

Abstract
The family of the mammalian small heat-shock proteins consists of 10 members (sHSPs/HSPBs: HSPB1–HSPB10) that all share a highly conserved C-terminal alpha-crystallin domain, important for the modulation of both their structural and functional properties. HSPB proteins are biochemically classified as molecular chaperones and participate in protein quality control, preventing the aggregation of unfolded or misfolded proteins and/or assisting in their degradation. Thus, several members of the HSPB family have been suggested to be protective in a number of neurodegenerative and neuromuscular diseases that are characterized by protein misfolding. However, the pro-refolding, anti-aggregation or pro-degradative properties of the various members of the HSPB family differ largely, thereby influencing their efficacy and protective functions. Such diversity depends on several factors, including biochemical and physical properties of the unfolded/misfolded client, the expression levels and the subcellular localization of both the chaperone and the client proteins. Furthermore, although some HSPB members are inefficient at inhibiting protein aggregation, they can still exert neuroprotective effects by other, as yet unidentified, manners; e.g. by maintaining the proper cellular redox state or/and by preventing the activation of the apoptotic cascade. Here, we will focus our attention on how the differences in the activities of the HSPB proteins can influence neurodegenerative and neuromuscular disorders characterized by accumulation of aggregate-prone proteins. Understanding their mechanism of action may allow us to target a specific member in a specific cell type/disease for therapeutic purposes.
Keywords: small heat-shock proteins, neurodegeneration, chaperones, heat-shock proteins
1. Introduction
The family of the mammalian small heat-shock proteins, also called the HSPB proteins, consists of 10 members (HSPB1–HSPB10, see alternative names of each member in table 1), all of a relatively low molecular weight (MW, ranging from 15 to 45 kDa) and sharing some structural similarities, like a highly conserved C-terminal alpha-crystallin domain (ACD, [52]). This ACD plays an important role in the modulation of both structural and functional properties of the HSPBs. Indeed, monomers of the HSPB proteins associate (partially via their ACDs) into dimers that are thought to act as basic units/building blocks, capable of generating oligomers ranging from ca 200 to 600 kDa [49,53]. The various HSPB monomers can form both homo- and hetero-dimers as well as homo- and hetero-oligomeric complexes [54,55]. The dynamic association/dissociation of the oligomers has been suggested to be key to the function of the HSPB proteins and is often regulated by their phosphorylation state [49,56]. HSPB proteins are biochemically classified as molecular chaperones and participate in protein quality control; in fact, several HSPB family members have been shown to be able to bind to (partially) unfolded or to misfolded, aggregation-prone proteins [57] preventing their aggregation. In conjunction with ATP-dependent chaperones (e.g. HSP70s/HSPAs), the HSPB-bound clients can either be refolded or degraded; the mechanisms for either refolding or degradation is not fully understood, but may depend both on the state of the client and on the specific HSPB member that is bound to it (see later).
Table 1.
The mammalian small heat-shock proteins. n.a., not analysed as far as we know; AD, Alzheimer's disease; ALD, Alexander disease; CJD, Creutzfeldt–Jakob disease; DMPK, dystrophia myotonica-protein kinase; DRG, dorsal root ganglia; MNDs, motor neuron diseases; NPD, Neuman Pick's disease; PD, Parkinson's disease; RT qPCR, real-time quantitative polymerase chain reaction.
| HSPB name (alt. name) | expression in |
stress-induced changes after brain injury/during neurodegeneration |
biochemical function (as detected in cells after upregulation) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| brain or sciatic nerves | cardiac muscles | skeletal muscles | ischaemia or nerve injury | misfolding diseases (neurons) | misfolding diseases (atrocytes/glia) | promoting refolding | anti-aggregation |
other | |||
| polyQ | ALS | other | |||||||||
| HSPB1 (hHsp27,mHsp25) | low/moderate in: cortex, cerebellum, striatum, hippocampus, spinal cord [1–3] | high [4] | high [4] | UP in neurons [5,6] | generally UNCHANGED UP in Purkinje cell (NPD) [7,8] | generally UP [9–16] extracellular in AD brain [17] | strong [3,18] | no [1,19] | SOD-1: +/- [20] TDP-43: no [21] | n.a. | cytoskelatal stabilization [22] anti-oxidant [23] |
| HSPB2 (MKBP) | low in: cortex, hippocampus, sciatic nerves [1,2] | high [4] | high [4] | n.a. | n.a. | extracellular in AD brain [17] | no [1] | no [1] | n.a. | n.a. | activation of DMPK [24]; maintaining myofibrillar integrity [25] |
| HSPB3 (HSPL27) | low in: cortex, cerebellum, spinal cord, sciatic nerve (RT qPCR) [1–3] | high [4] | high [4] | n.a. | n.a. | n.a. | no [1] | no [1] | n.a. | n.a. | maintaining myofibrillar integrity [25] |
| HSPB4 (αA-crystallin) | brain: not reported low in: sciatic nerve [2] | none | none | n.a. | n.a. | n.a. | ++ [1] | no [1] | n.a. | n.a. | maintaining the proper refractive index in the lens [26] |
| HSPB5 (α-B-crystallin) | high in: white matter, cortex, cerebellum, striatum, hippocampus, spinal cord, sciatic nerve [1–3] | high [4] | high and increasing with aging [4] | n.a. | UP in: neurons (in MNDs, NPD, AD, PD, CJD) [27–32] | UP in: reactive glial cells, microglia, oligodendrocytes [11,13,16,17, 33, 34] | ++ [1] | no [1] | no [21] | PD: ++[35] ALD: ++ [36] | cytoskeleton stabilization; maintaining the proper refractive index in the lens [37,38] |
| HSPB6 (Hsp20, p20) | high in: cortex, cerebellum cortex, choroid plexus of the ventricles, telencephalon, spinal cord, sciatic nerve [1–3] | high [4] | high [4] | UP after hypoxic stress (in neonatal hippocampus) [39,40] | n.a. | n.a. extracellularly in AD brain [17] | + [1] | + [1] | no [21] | n.a. | cytoskeleton stabilization [41] |
| HSPB7 (cvHsp) | low in brain [1,3] | high [4] | moderate/high increasing with aging [4] | n.a. | n.a. | n.a. | no [1] | +++ [1] | no [21] | n.a. | maintaining myofibrillar integrity [42] |
| HSPB8 (Hsp22, H11, E2IG1) | low in: cortex, cerebellum, striatum, hippocampus, spinal cord, sciatic nerve. low/moderate in motoneurons [1–3,43] | high [4] | high [4] | n.a. | UP in ALS in spinal cord and motoneurons [10,43] | UP in astrocytes in AD, PD, polyQ [44] and mildly UP in glial cells (not in microglia) in ALS [43,45] | no [1] | + [3, 46–48] | SOD-1: ++ [43] TDP-43: +++ [21] | n.a. | protein synthesis inhibitor; autophagy simulator [43,46–48] |
| HSPB9 (CT51) | n.a. (testis only?) | n.a. (testis) | n.a. (testis only?) | n.a. | n.a. | n.a. | no [49] | ++ [49] | +/− [21] | n.a. | cancer/testis antigen [50] |
| HSPB10 (ODFP, ODF1) | n.a. (testis only) | n.a. (testis only) | n.a. (testis only) | n.a. | n.a. | n.a. | no [49] | no [49] | n.a. | n.a. | cytoskeleton stabilization [51] |
The chaperone activity of small HSPs has been discovered and explored mainly in cell-free experiments with purified proteins [58–60] and it accounts for, for example, the role that HSPB4 plays in maintaining eye transparency [26]. Whether this chaperone function is also underlying other cellular functions of HSPB members is less clear. For example, some HSPBs members (e.g. HSPB1 and HSPB5) have the capability to modulate the assembly and stabilization of cytoskeleton components, such as actin and intermediate filaments [22,37,38,41,61–64], but how far these actions rely on their chaperone activity is unknown. It is also not clear whether other HSPB family members serve in cytoskeletal protection or whether different cytoskeletal and contractile elements may require different HSPB members. Other functional endpoints that have been shown to be affected by HSPB members include the maintenance of proper cellular redox state, protecting cells from oxidative stress conditions (HSPB1, [65]), a general anti-apoptotic function (HSPB1, [66,67]) and a role in skeletal muscle cell differentiation (HSPB2 and HSPB3, [25]). The biochemical mechanisms underlying these different cellular effects of the various HSPB members are often still elusive and not always directly linked to the in vitro-defined chaperone-like activities.
Several HSPB family members have been suggested to be protective in a number of neurodegenerative and neuromuscular diseases that are characterized by protein aggregation and axonal transport defects. This directly relates back to the two most postulated actions of these HSPBs: their chaperone action and cytoskeletal stabilizing function, respectively. On the other hand, mutations in some members of the HSPB family (namely, HSPB1, HSPB3, HSPB4, HSPB5 and HSPB8) have been associated with neurological and muscular alterations, suggesting that loss of their function as general chaperones or/and cytoskeletal protectors is crucial for maintaining neuronal and muscular cell function and viability. Here, we will discuss the implication of HSPB proteins in neurodegenerative and neuromuscular diseases, focusing our attention on how the differences in the HSPB activities influence their protective functions.
