

Abstract
In vivo measurements of lipid biomarkers are hampered by their low solubility in aqueous solution, which limits the choices for molecular separations. Here, we introduce non-aqueous microchip electrophoretic separations of lipid mixtures performed in three-dimensional hybrid nanofluidic/microfluidic polymeric devices. Electrokinetic injection is used to reproducibly introduce discrete femtolitre to picolitre volumes of charged lipids into a separation microchannel containing low (100 μM–10 mM) concentration tetraalkylammonium tetraphenylborate background electrolyte (BGE) in N-methylformamide, supporting rapid electro-osmotic fluid flow in polydimethylsiloxane microchannels. The quality of the resulting electrophoretic separations depends on the voltage and timing of the injection pulse, the BGE concentration and the electric field strength. Injected volumes increase with longer injection pulse widths and higher injection pulse amplitudes. Separation efficiency, as measured by total plate number, N, increases with increasing electric field and with decreasing BGE concentration. Electrophoretic separations of binary and ternary lipid mixtures were achieved with high resolution (Rs ∼ 5) and quality (N > 7.7 × 106 plates m−1). Rapid in vivo monitoring of lipid biomarkers requires high-quality separation and detection of lipids downstream of microdialysis sample collection, and the multilayered non-aqueous microfluidic devices studied here offer one possible avenue to swiftly process complex lipid samples. The resulting capability may make it possible to correlate oxidative stress with in vivo lipid biomarker levels.
Keywords: microfluidics, capillary electrophoresis, lipidomics, biomarkers
1. Introduction
Reactive oxygen species (ROS), produced by routine metabolic events in aerobic organisms, pose a threat to cellular components, including proteins, lipids and DNA. Although organisms produce antioxidants naturally in order to combat the potentially dangerous effects of ROS, when the antioxidant capacity is exceeded, oxidative stress results and recurring exposure may cause significant damage. The exact cause of ROS overproduction is not yet fully elucidated, partially because these harmful molecules display extremely short half-lives. One alternative is to analyse the degradative products of ROS-induced oxidation. The presence of these biomarkers has been successfully correlated to connect oxidative stress with a number of debilitating conditions, including Alzheimer's disease (AD), Parkinson's disease and amyotrophic lateral sclerosis [1]. For example, protein carbonyls, the product of amino acid oxidation by ROS, have been found in elevated levels in patients with diabetes, hypercholesterolaemia and juvenile arthritis [2].
Phospholipids are particularly susceptible to ROS. More specifically, peroxidation of phospholipid arachidonyl residues by ROS generates prostaglandins [3,4], a complex group of biomarkers found in biofluids, including urine, blood plasma, breath condensate, bile, seminal fluid, pericardial fluid and cerebrospinal fluid (CSF). Isoprostanes, a subset of prostaglandins, have been used as indicators of oxidative stress in cardiovascular disease, asthma, hepatic sclerosis, scleroderma and AD [5]. Mattsson et al. [6] observed elevated levels of F2-isoprostanes in the CSF of patients with multiple sclerosis. Using plasma F2-isoprostane levels as a metric, Riccobene et al. [7] determined that patients with chronic kidney disease are more likely to endure atherosclerotic processes brought about by oxidative stress.
The concentration of fluid-borne phospholipids and the associated products of lipid peroxidation are good indicators of the extent of damage brought about by ROS [8]. However, determinations of isoprostanes are typically performed using commercial immunoassay kits, which, despite pM limits of detection [9], are relatively expensive and can be slow. Thus, diagnostics capable of rapidly processing patient biofluids to resolve their complex molecular composition are needed. The work described here is part of a larger programme to develop in vivo microdialysis-based sampling for high-speed, on-site diagnostic assays.
Capillary electrophoresis (CE), a powerful separation technique, has been used to process DNA, RNA, protein, peptides and metabolite mixtures in minutes, using nanolitres of biological sample and microlitres of low-cost separation buffer [10]. Despite such minute quantities of material required, direct processing of patient biofluids via CE is very challenging owing to the vast number of distinct molecular entities involved. Nearly 400 different types of proteins are known to exist in human CSF alone [11]. Fortunately, trivial methods of extracting particular groups of biomarkers from human samples have been developed. For example, Zhao & Xu [12] recently formulated a facile method to extract lysophospholipids and phospholipids from blood samples. Unfortunately, neither conventional CE nor microchip electrophoresis (MCE) are well suited for lipid determinations, since common separation buffers consist of inorganic salts in aqueous media, in which lipids tend to aggregate. One solution, micellar electrokinetic chromatography (MEKC), which affords the ability to resolve molecules based not only on their electrophoretic mobility but also on their hydrophobicity [13], is well suited to lipid analysis and has been used to analyse hydrophobic mixtures in both capillaries and microchips [14–16]. However, MEKC cannot be directly coupled to mass spectrometric detection, because it requires high (millimoles) concentrations of surfactant, resulting in analyte signal suppression and contamination by separation additives [17]. Another promising alternative, non-aqueous capillary electrophoresis (NACE), exploits increased solubility of hydrophobic analytes in organic separation buffers [18] and has been applied to characterize biomarkers [19,20] and pharmaceutical compounds [21–23].
