of INZ and CIS or DOX is p53-dependent, but not evident in normal cells. These results demonstrate that INZ and DNA-damaging drugs can synergistically repress the growth of human cancer cell lines in a p53-dependent manner.
INZ and CIS or DOX Synergistically Induce p53-Dependent Apoptosis
To further address the p53-dependent synergistic antitumor effect of INZ and DNA-damaging drugs, we detected cell apoptosis, which is one of major mechanisms for p53 to inhibit tumor growth. The results fromFigures 1,2,3,4, andW1 also showed the increased amount of cleaved PARP, an indicator of cell apoptosis, by co-treatment of p53-possessing H460, HCT116, and A549 cells with INZ and CIS or DOX, suggesting that this combinational treatment might induce p53-dependent apoptosis. We performed fluorescence-activated cell sorter analyses (FACS) of H460 and HCT116p53+/+ cells after treatment with INZ and CIS or DOX for 48 hours (Figures 5 and6). In this assay, the molar ratios of compounds for co-treatment were still maintained the same as that in the cell survival assay as described above. As shown inFigures 5A and6A, either 2 µM CIS or 0.05 µM DOX exerted slight effects on apoptosis in H460 cells (∼3% of apoptosis), whereas 1 µM INZ induced approximately 6% to 7% of apoptotic cells. However, combinational treatment of H460 cells with the same dose of INZ and CIS (or DOX) significantly raised the apoptotic rate to 14% to 18%. For HCT116p53+/+ cells, either 28 µM CIS or 0.2 µM DOX only induced 3% to 5% of apoptosis, and 4 µM INZ induced 6% to 7% of apoptosis; however, combined treatment dramatically increased apoptosis to ∼24% and ∼15%, respectively (Figures 5B and6B). Even at lower doses of combination, additive effect was still exhibited in HCT116p53+/+ cells (Figures 5B and6B). Obviously, INZ sensitized these cancer cells to CIS or DOX, synergistically promoting apoptosis. This synergy on proapoptotic effect of these compounds was not seen in p53-null cells (as indicated by the level of cleaved PARP inFigure W2). Taken together, these results demonstrate that INZ and DNA-damaging drugs can synergistically suppress cell survival by inducing p53-dependent apoptosis.
Figure 5.

INZ significantly enhances the proapoptotic effect of CIS on H460 and HCT116 cells. H460 (A) and HCT116p53+/+ (B) were plated in 6-cm-diameter dishes and treated with INZ or/and CIS at the indicated concentrations for 48 hours. Cells were stained with PI and subjected to flow cytometry to determine the DNA content. The apoptotic cells, identified by sub-G1 DNA content, were presented as the M1 population. The data were expressed as means ± SEM (n = 4).
Figure 6.
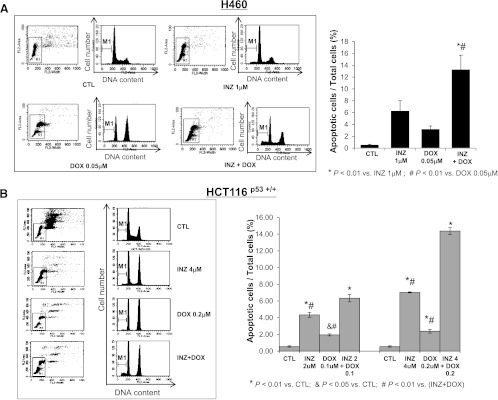
INZ significantly enhances the proapoptotic effect of DOX on H460 and HCT116 cells. H460 (A) and HCT116p53+/+ (B) were plated in 6-cm-diameter dishes and treated with INZ or/and DOX at the indicated concentrations for 48 hours. Apoptotic cells were analyzed as described above inFigure 5. The data were expressed as means ± SEM (n = 4).