2. The role of HSPBs in neurodegenerative and neuromuscular diseases
(a). Some HSPBs are upregulated in neurodegenerative diseases
The formation and accumulation of insoluble aggregates containing misfolded/mutated proteins is a pathological hallmark of many neurodegenerative and neuromuscular disorders with late onset, including amyotrophic lateral sclerosis (ALS), Alzheimer's disease (AD), Parkinson's disease (PD), polyglutamine (polyQ) diseases (e.g. Huntington disease (HD), spinal and bulbar muscular atrophy (SBMA), etc.), and Creutzfeldt–Jacob disease (CJD). These diseases include both sporadic and genetically inherited forms. Some inherited forms are linked to mutations in a specific protein that misfolds and/or is prone to aggregation. Notably, often the proteinaceous aggregates are found in the corresponding degenerated tissues. Protein aggregation is a multi-step nucleation-dependent process starting from the oligo merization of the self-associating misfolded protein, which generates pre-fibrillar aggregates (detergent-soluble). Pre-fibrillar aggregates can subsequently generate fibrillar structures (detergent-insoluble). During this aggregation process, the mutated protein interacts with and entraps different intracellular components. These include several HSPs (including several HSPB members; [68]), components of the proteasome system, elements involved in vesicular transport and transcription factors. Sequestration of HSPs and proteasomal components may reflect their failure to degrade the misfolded proteins as well as their failure to prevent their aggregation or their unsuccessful attempts to disaggregate the inclusions. As a consequence, the HSPs might be diverted away from their normal functions. Similarly, entrapping of vesicular transport components and transcription factors may lead to their reduced activities in essential neuronal processes. Finally, aggregates might physically impair cellular processes (e.g. axonal transport) [69]. Probably, a combination of these events contributes to disease progression and, therefore, aggregation has long been considered the key pathogenic mechanism. Nevertheless, the precise role of aggregates in neuronal cell death and disease progression is still largely debated. Several studies have suggested that not all forms of aggregates may be toxic [70–72]. In fact, large (amyloidic) aggregates that entrap these various key cellular elements have been found to be protective in certain systems and under certain conditions [70–75]. It has thus been proposed that smaller (amorphic) oligomeric and/or heteromeric species are more toxic as they are capable of freely moving around in the cellular milieu and perturb various neuronal functions. Besides the aforementioned effects of large aggregates (e.g. impairment of cellular proteostasis, alterations of the degradative systems, disruption of axonal transport, dysregulation of the transcription of specific genes), this could also include, for instance direct effects on membrane integrity and/or synaptic functioning [70,73,76,77]. However, irrespective of the presumed differential toxicity attributed to these different aggregates, it is clear that any factor/approach able to decrease mutated protein accumulation, to prevent the initiation of their aggregation or to facilitate the clearance of (early) aggregates will greatly contribute to restore (or maintain) the normal neuronal proteostasis and function, thus slowing down disease progression. This potentially can be achieved by boosting specific molecular chaperones/HSPs (e.g. HSP70s/HSPAs and HSP40s/DNAJs and some sHSPs/HSPBs), which will avoid or attenuate protein aggregation, or by stimulating the degradative pathways (i.e. autophagy), which will help to clear the aggregates. The efficacy of both approaches has been experimentally demonstrated using cellular and/or animal models of aggregate diseases (e.g. polyQ diseases, AD and PD) [19,43,46,78–91].
Here, we will focus on boosting the activity of the HSPB proteins as a potential approach to counteract protein toxicity in neurodegenerative diseases, keeping in mind that not all HSPB family members share the same functional properties [1,92]. Therefore, the putative protective efficacy of the 10 members may largely differ depending on the specific protein causing the aggregation disease.
In addition, the various members present very different tissue/cell-specific distribution. In basal conditions, only some HSPB family members are expressed in the central nervous system (CNS) and very few in neurons (some are mainly confined in glial components of the CNS; table 1). However, misfolded protein expression and the consequent cell stress during neurodegeneration might trigger both the overexpression of some HSPBs already present in target cells or of HSPBs normally silent in neurons (table 1). Thus even the HSPBs normally absent in the brain might become players in the intracellular response to mutant misfolded protein neurotoxicity [92]. Regarding the specific pattern of expression of the HSPB family members, HSPB2 and HSPB3 are mainly expressed in the skeletal muscle cells [25], while HSPB9 and HSPB10 are only found in testis [52]. HSPB7 was originally termed cvHSP and indeed is highly expressed in cardiac tissue, although it is also expressed at lower levels in several other tissues, including the brain [1,92]. HSPB6 is constitutively and highly expressed in smooth, cardiac and skeletal muscles and plays a role in muscle function [93]. The other members (HSPB1, HSPB5 and HSPB8) are highly expressed in muscle tissues, but are also expressed in many other tissues, including in the CNS, with peculiar cell-type-dependent differences in expression (table 1). For example, while HSPB1 is highly expressed in peripheral sensory neurons [94], HSPB8 is highly expressed in motor neurons [17,33,43,44,95]. As it will be discussed later, some members of the HSPB family can prevent aggregation of (some) disease-associated mutant proteins and that is though to be protective in neuronal cells. Thus, HSPB upregulation (both at the level of neuronal and glial cells) might represent a protective cellular response to neuronal damage. However, it is also possible that this over-induction occurs as a consequence of neuronal stress, without an active participation to the protective processes. While the presence of HSPB1 and HSPB5 in ballooned neurons (e.g. in AD, CJD, etc.) could suggest that they interact with aggregates in neurons (either actively engaged in protein quality control here or reflecting failed function or being trapped here), the upregulation of the same proteins in astrocytes is more difficult to understand. But several lines of evidence suggest that misfolded proteins may act in a non-cell autonomous way. For example, the primary toxicity can be exerted on the glial (and microglia) cells surrounding affected neurons, which may thus also be indirectly affected via release of neurotoxic factors from glia or reduced removal of neurotoxic agents by the glia, i.e. through an altered dialogue between these cells [96,97]. Indeed, recent evidence suggests that astrocytes, together with other glial cells (especially microglia), participate in the maintenance of the extracellular milieu containing debris and aggregates from dying neurons. A comparable non-cell autonomous mechanism may play a role in motor neuron diseases, involving interactions between motor neurons and the target muscle cells [98] in which muscle denervation from motor neurons rather than being causative to muscle degeneration also may result as consequence of initial muscle damage/death [99,100]. It can be hypothesized that HSPB8, for example, somehow is involved in this process: in anterior horn spinal cord of ALS mice, HSPB8 is highly induced not only in motor neuron (at very high levels in this primary target of mutant protein toxicity), but also in the glial cells (at lower levels) of the affected regions [43], and in muscle tissues (at the highest levels; P. Rusmini, V. Crippa, E. Giorgetti, A. Boncoraglio, R. Cristofani, A. Poletti 2013, unpublished data). Studies should therefore not only focus on elucidating the role of HSPBs in individual populations of cells, but also look at cell non-autonomous and cell–cell interactions.
Finally, low or even a complete lack of constitutive and induced expression in target cells of individual HSPB members that did reveal protective power in cell models of disease should not be a reason to imply that these members cannot exert protective functions in disease. Obviously, such members are not part of the canonic intrinsic (neuronal/glial/muscular) response to the diseased protein, but could be still considered potential therapeutic targets if one were able to induce them by pharmacological means, with the specific aim to assist mutant neurotoxic protein clearance.
3. Refolding and anti-aggregation are distinct properties of the HSPBs
As previously mentioned, a number of neurodegenerative diseases are characterized by the progressive accumulation of aggregation-prone proteins. Upregulation of specific members of the HSPB family could slow down or completely inhibit the aggregation process in cell models [43,46–48,92,95,101], with significant differences among the 10 HSPB family members. In particular, using mutated polyQ-containing proteins (e.g. huntingtin and ataxin-3) we showed that HSPB6, HSPB7, HSPB8 and HSPB9 overexpression inhibited protein aggregation and protected against its mediated toxicity, while overexpression of all the other members had no effect [1]. Curiously, HSPB6, HSPB7, HSPB8 and HSPB9 could not efficiently facilitate the refolding of denatured substrates (e.g. heat-denatured firefly luciferase), but rather were linked to protein degradation by either the proteasomal or the autophagic systems (table 1; see also later, [1,46]). Inversely, the strongly heat-stress-regulated HSPB1, HSPB4 and HSPB5 members, which were found to be very efficient in facilitating the refolding of heat-denatured substrates, both in cells and in vitro, showed poor anti-aggregation effects against polyQ proteins (table 1) [1]. These data suggest that the different HSPB members may either have different client specificity and/or have a different impact on client processing.
Despite the lack of anti-polyQ aggregation activity, HSPB1 and HSPB5 showed some protective effects in some cell models of polyQ disease. As HSPB1 and HSPB5 can increase the resistance of the cytoskeletal network [22,37,38,41,62–64] and are upregulated both in ballooned neurons and astrocytes [9,27,28], one may speculate that they protect axonal transport and vesicular trafficking against disruption by the protein aggregates and thus delay the consequences of aggregation, without affecting aggregate formation as such. Yet, when directly comparing the cytoprotective effects of, for example, HSPB5 to that of HSPB7 in cell models, we found HSPB5 protection was marginal ([1], figure 1c).
Figure 1.