Organic solvents used in NACE separations include methanol, ethanol, acetonitrile, formamide, dimethylformamide (DMF), N-methylformamide (NMF), dimethylsulfoxide and mixtures of these. Coupled with inorganic background electrolytes (BGEs), such as NaCl, phosphates and borate, high-efficiency separations have been achieved [24]. Unfortunately, the concentrations of some of these BGE/solvent solutions required for electrophoretic separations have been reported in the high millimolar range. To facilitate the separation and determination of biomarker lipids, a unique approach based on an organic BGE/solvent system is developed and studied here. NMF is chosen as the solvent, because it can solvate non-aqueous ions (high donor number), and it readily solvates commonly used electrolytes. Thus, tetraalkylammonium salts, commonly used in organic electrochemistry, produce excellent electro-osmotic flows (EOFs) appropriate for high-quality electrophoretic separations, even at micromolar concentrations.
Rapid in vivo diagnostic applications further require electrophoretic separations of femtolitre volumes, necessitating precision not available in bench-top CE systems. Microfluidic-based lab-on-a-chip systems address this requirement, as a number of automated fluidic manipulation schemes can be devised in both two- [25,26] and three-dimensional architectures [27,28]. In this work, we avoid the constraints of performing fluid manipulations in a single plane by using a hybrid three-dimensional microfluidic/nanofluidic polymeric device to swiftly separate lipid mixtures. The main advantages of this device include high precision, reproducible electrokinetic injections with outstanding peak integrity [29], the ability to perform quality separations in multiple fluidic planes [30], and the flexibility to accommodate a number of detection schemes [31]. NACE and MCE are combined here to perform rapid analysis of fluorescently tagged lipid mixtures in non-aqueous separation media. The resulting non-aqueous microchip electrophoresis (NAME) shows great promise for resolving complex biological samples in order to correlate oxidative stress conditions with disease.
2. Material and methods
2.1. Reagents
NBD-PA, 1-hexanoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]-hexanoyl]-sn-glycero-3-phosphate (ammonium salt); NBD-PG, 1-hexanoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]-hexanoyl]-sn-glycero-3-[phospho-rac-(1-glycerol)] (ammonium salt); and NBD-CoA, [N-[(7-nitro-2-1,3-benzoxadiazol-4-yl)-methyl]amino]-palmitoyl coenzyme A (ammonium salt) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). TBA-TPhB, tetrabutylammonium tetraphenylborate; TPhP-TPhB, tetraphenylphosphonium tetraphenylborate; and NMF were obtained from Sigma (St. Louis, MO, USA). All were used without further purification.
2.2. Background electrolyte/solvent preparation
Separation buffers were prepared from NMF solutions containing different (100 µM, 1 mM and 10 mM) concentrations of either TBA-TPhB or TPhP-TPhB. Analyte solutions containing NBD-PA, a mixture of NBD-PA and NBD-PG, or a mixture of NBD-PA, NBD-PG and NBD-CoA were formulated in separation buffer at 1 nM, 10 nM, 1 μM and 10 μM concentrations.
2.3. Microchip fabrication
Generally, the microchannels were aligned orthogonally with fluidic communication provided via the nanocapillary array membrane (NCAM) sandwiched between them. The assembled device (figure 1) consisted of four polymeric layers: two polydimethylsiloxane (PDMS) microchannel layers, one track-etched polycarbonate NCAM and one PDMS adhesive layer. The adhesive layer effectively seals the device and prevents unwanted leakage. The master mould for microchannel fabrication was constructed by Stanford Microfluidics Foundry (Stanford, CA, USA), and layers were produced using rapid prototyping [32]. The sealing procedure for the device was adapted from the work of Chueh et al. [33]. Briefly, uncured PDMS was spun onto a glass coverslip for 1 min at 12 000 r.p.m. The thin uncured PDMS coating, of the order of tens of nanometres thick, was then stamped onto the top microchannel layer. The NCAM, purchased from Osmonics (Minnetonka, MN, USA), was then positioned on the bottom microchannel layer just before both cured PDMS microchannel layers were brought into contact and pressed together firmly. Taking care to avoid pores located in the membrane area exposed to the orthogonal microchannels, the NCAM pores were filled with uncured PDMS. The device was then cured for 1 h at 75°C. The microchannels were 100 μm in width and height. The source microchannel (figure 1a) was 1.5 cm long and served as the sample reservoir. The receiving (separation) microchannel (figure 1b) was 4.25 cm long. The 6–10 μm thick NCAM contained an array (4 × 108 cm−2) of 100 nm diameter pores.
Figure 1.

(a,b) Schematic of a three-dimensional NAME separation device featuring two orthogonal microfluidic channels (teal) with an NCAM interconnect (yellow). Assembly requires a thin adhesion mortar (blue), which ensures leak-free bonding of the polymer layers. The adhesive is carefully placed to avoid incursion into the analyte and separation microchannels.