INZ and CIS Cooperatively Suppress Xenograft Tumor Growth
The above results show that combination of INZ and chemotherapeutic drugs is considerably effective in suppressing the growth of wild-type p53 containing cancer cells H460 and HCT116. To ranslate the cooperation of these compounds into biologic significance, we further tested if INZ could also synergistically enhance the anticancer effect of CIS by employing a xenograft tumor model system derived from H460 cells. Since our previous study showed that INZ at 30 mg/kg through i.p. injection reduced the HCT116p53+/+ tumor volume by 60% to 70% [16], or at 60 mg/kg reduced the H460 tumor volume by 50% to 60% (unpublished data), here we chose 20 mg/kg INZ for thein vivo study with low dose of CIS (3 mg/kg) as detailed in Materials and Methods section. As indicated inFigure 7, INZ or CIS alone reduced the average tumor volume by 25% to 30% or 45% to 50%, respectively. However, combination of the two compounds at the same doses significantly decreased the final tumor size by 80% to 85%. Furthermore, the final tumor weight also decreased to ∼20% of the control by combined treatment, which displayed statistical significance compared with the other three groups (Figure 7A). This result confirmed that INZ could indeed potentiate CIS-induced growth suppression of H460-derived xenograft tumorsin vivo. Consistent with this result, the two compounds synergistically induced p53 level and activity (Figure 7,B andC) as well as remarkably suppressed cell proliferation and induced apoptosis (Figure 7C). These results demonstrate that INZ and DNA-damaging chemotherapeutic drugs, such as CIS, can cooperatively activate p53 and suppress xenografted tumor growth in animals as well.
Figure 7.
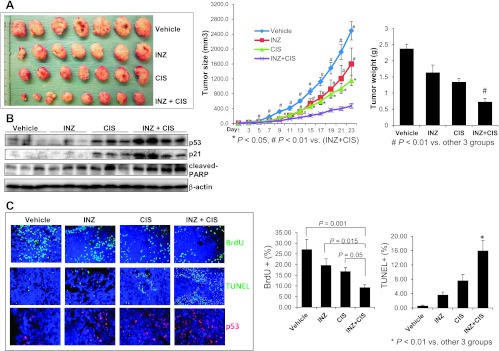
INZ significantly enhances CIS-induced p53 activation, apoptosis, and tumor suppression. (A) Combination of INZ and CIS markedly suppresses the growth of xenograft tumors derived from H460 cells. Mice bearing H460 xenografts were i.p. treated with INZ (20 mg/kg) everyday or/and CIS (3 mg/kg) once per week or vehicles for 21 days. Images of tumors isolated are presented at the left. The tumor growth is shown by the mean tumor volumes ± SEM (middle), and the final tumor weight is shown in columns (right;n = 6 mice per group; *P < .05). (B) WB analysis of proteins extracted from H460 tumor samples in A with antibodies as indicated on the right. (C) Representative images of TUNEL staining, BrdU, and p53 immunostaining of xenograft tumor sections are presented. Quantification of BrdU and TUNEL staining is expressed as mean ± SEM (n = 4 mice per group).
Discussion
Severe side effects and drug resistance are two major obstacles to limit the use of genotoxic agents, such as CIS and DOX, in systemic chemotherapy. Finding novel strategies to overcome the adverse effects and sensitize the response to existing antitumor drugs is an urgent and arduous challenge in the field of cancer chemotherapy and would have a significant clinical impact. In this study, we demonstrate that combination of INZ with low dose of CIS or DOX can synergize their abilities to significantly induce apoptosis and to inhibit growth of human wild-type p53 harboring non-small cell lung cancer cells and colon cancer cells.
