

Abstract
The familial nature of major depressive disorder (MDD) is now well recognized. We followed children and grandchildren of probands with and without MDD to examine transmission of depression over generations, and to identify early vulnerability markers prior to the onset of disease. The study now includes three generations and five completed assessment waves spanning 25 years, with a sixth wave underway. Beginning with the fourth wave, we collected measures of brain structure (magnetic resonance imaging, MRI) and physiology (electroencephalography, EEG) and DNA in order to examine at a biological level why the offspring of depressed parents were at higher risk. In this paper, we provide an overview of the study design, the main findings, including new data, and the role of the high-risk design in translational research. We demonstrate that offspring of depressed parents (‘high-risk’), when compared with those of non-depressed parents (‘low-risk’), were at increased risk for depressive and anxiety disorders, with anxiety appearing earlier and being a predisposing factor for MDD. Offspring with two generations previously affected were at greatest risk. Thinning of the cortical mantle (MRI) and reduced resting-state activity (EEG) within the right parieto-temporal hemisphere differentiated high- from low-risk offspring, regardless of whether the offspring had MDD, suggesting that these measures might serve as familial trait markers for depression and related syndromes. The high- and low-risk offspring also differed by serotonin transporter promoter length polymorphism genotypes, even though the same genotypes were not associated with the presence of MDD. The high-risk epidemiological design appears to be a particularly valuable asset in translational research as it allows targeting of biological processes that emerge prior to the onset of disease, and identifies individuals at high risk for the disorder who may carry the trait or marker but not yet be affected.
Keywords: major depressive disorder, high-risk, electroencephalography, imaging, genetics, endophenotype
1. Background: the familial nature of depression
Although the familial nature and early onset of major depression are well recognized, it is only recently that systematic data have been available. Until the 1970s, there were no epidemiological studies of psychiatric disorders in the USA, and very few studies conducted abroad. Little was known about prevalence, age of onset and populations at risk, and conventional wisdom held that major depression was a disorder primarily found in middle-aged and menopausal women and did not occur in children [1]. The 1980s introduced a revolution in psychiatric epidemiology, with the first large-scale community-based surveys [2–4] undertaken to determine rates and risks of psychiatric disorders within the community. These surveys used the same diagnostic criteria as did clinical research and practice, enabling the ensuing data on rates, age of onset and comorbidity to be more readily translatable into clinical research and health policy. Subsequent studies have extended these efforts to include large adult samples, minorities, and children and adolescents [5–7].
Numerous studies have since followed, confirming that major depressive disorder (MDD) runs in families [8], and that this familial aggregation is likely to have a genetic component [9]. Early onset MDD beginning in childhood and adolescence is the most familial form [10–16], with anxiety disorders often appearing earlier and predisposing an individual to later depression [17–21]. These studies however did not go beyond two generations, did not examine both parents and, with one exception [16], were not longitudinal. Conclusions about the sequence of disorder, early signs, specificity and stability of transmission were thus limited.
2. Three-generation family study
The familial nature of MDD and its early onset made the high-risk design, which studies offspring at risk for illness but prior to onset of disease, a particularly valuable approach. One of us (M.M.W.) initiated a longitudinal family study of depression in 1982 to study the patterns of transmission of MDD within generations of families. The project began with simultaneous recruitment of two sets of probands. The first, selected from outpatient specialty clinics in the New Haven, CT, area, had moderate to severe major depression with impairment. The second group, selected concurrently and from the same community, was required to have no lifetime history of psychiatric illness as determined through multiple interviews. Both proband groups (Generation 1) were followed prospectively over time, along with their biological children (Generation 2) and, subsequently, grandchildren (Generation 3). The offspring of the depressed probands formed the high-risk group, by virtue of their familial loading for depression, and those of the non-depressed probands formed the low-risk group. The overall study design is illustrated in figure 1 and further details can be found in numerous previous publications [22,23]. These families have now been followed through five completed assessment waves (baseline, 2, 10, 20 and 25 years), with a sixth wave currently underway (figure 2). The second generation has been followed for the longest time (more than 20 years) and has now passed the age of highest risk for most psychopathology, including MDD, thus enabling the study of the long-term trajectories that follow onset.
Figure 1.

Study design. The figure illustrates pedigree models from the (a) high- and (b) low-risk families. In the high-risk families, the index proband was required to have a diagnosis of MDD (filled in shapes) with impairment; in the low-risk families, the index proband, as well as their spouse, had no lifetime history of major depression (open shapes). No requirements were imposed on the children (Generation 2) or grandchildren (Generation 3) of these probands.
Figure 2.

Study waves and assessments. The chart illustrates the overall design of the study, and included six assessment waves over 30 years (columns) and three generations (rows). The original probands (Generation 1) were selected either from specialty clinics for the treatment of mood disorders (depressed probands) or from the community in the same geographical area (non-depressed probands). These probands were then followed longitudinally over time, along with their children (Generation 2) and, beginning with the third wave, grandchildren (Generation 3). Clinical assessment procedures, in blue, included a full diagnostic interview based on the age-appropriate version of the schedule for affective disorders and schizophrenia, as well as a battery of clinical, behavioural and family environmental assessments, as outlined in the text. Beginning with the fourth wave, biological measures (shown in green) were introduced into the study. Electroencephalographic recordings were included in the fourth, and structural and functional MRI assessments and DNA collection, in the fifth, waves. In the sixth wave of the study, which is ongoing, all three measures are being recollected.
The clinical findings from this study document a clear transmission of MDD across generations. Specifically, offspring of depressed parents were at increased relative risk for depression, anxiety and substance use disorders [23,24] (figure 3). The anxiety disorders began before puberty, with mood disorders emerging around puberty, especially in girls [23]. The fear-based anxiety disorders including panic disorder and the phobias further mediated the relationship between parental and offspring depression, whereas other anxiety disorders did not [25]. Parental alcohol abuse and earlier onset of MDD further added to the increased the risk for MDD in the offspring [26]. As the offspring aged, there was also a greater incidence of medical problems, particularly cardiovascular, in the high-risk group [23]. Finally, as the third generation aged in to the study, similar patterns were found. Parental depression severity increased the likelihood of a mood disorder in the offspring, and offspring from families with two generations—i.e. both a parent and a grandparent—previously affected were at greatest risk for psychopathology [22] (figure 4).
Figure 3.