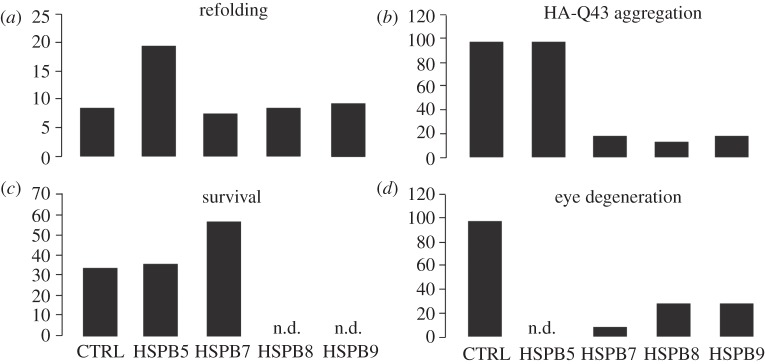
Comparison of effects of overexpression of HSPB5, HSPB7, HSPB8 and HSPB9 on the cellular capacity to refold heat-denatured luciferase (a) or to suppress protein aggregation (b) and toxicity (c) of polyQ proteins in cells or to reduced polyQ-mediated eye degeneration in flies (d). Data compiled and replotted from [1,101] and (M. P. Zijlstra, B. Kanon, H. H. Kampinga 2011, unpublished data).
Consistent with the cellular data, the three most potent suppressors of polyQ aggregation (HSPB7, HSPB8 and HSPB9) also reduced polyQ-mediated eye degeneration in a Drosophila melanogaster in vivo model [1,101] (M. P. Zijlstra, B. Kanon, H. H. Kampinga 2011, unpublished data; figure 1). These latter three HSPB members have been implied in polyQ degradation via the proteasome (HSPB9: M. P. Zijlstra, B. Kanon, H. H. Kampinga 2011, unpublished data), by supporting autophagic clearance of polyQ aggregates [1] or by enhancing autophagic flux [43,46,95,101,102], see below).
4. Anti-aggregation activity of HSPBs: stimulating degradative pathways
As mentioned earlier, HSPB7, HSPB8 and HSPB9 seem to have the potential to trigger (or facilitate) proteasomal and/or autophagic degradation of certain misfolded proteins. Among these HSPB members, HSPB8 is probably the most studied in this context. Overexpression of HSPB8 efficiently prevents aggregation of different polyQ-containing proteins (huntingtin, ataxin-3 and androgen receptor (AR), responsible for HD, spinocerebellar ataxia type 3 (SCA3) and SBMA, respectively) [1,47,101], as well as SOD1 (protein responsible for ALS) and various truncated forms of TDP-43s (associated with both ALS and frontotemporal dementia (FTD; table 1)) [43,95].
Figure 2 illustrates this for the mutant ARpolyQ that causes SBMA. Overexpression of HSPB8 in SBMA motor neurons leads to a substantial decrease in mutant ARpolyQ aggregates (IF, figure 2a) and large insoluble species (filter retardation assay, FRA; figure 2b,c), which in this disease are triggered by the AR ligand testosterone. Several data indicate that this anti-aggregation activity of HSPB8 is caused by a facilitation of autophagy-mediated degradation of the mutated proteins or their (initial) aggregates. In this process, HSPB8 collaborates with BAG3 [46] and HSPA8 (Hsc70) and CHIP [43,104]. Indeed, as shown in figure 2, the anti-aggregation effects of HSPB8 were greatly reduced in the presence of the autophagy inhibitors 3-methyladenine (3-MA) or bafilomycin (figure 2c).
Figure 2.

(a) High-resolution fluorescence microscopy analysis of NSC34 cells transfected with GFP-AR.Q48 and an empty vector (pCDNA3/untreated) or pCNEO-cMyc-HSPB8 (expressing rat HSPB8). Cells were treated with 10 nM of testosterone (+T) for 48 h and fixed using a 1 : 1 solution of 4% saccharose and 4% paraformaldeyde in 0.2 N PB (25 min at 37°C under weak agitation), and then in ice-cold methanol (10 min, RT). Nuclei were stained with DAPI (dark grey, blue). Images were obtained at 63× magnification. Fluorescence analysis showed that T-treated GFP-AR.Q48 formed intracellular aggregates that are completely removed by HSPB8; a massive decrease of mutant ARpolyQ protein levels was also detectable, suggesting that HSPB8 induces the removal of mutant ARpolyQ from immortalized motor neurons. Scale bar, 10 µM. (b) The effect of HSPB8 on the aggregation propensity of two AR mutants characterized by a polyQ of different length was tested using filter retardation assay (FRA). NSC34 cells co-transfected with plasmids coding for AR.Q46 or AR.112 and either an empty vector (pCDNA3) or pCNEO-cMyc-HSPB8 (expressing rat HSPB8) were treated with ethanol (−T) or with 10 nM of testosterone (+T) for 48 h. Cell lysates were prepared for FRA as previously described [86]. (c) The involvement of the autophagic pathway in the pro-degradative effects of HSPB8 on mutant AR.Q46 was tested by FRA after selective blockage of the autophagic flux. NSC34 cells were co-transfected with plasmids coding for AR.Q46 and an empty vector (pCDNA3) or pCNEO-cMyc-HSPB8 (expressing rat HSPB8) and were treated with ethanol (−T) or with 10 nM of testosterone (+T) for 48 h. Autophagic flux was modulated using treatments for 24 h with either 10 mM of 3-methyladenine (3-MA, acting on autophagosome assembly) or 10 nM of bafilomycin (BAF, acting on the process of fusion between autophagosome and lysosome as well as on the ATP-dependent H+-pump [103]). Cell lysates used for FRA were prepared as previously described [86]. (Online version in colour.)
Differently from many other members of the HSPB family (e.g. HSPB1 and HSPB7), HSPB8 forms a stable complex with the HSPA8 (Hsc70) co-chaperone BAG3, which may explain why HSPB8 is so efficient in autophagy-mediated degradation of misfolded polyQ substrates. We previously showed that not only HSPB8 stability, but also its anti-aggregation and pro-degradative functions, depend on its association with BAG3, whose knock-down prevented HSPB8 from exerting its protective role [46]. Also, within the complex, BAG3, but not HSPB8, is responsible for the stimulation of autophagy [46]. This suggests that HSPB8 might play a role in the recognition and delivery of the cargo, via BAG3, to the autophagosomes for degradation. In addition, we found that HSPB8 (in complex with BAG3), besides participating in autophagy-mediated degradation of misfolded proteins, is also involved in the translational shut-down mediated by the induced phosphorylation of eIF2 alpha that occurs during proteotoxic stress. Interestingly, induction of phospho-eIF2 alpha, which we observed upon overexpression of HSPB8, BAG3 or of the complex, leads to both protein synthesis inhibition (which decreases the total load of proteins to be refolded or degraded) and autophagy stimulation (which clears the aggregated proteins accumulating during proteotoxic stress) [48]. While in vitro translation experiments indeed have revealed that HSPB8 can cause translational shut-down, precise insight into how HSPB8 can modulate translation upon stress is still missing. Similarly, future studies will be needed to reveal the precise role of HSPB8 in the complex and if and how client recognition and targeting to autophagy is coupled to its action on translation.
5. The anti-aggregation power of HSPBs depends on several factors
The aggregation propensity of misfolded proteins depends on several factors, including the exposition of hydrophobic residues, the alteration of specific conformed domains, the capability to generate beta-plated sheets, and so on. These factors may affect the kinetics of aggregation, the biophysical nature of aggregates that are ultimately formed (e.g. insolubility and reversibility) and also the location inside the cells where these proteins may accumulate [105]. All of these may obviously also affect the possibilities of diverse HSPBs to interact and deal with these various proteins before or after they have aggregated. Even for apparently similar clients like the proteolytic polyQ-containing fragments, the length of the polyQ stretch and the kinetics of aggregation (the longer the polyQ repeat, the faster the aggregation [1]) were found to impede the possibility of the various HSPB members to suppress aggregation. While HSPB8 and HSPB9 were equally (or even a bit more) effective compared with HSPB7 on huntingtin fragments with relatively short expansions (43Q, figure 1 [1]) only HSPB7 was efficient in suppressing aggregation of the longest expansions tested (119Q; see [1]). For HSPB8, similar observations were made in our previous findings showing that HSPB8 efficiently decreases both soluble and insoluble levels of huntingtin fragments with relatively short expansions (43Q; [46,47]), but has no effect on long expansions (Q119; [1]). We also tested the selective effect of HSPB8 on ARpolyQ containing a stretch of different size (Q46 versus Q112), both being in the pathological range (even if no SBMA patients with Q112 have been described so far). The two types of ARpolyQs have a marked difference in their aggregation power, in response to the AR ligand [72,73]. While HSPB8 almost completely counteracts AR.Q46 aggregation, induced by testosterone, its effects on AR.Q112 are much lower, since there is only a relatively small decrease in the total amount of insoluble ARpolyQ (figure 2b).
These differences in the efficiency of HSPB7, on the one hand, and HSPB8 and HSPB9, on the other hand, to prevent aggregation of polyQ proteins with different expansion sizes clearly suggests that their modes of action to deal with these disease-related proteins must be largely different. HSPB8 and also HSPB9 (M. P. Zijlstra, B. Kanon, H. H. Kampinga 2011, unpublished data) seem to act by lowering the level of soluble species (non-aggregated or/and early aggregate intermediates) apparently maintaining these substrates in a state competent for degradation, thus disposing of them before they form large, insoluble species. In the case of HSPB8, the lowered levels of soluble mutant polyQ proteins might be due, at least in part, to its effects on eIF2 alpha phosphorylation and translational attenuation [48]. Also, HSPB8, together with the co-chaperone BAG3, the chaperone HSP70 and the ubiquitine-ligase CHIP, works through stimulating autophagic degradation [43,46,48,106], thus also taking care of early nucleating species. Concerning HSPB9, it was found to stimulate proteasomal degradation (M. P. Zijlstra, B. Kanon, H. H. Kampinga 2011, unpublished data), which may lower the number of nucleating species of the polyQ proteins; however, when nucleation is initiated, which occurs more readily with longer polyQs, HSPB9 might become ineffective. Since HSPB9 is only expressed in testis, HSPB9 upregulation should not play a role in neurodegenerative diseases. However, as stated before, it is important to underline that pharmacologically induced upregulation of specific members of the HSPB family (normally not expressed or upregulated) in a specific neuronal cell type might still result in protection, therefore representing a good therapeutic approach. For those HSPB members that are already expressed in target cells, drugs may be designed that stimulate their (chaperone-like) activity, e.g. by acting on the phosphorylation or oligomeric status of the HSPB members. Alternatively, or in case HSPB members are not already expressed in the target cells, analysis on expression regulation of the various HSPB members combined with drug screens using reporter constructs may identify routes towards boosting or inducing expression of individual disease-ameliorating members.