Activation of the p53 pathway has been known as one of the central mechanisms for the DNA-damaging agents to induce cell apoptosis and tumor inhibition [50]. Various DNA lesions, such as CIS-caused interstrand DNA cross-linking and DOX-caused DNA double-strand breaks, trigger the activation of ATM and ATR, followed by phosphorylating downstream proteins related to cell growth, proliferation, and survival, among which p53 is the prominent protein to be phosphorylated, stabilized, and activated [51–57]. In addition, phosphorylated ChK1/2 by ATM and ATR further phosphorylates MDMX, leading to the binding between p-MDMX and 14-3-3, and results in the disruption of the interaction between MDMX and p53, causing p53 stabilization and activation [58–61]. Maximizing the activity of p53 by concomitantly targeting different components in this pathway should be an effective approach to impede the growth of tumors harboring functional p53. INZ, as reported previously by our group, is a potent nongenotoxic p53 activator by inhibiting SIRT1-mediated deacetylation [16]. Combination of DNA-damaging agents with INZ would not only induce the phosphorylation of p53 in response to activation of ATM and ATR but also increase the acetylation and weaken the ubiquitination of p53 due to inactivated SIRT1. The sum outcome of this double treatment is, therefore, the more remarkable induction of p53 level and activity. Indeed, as expected, in both p53-containing H460 and HCT116 cells, p53 level and activity (indicated by the p21 level) exhibited a dramatic augment after combined treatment with INZ and CIS or DOX, even at relatively lower doses, compared with either of the single treatment (Figures 1,2, andW1). Our subsequent results further proved the p53-dependent cooperation between INZ and genotoxic drugs, particularly CIS, on cell growth inhibition, which might be attributed to the augment of apoptotic response, in p53 wild-type cells but not in p53-null cells (Figures 3,4,5,6, andW2).
One of the most strenuous challenges for the oncologists in current cancer chemotherapeutic practice is the cumulative and dose-dependent toxicity to normal tissues, such as nephrotoxicity and cardiomyopathy caused by CIS and DOX, respectively. Both CIS and DOX have been supposed to cause side effects by some mechanisms different from the typical DNA lesions that are primarily responsible for their suppression on tumor growth [3,5,62,63]. For instance, CIS may be metabolized to a reactive toxic thiol by some enzymes such as γ-glutamyl transpeptidase whose high level in renal proximal tubular cells sensitizes the cells to CIS toxicity and further target the mitochondria and endoplasmic reticulum of the proximal tubular cells [5,64–66]. Several approaches have been developed against the toxicity, including application of prodrugs and derivatives [67,68], combination with some specific protectors such as antioxidants [69–71], and improvement of drug delivery systems [72–74]. Lowering drug dosage by combination with agents with different mechanisms of action should be a simple, feasible, and effective means to minimize the side effects of DNA-interfering drugs, while retaining the antitumor effects. Again, our results prove that INZ possesses this kind of capability based on its specific p53 induction in tumors and minimal side effects on normal cells and tissues [16], as even much lower doses of CIS or INZ are required for combined therapy to achieve a satisfactory anticancer effect on tumor xenografts with wild-type p53 (Figure 7), but the same combination exerted little synergistic effect on normal cells (Figure W3). Even though more studies are needed to further illuminate the low toxicity of INZ in normal cells and tissues, INZ could be a promising component for adjuvant antitumor therapy.
Another major cause of treatment failure during tumor chemotherapy is drug resistance, which is a complicated multifactorial event. Taking CIS as an example again, the chemoresistance may involve many aspects, including loss of apoptotic response [41,75], overactivation of survival signals such as phosphatidylinositol 3-kinase (PI3K)/Akt [40,42,76–78], increased DNA repair [79–82], induction of multidrug resistance [83,84], increased efflux, and decreased influx of the drug [82]. A number ofin vitro, in vivo, and clinical studies support p53 as an important regulator during most of these processes to mediate the drug sensitivity [40–42,75,76,85–95]. Activation of prosurvival factor Akt [40,76], CBP/p300-interacting transactivator protein CITED2 [96], which is critical for cell growth and oncogenesis [96], oncogenic phosphatase protein PPM1D [41], or protein trafficking related protein NAPA [90] renders cancer cells resistant to chemotherapy, while suppression of their activity sensitizes cells to CIS, which requires the activation of wild-type p53. P53-HDAC2 complex represses the expression of multidrug resistance-related protein lung resistance-related protein (LRP) through binding to the LRP promoter, whereas loss of p53-mediated suppression on LRP may cause the chemoresistance in tumor cells [86]. Down-regulation of p53 in human breast cancer cells increases the CIS resistance mediated by variant, which is one of the factors in charge of DNA repair and telomere maintenance [97]. Restoration or reactivation of wild-type p53 dramatically improved the apoptotic response and sensitivity to CIS in tumor cells [49,85,98–100]. On the basis of these findings, it would be rational to sensitize the chemotherapy by specifically targeting p53, with very low possibility to induce a secondary pathway related to drug resistance. Our study as presented here indeed demonstrates that the specific p53 inducer INZ can sensitize cancer cells, particularly the lung cancer cell line H460 whose chemotherapy is mainly composed of CIS-based regimen, to low dose of CIS, leading to tumor suppression in a p53-dependent manner (Figures 3–7 andW2). Studies on chemoresistant cancer cell lines are necessary to further address the beneficial effects of combination with INZ and chemotherapy on drug sensitivity. Nevertheless, taken together, our current findings suggest that specifically targeting the p53 pathway promotes the sensitivity of cancer cells to traditional chemotherapeutic drugs, and combination of INZ with standard chemotherapy will provide an attractive anticancer protocol for the cancer patients with high sensitivity to side effects and refractory drug resistance in the future.