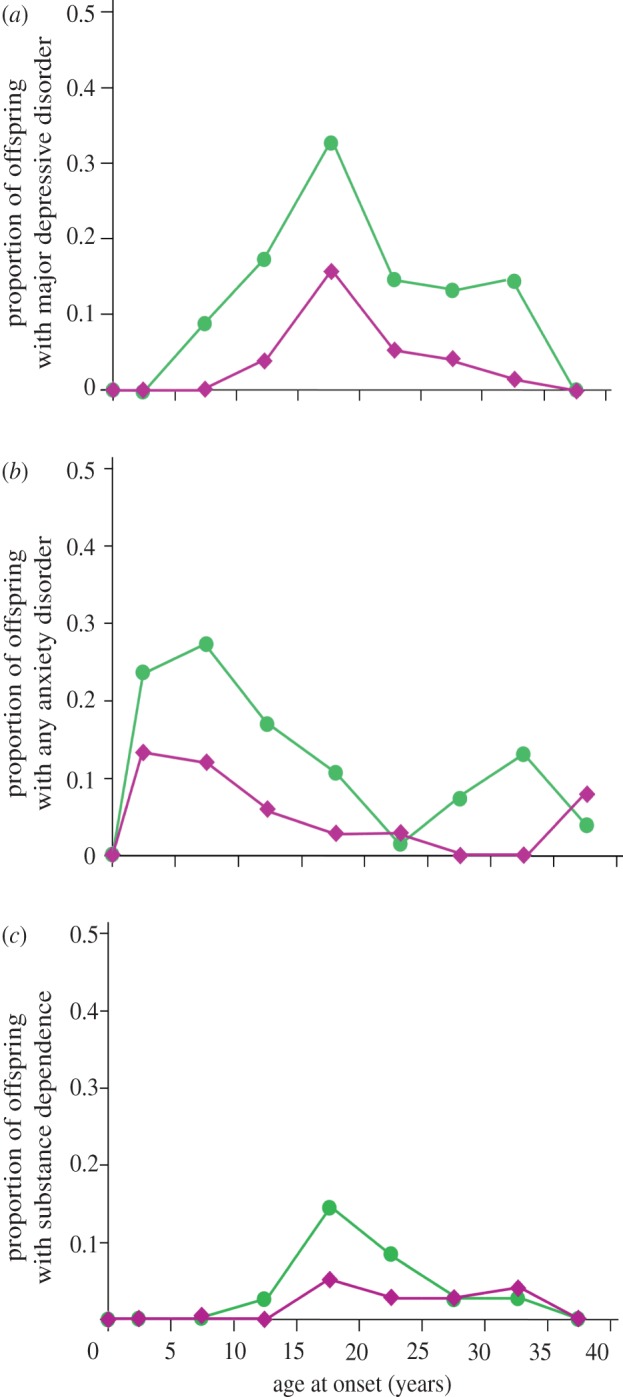
Psychopathology among second-generation (G2) offspring of depressed and non-depressed probands. (a) MDD, (b) any anxiety disorder and (c) substance dependence. Age-specific rates of MDD, anxiety disorders and substance dependence, over 20 years in second-generation offspring of depressed (green, one or more parents with major depressive disorder (n = 101)) and non-depressed (pink, no parents with major depressive disorder (n = 50)) parents. For each outcome, offspring of depressed parents had higher incidence, and onset of major depression followed that of anxiety disorders. Adapted with permission from Weissman et al. [23].
Figure 4.

Psychopathology among third-generation (G3) offspring of depressed and non-depressed probands. Rates of psychopathology among G3 offspring, based on whether they had one or more parent (G2), and one or more grandparent (G1) with major depression. As seen in the right-most column, offspring with both a depressed parent and grandparent had the highest rates of psychopathology, with 40% having any DSM-IV diagnosis.
Given the consistent patterns across waves and generations, we were interested in further identifying biological markers that may help explain why the offspring of depressed parents are at high risk. Biological markers (also referred to interchangeably as endophenotypes or intermediary phenotypes) not only help target aetiology and mechanisms, but can also help identify persons who are at increased risk, so that they can be targeted for appropriate intervention. The markers can also be used to strengthen classifications of clinical phenotypes, or to differentiate possible biological subtypes that may in turn have different clinical or treatment profiles. A majority of studies thus far however have targeted markers by comparing depressed with non-depressed, or sometimes non-ill, subjects. What these comparisons yield, however, are correlates of illness measured at an end state. A true endophenotype, however, should not be simply a disease correlate. The more rigorous definition requires it to lie in the causal pathway to disease (that is, it should not arise as a result of the disease); to be heritable; and to be state-independent (that is, it should be observed in subjects who are at risk but not currently symptomatic) [27]. Because the high-risk design (unlike case–control studies) conditions subjects based on their risk for rather than presence of the outcome of interest, it is particularly well suited to meeting these requirements.
We successively incorporated three biological protocols into our study, based on electrophysiological brain recordings (electroencephalography, EEG), structural and functional brain imaging (MRI) and genetics (DNA). EEG measurements were initially recorded in the fourth wave, and MRI and DNA collections in the fifth wave, as illustrated in figure 2 (green boxes). EEG and MRI recordings are now being re-administered in the sixth wave, allowing us not only to assess stability over time, but also to prospectively capture changes in these measures that may correspond to development of psychopathology. We illustrate these approaches in the sections that follow. We do not elaborate on methodological aspects, as these have largely been covered elsewhere [28–32], and are not our primary focus. Instead, we emphasize the translational elements of these studies, demonstrating how applying biological measures to the high-risk design can provide unique opportunities to examine risk factors for disease.
3. Electroencephalography
There is a convergence of evidence that EEG measures of brain asymmetry may provide neurophysiological markers of vulnerability to internalizing psychopathology. EEG studies have reported both higher overall alpha activity1 among depressed patients, when compared with controls [33–35], as well as alpha asymmetry among depressed adults [36–39] and infants of depressed mothers [40,41]. Studies have found depressed states to correspond with relative increases in right-hemispheric activity in the frontal cortex, and relative decreases in posterior sites [42–45]. Although the biology remains to be elucidated, these dual effects have been interpreted in terms of a two-dimensional model of emotions wherein a ‘valence’ dimension involves frontal systems, with pleasant or positive emotions associated with left frontal activity, and unpleasant or negative emotions associated with right frontal activity, and an ‘arousal’ dimension involving the right posterior system [46]. Depression, if applied to this framework, might be characterized by relatively greater right than left frontal activity associated with unpleasant affect and decreased right parieto-temporal activity associated with low emotional arousal.
We measured resting EEG in second- and third-generation offspring in order to evaluate a potential role for EEG alpha asymmetry—that is, the difference in recorded activity across the two hemispheres—as a biological marker of a phenotype of depression. We hypothesized that alpha asymmetry would vary as a function of familial loading of MDD. Specifically, offspring of depressed parents (i.e. the high-risk group) would show relatively greater right than left activity in frontal regions, and greater left than right activity in posterior regions, when compared with the offspring of non-depressed parents. We first examined offspring in the second generation (Gen 2), comparing those who had two parents with MDD with those who had one parent or neither parent with MDD. Offspring with both parents affected by MDD showed greater alpha asymmetry at medial sites (figure 5a) [28], compared with offspring having one or no parent with MDD. This asymmetry resulted from relatively less activity (i.e. greater alpha) over right central and parietal regions. Offspring having two parents with MDD also demonstrated the greatest anterior-to-posterior increase in alpha with eyes closed of the three groups. For third generation, because of the two preceding generations, we compared subjects who had both a parent and a grandparent with MDD with those with either a parent or grandparent, or neither. Consistent with the patterns from the previous generation, Generation 3 offspring with two generations of loading for depression demonstrated greater alpha asymmetry, with relatively less right- than left-hemisphere activity, compared to those with neither a depressed parent nor a depressed grandparent [29] (figure 5b). This difference was present in the parietal region only in the resting (eyes-closed) state, and not in the frontal regions for any condition.
