

Abstract
The serotonin (5-HT, 5-hydroxytryptamine) system has been implicated in the pathogenesis of major depressive disorder (MDD). The case for its contribution to the therapeutic efficacy of a wide variety of antidepressant treatments is, however, much stronger. All antidepressant strategies have been shown to enhance 5-HT transmission in the brain of laboratory animals. Catecholamines, norepinephrine (NE) and dopamine (DA) can also play a pivotal role in the mechanism of action of certain antidepressant strategies. The enhancement of 5-HT transmission by selective serotonin reuptake inhibitors, which leads to a dampening of the activity of NE and DA neurons, may account in part for the low remission rate achieved with these medications and/or the residuals symptoms after remission is achieved. The functional connectivity between the 5-HT, NE and DA systems can be used to understand the mechanism of action of a wide variety of augmentation strategies in treatment-resistant MDD. Proof-of-concept studies have shown that antidepressant medications with complementary mechanisms of action on monoaminergic systems can double the remission rate achieved in a trial of standard duration. Novel approaches are also being used to treat MDD, which also appear to involve the monoaminergic system(s) to a varying extent.
Keywords: serotonin, autoreceptor, neurotransmission, electrophysiology, drug combination, remission
1. Introduction
Major depressive disorder (MDD) is the most predominant illness among mental, neurological and substance-use disorders [1]. According to the World Health Organization [2], MDD is currently the leading cause of disability globally in middle- to high-income countries, measured in disability-adjusted life years. In addition, when MDD is present with other chronic medical disorders, such as asthma, epilepsy and diabetes, it worsens the morbidity of such chronic medical conditions [3]. Furthermore, mortality rates are increased several fold if MDD is present following a myocardial infarct [4]. These facts therefore make depression research a priority in the medical field.
There are numerous causal hypotheses for MDD, and they are not necessarily exclusive. The serotonin (5-HT, 5-hydroxytryptamine) hypothesis for depression has been intensely investigated since the 1967 seminal paper of Alec Coppen, laying down the foundation for such a hypothesis [5]. This exposé encompassed both aetiological and therapeutic considerations. These two types of issues need not, however, be linked. One possibility is that there may not be any anomaly in the 5-HT system in MDD, while a therapeutic approach may still work by enhancing 5-HT transmission above normal. An example for this therapeutic principle is the use of diuretics acting on kidneys to help treat chronic heart failure. Another possibility is that a deficient 5-HT system may indeed contribute to the manifestation of MDD, and an enhancement of 5-HT transmission by antidepressant strategies may restore euthymia. An example for this therapeutic principle is the use of dopamine (DA) precursors or agonists in the presence of DA neuronal cell loss in Parkinson's disease. The latter hypothesis for a causal role of 5-HT in MDD has often been dismissed over the years, because of a lack of consistency of observations on a decrease of 5-HT concentrations in post-mortem brain samples, and the fact that acute tryptophan depletion does not produce depression in healthy controls, tryptophan being the essential amino acid precursor of 5-HT [6]. Nevertheless, in unaffected individuals with a family history of depression, mild dysphoric symptoms can be triggered by this acute 5-HT-lowering challenge [7]. One can thus wonder whether a prolonged 5-HT lowering might not lead to MDD.
Although there is no single anomaly in the 5-HT system that is common to the majority of patients with MDD, one has to take into account that numerous neuronal factors control the synaptic concentration of 5-HT and its postsynaptic responses. These include tryptophan availability, genetic variants of tryptophan hydroxylase 2, cell body 5-HT1A receptors, terminal 5-HT1B autoreceptors, 5-HT transporters (5-HTT), different levels of monoamine oxidase (MAO) that inactivates 5-HT, and the polymorphism of a variety of postsynaptic 5-HT receptors (figures 1 and 2). If a single anomaly is present, it may only confer a slightly higher risk for developing MDD. For instance, the risk for MDD is directly proportional to the number of short alleles for the 5-HTT, conferring a lower 5-HTT efficiency, and the number of major life stressors [10]. Although this claim has been challenged [11], the analysis of all 54 studies, instead of subsamples, confirmed this important link [12]. It is possible that examining several of the abovementioned factors controlling 5-HT transmission would strengthen the direct link between a deficiency of the 5-HT system and MDD.
Figure 1.

Electrophysiological paradigm used to study in vivo the 5-HT system in the brain of laboratory animals. The firing activity of 5-HT neurons is recorded from the dorsal raphe nucleus either with single glass electrodes or microiontophoretic pipettes to test the sensitivity of 5-HT1A autoreceptors (yellow rectangles) with 5-HT or selective agonists. Serotonin axons are electrically stimulated in the ventromedial tegmentum where 5-HT fibres originating from both the dorsal and median raphe nuclei course. The responsiveness of postsynaptic 5-HT receptors (red and orange rectangles), as well as the effectiveness of the stimulations, can be assessed from recording neurons in the pyramidal layers of the hippocampus. The responsiveness of terminal 5-HT1B autoreceptors (blue rectangles) can be evaluated by varying the frequency of the stimulations [8]. The tonic activation of the postsynaptic 5-HT1A receptor following various antidepressant treatments can be evaluated in unstimulated conditions by injecting the selective 5-HT1A receptor WAY100635 and observing the increased firing rate of pyramidal neurons, which will be proportional to the degree of enhancement of 5-HT transmission [9].