Supplementary Material
Acknowledgments
We thank the members of the Lu Laboratory for active discussion.
Footnotes
This work was partially supported by National Institutes of Health-National Cancer Institute grants CA127724, CA095441, and CA129828 to H.L.
This article refers to supplementary materials, which are designated byFigures W1 toW3 and are available online atwww.neoplasia.com.
References
- 1.Dy GK, Adjei AA. Systemic cancer therapy: evolution over the last 60 years. Cancer. 2008;113:1857–1887. doi: 10.1002/cncr.23651. [DOI] [PubMed] [Google Scholar]
- 2.Boulikas T, Vougiouka M. Cisplatin and platinum drugs at the molecular level (Review) Oncol Rep. 2003;10:1663–1682. [PubMed] [Google Scholar]
- 3.Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol. 2003;93:105–115. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 4.Gonzales-Vitale JC, Hayes DM, Cvitkovic E, Sternberg SS. The renal pathology in clinical trials of cis-platinum (II) diamminedichloride. Cancer. 1977;39:1362–1371. doi: 10.1002/1097-0142(197704)39:4<1362::aid-cncr2820390403>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 5.Hanigan MH, Devarajan P. Cisplatin nephrotoxicity: molecular mechanisms. Cancer Ther. 2003;1:47–61. [PMC free article] [PubMed] [Google Scholar]
- 6.Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE, Sanders SP. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;324:808–815. doi: 10.1056/NEJM199103213241205. [DOI] [PubMed] [Google Scholar]
- 7.Zhang YW, Shi J, Li YJ, Wei L. Cardiomyocyte death in doxorubicininduced cardiotoxicity. Arch Immunol Ther Exp (Warsz) 2009;57:435–445. doi: 10.1007/s00005-009-0051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 9.Baird RD, Kaye SB. Drug resistance reversal—are we getting closer? Eur J Cancer. 2003;39:2450–2461. doi: 10.1016/s0959-8049(03)00619-1. [DOI] [PubMed] [Google Scholar]
- 10.Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2:a000893. doi: 10.1101/cshperspect.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boominathan L. The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network. Cancer Metastasis Rev. 2010;29:613–639. doi: 10.1007/s10555-010-9257-9. [DOI] [PubMed] [Google Scholar]
- 13.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 Mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 14.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 15.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q, Zeng SX, Zhang Y, Zhang Y, Ding D, Ye Q, Meroueh SO, Lu H. A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol Med. 2012;4:298–312. doi: 10.1002/emmm.201100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. Themdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 18.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 19.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299–309. doi: 10.1016/j.tcb.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. RING domain-mediated interaction is a requirement for MDM2's E3 ligase activity. Cancer Res. 2007;67:6026–6030. doi: 10.1158/0008-5472.CAN-07-1313. [DOI] [PubMed] [Google Scholar]
- 22.Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15:841–848. doi: 10.1038/sj.cdd.4402309. [DOI] [PubMed] [Google Scholar]
- 23.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 aut-oregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 24.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 27.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–1328. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 28.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, Safiran YJ, Oberoi P, Kenten JH, Phillips AC, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7:547–559. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 30.Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F, Mills N, Smithson DC, Regni CA, Bashford D, et al. Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem. 2010;285:10786–10796. doi: 10.1074/jbc.M109.056747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobet E, Zeng X, Zhu Y, Keller D, Lu H. MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc Natl Acad Sci USA. 2000;97:12547–12552. doi: 10.1073/pnas.97.23.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001;20:1331–1340. doi: 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 34.Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem. 2002;277:50607–50611. doi: 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- 35.Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, Nagy TR. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 37.Jung-Hynes B, Nihal M, Zhong W, Ahmad N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J Biol Chem. 2009;284:3823–3832. doi: 10.1074/jbc.M807869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNAdamage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 39.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, et al. Discovery,in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser M, Bai T, Tsang BK. Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int J Cancer. 2008;122:534–546. doi: 10.1002/ijc.23086. [DOI] [PubMed] [Google Scholar]
- 41.Ali AY, Abedini MR, Tsang BK. The oncogenic phosphatase PPM1D confers cisplatin resistance in ovarian carcinoma cells by attenuating checkpoint kinase 1 and p53 activation. Oncogene. 2012;31:2175–2186. doi: 10.1038/onc.2011.399. [DOI] [PubMed] [Google Scholar]
- 42.Fraser M, Leung BM, Yan X, Dan HC, Cheng JQ, Tsang BK. p53 is a determinant of X-linked inhibitor of apoptosis protein/Akt-mediated chemoresistance in human ovarian cancer cells. Cancer Res. 2003;63:7081–7088. [PubMed] [Google Scholar]
- 43.Song K, Cowan KH, Sinha BK. In vivo studies of adenovirusmediated p53 gene therapy for cis-platinum-resistant human ovarian tumor xenografts. Oncol Res. 1999;11:153–159. [PubMed] [Google Scholar]
- 44.Zhang Y, Zhang Q, Zeng SX, Mayo LD, Lu H. Inauhzin and Nutlin3 synergistically activate p53 and suppress tumor growth. Cancer Biol Ther. 2012;13:915–924. doi: 10.4161/cbt.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 46.Jafri SH, Glass J, Shi R, Zhang S, Prince M, Kleiner-Hancock H. Thymoquinone and cisplatin as a therapeutic combination in lung cancer:in vitro andin vivo. J Exp Clin Cancer Res. 2010;29:87. doi: 10.1186/1756-9966-29-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter CA, Chen C, Brink C, Vincent P, Maxuitenko YY, Gilbert KS, Waud WR, Zhang X. Sorafenib is efficacious and tolerated in combination with cytotoxic or cytostatic agents in preclinical models of human non-small cell lung carcinoma. Cancer Chemother Pharmacol. 2007;59:183–195. doi: 10.1007/s00280-006-0257-y. [DOI] [PubMed] [Google Scholar]
- 48.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 49.Gutekunst M, Oren M, Weilbacher A, Dengler MA, Markwardt C, Thomale J, Aulitzky WE, van der Kuip H. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS One. 2011;6:e19198. doi: 10.1371/journal.pone.0019198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 51.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 52.Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–7779. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 53.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 54.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 56.Hurley PJ, Bunz F. ATM and ATR: components of an integrated circuit. Cell Cycle. 2007;6:414–417. doi: 10.4161/cc.6.4.3886. [DOI] [PubMed] [Google Scholar]