HSPB6, which only effectively suppresses shorter polyQs [1,107], also plays a role in modulating autophagy [108]. By contrast, HSPB7 does not change the rate of proteasomal degradation and does not increases the autophagic flux; rather, it appears to prevent early aggregates from nucleating into inclusions with sizes that are too large to be handled by the autophagic machinery, probably by marking these early seeds, which enables their shuttling into the autophagosomes [1,57,109,110]. This HSPB7 action thus does not rely on the speed of seed formation but rather on the rate at which these seeds grow (and thus is less dependent on the size of the expansion).
Another aspect that might influence the efficacy of the diverse HSPBs to prevent mutated protein aggregation is represented by specific physical properties of the mutant protein and aggregate itself. Indeed, the effects of HSPB members on different aggregation-causing mutants seem to differ widely (table 1). It has been shown that, while polyQ proteins form aggregates with a core that is inaccessible to nascent proteins, mutated SOD1 (G85R/G93A), associated with ALS, forms a porous aggregate, through which nascent proteins can diffuse [110]. As stated above, we have already shown that overexpression of HSPB8 efficiently prevented the aggregation and facilitated the autophagy-mediated degradation of mutated SOD1 and of various mutated forms of TDP43, which is associated with both ALS and FTD [43] (figure 3a). When we compared the HSPB8 activity to that of other HSPB family members with anti-aggregation activity towards mutated polyQ proteins, namely HSPB6, HSPB7 and HSPB9, none of these blocked the aggregation of the truncated mutant form of TDP43 (TDP43 ΔC; figure 3b: HSPB1-5-6-7-9; table 1). HSPB1 and HSPB5, which mainly showed pro-refolding activity, were also unable to prevent aggregation of TDP43 ΔC (figure 3b). HSPB9 seems to have a mild effect on TDP-43 ΔC. Why HSPB6 and HSPB7 showed no anti-aggregation activity towards TDP43 ΔC, while being very active towards mutated polyQ proteins [1,107] (table 1), is still unclear and will be investigated in the future.
Figure 3.

HSPB8 blocks the aggregation of several disease-associated mutant forms of TDP43. (a) HEK293 cells were transfected with vectors encoding for different disease-associated mutant forms of TDP43 (ΔC, N352S, G348V or N345K, kindly provided by Dr E. Buratti) and either an empty vector or a vector encoding for human HSPB8. Cellular lysates were prepared 72 h post-transfection and the aggregation of TDP43 mutants was analysed by FTA (as previously described; [43]). (b) HEK293 cells were transfected with a vector encoding for ΔC TDP43 and either an empty vector or vectors encoding for various members of the HSPB family (V5-HSPB1, V5-HSPB5, V5-HSPB6, V5-HSPB7 and V5-HSPB9). Aggregation of ΔC TDP43 was analysed as described in (a).
Finally, another factor that can explain the differential efficacy of the various members of the HSPB proteins in inhibiting protein aggregation is the stage at which they act. As previously mentioned, many neurodegenerative diseases, including AD, polyQ diseases and PD, are characterized by the accumulation of fibrillar proteinaceous aggregates in specific neuronal types [111–117]. The formation of these fibrillar aggregates consists of a multi-step process involving a nucleation step and the subsequent elongation of the fibrils [118]. Interestingly, it has been shown that some HSPBs can only prevent protein aggregation at a specific stage. For example, HSPB5, which showed very high refolding capacity in cells and in vitro but no anti-aggregation activity towards mutated polyQ proteins [1], inhibits fibrillar aggregate formation of mutated alpha-synuclein, associated with PD, only at early stages. HSPB5 binds to partially folded monomers of mutated alpha-synuclein, thereby preventing mature fibril formation and shifting the equilibrium to monomer fibrils, which can be easily disposed/degraded [35,119,120]. A similar mechanism has been shown for ataxin-3. HSPB5 can significantly inhibit the first stage of ataxin-3 aggregation [121], by directly interacting with the Josephin domain of ataxin-3, which has an intrinsic tendency to aggregate and form fibrils [122]; however, HSPB5 is ineffective on already formed SDS-insoluble fibrils of mutated ataxin-3. All together, these data strongly suggest that HSPB5 acts as a chaperone specifically towards growing fibrils at an early stage and may have only limited protective powers because it cannot block protein aggregation at later stages and does not seem to target its bound substrate to the proteolytic pathways. They also suggest that, in order to be efficient in inhibiting protein aggregation, upregulation of HSPB5 should either take place at a very early stage of disease or occur concomitantly with the upregulation of other chaperones able to bind to and target intermediate species and/or fibrils to degradation.
6. Subcellular localization of the aggregate-prone species and HSPBs
Finally, subcellular localization can influence the efficiency of the various members of the HSPB family in inhibiting the aggregation of a specific misfolded substrate involved in neurodegenerative diseases. In fact, while the ubiquitin proteasome system is present and active both in the nucleus and in the cytoplasm, autophagy (which can be facilitated by some HSPB members; see earlier) is confined to the cytoplasm [123,124]. HSPB8 and HSPB6, which mainly rely on autophagy, are expected to mainly act on cytoplasmic aggregating proteins. Indeed, HSPB8 could only efficiently block the aggregation and facilitate the autophagy-mediated clearance of ARQ46, which is located in the cytosol, but had very limited effect on ARQ112, which is aggregating inside the nucleus. Considering that the cytoplasmic retention of a mutated form of AR containing a Q112 tract ameliorates disease via autophagy [125], strategies that allow keeping the mutated proteins in the cytosol, where several HSPBs (and other molecular chaperones) can participate directly or indirectly in their targeting to the autophagosomes for degradation, will slow-down disease progression. Whether some HSPBs can also participate in modulating the shuttling of the mutated proteins from the nucleus to the cytoplasm and whether this may contribute to their protective effects is still unknown.
7. HSPB-mediated protection can also be independent of anti-aggregation/pro-refolding activities and/or autophagy facilitation
HSPB1, which shows good refolding capacity, had no anti-aggregation activity in cells against mutated huntingtin, both with short and long polyQ stretches [1] (table 1). However, overexpression of HSPB1 could inhibit the aggregation of mutated SOD1 [20] (table 1) and could also prevent the toxicity mediated by several polyQ-containing proteins [19]. This may depend on the different physical properties of the aggregating substrates (e.g. immobile versus mobile aggregates), as stated before, although so far no experimental proof supporting such a hypothesis exists. However, in the case of HSPB1, the protective effects may also be due to other specific functions, which are not related to refolding or inhibition of aggregation, but rather to its ability to prevent the activation of APAF-1 by the cytochrome C released from the mitochondria, that will trigger the apoptotic caspase cascade, as well as on its anti-oxidants effects [67]. In fact, overexpression of HSPB1 exerts a protective effect and significantly decreases cell death in cellular models of HD, characterized by high oxidative stress, by maintaining the redox state of the cell without showing effects on aggregation [19]. Moreover, in neuronal-like cells, HSPB1 could also protect against the toxicity mediated by mutated ataxin-3, associated with SCA3, where increased oxidative stress has been suggested to play a role in disease pathogenesis, again without effects on aggregates. Interestingly, overexpression of mutated ataxin-3 correlated with a reduction in the expression levels of HSPB1, suggesting that a decrease in the function of HSPB1 (not directly related to effects on the diseased protein as such) may play a role in disease progression. Such a decrease in HSPB1 expression has also been documented in other forms of SCA diseases, like SCA7 [126,127] and in transgenic mice models of SCA-17 [128]. This would imply that re-introduction of HSPB1 can compensate for such a loss of function of HSPB1 as a factor contributing to disease progression. However, data in HD mice did not reveal such an effect [129], suggesting that this may not apply to all polyQ diseases.
8. Conclusions and perspectives
Although similar in terms of primary sequence, the various members of the mammalian HSPB family are differentially expressed in tissues and cells, have different abilities to form homo- and hetero-oligomers, show other non-HSPB partner interactions and display different functions. The tissue-selective expression pattern of the members probably reflects a highly specific need of a given HSPB to assure proper function and viability of that cell type/tissue, and a particular HSPB may exert a protective function in a specific cell type. Some functional redundancy does exist between the various members, as evidenced by findings that several HSPB members can handle the same (un- or misfolded) protein equally (e.g. assist their (re)folding). However, HSPB members posses different affinity and specificity for clients and may handle the same client differently (e.g. routing them to proteasomal or autophagosomal degradation). Thus, depending on their client specificity and mechanism of action, only upregulation of specific HSPB members would exert protective functions against neurodegeneration. Moreover, impaired HSPB function may harm certain tissues more than others and explain why certain HSPB mutants have been linked to tissue-specific (e.g. motor neurons and muscle cells) degeneration. In addition, the same HSPB may have different biochemical activities: depending on their oligomeric status, they may independently function as chaperones for soluble proteins, as stabilizers/chaperones for cytoskeletal elements, or as modifiers of the cellular redox state. As a consequence of these properties, the same HSPB member may protect more or less efficiently and certain HSPB members may be better targets than others.