Figure 5.
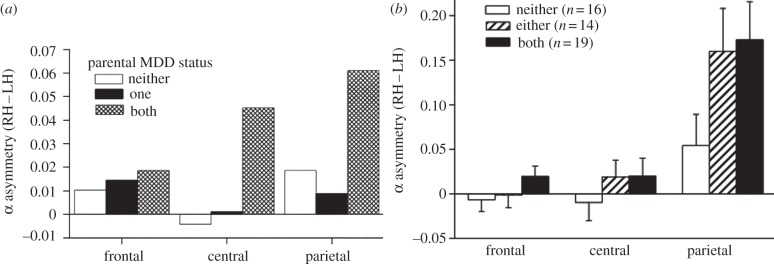
EEG alpha asymmetry among second- and third-generation offspring of depressed and non-depressed probands. (a) Second-generation offspring (G2). Adapted with permission from Bruder et al. [28]. (b) Third-generation offspring (G3). Adapted with permission from Bruder et al. [29]. Graphs illustrate mean alpha asymmetry (that is, difference between the right-hemispheric and left-hemispheric log alpha power) at frontal, central and parietal sites during eyes-closed conditions. Among G2 (a), offspring who had two depressed parents had significantly reduced activity in right-hemispheric central and parietal regions when compared with offspring with either one or no depressed parents. Among G3 (b), grandchildren with two preceding generations with MDD (i.e. a parent and a grandparent) as well as those with only one preceding generation (i.e. a parent or a grandparent but not both) had significantly reduced activity in right-hemispheric parietal regions than offspring with no history of depression in either preceding generation.
4. Magnetic resonance imaging
Although a number of previous imaging studies have implicated frontal and limbic dysfunction in MDD, findings have been difficult to interpret [47], partly because the studies compared depressed subjects with non-depressed or non-ill subjects cross-sectionally. It is therefore difficult to interpret identified abnormalities in brain structure or function as cause of the illness, a compensatory process resulting from the illness, or an epiphenomenon [48]. The high-risk design targets biological vulnerabilities prior to the onset of illness, and thus enables the identification of brain-based phenotypes that are more likely to represent true causal processes.
Beginning with the fifth wave of the study, we imaged 131 subjects from the second and third generation using a 1.5 T MRI scanner, and compared the measures of thickness of the cortical mantle between the brains of offspring from the high- and low-risk families. The cortical mantle is approximately a 6 mm layer of grey matter that forms the surface of the brain. It contains the greatest concentration of neurons and it is here where the most computation functions of the brain are performed [32]. As illustrated in figure 6, we detected large expanses of cortical thinning across the lateral surface of the right cerebral hemisphere in persons at high risk for depression. The thinning was very substantial, averaging 30 per cent reduction in thickness. As observed with the patterns of EEG alpha asymmetry, the thinning was present in high-risk individuals who were neither affected by MDD nor an anxiety disorder in their lifetimes. We therefore surmised that the thinning cannot simply be a consequence of being depressed, or of being treated for depression, and more likely reflects some familial trait vulnerability for the development of depression-related clinical phenotypes. There was some thickening observed in the medial wall of the right cingulate cortex among depressed offspring. In a follow-up analysis, further white matter reductions, particularly in the superior longitudinal fasciculus tract that connects the frontal to parietal regions, was found in high-risk subjects [32]. Cortical mantle thinning and white matter hypoplasia were highly correlated, suggesting that both may arise from a common underlying disturbance. Cortical thinning was also correlated with multiple measures of current (but not lifetime) symptom severity, inattention (staying focused on tasks, filtering out non-essential stimuli) and visual memory for social stimuli (face identification), and mediated the associations of familial risk with these measures. Finally, among offspring who developed the illness, there was additional cortical thinning observed in homologous left-hemisphere parietal regions, suggesting that while right-hemispheric abnormalities may predispose to MDD, additional left-hemisphere assault is required to produce active psychopathology. Whilst intriguing, these findings require replication in an independent sample.
Figure 6.
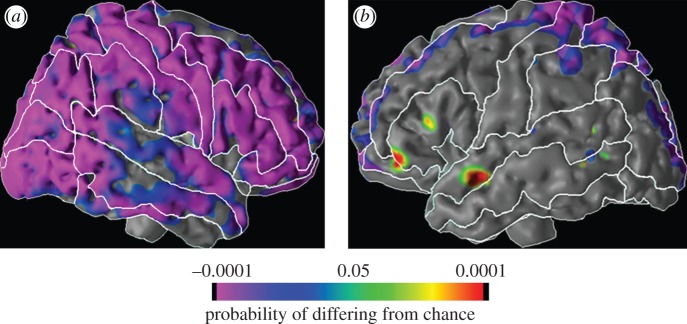
Morphological differences among second- and third-generation offspring of depressed and non-depressed probands (n = 131). The figure illustrates the surface of the (a) right and (b) left hemispheres of the cerebral cortex. At each point on the cerebral surface, the colours overlaid represent the statistical significance of differences in cortical thickness across the offspring of high- and low-risk groups. As illustrated in the colour bar, orange represents thickening of the cortical mantle in the high, when compared with low-risk group; purple represents the reverse. As illustrated above, G2 and G3 offspring who were descended from the G1 probands with major depression had substantial (greater than 30%) and non-specific cortical thinning when compared with offspring from non-depressed probands. This thinning was predominantly in the right cortical hemisphere, and was also observed among offspring with no lifetime history of any anxiety or depressive disorder, making these morphological changes unlikely to be a result of illness. Further details can be found in the study of Peterson et al. [31].
Given the co-localization of alpha asymmetry and cortical mantle thinning in the right parieto-temporal cortices, we sought to quantify how closely the two measures were coupled, and whether having a thinner cortical mantle would translate directly into less cortical activity in these regions. Conjoining data across multiple modalities serves several functions. First, observing converging effects using two methodologically distinct tools enhances validity and minimizes the chance of method-related bias; secondly, the approaches are complementary in terms of resolution: combining the superior temporal resolution of EEG with the spatial resolution of MRI can provide greater precision. And finally, it allows us to examine the extent to which markers identified by each method serve as proxies for each other. If brain-based endophenotypes make their way into clinical practice, EEG would provide a significantly cheaper and more accessible diagnostic tool than MRI, as long as it is demonstrated that these methods tap into the same biological substrates.