- 57.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee JH, Jin Y, He G, Zeng SX, Wang YV, Wahl GM, Lu H. Hypoxia activates tumor suppressor p53 by inducing ATR-Chk1 kinase cascade-mediated phosphorylation and consequent 14-3-3γ inactivation of MDMX protein. J Biol Chem. 2012;287:20898–20903. doi: 10.1074/jbc.M111.336875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin Y, Dai MS, Lu SZ, Xu Y, Luo Z, Zhao Y, Lu H. 14-3-3γ binds to MDMX that is phosphorylated by UV-activated Chk1, resulting in p53 activation. EMBO J. 2006;25:1207–1218. doi: 10.1038/sj.emboj.7601010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.LeBron C, Chen L, Gilkes DM, Chen J. Regulation of MDMX nuclear import and degradation by Chk2 and 14-3-3. EMBO J. 2006;25:1196–1206. doi: 10.1038/sj.emboj.7601032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005;24:3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iarussi D, Indolfi P, Casale F, Coppolino P, Tedesco MA, Di Tullio MT. Recent advances in the prevention of anthracycline cardiotoxicity in childhood. Curr Med Chem. 2001;8:1649–1660. doi: 10.2174/0929867013371888. [DOI] [PubMed] [Google Scholar]
- 63.Neilan TG, Blake SL, Ichinose F, Raher MJ, Buys ES, Jassal DS, Furutani E, Perez-Sanz TM, Graveline A, Janssens SP, et al. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation. 2007;116:506–514. doi: 10.1161/CIRCULATIONAHA.106.652339. [DOI] [PubMed] [Google Scholar]
- 64.Hanigan MH, Gallagher BC, Townsend DM, Gabarra V. γ-Glutamyl transpeptidase accelerates tumor growth and increases the resistance of tumors to cisplatinin vivo. Carcinogenesis. 1999;20:553–559. doi: 10.1093/carcin/20.4.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hanigan MH, Lykissa ED, Townsend DM, Ou CN, Barrios R, Lieberman MW. γ-glutamyl transpeptidase-deficient mice are resistant to the nephrotoxic effects of cisplatin. Am J Pathol. 2001;159:1889–1894. doi: 10.1016/s0002-9440(10)63035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wainford RD, Weaver RJ, Stewart KN, Brown P, Hawksworth GM. Cisplatin nephrotoxicity is mediated by gamma glutamyltranspeptidase, not via a C-S lyase governed biotransformation pathway. Toxicology. 2008;249:184–193. doi: 10.1016/j.tox.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 67.Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapyin vivo. Proc Natl Acad Sci USA. 2011;108:1850–1855. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Kratz F, Walker UA. The 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH) is superior to free doxorubicin with respect to cardiotoxicity and mitochondrial damage. Int J Cancer. 2007;120:927–934. doi: 10.1002/ijc.22409. [DOI] [PubMed] [Google Scholar]
- 69.Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Walker UA. Dexrazoxane prevents doxorubicin-induced long-term cardiotoxicity and protects myocardial mitochondria from genetic and functional lesions in rats. Br J Pharmacol. 2007;151:771–778. doi: 10.1038/sj.bjp.0707294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hensley ML, Hagerty KL, Kewalramani T, Green DM, Meropol NJ, Wasserman TH, Cohen GI, Emami B, Gradishar WJ, Mitchell RB, et al. American Society of Clinical Oncology 2008 clinical practice guideline update: use of chemotherapy and radiation therapy protectants. J Clin Oncol. 2009;27:127–145. doi: 10.1200/JCO.2008.17.2627. [DOI] [PubMed] [Google Scholar]
- 71.Asna N, Lewy H, Ashkenazi IE, Deutsch V, Peretz H, Inbar M, Ron IG. Time dependent protection of amifostine from renal and hematopoietic cisplatin induced toxicity. Life Sci. 2005;76:1825–1834. doi: 10.1016/j.lfs.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 72.Kuang Y, Liu J, Liu Z, Zhuo R. Cholesterol-based anionic longcirculating cisplatin liposomes with reduced renal toxicity. Biomaterials. 2012;33:1596–1606. doi: 10.1016/j.biomaterials.2011.10.081. [DOI] [PubMed] [Google Scholar]