In general, a better understanding of the client specificity and functional diversity of the various HSPBs will be required in order to target a specific member or functionality thereof for therapeutic purposes.
Acknowledgements
This study is supported by Marie Curie International Reintegration grant (PIRG-03-GA-2008-230908, to S.C.), Prinses Beatrix Fonds/Dutch Huntington Association (WAR09-23, to S.C. and H.H.K.), Association Française contre les Myopathies Trampoline grant (2010, to S.C.), Rita Levi Montalcini Prize (2011, to S.C.), Telethon, Italy (GGP06063 and GGP07063 to A.P.), Fondazione CARIPLO (2008-2307, to A.P.), ARISLA, Italy (ALS_HSPB8, to A.P. and S.C.), Italian Ministry of Labour, Health and Social Affairs (2007-36; 2008-15), Regione Lombardia (to A.P.) and convenzione Fondazione Mondino/UNIMI (to A.P.), Fondation Thierry Latran, France (FLT AAP091102, to A.P.), Università degli Studi di Milano (Italy, to A.P.). The cDNAs encoding for FLAG-tagged deltaC TDP43 (ΔC), N352S TDP43, G348V TDP-43 and N345K TDP43 were a kind gift from Dr E. Buratti.
References
- 1.Vos MJ, Zijlstra MP, Kanon B, van Waarde-Verhagen MA, Brunt ER, Oosterveld-Hut HM, Carra S, Sibon OC, Kampinga HH. 2010. HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet. 19, 4677–4693 10.1093/hmg/ddq398 (doi:10.1093/hmg/ddq398) [DOI] [PubMed] [Google Scholar]
- 2.Kirbach BB, Golenhofen N. 2011. Differential expression and induction of small heat shock proteins in rat brain and cultured hippocampal neurons. J. Neurosci. Res. 89, 162–175 10.1002/jnr.22536 (doi:10.1002/jnr.22536) [DOI] [PubMed] [Google Scholar]
- 3.Quraishe S, Asuni A, Boelens WC, O'Connor V, Wyttenbach A. 2008. Expression of the small heat shock protein family in the mouse CNS: differential anatomical and biochemical compartmentalization. Neuroscience 153, 483–491 10.1016/j.neuroscience.2008.01.058 (doi:10.1016/j.neuroscience.2008.01.058) [DOI] [PubMed] [Google Scholar]
- 4.Boncoraglio A, Minoia M, Carra S. 2012. The family of mammalian small heat shock proteins (HSPBs): implications in protein deposit diseases and motor neuropathies. Int. J. Biochem. Cell Biol. 14, 1657–1669 10.1016/j.biocel.2012.03.011 (doi:10.1016/j.biocel.2012.03.011) [DOI] [PubMed] [Google Scholar]
- 5.Franklin TB, Krueger-Naug AM, Clarke DB, Arrigo AP, Currie RW. 2005. The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system. Int. J. Hyperthermia 21, 379–392 10.1080/02656730500069955 (doi:10.1080/02656730500069955) [DOI] [PubMed] [Google Scholar]
- 6.Kim DS, Lee SJ, Park SY, Yoo HJ, Kim SH, Kim KJ, Cho HJ. 2001. Differentially expressed genes in rat dorsal root ganglia following peripheral nerve injury. Neuroreport 12, 3401–3405 10.1097/00001756-200110290-00050 (doi:10.1097/00001756-200110290-00050) [DOI] [PubMed] [Google Scholar]
- 7.Armstrong CL, Duffin CA, McFarland R, Vogel MW. 2011. Mechanisms of compartmental Purkinje cell death and survival in the Lurcher mutant mouse. Cerebellum 10, 504–514 10.1007/s12311-010-0231-4 (doi:10.1007/s12311-010-0231-4) [DOI] [PubMed] [Google Scholar]
- 8.Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R. 2003. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 456, 279–291 10.1002/cne.10522 (doi:10.1002/cne.10522) [DOI] [PubMed] [Google Scholar]
- 9.Brzyska M, Stege GJ, Renkawek K, Bosman GJ. 1998. Heat shock, but not the reactive state per se, induces increased expression of the small stress proteins HSP25 and α B-crystallin in glial cells in vitro. Neuroreport 9, 1549–1552 10.1097/00001756-199805110-00056 (doi:10.1097/00001756-199805110-00056) [DOI] [PubMed] [Google Scholar]
- 10.Anagnostou G, Akbar MT, Paul P, Angelinetta C, Steiner TJ, de Belleroche J. 2010. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 31, 969–985 10.1016/j.neurobiolaging.2008.07.005 (doi:10.1016/j.neurobiolaging.2008.07.005) [DOI] [PubMed] [Google Scholar]
- 11.Fukada Y, Yasui K, Kitayama M, Doi K, Nakano T, Watanabe Y, Nakashima K. 2007. Gene expression analysis of the Murine model of amyotrophic lateral sclerosis: studies of the Leu126delTT mutation in SOD1. Brain Res. 1160, 1–10 10.1016/j.brainres.2007.05.044 (doi:10.1016/j.brainres.2007.05.044) [DOI] [PubMed] [Google Scholar]
- 12.Maatkamp A, Vlug A, Haasdijk E, Troost D, French PJ, Jaarsma D. 2004. Decrease of Hsp25 protein expression precedes degeneration of motoneurons in ALS-SOD1 mice. Eur. J. Neurosci. 20, 14–28 10.1111/j.1460-9568.2004.03430 (doi:10.1111/j.1460-9568.2004.03430) [DOI] [PubMed] [Google Scholar]
- 13.Nakamoto H, Vigh L. 2007. The small heat shock proteins and their clients. Cell Mol. Life Sci. 64, 294–306 10.1007/s00018-006-6321-2 (doi:10.1007/s00018-006-6321-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ninkina N, Peters O, Millership S, Salem H, van der Putten H, Buchman VL. 2009. γ-Synucleinopathy: neurodegeneration associated with overexpression of the mouse protein. Hum. Mol. Genet. 18, 1779–1794 10.1093/hmg/ddp090 (doi:10.1093/hmg/ddp090) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pieri I, Cifuentes-Diaz C, Oudinet JP, Blondet B, Rieger F, Gonin S, Arrigo AP, Thomas Y. 2001. Modulation of HSP25 expression during anterior horn motor neuron degeneration in the paralyse mouse mutant. J. Neurosci. Res. 65, 247–253 10.1002/jnr.1148 (doi:10.1002/jnr.1148) [DOI] [PubMed] [Google Scholar]
- 16.Vleminckx V, Van Damme P, Goffin K, Delye H, Van Den Bosch L, Robberecht W. 2002. Upregulation of HSP27 in a transgenic model of ALS. J. Neuropathol. Exp. Neurol. 61, 968–974 10.1046/j.0179-1613.2003.00938.x (doi:10.1046/j.0179-1613.2003.00938.x) [DOI] [PubMed] [Google Scholar]
- 17.Wilhelmus MM, Otte-Holler I, Wesseling P, de Waal RM, Boelens WC, Verbeek MM. 2006. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer's disease brains. Neuropathol. Appl. Neurobiol. 32, 119–130 10.1111/j.1365-2990.2006.00689.x (doi:10.1111/j.1365-2990.2006.00689.x) [DOI] [PubMed] [Google Scholar]
- 18.Bryantsev AL, Kurchashova SY, Golyshev SA, Polyakov VY, Wunderink HF, Kanon B, Budagova KR, Kabakov AE, Kampinga HH. 2007. Regulation of stress-induced intracellular sorting and chaperone function of Hsp27 (HspB1) in mammalian cells. Biochem. J. 407, 407–417 10.1042/BJ20070195 (doi:10.1042/BJ20070195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. 2002. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 11, 1137–1151 10.1093/hmg/11.9.1137 (doi:10.1093/hmg/11.9.1137) [DOI] [PubMed] [Google Scholar]
- 20.An JJ, et al. 2009. Transduced HSP27 protein protects neuronal cell death by enhancing FALS-associated SOD1 mutant activity. BMB Rep. 42, 136–141 10.5483/BMBRep.2009.42.3.136 (doi:10.5483/BMBRep.2009.42.3.136) [DOI] [PubMed] [Google Scholar]
- 21.Carra S, et al. 2013. Different anti-aggregation and pro-degradative functions of the members of the mammalian sHSP family in neurological disorders. Phil. Trans. R. Soc. B 368, 20110409. 10.1098/rstb.2011.0409 (doi:10.1098/rstb.2011.0409) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavoie JN, Hickey E, Weber LA, Landry J. 1993. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J. Biol. Chem. 268, 24 210–24 214 10.1890/08-1540.1 (doi:10.1890/08-1540.1) [DOI] [PubMed] [Google Scholar]
- 23.Arrigo AP, Virot S, Chaufour S, Firdaus W, Kretz-Remy C, Diaz-Latoud C. 2005. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid. Redox Signal. 7, 414–422 10.1089/ars.2005.7.414 (doi:10.1089/ars.2005.7.414) [DOI] [PubMed] [Google Scholar]
- 24.Suzuki A, Sugiyama Y, Hayashi Y, Nyu-i N, Yoshida M, Nonaka I, Ishiura S, Arahata K, Ohno S. 1998. MKBP, a novel member of the small heat shock protein family, binds and activates the myotonic dystrophy protein kinase. J. Cell Biol. 140, 1113–1124 10.1083/jcb.140.5.1113 (doi:10.1083/jcb.140.5.1113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugiyama Y, Suzuki A, Kishikawa M, Akutsu R, Hirose T, Waye MM, Tsui SK, Yoshida S, Ohno S. 2000. Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J. Biol. Chem. 275, 1095–1104 10.1074/jbc.275.2.1095 (doi:10.1074/jbc.275.2.1095) [DOI] [PubMed] [Google Scholar]