Figure 2.

Functional interactions between the 5-HT, NE and DA systems and their postsynaptic targets. The circles crossed by an arrow represent reuptake transporters. The small circles with+and – signs represent the excitatory and inhibitory effects, respectively, of the receptors on the firing rate of the neurons. Note the presence of α2-adrenoceptors on 5-HT terminals. (Online version in colour.)
2. Commonality of antidepressant strategies on the 5-HT system
Extensive in vivo electrophysiological studies of various antidepressant strategies carried out in the rat brain have revealed a striking commonality of action on the 5-HT system (table 1). The approach taken has basically been the one mentioned above: rather than looking at a single parameter controlling 5-HT transmission possibly altered by several types of treatments, a variety of neuronal elements have been examined. Most importantly, overall synaptic transmission has been assessed to determine whether the net effects of such alteration(s) led to increased transmission (figure 1). Initially, it was observed that long-term administration of tricyclic antidepressants (TCAs) with various action(s) on 5-HT and norepinephrine (NE) reuptake sensitized postsynaptic 5-HT receptor responsiveness in forebrain structures [13–16]. Such a possible unifying theory gained ground when it was observed that repeated, but not a single electroconvulsive shock (ECS) produced the same effect in the hippocampus [17]. These two distinct treatments were subsequently shown to enhance net 5-HT transmission by stimulating the 5-HT pathway at physiological firing frequencies for 5-HT neurons and increasing the response on postsynaptic neurons in the hippocampus [8,18]. In 1983, it was first reported that the selective serotonin reuptake inhibitor (SSRI) zimelidine initially decreased the firing rate of 5-HT neurons with repeated injections, but that the discharge frequency returned to normal after 14 daily injections due to the desensitization of the cell body 5-HT autoreceptor. The stimulation of the 5-HT pathway led to a greater effect in the hippocampus after a two-week zimelidine regimen [19]. It was also concluded that this enhancement was not due to mere reuptake inhibition, because acute injection of the SSRI citalopram did not produce this effect, but that the terminal 5-HT1B autoreceptor controlling 5-HT release was desensitized like its cell body counterpart that controls firing activity [20]. Identical results were obtained with the SSRIs fluoxetine, paroxetine and fluvoxamine [8,21,22]. SSRIs therefore seemed to act by enhancing the function of 5-HT neurons (after they regain their normal firing rate), while leaving intact the sensitivity of postsynaptic neurons in the hippocampus, unlike TCAs and ECS. The mechanism of action of MAO inhibitors on the 5-HT system is similar, in some aspects, to that of SSRIs. Initially, they produce a decreased firing of 5-HT neurons, followed by a recovery after a three-week regimen [23]. In contrast, MAO inhibitors do not desensitize terminal 5-HT autoreceptors, but they still lead to enhanced 5-HT release [9]. By the mid-1980s, there was direct evidence that long-term, but not short-term, administration of TCAs, ECS, SSRIs and MAO inhibitors enhanced 5-HT transmission in the rat brain.
Table 1.
Summary of the effects of various antidepressant strategies on the 5-HT system as assessed in the brain of laboratory animals. SSRI, selective serotonin reuptake inhibitor; SNRI, serotonin and norepinephrine reuptake inhibitor; MAOI, monoamine oxidase inhibitor; ECS, electroconvulsive shocks. Arrows pointing down represent decreased responsiveness, arrows pointing upward represent an increase in function, open circles represent no change, and n.d. not determined. The net effect on transmission was assessed either by stimulating the 5-HT pathway or by determining the degree of activation of postsynaptic 5-HT1A receptors using the 5-HT1A receptor antagonist WAY100635. All regimens were administered for periods of 14–21 days.
| cell body 5-HT1A autoreceptor responsiveness | terminal 5-HT1B autoreceptor responsiveness | terminal α2-adrenoceptor responsiveness on 5-HT terminals | postsynaptic 5-HT1A receptor responsiveness | net effect on 5-HT transmission | |
|---|---|---|---|---|---|
| SSRI | ↓ | ↓ | ○ | ○ | ↑ |
| SNRI | ↓ | ○ | ↓ | ○ | ↑ |
| MAOI | ↓ | ○ | ↓ | ○ or ↓ | ↑ |
| 5-HT1A agonists | ↓ | ○ | n.d. | ○ | ↑ |
| tricyclics | ○ | ○ | ↓ | ↑ | ↑ |
| ECS | ○ | ○ | ○ | ↑ | ↑ |
| mirtazapinea | ↓ | ○ | ↓ | ○ | ↑ |
| bupropiona | ↓ | ○ | ↓ | ○ | ↑ |
| vagus nerve st.a | ○ | n.d. | n.d. | ○ | ↑ |
| agomelatinea | ○ | n.d. | n.d. | ○ | ↑ |
aThese treatments increase the firing of 5-HT neurons above their normal baseline levels.