- 73.Rigacci L, Mappa S, Nassi L, Alterini R, Carrai V, Bernardi F, Bosi A. Liposome-encapsulated doxorubicin in combination with cyclophosphamide, vincristine, prednisone and rituximab in patients with lymphoma and concurrent cardiac diseases or pre-treated with anthracyclines. Hematol Oncol. 2007;25:198–203. doi: 10.1002/hon.827. [DOI] [PubMed] [Google Scholar]
- 74.Yildirim Y, Gultekin E, Avci ME, Inal MM, Yunus S, Tinar S. Cardiac safety profile of pegylated liposomal doxorubicin reaching or exceeding lifetime cumulative doses of 550 mg/m2 in patients with recurrent ovarian and peritoneal cancer. Int J Gynecol Cancer. 2008;18:223–227. doi: 10.1111/j.1525-1438.2007.00992.x. [DOI] [PubMed] [Google Scholar]
- 75.Sato S, Kigawa J, Minagawa Y, Okada M, Shimada M, Takahashi M, Kamazawa S, Terakawa N. Chemosensitivity and p53-dependent apoptosis in epithelial ovarian carcinoma. Cancer. 1999;86:1307–1313. [PubMed] [Google Scholar]
- 76.Yang X, Fraser M, Moll UM, Basak A, Tsang BK. Akt-mediated cisplatin resistance in ovarian cancer: modulation of p53 action on caspasedependent mitochondrial death pathway. Cancer Res. 2006;66:3126–3136. doi: 10.1158/0008-5472.CAN-05-0425. [DOI] [PubMed] [Google Scholar]
- 77.Lee S, Choi EJ, Jin C, Kim DH. Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol. 2005;97:26–34. doi: 10.1016/j.ygyno.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 78.Riedel RF, Porrello A, Pontzer E, Chenette EJ, Hsu DS, Balakumaran B, Potti A, Nevins J, Febbo PG. A genomic approach to identify molecular pathways associated with chemotherapy resistance. Mol Cancer Ther. 2008;7:3141–3149. doi: 10.1158/1535-7163.MCT-08-0642. [DOI] [PubMed] [Google Scholar]
- 79.Barakat K, Gajewski M, Tuszynski JA. DNA repair inhibitors: the next major step to improve cancer therapy. Curr Top Med Chem. 2012);12:1376–1390. doi: 10.2174/156802612801319070. [DOI] [PubMed] [Google Scholar]
- 80.Pan Y, Zhang Q, Atsaves V, Yang H, Claret FX. Suppression of Jab1/CSN5 induces radio- and chemo-sensitivity in nasopharyngeal carcinoma through changes to the DNA damage and repair pathways. Oncogene. 2012:1–11. doi: 10.1038/onc2012.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wilk A, Waligorska A, Waligorski P, Ochoa A, Reiss K. Inhibition of ERβ induces resistance to cisplatin by enhancing Rad51-mediated DNA repair in human medulloblastoma cell lines. PLoS One. 2012;7:e33867. doi: 10.1371/journal.pone.0033867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chu G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J Biol Chem. 1994;269:787–790. [PubMed] [Google Scholar]
- 83.Itoh Y, Tamai M, Yokogawa K, Nomura M, Moritani S, Suzuki H, Sugiyama Y, Miyamoto K. Involvement of multidrug resistance-associated protein 2 inin vivo cisplatin resistance of rat hepatoma AH66 cells. Anticancer Res. 2002;22:1649–1653. [PubMed] [Google Scholar]
- 84.To KK, Yu L, Liu S, Fu J, Cho CH. Constitutive AhR activation leads to concomitant ABCG2-mediated multidrug resistance in cisplatin-resistant esophageal carcinoma cells. Mol Carcinog. 2012;51:449–464. doi: 10.1002/mc.20810. [DOI] [PubMed] [Google Scholar]
- 85.Guntur VP, Waldrep JC, Guo JJ, Selting K, Dhand R. Increasing p53 protein sensitizes non-small cell lung cancer to paclitaxel and cisplatinin vitro. Anticancer Res. 2010;30:3557–3564. [PubMed] [Google Scholar]
- 86.Tian B, Liu J, Liu B, Dong Y, Song Y, Sun Z. p53 Suppresses lung resistance-related protein expression through Y-box binding protein 1 in the MCF-7 breast tumor cell line. J Cell Physiol. 2011;226:3433–3441. doi: 10.1002/jcp.22700. [DOI] [PubMed] [Google Scholar]
- 87.Han JY, Lee GK, Jang DH, Lee SY, Lee JS. Association of p53 codon 72 polymorphism and MDM2 SNP309 with clinical outcome of advanced nonsmall cell lung cancer. Cancer. 2008;113:799–807. doi: 10.1002/cncr.23668. [DOI] [PubMed] [Google Scholar]