- 26.Bhagyalaxmi SG, Srinivas P, Barton KA, Kumar KR, Vidyavathi M, Petrash JM, Bhanuprakash Reddy G, Padma T. 2009. A novel mutation (F71L) in αA-crystallin with defective chaperone-like function associated with age-related cataract. Biochim. Biophys. Acta 1792, 974–981 10.1016/j.bbadis.2009.06.011 (doi:10.1016/j.bbadis.2009.06.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato S, Hirano A, Llena JF, Ito H, Yen SH. 1992. Ultrastructural identification of neurofibrillary tangles in the spinal cords in Guamanian amyotrophic lateral sclerosis and Parkinsonism-dementia complex on Guam. Acta Neuropathol. 83, 277–282 10.1007/BF00296790 (doi:10.1007/BF00296790) [DOI] [PubMed] [Google Scholar]
- 28.Lowe J, Errington DR, Lennox G, Pike I, Spendlove I, Landon M, Mayer RJ. 1992. Ballooned neurons in several neurodegenerative diseases and stroke contain αB crystallin. Neuropathol. Appl. Neurobiol. 18, 341–350 10.1111/j.1365-2990.1992.tb00796.x (doi:10.1111/j.1365-2990.1992.tb00796.x) [DOI] [PubMed] [Google Scholar]
- 29.Braak H, Del Tredici K, Sandmann-Kiel D, Rub U, Schultz C. 2001. Nerve cells expressing heat-shock proteins in Parkinson's disease. Acta Neuropathol. 102, 449–454 [DOI] [PubMed] [Google Scholar]
- 30.Lowe J, Landon M, Pike I, Spendlove I, McDermott H, Mayer RJ. 1990. Dementia with beta-amyloid deposition: involvement of αB-crystallin supports two main diseases. Lancet 336, 515–516 10.1016/0140-6736(90)92075-S (doi:10.1016/0140-6736(90)92075-S) [DOI] [PubMed] [Google Scholar]
- 31.Lowe J, McDermott H, Pike I, Spendlove I, Landon M, Mayer RJ. 1992. α B crystallin expression in non-lenticular tissues and selective presence in ubiquitinated inclusion bodies in human disease. J. Pathol. 166, 61–68 10.1002/path.1711660110 (doi:10.1002/path.1711660110) [DOI] [PubMed] [Google Scholar]
- 32.Sun Y, Makarava N, Lee CI, Laksanalamai P, Robb FT, Baskakov IV. 2008. Conformational stability of PrP amyloid fibrils controls their smallest possible fragment size. J. Mol. Biol. 376, 1155–1167 10.1016/j.jmb.2007.12.053 (doi:10.1016/j.jmb.2007.12.053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Renkawek K, Voorter CE, Bosman GJ, van Workum FP, de Jong WW. 1994. Expression of α B-crystallin in Alzheimer's disease. Acta Neuropathol. 87, 155–160 10.1007/BF00296185 (doi:10.1007/BF00296185) [DOI] [PubMed] [Google Scholar]
- 34.Shinohara H, Inaguma Y, Goto S, Inagaki T, Kato K. 1993. α B crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer's disease. J. Neurol. Sci. 119, 203–208 10.1016/0022-510X(93)90135-L (doi:10.1016/0022-510X(93)90135-L) [DOI] [PubMed] [Google Scholar]
- 35.Rekas A, et al. 2004. Interaction of the molecular chaperone αB-crystallin with alpha-synuclein: effects on amyloid fibril formation and chaperone activity. J. Mol. Biol. 340, 1167–1183 10.1016/j.jmb.2004.05.054 (doi:10.1016/j.jmb.2004.05.054) [DOI] [PubMed] [Google Scholar]
- 36.Quinlan RA, Brenner M, Goldman JE, Messing A. 2007. GFAP and its role in Alexander disease. Exp. Cell Res. 313, 2077–2087 10.1016/j.yexcr.2007.04.004 (doi:10.1016/j.yexcr.2007.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwaki T, Iwaki A, Tateishi J, Goldman JE. 1994. Sense and antisense modification of glial α B-crystallin production results in alterations of stress fiber formation and thermoresistance. J. Cell Biol. 125, 1385–1393 10.1083/jcb.125.6.1385 (doi:10.1083/jcb.125.6.1385) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholl ID, Quinlan RA. 1994. Chaperone activity of alpha-crystallins modulates intermediate filament assembly. EMBO J. 13, 945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.David JC, Boelens WC, Grongnet JF. 2006. Up-regulation of heat shock protein HSP 20 in the hippocampus as an early response to hypoxia of the newborn. J. Neurochem. 99, 570–581 10.1111/j.1471-4159.2006.04071.x (doi:10.1111/j.1471-4159.2006.04071.x) [DOI] [PubMed] [Google Scholar]
- 40.Edwards HV, Cameron RT, Baillie GS. 2011. The emerging role of HSP20 as a multifunctional protective agent. Cell Signal. 23, 1447–1454 10.1016/j.cellsig.2011.05.009 (doi:10.1016/j.cellsig.2011.05.009) [DOI] [PubMed] [Google Scholar]
- 41.Dreiza CM, et al. 2005. Transducible heat shock protein 20 (HSP20) phosphopeptide alters cytoskeletal dynamics. FASEB J. 19, 261–263 10.1096/fj.04-2911fje (doi:10.1096/fj.04-2911fje) [DOI] [PubMed] [Google Scholar]
- 42.Golenhofen N, Perng MD, Quinlan RA, Drenckhahn D. 2004. Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem. Cell Biol. 122, 415–425 10.1007/s00418-004-0711-z (doi:10.1007/s00418-004-0711-z) [DOI] [PubMed] [Google Scholar]
- 43.Crippa V, et al. 2010. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 19, 3440–3456 10.1093/hmg/ddq257 (doi:10.1093/hmg/ddq257) [DOI] [PubMed] [Google Scholar]
- 44.Seidel K, et al. 2011. The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol. Appl. Neurobiol. 38, 39–53 10.1111/j.1365-2990.2011.01198.x (doi:10.1111/j.1365-2990.2011.01198.x) [DOI] [PubMed] [Google Scholar]
- 45.Laskowska E, Matuszewska E, Kuczynska-Wisnik D. 2010. Small heat shock proteins and protein-misfolding diseases. Curr. Pharm. Biotechnol. 11, 146–157 10.2174/138920110790909669 (doi:10.2174/138920110790909669) [DOI] [PubMed] [Google Scholar]
- 46.Carra S, Seguin SJ, Lambert H, Landry J. 2008. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem. 283, 1437–1444 10.1074/jbc.M706304200 (doi:10.1074/jbc.M706304200) [DOI] [PubMed] [Google Scholar]
- 47.Carra S, Sivilotti M, Chavez Zobel AT, Lambert H, Landry J. 2005. HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum. Mol. Genet. 14, 1659–1669 10.1093/hmg/ddi174 (doi:10.1093/hmg/ddi174) [DOI] [PubMed] [Google Scholar]
- 48.Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH. 2009. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2α phosphorylation. J. Biol. Chem. 284, 5523–5532 10.1074/jbc.M807440200 (doi:10.1074/jbc.M807440200) [DOI] [PubMed] [Google Scholar]
- 49.Lambert H, Charette SJ, Bernier AF, Guimond A, Landry J. 1999. HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J. Biol. Chem. 274, 9378–9385 10.1074/jbc.274.14.9378 (doi:10.1074/jbc.274.14.9378) [DOI] [PubMed] [Google Scholar]
- 50.de Wit NJ, Verschuure P, Kappe G, King SM, de Jong WW, van Muijen GN, Boelens WC. 2004. Testis-specific human small heat shock protein HSPB9 is a cancer/testis antigen, and potentially interacts with the dynein subunit TCTEL1. Eur. J. Cell Biol. 83, 337–345 10.1078/0171-9335-00396 (doi:10.1078/0171-9335-00396) [DOI] [PubMed] [Google Scholar]
- 51.Fontaine JM, Rest JS, Welsh MJ, Benndorf R. 2003. The sperm outer dense fiber protein is the 10th member of the superfamily of mammalian small stress proteins. Cell Stress Chaperones 8, 62–69 10.1379/1466-1268 (doi:10.1379/1466-1268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kappe G, Franck E, Verschuure P, Boelens WC, Leunissen JA, de Jong WW. 2003. The human genome encodes 10 α-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones 8, 53–61 10.1379/1466-1268 (doi:10.1379/1466-1268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cobb BA, Petrash JM. 2000. Characterization of alpha-crystallin-plasma membrane binding. J. Biol. Chem. 275, 6664–6672 10.1074/jbc.275.9.6664 (doi:10.1074/jbc.275.9.6664) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Montfort RL, Basha E, Friedrich KL, Slingsby C, Vierling E. 2001. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 8, 1025–1030 10.1038/nsb722 (doi:10.1038/nsb722) [DOI] [PubMed] [Google Scholar]
- 55.Van Montfort R, Slingsby C, Vierling E. 2001. Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv. Protein Chem. 59, 105–156 10.1016/S0065-3233(01)59004.X (doi:10.1016/S0065-3233(01)59004.X) [DOI] [PubMed] [Google Scholar]
- 56.Ito H, Kamei K, Iwamoto I, Inaguma Y, Nohara D, Kato K. 2001. Phosphorylation-induced change of the oligomerization state of α B-crystallin. J. Biol. Chem. 276, 5346–5352 10.1074/jbc.M009004200 (doi:10.1074/jbc.M009004200) [DOI] [PubMed] [Google Scholar]
- 57.Vos MJ, Zijlstra MP, Carra S, Sibon OC, Kampinga HH. 2011. Small heat shock proteins, protein degradation and protein aggregation diseases. Autophagy 7, 101–103 10.4161/auto.7.1.13935 (doi:10.4161/auto.7.1.13935) [DOI] [PubMed] [Google Scholar]
- 58.Horwitz J. 1992. α-Crystallin can function as a molecular chaperone. Proc. Natl Acad. Sci. USA 89, 10 449–10 453 10.1073/pnas.89.21.10449 (doi:10.1073/pnas.89.21.10449) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leroux MR, Melki R, Gordon B, Batelier G, Candido EP. 1997. Structure-function studies on small heat shock protein oligomeric assembly and interaction with unfolded polypeptides. J. Biol. Chem. 272, 24 646–24 656 10.1074/jbc.272.39.24646 (doi:10.1074/jbc.272.39.24646) [DOI] [PubMed] [Google Scholar]