Over the years, further electrical and pharmacological treatments with antidepressant properties have been studied and all shown to enhance overall 5-HT transmission in the rat hippocampus (table 1). These include the 5-HT1A receptor agonist gepirone (for which there are three positive placebo-controlled studies in MDD), the α2-adrenoceptor antagonist mirtazapine, the catecholamine releaser bupropion, the melatonin receptor agonist/5-HT2C antagonist agomelatine and vagus nerve stimulation [9,24–26]. The DA D3/D2 receptor agonist pramipexole, which has been shown to be an antidepressant in patients with Parkinson's disease and in unaffected individuals with MDD, leads to delayed enhancement of the firing of 5-HT and a desensitization of the 5-HT1A autoreceptor and a net increase in 5-HT transmission [27,28]. The atypical antipsychotic quetiapine, which is an antidepressant in monotherapy at subtherapeutic doses for schizophrenia, increases net 5-HT and NE transmission in the rat hippocampus [29]. It is important to mention that the nomenclature/denomination of certain psychotropic medications usually reflects their index indications (i.e. anticonvulsants and antipsychotics) rather than their distinct properties or mechanisms of action, contributing to further confusion and stigmatization.
3. The 5-HT system and the antidepressant response: important but not exclusive
All antidepressant strategies thus far tested enhance 5-HT transmission at least in normal rats (table 1). Nevertheless, it is obvious that other neuronal systems can also play a role in the antidepressant response. For instance, TCAs also sensitize postsynaptic α1- and/or α2-adrenoceptors in regions such as the thalamus, the amygdala and the facial motor nucleus, and most TCAs block NE reuptake [14–17,30]. The clinical parallel to these basic observations is that catecholamine depletion, but not 5-HT depletion, worsens depressive symptoms in patients who had responded to an NE reuptake inhibitor [31]. Similarly, tryptophan depletion does not cause a re-emergence of depressive symptoms in patients who responded to ECS [32]. In contrast, patients who responded to mirtazapine were observed to have a recurrence of their depressive symptoms with both tryptophan depletion and catecholamine depletion [33].
4. Enhancement of 5-HT and the antidepressant response: sometimes a limiting factor
Long-term administration of SSRIs enhances 5-HT transmission in the locus coeruleus [34]. As 5-HT exerts an inhibitory action on NE neurons through enhanced γ-aminobutyric acid (GABA) release, as a result of increased activation of excitatory 5-HT2A receptors on GABA neurons, the firing activity of NE neurons is dampened by sustained regimens of the SSRIs such as citalopram, escitalopram, paroxetine and even a low dose of venlafaxine that inhibits 5-HT but not NE reuptake [34–37]. It was subsequently shown that the same regimen of citalopram that attenuates NE firing also decreased basal and evoked extracellular levels of NE in the amygdala [38]. It is thus conceivable that the lack of therapeutic benefits of SSRIs in some depressed patients may result from an attenuation of NE transmission in the presence of enhanced 5-HT levels (figure 3).
Figure 3.
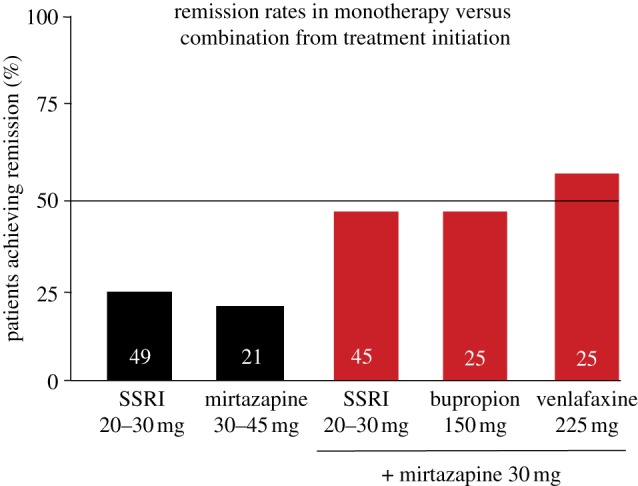
Summary of two double-blind controlled studies of six weeks duration comparing the effects of single drugs versus their combination from treatment initiation. The SSRIs used were paroxetine 20–30 mg d−1 and fluoxetine 20 mg d−1. The numbers within the histograms represent the number of patients. The remission rates are expressed as patients achieving a Montgomery Asberg Depression Rating Scale score of 10 or less in Blier et al. [39] and a Hamilton Depression-17 item score of 7 or less in Blier et al. [40]. (Online version in colour.)
Serotonin neurons exert an inhibitory influence on DA neurons in the ventral tegmental area (VTA), which give rise to mesolimbic and mesocortical projections [41]. The SSRI escitalopram suppresses the mean firing rate of VTA DA neurons through enhanced activation of 5-HT2C receptors, likely on GABA neurons [42]. It is thus also conceivable that the lack of therapeutic benefits of SSRIs in some depressed patients may result from an attenuation of DA transmission in the presence of enhanced 5-HT levels. The inhibitory action of SSRIs on catecholamine neurons may also account for residual symptoms often observed after remission has been achieved with SSRIs.
Attempts to reverse the dampening action of enhanced 5-HT levels produced by SSRIs on catecholamine neurons have led to effective augmentation strategies for MDD. For instance, adding an NE reuptake inhibitor to the regimen of SSRI-resistant patients, or adding DA agonists in treatment-resistant patients [43,44] can be beneficial. This basic approach underlies the common enhancing action of low doses of atypical antipsychotics in patients not responding to SSRIs. Indeed, at subtherapeutic doses of atypical antipsychotics for schizophrenia, 5-HT2A and 5-HT2C receptors are potently blocked. In the rat brain, 5-HT2A and 5-HT2C receptor antagonists reverse the inhibitory effect of SSRIs on NE and DA neurons, respectively, including atypical antipsychotics [29,35,44,45].