We thus overlaid EEG recordings onto a template brain and compared alpha power with the thickness of the cortical mantle, on a voxel-by-voxel basis across the entire brain [30]. This was done in the subset of 75 subjects, each of whom had both usable MRI and EEG data. We found expanses of significant correlations between alpha asymmetry and cortical thickness in both the high- and low-risk groups. The correlations were strongest in medial parietal sites, where alpha asymmetry was greatest. We did not find sufficient evidence that cortical thinning mediated the association between familial risk for depression and alpha asymmetry, suggesting that the structural and functional abnormalities may not lie along entirely overlapping pathways.
5. The role of the right hemisphere
The right hemisphere is no stranger to studies of depression. Previous work has reported decreased brain activity over the right posterior cortex in individuals with both current and remitted depression [44,49–51]. Depressed individuals also appear to demonstrate selective impairment when performing tasks that require right hemisphere function [52,53]. Right parieto-temporal activity has been associated with both autonomic and behavioural aspects of arousal, and the impairments seen in depressed subjects may be indicative of lower emotional arousal. The right parietal cortex is known to subserve perception of emotion, and cortical activity over this region during emotional perception is reduced in depressed patients [50,54]. Children who have relatively low cortical activity in the right parieto-temporal region may be at increased risk for depression due to diminished abilities to perceive, process and respond to emotional information. This is supported by the MRI findings where attentional and arousal processes mediated the risk for developing an MDD [31].
Taken together, the data suggest that right-hemispheric parieto-temporal dysfunction, whether indexed by alpha asymmetry or cortical thinning, may serve as a familial trait marker of vulnerability to MDD. The extent to which these measures might serve, either alone or in combination, as markers for predicting subsequent depressive disorders is currently under investigation. Two points should be considered, however, when evaluating the role of these markers. First, as subjects were scanned at a single time-point, the precise temporal sequence of when the thinning occurred cannot be determined. Likewise, we cannot conclude when signs of alpha asymmetry first emerged. Given that these determinants were observed across a wide-age range that included young children, and among offspring with no history of illness, it is likely that they operate early in development, possibly in utero. Following younger children or infants, both of which are beyond the scope of the present study, would be required to further explore this. Secondly, because the study design was conditioned on familial risk for depression, we cannot determine whether similar markers would be observed for other psychiatric illnesses.
6. The high-risk design
The role of the high-risk design in this series of studies should also be explicitly recognized. Had we simply compared depressed with non-ill subjects using a case–control design, these markers would have likely eluded us. By identifying subjects based on risk for, rather than, presence of the outcome of interest, the high-risk design allowed us to target biological processes prior to the onset of disease. This is an asset when searching for endophenotypes, which should be observed in subjects who are at risk but not themselves ill [27]. The utility of the high-risk approach is certainly not restricted to depressive illness. One of the most fruitful applications has been the Edinburgh High Risk Study of Schizophrenia. Instead of recruiting cases and controls, the study instead targeted non-ill individuals who were at high risk by virtue of having at least two relatives who had the full syndrome. They then followed up these individuals longitudinally, as a portion went on to develop active psychoses. As with our study, they progressively introduced biological measures and DNA collection. The study has now yielded a repository of elegant findings, including identification of structural and functional abnormalities in the brain that predispose individuals to the disorder, as well as genetic moderators of these abnormalities [55–57]. Identification of at-risk individuals, either through history or biomarkers, may have substantial impact on prevention, which may be presumably more efficient than treatment of disease. For example, identification of youth at risk for depression followed by group cognitive therapy is associated with lower rates of subsequent depression [58].
These designs are however not without limitations. Most importantly, a majority of offspring who we define as being at high risk will never go on to develop the full syndrome. This has not only practical implications (the lower the likelihood of onset, the larger sample would be required to obtain sufficient cases) but is also aetiologically relevant, as it suggests that exposure to the risk (here, having a depressed parent) is insufficient to explain the onset of disease. Environmental factors, and by extension, gene-by-environment interactions, may help explain why risk factors may have different effects in different situations. But in high risk, or other family-based designs, genetic and environmental variations may be entangled with each other in ways that are neither explicit nor separable. Another related limitation, although applicable more broadly than to high-risk studies, is that although the measured risk may be precise (here, parental depression can only equal yes or no), the underlying risk may be far less so, since neither MDD, nor its transmission across generations, operate uniformly across individuals or populations. What is thus being measured is likely an arithmetic composite of multiple heterogeneous effects.
Family-based designs gradually fell out of favour over the past two decades, given their complexities and the increasing focus on direct molecular genetics [11,59,60]. But the evidence for high familial loading for a number of psychiatric disorders [61] suggests that these designs may be able to increase tractability in translational research as well [62]. In the case of MDD, for example, we had previously noted that offspring who had childhood or adolescent onset of depression and a history of parental depression had the longest course of chronic depressive illness in adulthood [63]. This epidemiologically defined subtype was translated into a series of genetic studies (Genetics of Early Onset Recurrent Depression (GenRED)), with the view that targeting cases with the earliest onset and most severe course may increase the power to identify genetic correlates of the disorder [64–66].
7. Genetic studies
Given the implications of the above discussion for genetic research, we conclude with some preliminary data on a study on role of genetic variation in the serotonin transporter in the transmission of MDD. A substantial body of work has targeted the role of serotonin in the aetiology of depression and anxiety, as reflected in the theme of the symposium from which this article derives. Much of this work has focused on the serotonin transporter (SLC6A4), a synaptic protein which removes serotonin from the synaptic cleft and is the site of action for the selective serotonin reuptake inhibitor (SSRI) class of antidepressants. A complex repeat polymorphism within the promoter region (5HTTLPR) that functionally alters transcription levels has received the greatest attention, although its precise role in depression remains obscure given numerous failures to replicate, findings in inconsistent directions, and observations of moderating rather than direct effects [67,68].
We have begun to test the role of SLC6A4 in the transmission of risk for depression in our three generation study, which is ethnically homogenous. We predicted that if the short—that is, the transcription-lowering and thus less efficient—variant predisposes persons to depression, then offspring from the high-risk families should have higher rates of this variant than offspring from the low-risk families. As shown in figure 7, 5HTTLPR genotype did not vary by major depression status (a), but it did by risk status (b): specifically, high-risk offspring had more than fourfold higher rates of the SS genotype than their low-risk counterparts (p = 0.01). However, there was an unexpected nuance: the rates in the high-risk group were similar to those generally observed in other Caucasian populations [69]. The rates of the SS genotype among the low-risk group instead were much lower (6%) than population rates, suggesting that it was not offspring of depressed parents who were being enriched with genetic risk over time, but rather, the offspring of non-depressed parents who were being depleted. When we stratified further by generation, similar patterns were obtained in the two generations (see figure 7, inset), although the sample size precluded formal testing of genotype-by-generation effects.
Figure 7.