- 60.Morrow G, Heikkila JJ, Tanguay RM. 2006. Differences in the chaperone-like activities of the four main small heat shock proteins of Drosophila melanogaster. Cell Stress Chaperones. 11, 51–60 10.1379/CSC-166.1 (doi:10.1379/CSC-166.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Evgrafov OV, et al. 2004. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 36, 602–606 10.1038/ng1354 (doi:10.1038/ng1354) [DOI] [PubMed] [Google Scholar]
- 62.Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. 1995. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol. Cell. Biol. 15, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang P, MacRae TH. 1997. Molecular chaperones and the cytoskeleton. J. Cell Sci. 110, 1431–1440 [DOI] [PubMed] [Google Scholar]
- 64.Perng MD, Cairns L, van den IJssel P, Prescott A, Hutcheson AM, Quinlan RA. 1999. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J. Cell Sci. 112, 2099–2112 [DOI] [PubMed] [Google Scholar]
- 65.Arrigo AP. 2001. Hsp27: novel regulator of intracellular redox state. IUBMB Life. 52, 303–307 10.1080/152165401317291165 (doi:10.1080/152165401317291165) [DOI] [PubMed] [Google Scholar]
- 66.Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo AP. 2002. Hsp27 as a negative regulator of cytochrome C release. Mol. Cell. Biol. 22, 816–834 10.1128/MCB.22.3.816-834.2002 (doi:10.1128/MCB.22.3.816-834.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paul C, Simon S, Gibert B, Virot S, Manero F, Arrigo AP. 2010. Dynamic processes that reflect anti-apoptotic strategies set up by HspB1 (Hsp27). Exp. Cell. Res. 316, 1535–1552 10.1016/j.yexcr.2010.03.006 (doi:10.1016/j.yexcr.2010.03.006) [DOI] [PubMed] [Google Scholar]
- 68.Muchowski PJ, Wacker JL. 2005. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 6, 11–22 10.1038/nrn1587 (doi:10.1038/nrn1587) [DOI] [PubMed] [Google Scholar]
- 69.Sau D, Rusmini P, Crippa V, Onesto E, Bolzoni E, Ratti A, Poletti A. 2011. Dysregulation of axonal transport and motorneuron diseases. Biol. Cell 103, 87–107 10.1042/BC20100093 (doi:10.1042/BC20100093) [DOI] [PubMed] [Google Scholar]
- 70.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810 10.1038/nature02998 (doi:10.1038/nature02998) [DOI] [PubMed] [Google Scholar]
- 71.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. 1998. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95, 41–53 10.1016/S0092-8674(00)81781-X (doi:10.1016/S0092-8674(00)81781-X) [DOI] [PubMed] [Google Scholar]
- 72.Simeoni S, Mancini MA, Stenoien DL, Marcelli M, Weigel NL, Zanisi M, Martini L, Poletti A. 2000. Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum. Mol. Genet. 9, 133–144 10.1093/hmg/9.1.133 (doi:10.1093/hmg/9.1.133) [DOI] [PubMed] [Google Scholar]
- 73.Rusmini P, Sau D, Crippa V, Palazzolo I, Simonini F, Onesto E, Martini L, Poletti A. 2007. Aggregation and proteasome: the case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol. Aging 28, 1099–1111 10.1016/j.neurobiolaging.2006.05.015 (doi:10.1016/j.neurobiolaging.2006.05.015) [DOI] [PubMed] [Google Scholar]
- 74.Saudou F, Finkbeiner S, Devys D, Greenberg ME. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66 10.1016/S0092-8674(00)81782-1 (doi:10.1016/S0092-8674(00)81782-1) [DOI] [PubMed] [Google Scholar]
- 75.Tagawa K, et al. 2004. Distinct aggregation and cell death patterns among different types of primary neurons induced by mutant huntingtin protein. J. Neurochem. 89, 974–987 10.1111/j.1471-4159.2004.02372.x (doi:10.1111/j.1471-4159.2004.02372.x) [DOI] [PubMed] [Google Scholar]
- 76.Selkoe DJ. 2004. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat. Cell Biol. 6, 1054–1061 10.1038/ncb1104-1054 (doi:10.1038/ncb1104-1054) [DOI] [PubMed] [Google Scholar]
- 77.Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. 2003. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 12, 749–757 10.1093/hmg/ddg074 (doi:10.1093/hmg/ddg074) [DOI] [PubMed] [Google Scholar]
- 78.Adachi H, et al. 2003. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J. Neurosci. 23, 2203–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bailey CK, Andriola IF, Kampinga HH, Merry DE. 2002. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 11, 515–523 10.1093/hmg/11.5.515 (doi:10.1093/hmg/11.5.515) [DOI] [PubMed] [Google Scholar]
- 80.Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. 1999. Analysis of the role of heat shock protein (HSP) molecular chaperones in polyglutamine disease. J. Neurosci. 19, 10 338–10 347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. 2001. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum. Mol. Genet. 10, 1511–1518 10.1093/hmg/10.14.1511 (doi:10.1093/hmg/10.14.1511) [DOI] [PubMed] [Google Scholar]
- 82.Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. 2005. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc. Natl Acad. Sci. USA 102, 16 801–16 806 10.1073/pnas.0506249102 (doi:10.1073/pnas.0506249102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kobayashi Y, Sobue G. 2001. Protective effect of chaperones on polyglutamine diseases. Brain Res. Bull. 56, 165–168 10.1016/S0361-9230(01)00593-7 (doi:10.1016/S0361-9230(01)00593-7) [DOI] [PubMed] [Google Scholar]
- 84.Koyama S, et al. 2006. Alteration of familial ALS-linked mutant SOD1 solubility with disease progression: its modulation by the proteasome and Hsp70. Biochem. Biophys. Res. Commun. 343, 719–730 10.1016/j.bbrc.2006.02.170 (doi:10.1016/j.bbrc.2006.02.170) [DOI] [PubMed] [Google Scholar]
- 85.Patel YJ, Payne Smith MD, de Belleroche J, Latchman DS. 2005. Hsp27 and Hsp70 administered in combination have a potent protective effect against FALS-associated SOD1-mutant-induced cell death in mammalian neuronal cells. Brain Res. Mol. Brain Res. 134, 256–274 10.1016/j.molbrainres.2004.10.028 (doi:10.1016/j.molbrainres.2004.10.028) [DOI] [PubMed] [Google Scholar]
- 86.Rusmini P, Simonini F, Crippa V, Bolzoni E, Onesto E, Cagnin M, Sau D, Ferri N, Poletti A. 2011. 17-AAG increases autophagic removal of mutant androgen receptor in spinal and bulbar muscular atrophy. Neurobiol. Dis. 41, 83–95 10.1016/j.nbd.2010.08.023 (doi:10.1016/j.nbd.2010.08.023) [DOI] [PubMed] [Google Scholar]
- 87.Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. 1999. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 23, 425–428 10.1038/70532 (doi:10.1038/70532) [DOI] [PubMed] [Google Scholar]
- 88.Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 2005. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 11, 1088–1095 10.1038/nm1298 (doi:10.1038/nm1298) [DOI] [PubMed] [Google Scholar]
- 89.Waza M, Adachi H, Katsuno M, Minamiyama M, Tanaka F, Doyu M, Sobue G. 2006. Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J. Mol. Med. 84, 635–646 10.1007/s00109-006-0066-0 (doi:10.1007/s00109-006-0066-0) [DOI] [PubMed] [Google Scholar]
- 90.Yamashita H, Kawamata J, Okawa K, Kanki R, Nakamizo T, Hatayama T, Yamanaka K, Takahashi R, Shimohama S. 2007. Heat-shock protein 105 interacts with and suppresses aggregation of mutant Cu/Zn superoxide dismutase: clues to a possible strategy for treating ALS. J. Neurochem. 102, 1497–1505 10.1111/j.1471-4159.2007.04534.x (doi:10.1111/j.1471-4159.2007.04534.x) [DOI] [PubMed] [Google Scholar]
- 91.Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HM, Lubsen NH, Kampinga HH. 2010. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369 10.1016/j.molcel.2010.01.001 (doi:10.1016/j.molcel.2010.01.001) [DOI] [PubMed] [Google Scholar]
- 92.Vos MJ, Hageman J, Carra S, Kampinga HH. 2008. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 47, 7001–7011 10.1021/bi800639z (doi:10.1021/bi800639z) [DOI] [PubMed] [Google Scholar]
- 93.Dreiza CM, Komalavilas P, Furnish EJ, Flynn CR, Sheller MR, Smoke CC, Lopes LB, Brophy CM. 2010. The small heat shock protein, HSPB6, in muscle function and disease. Cell Stress Chaperones 15, 1–11 10.1007/s12192-009-0127-8 (doi:10.1007/s12192-009-0127-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Benn SC, Perrelet D, Kato AC, Scholz J, Decosterd I, Mannion RJ, Bakowska JC, Woolf CJ. 2002. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 36, 45–56 10.1016/S0896-6273(02)00941-8 (doi:10.1016/S0896-6273(02)00941-8) [DOI] [PubMed] [Google Scholar]