5. The forebrain structures involved in the antidepressant response
Patients with MDD generally exhibit hyperactivity of the hippocampus, the amygdala and the subgenual anterior cingulate cortex; when they respond to antidepressant strategies, this hyperactivity is diminished [46–49]. In such structures, or their equivalent in the rat brain, 5-HT, NE and DA generally exert an inhibitory action on neuronal firing as shown by their direct microiontophoretic application in vivo [13,16,28]. Consequently, the antidepressant response may be triggered by medications potentiating inhibitory transmission in limbic structures through one or more of these monoaminergic systems. The effectiveness of combination of antidepressant medications in treatment-resistant patients may thus be better understood through a greater potentiation of inhibitory transmission in such structures.
In the dorsolateral frontal cortex, the work of Mayberg documented an increase in activity in MDD patients responding to antidepressants [48,49]. Interestingly, 5-HT classically exerts a net excitatory effect on pyramidal neurons in this area of the frontal cortex [50]. It is thus conceivable that enhanced 5-HT transmission at such sites may contribute to the therapeutic action of some antidepressant treatments.
6. Combination of 5-HT targets to improve effectiveness of treatments
The first-line treatment for MDD is now selective reuptake inhibition. This approach was obtained by extracting and enhancing one of the properties of the prototypical agent imipramine. In so doing, the SSRIs have become a class of drugs that can no longer be used to commit suicide, in contrast to TCAs, and are much more tolerable. They can still produce cumbersome side effects, like sexual dysfunctions, which is often a significant hurdle for their long-term use. One strategy to minimize the latter problem has been to use compounds with low potency at the 5-HTT and acting on other 5-HT receptor targets. Trazodone is such a low-potency 5-HTT inhibitor, a 5-HT2A/2C antagonist and a 5-HT1A agonist; furthermore, in its new slow-release preparation daytime sedation has been minimized [51,52]. Vortioxetine (Lu AA21004) is another low-potency 5-HTT inhibitor that acts as an agonist on 5-HT1A and 5-HT1B receptors, and as an antagonist of 5-HT3 and 5-HT7 receptors. Interestingly, vortioxetine has been shown to be as effective as a regimen of venlafaxine that blocks both 5-HT and NE reuptake [53,54].
Such new and old medications have not, however, significantly enhanced the standard remission rate that hovers around 30–40%. Even the rapidly acting ketamine infusions still produce such a low success rate (see below; [55–57]). Drug combinations at adequate regimens have nevertheless pushed up the remission rate when used from treatment initiation in proof of concept studies. These include combinations of SSRIs or the serotonin and norepinephrine reuptake inhibitor venlafaxine with the noradrenergic TCA desipramine or mirtazapine [39,40,43]. While such combinations act on more than one 5-HT neuronal target, they also exert noradrenergic effects, such as blocking the NE reuptake or presynaptic α2-adrenoceptors. In the case of the mirtazapine combinations, the remission rate after six weeks was approximately doubled compared with SSRIs used alone [39,40] (figure 3). In this context, it is important to emphasize that clinical studies in MDD may be flawed by trial design, the selection of patients with reliable symptomatology and high dropout rates. Nevertheless, clinical results using combinations of drugs clearly indicate that when multiple targets are solicited, a greater proportion of patients with MDD can be brought to remission [58].
7. Beyond serotonin and other monoamines to an antidepressant response
There are several strategies that may not directly target monoamine systems and trigger an antidepressant response. Sleep deprivation, for instance, may produce a rapid antidepressant response, albeit very transient [59]. This strategy, like several others, can indirectly affect the function of monoamine systems. In the case of sleep deprivation, it was reported that in freely moving cats, the firing frequency of 5-HT neurons is enhanced one day after the deprivation, a time when the antidepressant benefits are seen in patients with MDD [60].
A novel antidepressant strategy producing strikingly rapid therapeutic benefits in MDD has provided credence to the theory that neuronal hyperactivity in limbic structures may account in large part for the pathology of MDD. The injection of a subanaesthetic dose of the glutamate N-methyl-d-aspartate (NMDA) antagonist ketamine can produce, within hours, a robust antidepressant response, presumably in part by blocking excitatory NMDA receptors [55–57]. In other words, within hours ketamine may produce a dampening of the excessive neuronal activity in limbic structures that antidepressants take weeks to build up by enhancing inhibitory monoaminergic transmission. It is also noteworthy that even before the first controlled observations of the antidepressant action of ketamine infusion, it had been reported that a variety of classical antidepressants induce a decreased function of NMDA receptors, probably through their monoaminergic properties [61].
Deep brain stimulation is another novel, yet still experimental, approach to treat refractory MDD. Thus far, stimulations at high frequencies of the subgenual anterior cingulate cortex have been the most studied [62]. Consistent with the attenuated hyperactivity that decreases with effective antidepressant treatments, deep brain stimulation also produces the same phenomenon. Similar stimulations applied to the equivalent region in the rat brain leads to an enhancement of the firing rate of 5-HT neurons in the dorsal raphe nucleus and an increase in the extracellular levels of 5-HT in the hippocampus [63,64]. Furthermore, the antidepressant-like action of deep brain stimulation is abolished in rats in which 5-HT neurons have been lesioned [64]. This is yet another example of the important connectivity between various brain structures involved in mood disorders.