Differences in serotonin transporter genotypes among second- and third-generation offspring of depressed and non-depressed probands. Serotonin transporter genotype was not associated with major depression (a): 23% of offspring with a lifetime diagnosis of MDD had both risk-encoding alleles (that is, the ‘SS’ genotype) at the 5HTTLPR polymorphism, when compared with 21% of those with no lifetime diagnosis. These rates are similar to those observed in other Caucasian populations. However, upon reclassification of the same sample by familial risk for depression (by virtue of grandparental status) (b), the offspring in the high-risk group had over threefold rates of the SS genotype than offspring in the low-risk group. The genotypes in the high-risk group were similar to those observed in the population; rates of SS among the low-risk offspring were however substantially lower (4%). When we further stratified the offspring by generation (inset), there appeared a successive depletion of the SS genotype over generation.
Although a number of studies and meta-analyses have found 5HTTLPR to moderate the effects of stress on psychiatric illness, most have not found a direct genetic relationship [67,68]. Importantly, we also did not find a direct relationship between 5HTTLPR and depression (figure 7a). Thus, it may be that the high-risk offspring carry no additional genetic susceptibility than the general population does, but rather are exposed to more stressors (e.g. those associated with having a depressed parent) that accelerate adverse outcomes. The low-risk group, in contrast, may not only endure relatively fewer stressful events, but lower genetic risks may also provide resilience to the effects of such stressors when they do occur. This may be a simplistic view, as genetic variation, stressful life events and resilience to them, may not be independent from each other, further underscoring the fluidity with which genetic risks may operate in vivo. The reasons for the apparent depletion in the low-risk group is currently under further investigation, as we are collecting and genotyping additional family members, including the parental generation (Generation 1) as well as spouses of the second and third generations. Until further investigation, these observations should be interpreted provisionally, because the sample is modest and restricted to second- and third-generation offspring. Furthermore, we caution against over-interpreting effects of single genetic variants. Genetic polymorphisms may be static but expression and function are highly vulnerable to interactions with other genes and environment. As additional genetic data emerge, we will examine the effects of genetic variation related to serotonergic function and the role it plays, not only in the development of major depressive and associated disorders, but also in the cortical abnormalities that might underlie them.
8. Conclusions
Translating the epidemiologic observations of the early age of onset of MDD and its high familial loading led naturally to clinical studies of high- and low-risk families. The important clinical findings regarding risk, sequence and course of illness had clinical implications, but did not lead to an understanding of mechanism. For that, we have undertaken a series of biological studies involving MRI, EEG and genetics. The process is an example of translational epidemiology [70], linking population to laboratory studies, and is part of the long journey to understanding the course of major depression.
Acknowledgements
The authors wish to thank Annie Rabinovitch, BA, and Jake Kaufman (B. S. candidate) for their assistance in the preparation of this manuscript. This work is funded by grants from the National Institute of Mental Health (2 R01 MH36197, Weissman PI) (1P50MH090966, Gingrich PI); the National Institute of Drug Abuse (1 K01 DA029598-01, Talati, PI); the Sackler Institute for Developmental Biology at Columbia University (M.M.W.), NARSAD Senior Investigator (M.M.W.) and Junior Investigator (A.T.) Awards, and the Research Foundation for Mental Hygiene, Inc. (A.T.). In the past two years, Dr Weissman has received royalties from the Oxford University Press, Perseus Press, the American Psychiatric Association Press and MultiHealth Systems. None of these present a conflict of interest with this manuscript. Dr Talati and Dr Hamilton have no financial disclosures or conflicts.
Endnote
The most informative index of regional hemispheric activity is alpha power. Alpha is an inverse reflection of cortical activity, that is, alpha power is maximum when subjects are in a restful, awake state with their eyes closed. Increase in cortical activity—such as when the eyes are opened or stimuli are sensed—will decrease alpha power.
References
- 1.Lefkowitz MM, Burton N. 1978. Childhood depression: a critique of the concept. Psychol. Bull. 85, 716–726 10.1037/0033-2909.85.4.716 (doi:10.1037/0033-2909.85.4.716) [DOI] [PubMed] [Google Scholar]
- 2.Alegria M, Mulvaney-Day N, Torres M, Polo A, Cao Z, Canino G. 2009. Prevalence of psychiatric disorders across Latino subgroups in the United States. Am. J. Public Health. 97, 68–75 10.2105/AJPH.2006.087205 (doi:10.2105/AJPH.2006.087205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kessler RC, et al. 2004. The US National Comorbidity Survey Replication (NCS-R): design and field procedures. Int. J. Methods Psychiat. Res. 13, 69–92 10.1002/mpr.167 (doi:10.1002/mpr.167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robins L, Regier DA. 1991. Psychiatric disorders in America: the epidemiologic catchment area study. New York, NY: the Free Press [Google Scholar]