- 95.Crippa V, Carra S, Rusmini P, Sau D, Bolzoni E, Bendotti C, De Biasi S, Poletti A. 2010. A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy 6, 958–960 10.4161/auto.6.7.13042 (doi:10.4161/auto.6.7.13042) [DOI] [PubMed] [Google Scholar]
- 96.Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. 2006. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392 10.1126/science.1123511 (doi:10.1126/science.1123511) [DOI] [PubMed] [Google Scholar]
- 97.Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. 1999. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat. Neurosci. 2, 427–433 10.1038/8091 (doi:10.1038/8091) [DOI] [PubMed] [Google Scholar]
- 98.Onesto E, Rusmini P, Crippa V, Ferri N, Zito A, Galbiati M, Poletti A. 2011. Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J. Neurochem. 118, 266–280 10.1111/j.1471-4159.2011.07298.x (doi:10.1111/j.1471-4159.2011.07298.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dobrowolny G, et al. 2008. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 8, 425–436 10.1016/j.cmet.2008.09.002 (doi:10.1016/j.cmet.2008.09.002) [DOI] [PubMed] [Google Scholar]
- 100.Wong M, Martin LJ. 2010. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 19, 2284–2302 10.1093/hmg/ddq106 (doi:10.1093/hmg/ddq106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Carra S, et al. 2010. Identification of the Drosophila ortholog of HSPB8: implication of HSPB8 loss of function in protein folding diseases. J. Biol. Chem. 285, 37 811–37 822 10.1074/jbc.M110.127498 (doi:10.1074/jbc.M110.127498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carra S. 2009. The stress-inducible HspB8-Bag3 complex induces the eIF2α kinase pathway: implications for protein quality control and viral factory degradation? Autophagy 5, 428–429 10.4161/auto.5.3.7894 (doi:10.4161/auto.5.3.7894) [DOI] [PubMed] [Google Scholar]
- 103.Klionsky DJ, et al. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arndt V, et al. 2010. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 20, 143–148 10.1016/j.cub.2009.11.022 (doi:10.1016/j.cub.2009.11.022) [DOI] [PubMed] [Google Scholar]
- 105.Kaganovich D, Kopito R, Frydman J. 2008. Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095 10.1038/nature07195 (doi:10.1038/nature07195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carra S, Crippa V, Rusmini P, Boncoraglio A, Minoia M, Giorgetti E, Kampinga HH, Poletti A. 2012. Alteration of protein folding and degradation in motor neuron diseases: implications and protective functions of small heat shock proteins. Prog. Neurobiol. 97, 83–100 10.1016/j.pneurobio.2011.09.009 (doi:10.1016/j.pneurobio.2011.09.009) [DOI] [PubMed] [Google Scholar]
- 107.Fuchs M, Poirier DJ, Seguin SJ, Lambert H, Carra S, Charette SJ, Landry J. 2010. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem. J. 425, 245–255 10.1042/BJ20090907 (doi:10.1042/BJ20090907) [DOI] [PubMed] [Google Scholar]
- 108.Qian J, Ren X, Wang X, Zhang P, Jones WK, Molkentin JD, Fan GC, Kranias EG. 2009. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ. Res. 105, 1223–1231 10.1161/CIRCRESAHA.109.200378 (doi:10.1161/CIRCRESAHA.109.200378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Filimonenko M, et al. 2010. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 38, 265–279 10.1016/j.molcel.2010.04.007 (doi:10.1016/j.molcel.2010.04.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Matsumoto G, Kim S, Morimoto RI. 2006. Huntingtin and mutant SOD1 form aggregate structures with distinct molecular properties in human cells. J. Biol. Chem. 281, 4477–4485 10.1074/jbc.M509201200 (doi:10.1074/jbc.M509201200) [DOI] [PubMed] [Google Scholar]
- 111.Chen S, Ferrone FA, Wetzel R. 2002. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl Acad. Sci. USA 99, 11 884–11 889 10.1073/pnas.182276099 (doi:10.1073/pnas.182276099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ellisdon AM, Pearce MC, Bottomley SP. 2007. Mechanisms of ataxin-3 misfolding and fibril formation: kinetic analysis of a disease-associated polyglutamine protein. J. Mol. Biol. 368, 595–605 10.1016/j.jmb.2007.02.058 (doi:10.1016/j.jmb.2007.02.058) [DOI] [PubMed] [Google Scholar]
- 113.Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 10.1126/science.1072994 (doi:10.1126/science.1072994) [DOI] [PubMed] [Google Scholar]
- 114.Ignatova Z, Gierasch LM. 2006. Extended polyglutamine tracts cause aggregation and structural perturbation of an adjacent beta barrel protein. J. Biol. Chem. 281, 12 959–12 967 10.1074/jbc.M511523200 (doi:10.1074/jbc.M511523200) [DOI] [PubMed] [Google Scholar]
- 115.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. 1995. The precursor protein of non-A β component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475 10.1016/0896-6273(95)90302-X (doi:10.1016/0896-6273(95)90302-X) [DOI] [PubMed] [Google Scholar]
- 116.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. 2000. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269 10.1126/science.287.5456.1265 (doi:10.1126/science.287.5456.1265) [DOI] [PubMed] [Google Scholar]
- 117.Scherzinger E, et al. 1997. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 10.1016/S0092-8674(00)80514-0 (doi:10.1016/S0092-8674(00)80514-0) [DOI] [PubMed] [Google Scholar]
- 118.Ross CA, Poirier MA. 2004. Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl.), S10–S17 10.1038/nm1066 (doi:10.1038/nm1066) [DOI] [PubMed] [Google Scholar]
- 119.Waudby CA, et al. 2010. The interaction of alphaB-crystallin with mature alpha-synuclein amyloid fibrils inhibits their elongation. Biophys. J. 98, 843–851 10.1016/j.bpj.2009.10.056 (doi:10.1016/j.bpj.2009.10.056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rekas A, Jankova L, Thorn DC, Cappai R, Carver JA. 2007. Monitoring the prevention of amyloid fibril formation by alpha-crystallin. Temperature dependence and the nature of the aggregating species. FEBS J. 274, 6290–6304 10.1111/j.1742-4658.2007.06144.x (doi:10.1111/j.1742-4658.2007.06144.x) [DOI] [PubMed] [Google Scholar]
- 121.Robertson AL, Headey SJ, Saunders HM, Ecroyd H, Scanlon MJ, Carver JA, Bottomley SP. 2010. Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. Proc. Natl Acad. Sci. USA 107, 10 424–10 429 10.1073/pnas.0914773107 (doi:10.1073/pnas.0914773107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Masino L, Nicastro G, Menon RP, Dal Piaz F, Calder L, Pastore A. 2004. Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J. Mol. Biol. 344, 1021–1035 10.1016/j.jmb.2004.09.065 (doi:10.1016/j.jmb.2004.09.065) [DOI] [PubMed] [Google Scholar]
- 123.Brooks P, et al. 2000. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem. J. 346, 155–161 10.1042/0264-6021:3460155 (doi:10.1042/0264-6021:3460155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xie Z, Klionsky DJ. 2007. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109 10.1038/ncb1007-1102 (doi:10.1038/ncb1007-1102) [DOI] [PubMed] [Google Scholar]
- 125.Montie HL, Cho MS, Holder L, Liu Y, Tsvetkov AS, Finkbeiner S, Merry DE. 2009. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 18, 1937–1950 10.1093/hmg/ddp115 (doi:10.1093/hmg/ddp115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsai HF, Lin SJ, Li C, Hsieh M. 2005. Decreased expression of Hsp27 and Hsp70 in transformed lymphoblastoid cells from patients with spinocerebellar ataxia type 7. Biochem. Biophys. Res. Comm. 334, 1279–1286 10.1016/j.bbrc.2005.06.207 (doi:10.1016/j.bbrc.2005.06.207) [DOI] [PubMed] [Google Scholar]
- 127.Hsieh M, Tsai HF, Chang WH. 2005. HSP27 and cell death in spinocerebellar ataxia type 3. Cerebellum 4, 31–36 10.1080/14734220410026248 (doi:10.1080/14734220410026248) [DOI] [PubMed] [Google Scholar]
- 128.Friedman MJ, Li S, Li XJ. 2009. Activation of gene transcription by heat shock protein 27 may contribute to its neuronal protection. J. Biol. Chem. 284, 27 944–27 951 10.1074/jbc.M109.037937 (doi:10.1074/jbc.M109.037937) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zourlidou A, et al. 2007. Hsp27 overexpression in the R6/2 mouse model of Huntington's disease: chronic neurodegeneration does not induce Hsp27 activation. Hum. Mol. Genet. 16, 1078–1090 10.1093/hmg/ddm057 (doi:10.1093/hmg/ddm057) [DOI] [PubMed] [Google Scholar]