8. Conclusion
In closing, the 5-HT system still appears to play an important role both in the pathophysiology and in the treatment of MDD. Above all, it has been a remarkable target for the development of antidepressant strategies. Clinically meaningful progress has been achieved by exploiting the capacity of various 5-HT neuronal elements to modulate overall synaptic 5-HT transmission, and more is still to come. Nevertheless, novel targets have been identified and more are eagerly awaited so that more patients with MDD can be brought to remission and more rapidly. Given the functional connectivity between monoamine systems and other neurotransmitters, it appears unlikely that even novel antidepressant strategies will exert their antidepressant activity totally independent of these monoamine systems.
References
- 1.Collins PY, Patel V, Joestl SS, March D, Insel TR, Daar AS. 2011. Grand challenges in global mental health. Nature 475, 27–30 10.1038/475027a (doi:10.1038/475027a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. 2008. The global burden of diseases. 2004 Update, part 4, pp. 40–51. Geneva, Switzerland: WHO Press [Google Scholar]
- 3.Moussavi S, Chatterji S, Verdes E, Tandon A, Patel V, Ustun B. 2007. Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet 370, 851–858 10.1016/S0140-6736(07)61415-9 (doi:10.1016/S0140-6736(07)61415-9) [DOI] [PubMed] [Google Scholar]
- 4.Frasure-Smith N, Lespérance F, Gravel G, Masson G, Juneau M, Talajic M, Bourassa MG. 2000. Social support, depression, and mortality during the first year after myocardial infarction. Circulation 101, 1919–1924 10.1161/01.CIR.101.16.1919 (doi:10.1161/01.CIR.101.16.1919) [DOI] [PubMed] [Google Scholar]
- 5.Coppen A. 1967. The biochemistry of affective disorders. Br. J. Pharmacol. 113, 1237–1264 10.1192/bjp.113.504.1237 (doi:10.1192/bjp.113.504.1237) [DOI] [PubMed] [Google Scholar]
- 6.Delgado P, Charney DS, Price LH, Landis H, Heninger GS. 1989. Neuroendocrine and behavioral effects of dietary tryptophan restriction in healthy subjects. Life Sci. 45, 2323–2332 10.1016/0024-3205(89)90114-8 (doi:10.1016/0024-3205(89)90114-8) [DOI] [PubMed] [Google Scholar]
- 7.Benkelfat C, Ellenbogen MA, Dean P, Palmour R, Young S. 1994. Mood-lowering effect of tryptophan depletion. Enhanced susceptibility in young men at genetic risk for major affective disorders. Arch. Gen. Psychiat. 51, 687–697 10.1001/archpsyc.1994.03950090019003 (doi:10.1001/archpsyc.1994.03950090019003) [DOI] [PubMed] [Google Scholar]
- 8.Chaput Y, de Montigny C, Blier P. 1991. Presynaptic and postsynaptic modifications of the serotonin system by long-term administration of antidepressant treatments: an in vivo electrophysiologic study in the rat. Neuropsychopharmacology 5, 219–229 [PubMed] [Google Scholar]
- 9.Haddjeri N, Blier P, de Montigny C. 1998. Long-term antidepressant treatments result in a tonic activation of forebrain 5-HT1A receptors. J. Neurosci. 18, 10 150–10 156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caspi A. et al 2003. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301, 386–389 10.1126/science.1083968 (doi:10.1126/science.1083968) [DOI] [PubMed] [Google Scholar]
- 11.Risch N, et al. 2009. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. J. Am. Med. Assoc. 301, 2462–2471 10.1001/jama.2009.878 (doi:10.1001/jama.2009.878) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kragh K, Burmeister M, Shedden K, Sen S. 2011. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch. Gen. Psychiat. 68, 444–454 10.1001/archgenpsychiatry.2010.189 (doi:10.1001/archgenpsychiatry.2010.189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Montigny C, Aghajanian GK. 1978. Tricyclic antidepressants: long term treatment increases responsivity of rat forebrain neurons to serotonin. Science 202, 1301–1306 10.1126/science.725608 (doi:10.1126/science.725608) [DOI] [PubMed] [Google Scholar]
- 14.Menkes DB, Aghajanian GK. 1980. Chronic antidepressant treatment enhances α-adrenergic and serotonergic responses in the facial motor nucleus. Life Sci. 27, 45–55 10.1016/0024-3205(80)90018-1 (doi:10.1016/0024-3205(80)90018-1) [DOI] [PubMed] [Google Scholar]
- 15.Menkes DB, Aghajanian GK. 1981. α1-adrenoceptor-mediated responses in the lateral geniculate nucleus are enhanced by chronic antidepressant treatment. Eur. J. Pharmacol. 74, 27–35 10.1016/0014-2999(81)90319-8 (doi:10.1016/0014-2999(81)90319-8) [DOI] [PubMed] [Google Scholar]