- 5.Gavin AR, Walton E, Chae DH, Alegria M, Jackson JS, Takeuchi D. 2010. The associations between socio-economic status and major depressive disorder among Blacks, Latinos, Asians and non-Hispanic Whites: findings from the Collaborative Psychiatric Epidemiology Studies. Psychol. Med. 40, 51–61 10.1017/S0033291709006023 (doi:10.1017/S0033291709006023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson JS, Torres M, Caldwell CH, Neighbors HW, Nesse RM, Taylor RJ, Trierweiler SJ, Williams DR. 2004. The National Survey of American Life: a study of racial, ethnic and cultural influences on mental disorders and mental health. Int. J. Methods Psychiat. Res. 13, 196–207 10.1002/mpr.177 (doi:10.1002/mpr.177) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szatmari P. 2009. More than counting: milestones in child and adolescent psychiatric epidemiology. J. Am. Acad. Child Adolesc. Psychiat. 48, 353–355 10.1097/CHI.0b013e31819ac03b (doi:10.1097/CHI.0b013e31819ac03b) [DOI] [PubMed] [Google Scholar]
- 8.Gershon ES, et al. 1982. A family study of schizoaffective, bipolar I, bipolar II, unipolar, and normal control probands. Arch. Gen. Psychiat. 39, 1157–1167 10.1001/archpsyc.1982.04290100031006 (doi:10.1001/archpsyc.1982.04290100031006) [DOI] [PubMed] [Google Scholar]
- 9.Sullivan PF, Neale MC, Kendler KS. 2000. Genetic epidemiology of major depression: review and meta-analysis. Am. J. Psychiat. 157, 1552–1562 10.1176/appi.ajp.157.10.1552 (doi:10.1176/appi.ajp.157.10.1552) [DOI] [PubMed] [Google Scholar]
- 10.Williamson DE, Ryan ND, Birmaher B, Dahl RE, Kaufman J, Rao U, Puig-Antich J. 1995. A case-control family history study of depression in adolescents. J. Am. Acad. Child Adolesc. Psychiat. 34, 1596–1607 10.1097/00004583-199512000-00010 (doi:10.1097/00004583-199512000-00010) [DOI] [PubMed] [Google Scholar]
- 11.Weissman MM, Wickramaratne P, Merikangas KR, Leckman JF, Prusoff BA, Caruso KA, Kidd KK, Gammon GD. 1984. Onset of major depression in early adulthood. Increased familial loading and specificity. Arch. Gen. Psychiat. 41, 1136–1143 10.1001/archpsyc.1984.01790230022003 (doi:10.1001/archpsyc.1984.01790230022003) [DOI] [PubMed] [Google Scholar]
- 12.Weissman MM, Gammon GD, John K, Merikangas KR, Warner V, Prusoff BA, Sholomskas D. 1987. Children of depressed parents. Increased psychopathology and early onset of major depression. Arch. Gen. Psychiat. 44, 847–853 10.1001/archpsyc.1987.01800220009002 (doi:10.1001/archpsyc.1987.01800220009002) [DOI] [PubMed] [Google Scholar]
- 13.Orvaschel H, Walsh-Allis G, Ye WJ. 1988. Psychopathology in children of parents with recurrent depression. J. Abnorm. Child Psychol. 16, 17–28 10.1007/BF00910497 (doi:10.1007/BF00910497) [DOI] [PubMed] [Google Scholar]
- 14.Kovacs M, Devlin B, Pollock M, Richards C, Mukerji P. 1997. A controlled family history study of childhood-onset depressive disorder. Arch. Gen. Psychiat. 54, 613–623 10.1001/archpsyc.1997.01830190033004 (doi:10.1001/archpsyc.1997.01830190033004) [DOI] [PubMed] [Google Scholar]
- 15.Keller MB, Beardslee WR, Dorer DJ, Lavori PW, Samuelson H, Klerman GR. 1986. Impact of severity and chronicity of parental affective illness on adaptive functioning and psychopathology in children. Arch. Gen. Psychiat. 43, 930–937 10.1001/archpsyc.1986.01800100020004 (doi:10.1001/archpsyc.1986.01800100020004) [DOI] [PubMed] [Google Scholar]
- 16.Hammen C, Burge D, Burney E, Adrian C. 1990. Longitudinal study of diagnoses in children of women with unipolar and bipolar affective disorder. Arch. Gen. Psychiat. 47, 1112–1117 10.1001/archpsyc.1990.01810240032006 (doi:10.1001/archpsyc.1990.01810240032006) [DOI] [PubMed] [Google Scholar]
- 17.Cole DA, Peeke LG, Martin JM, Truglio R, Seroczynski AD. 1998. A longitudinal look at the relation between depression and anxiety in children and adolescents. J. Consult. Clin. Psychol. 66, 451–460 10.1037/0022-006X.66.3.451 (doi:10.1037/0022-006X.66.3.451) [DOI] [PubMed] [Google Scholar]
- 18.Kovacs M, Gatsonis C, Paulauskas SL, Richards C. 1989. Depressive disorders in childhood. IV. A longitudinal study of comorbidity with and risk for anxiety disorders. Arch. Gen. Psychiat. 46, 776–782 10.1001/archpsyc.1989.01810090018003 (doi:10.1001/archpsyc.1989.01810090018003) [DOI] [PubMed] [Google Scholar]
- 19.Lewinsohn PM, Gotlib IH, Seeley JR. 1995. Adolescent psychopathology. IV. Specificity of psychosocial risk factors for depression and substance abuse in older adolescents. J. Am. Acad. Child Adolesc. Psychiat. 34, 1221–1229 10.1097/00004583-199509000-00021 (doi:10.1097/00004583-199509000-00021) [DOI] [PubMed] [Google Scholar]
- 20.Pine DS, Cohen P, Gurley D, Brook J, Ma Y. 1998. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch. Gen. Psychiat. 55, 56–64 10.1001/archpsyc.55.1.56 (doi:10.1001/archpsyc.55.1.56) [DOI] [PubMed] [Google Scholar]
- 21.Rohde P, Lewinsohn PM, Seeley JR. 1991. Comorbidity of unipolar depression. II. Comorbidity with other mental disorders in adolescents and adults. J. Abnorm. Psychol. 100, 214–222 10.1037/0021-843X.100.2.214 (doi:10.1037/0021-843X.100.2.214) [DOI] [PubMed] [Google Scholar]
- 22.Weissman MM, Wickramaratne P, Nomura Y, Warner V, Verdeli H, Pilowsky DJ, Grillon C, Bruder G. 2005. Families at high and low risk for depression: a 3-generation study. Arch. Gen. Psychiat. 62, 29–36 10.1001/archpsyc.62.1.29 (doi:10.1001/archpsyc.62.1.29) [DOI] [PubMed] [Google Scholar]
- 23.Weissman MM, Wickramaratne P, Nomura Y, Warner V, Pilowsky D, Verdeli H. 2006. Offspring of depressed parents: 20 years later. Am. J. Psychiat. 163, 1001–1008 10.1176/appi.ajp.163.6.1001 (doi:10.1176/appi.ajp.163.6.1001) [DOI] [PubMed] [Google Scholar]
- 24.Weissman MM, Warner V, Wickramaratne P, Moreau D, Olfson M. 1997. Offspring of depressed parents. 10 Years later. Arch. Gen. Psychiat. 54, 932–940 10.1001/archpsyc.1997.01830220054009 (doi:10.1001/archpsyc.1997.01830220054009) [DOI] [PubMed] [Google Scholar]
- 25.Warner V, Wickramaratne P, Weissman MM. 2008. The role of fear and anxiety in the familial risk for major depression: a three-generation study. Psychol. Med. 38, 1543–1556 10.1017/S0033291708002894 (doi:10.1017/S0033291708002894) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warner V, Mufson L, Weissman MM. 1995. Offspring at high and low risk for depression and anxiety: mechanisms of psychiatric disorder. J. Am. Acad. Child Adolesc. Psychiat. 34, 786–797 10.1097/00004583-199506000-00020 (doi:10.1097/00004583-199506000-00020) [DOI] [PubMed] [Google Scholar]