- 88.Ikuta K, Takemura K, Kihara M, Naito S, Lee E, Shimizu E, Yamauchi A. Defects in apoptotic signal transduction in cisplatin-resistant non-small cell lung cancer cells. Oncol Rep. 2005;13:1229–1234. [PubMed] [Google Scholar]
- 89.Kandioler D, Stamatis G, Eberhardt W, Kappel S, Zochbauer-Muller S, Kuhrer I, Mittlbock M, Zwrtek R, Aigner C, Bichler C, et al. Growing clinical evidence for the interaction of the p53 genotype and response to induction chemotherapy in advanced non-small cell lung cancer. J Thorac Cardiovasc Surg. 2008;135:1036–1041. doi: 10.1016/j.jtcvs.2007.10.072. [DOI] [PubMed] [Google Scholar]
- 90.Wu ZZ, Chao CC. Knockdown of NAPA using short-hairpin RNA sensitizes cancer cells to cisplatin: implications to overcome chemoresistance. Biochem Pharmacol. 2010;80:827–837. doi: 10.1016/j.bcp.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 91.Hovelmann S, Beckers TL, Schmidt M. Molecular alterations in apoptotic pathways after PKB/Akt-mediated chemoresistance in NCI H460 cells. Br J Cancer. 2004;90:2370–2377. doi: 10.1038/sj.bjc.6601876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu ZZ, Sun NK, Chien KY, Chao CC. Silencing of the SNARE protein NAPA sensitizes cancer cells to cisplatin by inducing ERK1/2 signaling, synoviolin ubiquitination and p53 accumulation. Biochem Pharmacol. 2011;82:1630–1640. doi: 10.1016/j.bcp.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 93.Lin X, Howell SB. DNA mismatch repair and p53 function are major determinants of the rate of development of cisplatin resistance. Mol Cancer Ther. 2006;5:1239–1247. doi: 10.1158/1535-7163.MCT-05-0491. [DOI] [PubMed] [Google Scholar]
- 94.Hayashi S, Ozaki T, Yoshida K, Hosoda M, Todo S, Akiyama S, Nakagawara A. p73 and MDM2 confer the resistance of epidermoid carcinoma to cisplatin by blocking p53. Biochem Biophys Res Commun. 2006;347:60–66. doi: 10.1016/j.bbrc.2006.06.095. [DOI] [PubMed] [Google Scholar]
- 95.Bauer JA, Kumar B, Cordell KG, Prince ME, Tran HH, Wolf GT, Chepeha DB, Teknos TN, Wang S, Eisbruch A, et al. Targeting apoptosis to overcome cisplatin resistance: a translational study in head and neck cancer. Int J Radiat Oncol Biol Phys. 2007;69:S106–S108. doi: 10.1016/j.ijrobp.2007.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu ZZ, Sun NK, Chao CC. Knockdown of CITED2 using short-hairpin RNA sensitizes cancer cells to cisplatin through stabilization of p53 and enhancement of p53-dependent apoptosis. J Cell Physiol. 2011;226:2415–2428. doi: 10.1002/jcp.22589. [DOI] [PubMed] [Google Scholar]
- 97.Nadkarni A, Rajesh P, Ruch RJ, Pittman DL. Cisplatin resistance conferred by the RAD51D (E233G) genetic variant is dependent upon p53 status in human breast carcinoma cell lines. Mol Carcinog. 2009;48:586–591. doi: 10.1002/mc.20545. [DOI] [PubMed] [Google Scholar]
- 98.Lax SA, Chia MC, Busson P, Klamut HJ, Liu FF. Adenovirus-p53 gene therapy in human nasopharyngeal carcinoma xenografts. Radiother Oncol. 2001;61:309–312. doi: 10.1016/s0167-8140(01)00398-x. [DOI] [PubMed] [Google Scholar]
- 99.Roh JL, Ko JH, Moon SJ, Ryu CH, Choi JY, Koch WM. The p53-reactivating small-molecule RITA enhances cisplatin-induced cytotoxicity and apoptosis in head and neck cancer. Cancer Lett. 2012;325:35–41. doi: 10.1016/j.canlet.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 100.Li Q, Kawamura K, Yamanaka M, Okamoto S, Yang S, Yamauchi S, Fukamachi T, Kobayashi H, Tada Y, Takiguchi Y, et al. Upregulated p53 expression activates apoptotic pathways in wild-type p53-bearing mesothelioma and enhances cytotoxicity of cisplatin and pemetrexed. Cancer Gene Ther. 2012;19:218–228. doi: 10.1038/cgt.2011.86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.