- 16.Wang RX, Aghajanian GK. 1980. Enhanced sensitivity of amygdaloid neurons to serotonin and norepinephrine after chronic antidepressant treatment. Commun. Psychopharmacol. 4, 83–90 [PubMed] [Google Scholar]
- 17.de Montigny C. 1984. Electroconvulsive shock treatments enhance responsiveness of forebrain neurons to serotonin. J. Pharmacol. Exp. Ther. 228, 230–234 [PubMed] [Google Scholar]
- 18.Blier P, de Montigny C, Chaput Y. 1987. Modification of the serotonin system by antidepressant treatments: implications for the therapeutic response in major depression. J. Clin. Psychopharmacol. 7(Suppl.), 24–35 [PubMed] [Google Scholar]
- 19.Blier P, de Montigny C. 1983. Electrophysiological investigations on the effect of repeated zimelidine administration on serotonergic neurotransmission in the rat. J. Neurosci. 3, 1270–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaput Y, de Montigny C, Blier P. 1986. Effects of a selective 5-HT reuptake blocker, citalopram, on the sensitivity of 5-HT autoreceptors: electrophysiological studies in the rat brain. Naunyn Schmiedebergs Arch. Pharmacol. 333, 342–348 10.1007/BF00500007 (doi:10.1007/BF00500007) [DOI] [PubMed] [Google Scholar]
- 21.Blier P, de Montigny C. 1980. Effect of chronic tricyclic antidepressant treatment on the serotoninergic autoreceptor. A microiontophoretic study in the rat. Naunyn Schmiedebergs Arch. Pharmacol. 314, 123–128 10.1007/BF00504527 (doi:10.1007/BF00504527) [DOI] [PubMed] [Google Scholar]
- 22.Dong J, de Montigny C, Blier P. 1999. Assessment of the serotonin reuptake blocking property of YM992: electrophysiological studies in the rat hippocampus and dorsal raphe. Synapse 34, 277–289 (doi:10.1002/(SICI)1098-2396(19991215)34:4<277::AID-SYN4>3.0.CO;2-W) [DOI] [PubMed] [Google Scholar]
- 23.Blier P, de Montigny C. 1984. Serotonergic but not noradrenergic neurons in rat central nervous system adapt to long-term treatment with monoamine oxidase inhibitors. Neuroscience 16, 949–955 10.1016/0306-4522(85)90107-1 (doi:10.1016/0306-4522(85)90107-1) [DOI] [PubMed] [Google Scholar]
- 24.Ghanbari R, El Mansari M, Blier P. 2011. Enhancement of serotonergic and noradrenergic neurotransmission in the rat hippocampus by sustained administration of bupropion. Psychopharmacology 217, 61–73 10.1007/s00213-011-2260-1 (doi:10.1007/s00213-011-2260-1) [DOI] [PubMed] [Google Scholar]
- 25.Chenu F, El Mansari M, Blier P. 2013. Electrophysiological effects of repeated administration of agomelatine on the dopamine, norepinephrine, and serotonin systems in the rat brain. Neuropsychopharmacology 38, 275–284 10.1038/npp.2012.140 (doi:10.1038/npp.2012.140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manta S, Dong J, Debonnel G, Blier P. 2009. Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J. Psychiat. Neurosci. 34, 272–280 [PMC free article] [PubMed] [Google Scholar]
- 27.Chernoloz O, El Mansari M, Blier P. 2009. Sustained administration of pramipexole modifies the spontaneous firing of dopamine, norepinephrine and serotonin neurons in the rat brain. Neuropsychopharmacology 34, 651–661 10.1038/npp.2008.114 (doi:10.1038/npp.2008.114) [DOI] [PubMed] [Google Scholar]
- 28.Chernoloz O, El Mansari M, Blier P. 2011. Long term administration of the dopamine D3/2 receptor agonist pramipexole increases dopamine and serotonin neurotransmission in the male rat forebrain. J. Psychiat. Neurosci. 37, 113–121 10.1503/jpn.110038 (doi:10.1503/jpn.110038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chernoloz O, El Mansari M, Blier P. 2012. Effects of sustained administration of quetiapine alone and in combination with a serotonin reuptake inhibitor on norepinephrine and serotonin transmission. Neuropsychopharmacology 37, 1717–1728 10.1038/npp.2012.18 (doi:10.1038/npp.2012.18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatsumi M, Groshan K, Blakely R, Richelson E. 1999. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 340, 249–258 10.1016/S0014-2999(97)01393-9 (doi:10.1016/S0014-2999(97)01393-9) [DOI] [PubMed] [Google Scholar]
- 31.Delgado PL, Miller HL, Salomon RM, Licinio J, Krystal JH, Moreno FA, Heninger GR, Charney DS. 1999. Tryptophan-depletion challenge in depressed patients treated with desipramine or fluoxetine: implications for the role of serotonin in the mechanism of antidepressant action. Biol. Psychiat. 46, 212–220 10.1016/S0006-3223(99)00014-1 (doi:10.1016/S0006-3223(99)00014-1) [DOI] [PubMed] [Google Scholar]