- 27.Gottesman II, Gould TD. 2003. The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiat. 160, 636–645 10.1176/appi.ajp.160.4.636 (doi:10.1176/appi.ajp.160.4.636) [DOI] [PubMed] [Google Scholar]
- 28.Bruder GE, Tenke CE, Warner V, Nomura Y, Grillon C, Hille J, Leite P, Weissman MM. 2005. Electroencephalographic measures of regional hemispheric activity in offspring at risk for depressive disorders. Biol. Psychiat. 57, 328–335 10.1016/j.biopsych.2004.11.015 (doi:10.1016/j.biopsych.2004.11.015) [DOI] [PubMed] [Google Scholar]
- 29.Bruder GE, Tenke CE, Warner V, Weissman MM. 2007. Grandchildren at high and low risk for depression differ in EEG measures of regional brain asymmetry. Biol. Psychiat. 62, 1317–1323 10.1016/j.biopsych.2006.12.006 (doi:10.1016/j.biopsych.2006.12.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruder GE, Bansal R, Tenke CE, Liu J, Hao X, Warner V, Peterson BS, Weissman MM. 2011. Relationship of resting EEG with anatomical MRI measures in individuals at high and low risk for depression. Hum. Brain Mapp. 33, 1325–1333 10.1002/hbm.21284 (doi:10.1002/hbm.21284) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peterson BS, et al. 2009. Cortical thinning in persons at increased familial risk for major depression. Proc. Natl Acad. Sci. USA 106, 6273–6278 10.1073/pnas.0805311106 (doi:10.1073/pnas.0805311106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peterson BS, Weissman MM. 2011. A brain-based endophenotype for major depressive disorder. Annu. Rev. Med. 62, 461–474 10.1146/annurev-med-010510-095632 (doi:10.1146/annurev-med-010510-095632) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shagass C. 1972. Electrophysiological studies of psychiatric problems. Rev. Can. Biol. 31, 77–95 [PubMed] [Google Scholar]
- 34.Pollock VE, Schneider LS. 1990. Topographic quantitative EEG in elderly subjects with major depression. Psychophysiology 27, 438–444 10.1111/j.1469-8986.1990.tb02340.x (doi:10.1111/j.1469-8986.1990.tb02340.x) [DOI] [PubMed] [Google Scholar]
- 35.Shagass C, Roemer RA, Josiassen RC. 1988. Some quantitative EEG findings in unmedicated and medicated major depressives. Neuropsychobiology 19, 169–175 10.1159/000118455 (doi:10.1159/000118455) [DOI] [PubMed] [Google Scholar]
- 36.Henriques JB, Davidson RJ. 1991. Left frontal hypoactivation in depression. J. Abnorm. Psychol. 100, 535–545 10.1037/0021-843X.100.4.535 (doi:10.1037/0021-843X.100.4.535) [DOI] [PubMed] [Google Scholar]
- 37.Gotlib IH, Ranganath C, Rosenfeld P. 1998. Frontal EEG alpha asymmetry, depression, and cognitive functioning. Cogn. Emotion 12, 449–478 10.1080/026999398379673 (doi:10.1080/026999398379673) [DOI] [Google Scholar]
- 38.Davidson RJ, Chapman JP, Chapman LJ. 1987. Task-dependent EEG asymmetry discriminates between depressed and non-depressed subjects. Psychophysiology 24, 585 [Google Scholar]
- 39.Bell IR, Schwartz GE, Hardin EE, Baldwin CM, Kline JP. 1998. Differential resting quantitative electroencephalographic alpha patterns in women with environmental chemical intolerance, depressives, and normals. Biol. Psychiat. 43, 376–388 10.1016/S0006-3223(97)00245-X (doi:10.1016/S0006-3223(97)00245-X) [DOI] [PubMed] [Google Scholar]
- 40.Field T, Fox NA, Pickens J, Nawrocki T. 1995. Relative right frontal EEG activation in 3- to 6- month-old infants of ‘depressed’ mothers. Dev. Psychol. 31, 358–363 10.1037/0012-1649.31.3.358 (doi:10.1037/0012-1649.31.3.358) [DOI] [Google Scholar]
- 41.Dawson G, Frey K, Panagiotides H, Osterling J, Hessl D. 1997. Infants of depressed mothers exhibit atypical frontal brain activity: a replication and extension of previous findings. J. Child Psychol. Psychiat. 38, 179–186 10.1111/j.1469-7610.1997.tb01852.x (doi:10.1111/j.1469-7610.1997.tb01852.x) [DOI] [PubMed] [Google Scholar]
- 42.Reid SA, Duke LM, Allen JJ. 1998. Resting frontal electroencephalographic asymmetry in depression: inconsistencies suggest the need to identify mediating factors. Psychophysiology 35, 389–404 10.1111/1469-8986.3540389 (doi:10.1111/1469-8986.3540389) [DOI] [PubMed] [Google Scholar]
- 43.Kentgen LM, Tenke CE, Pine DS, Fong R, Klein RG, Bruder GE. 2000. Electroencephalographic asymmetries in adolescents with major depression: influence of comorbidity with anxiety disorders. J. Abnorm. Psychol. 109, 797–802 10.1037/0021-843X.109.4.797 (doi:10.1037/0021-843X.109.4.797) [DOI] [PubMed] [Google Scholar]
- 44.Henriques JB, Davidson RJ. 1990. Regional brain electrical asymmetries discriminate between previously depressed and healthy control subjects. J. Abnorm. Psychol. 99, 22–31 10.1037/0021-843X.99.1.22 (doi:10.1037/0021-843X.99.1.22) [DOI] [PubMed] [Google Scholar]
- 45.Bruder GE, Fong R, Tenke CE, Leite P, Towey JP, Stewart JE, McGrath PJ, Quitkin FM. 1997. Regional brain asymmetries in major depression with or without an anxiety disorder: a quantitative electroencephalographic study. Biol. Psychiat. 41, 939–948 10.1016/S0006-3223(96)00260-0 (doi:10.1016/S0006-3223(96)00260-0) [DOI] [PubMed] [Google Scholar]
- 46.Heller W, Nitschke JB. 1997. Regional brain activity in emotion: a framework for understanding cognition in depression. Cogn. Emotion 11, 637–661 10.1080/026999397379845a (doi:10.1080/026999397379845a) [DOI] [Google Scholar]
- 47.Price JL, Drevets WC. 2010. Neurocircuitry of mood disorders. Neuropsychopharmacology 35, 192–216 10.1038/npp.2009.104 (doi:10.1038/npp.2009.104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson BS. 2003. Conceptual, methodological, and statistical challenges in brain imaging studies of developmentally based psychopathologies. Dev. Psychopathol. 15, 811–832 10.1017/S0954579403000385 (doi:10.1017/S0954579403000385) [DOI] [PubMed] [Google Scholar]
- 49.Moratti S, Rubio G, Campo P, Keil A, Ortiz T. 2008. Hypofunction of right temporoparietal cortex during emotional arousal in depression. Arch. Gen. Psychiat. 65, 532–541 10.1001/archpsyc.65.5.532 (doi:10.1001/archpsyc.65.5.532) [DOI] [PubMed] [Google Scholar]