- 32.Cassidy F, Weiner RD, Cooper TB, Carroll BJ. 2010. Combined catecholamine and indolamine depletion following response to ECT. Br. J. Psychiat. 196, 493–494 10.1192/bjp.bp.109.070573 (doi:10.1192/bjp.bp.109.070573) [DOI] [PubMed] [Google Scholar]
- 33.Delgado PL, Moreno FA, Onate L, Gelenberg AJ. 2002. Sequential catecholamine and serotonin depletion in mirtazapine-treated depressed patients. Int. J. Neuropsychopharmacol. 5, 63–66 10.1017/S1461145702002778 (doi:10.1017/S1461145702002778) [DOI] [PubMed] [Google Scholar]
- 34.Szabo ST, de Montigny C, Blier P. 1997. Modulation of noradrenergic neuronal firing by selective serotonin reuptake blockers. Br. J. Pharmacol. 126, 568–571 10.1038/sj.bjp.0702343 (doi:10.1038/sj.bjp.0702343) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dremencov E, El Mansari M, Blier P. 2007. Noradrenergic augmentation of escitalopram response by risperidone: electrophysiologic studies in the rat brain. Biol. Psychiat. 61, 671–678 10.1016/j.biopsych.2006.05.015 (doi:10.1016/j.biopsych.2006.05.015) [DOI] [PubMed] [Google Scholar]
- 36.Beique JC, de Montigny C, Blier P, Debonnel G. 1999. Venlafaxine: discrepancy between in vivo 5-HT and NE reuptake blockade and affinity for reuptake sites. Synapse 32, 198–211 (doi:10.1002/(SICI)1098-2396(19990601)32:3<198::AID-SYN6>3.0.CO;2-2) [DOI] [PubMed] [Google Scholar]
- 37.West CH, Ritchie JC, Boss-Williams KA, Weiss JA. 2009. Antidepressant drugs with differing pharmacological actions decrease activity of locus coeruleus neurons. Int. J. Neuropsychopharmacol. 12, 627–641 10.1017/S1461145708009474 (doi:10.1017/S1461145708009474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawahara Y, Kawahara H, Kaneko F, Tanaka M. 2007. Long-term administration of citalopram reduces basal and stress-induced extracellular noradrenaline levels in rat brain. Psychopharmacology 194, 73–81 10.1007/s00213-007-0826-8 (doi:10.1007/s00213-007-0826-8) [DOI] [PubMed] [Google Scholar]
- 39.Blier P, Gobbi G, Turcotte JE, de Montigny C, Boucher N, Hébert C, Debonnel G. 2009. Mirtazapine and paroxetine in major depression: comparison of monotherapy versus their combination from treatment initiation. Eur. Neuropsychopharmacol. 19, 457–465 10.1016/j.euroneuro.2009.01.015 (doi:10.1016/j.euroneuro.2009.01.015) [DOI] [PubMed] [Google Scholar]
- 40.Blier P, Ward HE, Tremblay P, Laberge L, Hébert C, Bergeron R. 2010. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am. J. Psychiat. 167, 281–288 10.1176/appi.ajp.2009.09020186 (doi:10.1176/appi.ajp.2009.09020186) [DOI] [PubMed] [Google Scholar]
- 41.Ugedo L, Grenhoff J, Svensson TH. 1989. Ritanserin, a 5-HT2 receptor antagonist, activates midbrain dopamine neurons by blocking serotonergic inhibition. Psychopharmacology 98, 45–50 10.1007/BF00442004 (doi:10.1007/BF00442004) [DOI] [PubMed] [Google Scholar]
- 42.Dremencov E, El Mansari M, Blier P. 2009. Effects of sustained serotonin reuptake inhibition on the firing of dopamine neurons in the rat ventral tegmental area. J. Psychiat. Neurosci. 34, 223–229 [PMC free article] [PubMed] [Google Scholar]
- 43.Nelson JC, Mazure CM, Jatlow PI, Bowers MB, Price LH. 2004. Combining norepinephrine and serotonin reuptake inhibition mechanisms for treatment of depression: a double-blind, randomized study. Biol. Psychiat. 55, 296–300 10.1016/j.biopsych.2003.08.007 (doi:10.1016/j.biopsych.2003.08.007) [DOI] [PubMed] [Google Scholar]
- 44.Seager MA, Barth VN, Phebus LA, Rasmussen K. 2005. Chronic coadministration of olanzapine and fluoxetine activates locus coeruleus neurons in rats: implications for bipolar disorder. Psychopharmacology 181, 126–133 10.1007/s00213-005-2198-2 (doi:10.1007/s00213-005-2198-2) [DOI] [PubMed] [Google Scholar]
- 45.Chernoloz O, El Mansari M, Blier P. 2009. Electrophysiological studies in the rat brain on the basis for aripiprazole augmentation of antidepressants in major depressive disorder. Psychopharmacology 206, 335–344 10.1007/s00213-009-1611-7 (doi:10.1007/s00213-009-1611-7) [DOI] [PubMed] [Google Scholar]
- 46.Drevets WC. 1992. A functional anatomical study of unipolar depression. J. Neurosci. 12, 3628–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drevets WC. 2003. Neuroimaging abnormalities in the amygdala in mood disorders. Ann. NY Acad. Sci. 985, 420–424 10.1111/j.1749-6632.2003.tb07098.x (doi:10.1111/j.1749-6632.2003.tb07098.x) [DOI] [PubMed] [Google Scholar]
- 48.Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S, Jerabek PA. 2000. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol. Psychiat. 48, 830–843 10.1016/S0006-3223(00)01036-2 (doi:10.1016/S0006-3223(00)01036-2) [DOI] [PubMed] [Google Scholar]
- 49.Kennedy SH, Konarski JZ, Segal ZV, Lau MA, Bieling PJ, McIntyre RS, Mayberg HS. 2007. Differences in brain glucose metabolism between responders to CBT and venlafaxine in a 16-week randomized controlled trial. Am. J. Psychiat. 164, 778–788 10.1176/appi.ajp.164.5.778 (doi:10.1176/appi.ajp.164.5.778) [DOI] [PubMed] [Google Scholar]
- 50.Marek GJ, Aghajanian GK. 1999. 5-HT2A receptor or alpha1-adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur. J. Pharmacol. 367, 197–206 10.1016/S0014-2999(98)00945-5 (doi:10.1016/S0014-2999(98)00945-5) [DOI] [PubMed] [Google Scholar]
- 51.Ghanbari R, El Mansari M, Blier P. 2010. Sustained administration of trazodone enhances serotonergic neurotransmission: in vivo electrophysiological study in the rat brain. J. Pharmacol. Exp. Ther. 305, 197–206 10.1124/jpet.110.169417 (doi:10.1124/jpet.110.169417) [DOI] [PubMed] [Google Scholar]
- 52.Sheehan DV, Croft HA, Gossen ER, Levitt RJ, Brullé C, Bouchard S, Rozova A. 2009. Effects of sustained administration of quetiapine alone and in combination with a serotonin reuptake inhibitor on norepinephrine and serotonin transmission. Psychiatry 6, 20–3319724732 [Google Scholar]
- 53.Mørk A, et al. 2012. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. Pharmacol. Exp. Ther. 340, 666–675 10.1124/jpet.111.189068 (doi:10.1124/jpet.111.189068) [DOI] [PubMed] [Google Scholar]
- 54.Alvarez E, Perez V, Dragheim M, Loft H, Artigas F. 2012. A double-blind, randomized, placebo-controlled, active reference study of LuAA21004 in patients with major depressive disorder. Int. J. Neuropsychopharmacol. 15, 589–600 10.1017/S1461145711001027 (doi:10.1017/S1461145711001027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. 2000. Antidepressant effects of ketamine in depressed patients. Biol. Psychiat. 47, 351–354 10.1016/S0006-3223(99)00230-9 (doi:10.1016/S0006-3223(99)00230-9) [DOI] [PubMed] [Google Scholar]
- 56.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. 2006. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiat. 63, 856–864 10.1001/archpsyc.63.8.856 (doi:10.1001/archpsyc.63.8.856) [DOI] [PubMed] [Google Scholar]
- 57.Diazgranados N, et al. 2010. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiat. 67, 793–802 10.1001/archgenpsychiatry.2010.90 (doi:10.1001/archgenpsychiatry.2010.90) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rush AJ, et al. 2006. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a Star*D report. Am. J. Psychiat. 163, 1905–1917 10.1176/appi.ajp.163.11.1905 (doi:10.1176/appi.ajp.163.11.1905) [DOI] [PubMed] [Google Scholar]
- 59.Bunney BG, Bunney WE. 2012. Rapid-acting antidepressant strategies: mechanism of action. Int. J. Neuropsychopharmacol. 15, 695–713 10.1017/S1461145711000927 (doi:10.1017/S1461145711000927) [DOI] [PubMed] [Google Scholar]
- 60.Gardner JP, Fornal CA, Jacobs BL. 1997. Effects of sleep deprivation on serotonergic neuronal activity in the dorsal raphe nucleus of the freely moving cat. Neuropsychopharmacology 17, 72–81 10.1016/S0893-133X(97)00025-0 (doi:10.1016/S0893-133X(97)00025-0) [DOI] [PubMed] [Google Scholar]
- 61.Paul IA, Nowak G, Layer RT, Popik P, Skolnick P. 1993. Adaptation of the N-methyl-D-aspartate receptor complex following chronic antidepressant treatments. J. Pharmacol. Exp. Ther. 269, 95–102 [PubMed] [Google Scholar]
- 62.Holtzheimer PE, Mayberg HS. 2011. Deep brain stimulation for psychiatric disorders. Ann. Rev. Neurosci. 34, 289–307 10.1146/annurevneuro-061010-113638 (doi:10.1146/annurevneuro-061010-113638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Etiévant A, Oosterhof CA, Bétry C, Abrial E, Lambas-Senas L, Scarna H, Lucas G, Haddjeri N. 2011. Antidepressant-like action of medial prefrontal cortex deep brain stimulation is modulated by glial system. Eur. Neuropsychopharmacol. 21(Suppl. 2), 405–406 10.1016/S0924-977X(11)70654-X (doi:10.1016/S0924-977X(11)70654-X) [DOI] [Google Scholar]
- 64.Hamani C, Diwan M, Macedo CE, Brandao MI, Shumake J, Gonzalez-Lima F, Raymond R, Lozano AM, Fletcher PJ. 2010. Antidepressant-like effects of medial prefrontal cortex deep brain stimulation in rats. Biol. Psychiat. 67, 117–124 10.1016/j.biopsych.2009.08.025 (doi:10.1016/j.biopsych.2009.08.025) [DOI] [PubMed] [Google Scholar]