- 50.Deldin PJ, Keller J, Gergen JA, Miller GA. 2000. Right-posterior face processing anomaly in depression. J. Abnorm. Psychol. 109, 116–121 10.1037/0021-843X.109.1.116 (doi:10.1037/0021-843X.109.1.116) [DOI] [PubMed] [Google Scholar]
- 51.Post RM, DeLisi LE, Holcomb HH, Uhde TW, Cohen R, Buchsbaum MS. 1987. Glucose utilization in the temporal cortex of affectively ill patients: positron emission tomography. Biol. Psychiat. 22, 545–553 10.1016/0006-3223(87)90182-X (doi:10.1016/0006-3223(87)90182-X) [DOI] [PubMed] [Google Scholar]
- 52.Rubinow DR, Post RM. 1992. Impaired recognition of affect in facial expression in depressed patients. Biol. Psychiat. 31, 947–953 10.1016/0006-3223(92)90120-O (doi:10.1016/0006-3223(92)90120-O) [DOI] [PubMed] [Google Scholar]
- 53.Bruder GE, Quitkin FM, Stewart JW, Martin C, Woglmaier MM, Harrison WM. 1989. Cerebral laterality and depression: differences in perceptual asymmetry among diagnostic subtypes. J. Abnorm. Psychol. 98, 177–186 10.1037/0021-843X.98.2.177 (doi:10.1037/0021-843X.98.2.177) [DOI] [PubMed] [Google Scholar]
- 54.Kayser J, Bruder GE, Tenke CE, Stewart JE, Quitkin FM. 2000. Event-related potentials (ERPs) to hemifield presentations of emotional stimuli: differences between depressed patients and healthy adults in P3 amplitude and asymmetry. Int. J. Psychophysiol. 36, 211–236 10.1016/S0167-8760(00)00078-7 (doi:10.1016/S0167-8760(00)00078-7) [DOI] [PubMed] [Google Scholar]
- 55.Hall J, Whalley HC, Moorhead TW, Baig BJ, McIntosh AM, Job DE, Owens DG, Lawrie SM, Johnstone EC. 2008. Genetic variation in the DAOA (G72) gene modulates hippocampal function in subjects at high risk of schizophrenia. Biol. Psychiat. 64, 428–433 10.1016/j.biopsych.2008.03.009 (doi:10.1016/j.biopsych.2008.03.009) [DOI] [PubMed] [Google Scholar]
- 56.Lawrie SM, McIntosh AM, Hall J, Owens DG, Johnstone EC. 2008. Brain structure and function changes during the development of schizophrenia: the evidence from studies of subjects at increased genetic risk. Schizophr. Bull. 34, 330–340 10.1093/schbul/sbm158 (doi:10.1093/schbul/sbm158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lieb R, Isensee B, Hofler M, Pfister H, Wittchen HU. 2002. Parental major depression and the risk of depression and other mental disorders in offspring: a prospective-longitudinal community study. Arch. Gen. Psychiat. 59, 365–374 10.1001/archpsyc.59.4.365 (doi:10.1001/archpsyc.59.4.365) [DOI] [PubMed] [Google Scholar]
- 58.Clarke GN, Hornbrook M, Lynch F, Polen M, Gale J, Beardslee W, O'Connor E, Seeley J. 2001. A randomized trial of a group cognitive intervention for preventing depression in adolescent offspring of depressed parents. Arch. Gen. Psychiat. 58, 1127–1134 10.1001/archpsyc.58.12.1127 (doi:10.1001/archpsyc.58.12.1127) [DOI] [PubMed] [Google Scholar]
- 59.Kendler KS, Gruenberg AM, Tsuang MT. 1985. Psychiatric illness in first-degree relatives of schizophrenic and surgical control patients. A family study using DSM-III criteria. Arch. Gen. Psychiat. 42, 770–779 10.1001/archpsyc.1985.01790310032004 (doi:10.1001/archpsyc.1985.01790310032004) [DOI] [PubMed] [Google Scholar]
- 60.Tsuang MT, Winokur G, Crowe RR. 1980. Morbidity risks of schizophrenia and affective disorders among first degree relatives of patients with schizophrenia, mania, depression and surgical conditions. Br. J. Psychiat. 137, 497–504 10.1192/bjp.137.6.497 (doi:10.1192/bjp.137.6.497) [DOI] [PubMed] [Google Scholar]
- 61.Weissman MM, Merikangas KR, John K, Wickramaratne P, Prusoff BA, Kidd KK. 1986. Family-genetic studies of psychiatric disorders. Developing technologies. Arch. Gen. Psychiat. 43, 1104–1116 10.1001/archpsyc.1986.01800110090012 (doi:10.1001/archpsyc.1986.01800110090012) [DOI] [PubMed] [Google Scholar]
- 62.Hopper JL. 2003. Commentary: Case-control-family designs: a paradigm for future epidemiology research? Int. J. Epidemiol. 32, 48–50 10.1093/ije/dyg114 (doi:10.1093/ije/dyg114) [DOI] [PubMed] [Google Scholar]
- 63.Wickramaratne PJ, Warner V, Weissman MM. 2000. Selecting early onset MDD probands for genetic studies: results from a longitudinal high-risk study. Am. J. Med. Genet. 96, 93–101 (doi:10.1002/(SICI)1096-8628(20000207)96:1<93::AID-AJMG19>3.0.CO;2-5) [DOI] [PubMed] [Google Scholar]
- 64.Shi J, et al. 2011. Genome-wide association study of recurrent early-onset major depressive disorder. Mol. Psychiat. 16, 193–201 10.1038/mp.2009.124 (doi:10.1038/mp.2009.124) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holmans P, et al. 2007. Genetics of recurrent early-onset major depression (GenRED): final genome scan report. Am. J. Psychiat. 164, 248–258 10.1176/appi.ajp.164.2.248 (doi:10.1176/appi.ajp.164.2.248) [DOI] [PubMed] [Google Scholar]
- 66.Levinson DF, et al. 2003. Genetics of recurrent early-onset depression (GenRED): design and preliminary clinical characteristics of a repository sample for genetic linkage studies. Am. J. Med. Genet. B Neuropsychiat. Genet. 119B, 118–130 10.1002/ajmg.b.20009 (doi:10.1002/ajmg.b.20009) [DOI] [PubMed] [Google Scholar]
- 67.Uher R, McGuffin P. 2010. The moderation by the serotonin transporter gene of environmental adversity in the etiology of depression: 2009 update. Mol. Psychiat. 15, 18–22 10.1038/mp.2009.123 (doi:10.1038/mp.2009.123) [DOI] [PubMed] [Google Scholar]
- 68.Karg K, Burmeister M, Shedden K, Sen S. 2011. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch. Gen. Psychiat. 68, 444–454 10.1001/archgenpsychiatry.2010.189 (doi:10.1001/archgenpsychiatry.2010.189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clarke H, Flint J, Attwood AS, Munafo MR. 2010. Association of the 5- HTTLPR genotype and unipolar depression: a meta-analysis. Psychol. Med. 40, 1767–1778 10.1017/S0033291710000516 (doi:10.1017/S0033291710000516) [DOI] [PubMed] [Google Scholar]
- 70.Weissman MM, Brown AS, Talati A. 2011. Translational epidemiology in psychiatry: linking population to clinical and basic sciences. Arch. Gen. Psychiat. 68, 600–608 10.1001/archgenpsychiatry.2011.47 (doi:10.1001/archgenpsychiatry.2011.47) [DOI] [PMC free article] [PubMed] [Google Scholar]