2.4. Instrumentation
Fluidic control in the microchip was established using two high-voltage DC power supplies (602C-30P) from Spellman High Voltage Electronics Corp. (Hauppauge, NY, USA), specially constructed relay and switch boxes (University of Illinois, Urbana, IL, USA), and a PCI data acquisition card (PCI-6221) from National Instruments (Austin, TX, USA). A LabView (National Instruments) program controlled the voltage applied to each of the four Pd electrodes that drive electrokinetic flow. Analyte transport was observed using an Olympus IX-71 (Center Valley, PA, USA) epifluorescence microscope featuring a 41001 fluorescein filter set (Chroma Technology Inc., Rockingham, VT, USA). Illumination was obtained from a 100 W light source (X-Cite 120 PC) from Lumen Dynamics (Mississauga, Ontario, Canada). Images were recorded at six frames per second using a PhotonMax512 EMCCD camera (Princeton Instruments, Trenton, NJ, USA).
2.5. Procedures
Fabricated devices were first vacuum-filled with solutions; analyte mixtures were loaded in the source microchannel, and separation buffer in the receiving microchannel. Microchips were then mounted onto the microscope stage, where microfluidic channels were positioned above a 10× objective lens. Pd electrodes were placed in each of the four fluid reservoirs. Taking advantage of the transparency of PDMS, fluorescence intensity was observed along the length of the microfluidic channels. Following each 5–10 min experiment, microchannels were rinsed with ca 100 channel volumes of analyte solution or separation buffer by vacuum filling. Each of the experiments conducted in this work was performed in a new device. Although the magnitude of EOF in these PDMS-based structures varied significantly across devices, relative analyte electrophoresis behaviour remained consistent from device to device.
2.6. Solvent and device compatibility
PDMS is known to swell in the presence of organic solvents. Chemically, NMF is closely related to DMF, a formamide that has been shown by previous work to swell PDMS minimally [34]. Additionally, the PDMS layers and polycarbonate NCAMs showed no signs of degradation or chemical breakdown when left suspended in a bulk volume non-aqueous solvent for times as long as 48 h.
3. Results and discussion
3.1. Lipid injections
The spatially separated microchannels featured in this device are bridged by an array of high aspect ratio nanocapillaries that simultaneously restrict free diffusion of analyte [35] and facilitate electrokinetic injection. Just as with aqueous systems, relatively small potentials effect reproducible sample plug introduction. In separation experiments, a small (less than 1 nl) volume of fluorescently tagged lipids is first injected from the source channel, across the NCAM, into the separation channel and then transported downstream [36]. The bias applied across the NCAM to achieve sample injection is defined by
| 3.1 |
where Vrecieving and Vsource represent the relative potential of the separation microchannel and analyte reservoirs, respectively. Figure 2 depicts how material is electrokinetically injected, where Vinj and Vsep represent the magnitude of the potential applied along the separation microchannel to drive either an injection or a separation, respectively. A brief (t < 1 s) voltage pulse across the NCAM electrophoretically injects lipid-containing solution into the separation microchannel. Floating the source microchannel electrodes then disengages the electric field across the NCAM, and a potential is applied along the length of the separation channel to complete the transfer of the injected sample into the separation region of the device via cation-driven EOF, and begin the electrophoretic separation.
Figure 2.

Top view fluorescence images depicting gated injection of a phospholipid (10 μM NBD-PA in 1 mM TBA-TPhB/NMF) from a vertical channel (sample reservoir in figure 1) to a horizontal channel (separation microchannel in figure 1) across an array of 100 nm pores, and the corresponding driving potential configuration at each stage: (a) before injection (t = 0 s), (b) after electrophoretic injection (Vinj = 10 V, t = 1 s), and (c) at the onset of separation where transport downstream moves from right to left (Vsep = 900 V, t = 3 s). B, BW, S and SW represent, respectively, the buffer, buffer waste, sample and sample waste reservoir assignments for each of the three stages of operation.
Figure 3 shows the effect of the gate pulse duration, Δtinj, and amplitude, ΔVinj, on the quantity of material injected. Each peak represents a fluidic volume of material injected for a given time and then transported downstream ((c) to (a) in figure 2). The volumetric flow rate, F, of material injected through the NCAM can be written as
| 3.2 |
where μobs, Eapp and Apore represent the observed mobility, applied electric field and effective cross-sectional area, respectively. Based on the area beneath each peak, the data shown in figure 3 are consistent with equation (3.2). Positive linear relationships are observed between both Δtinj and ΔVinj and the quantity of lipid transferred across the NCAM. The ability to tune the volume injected permits trade-offs between sensitivity and resolution. For example, for mass-limited samples, large values of Δtinj and ΔVinj can be used to enhance the sensitivity at the expense of a modest degradation in resolution. In addition, the reproducibility of injections depicted in figure 3 using a three-dimensional hybrid architecture is commensurate with similar injections performed on aqueous systems.
Figure 3.
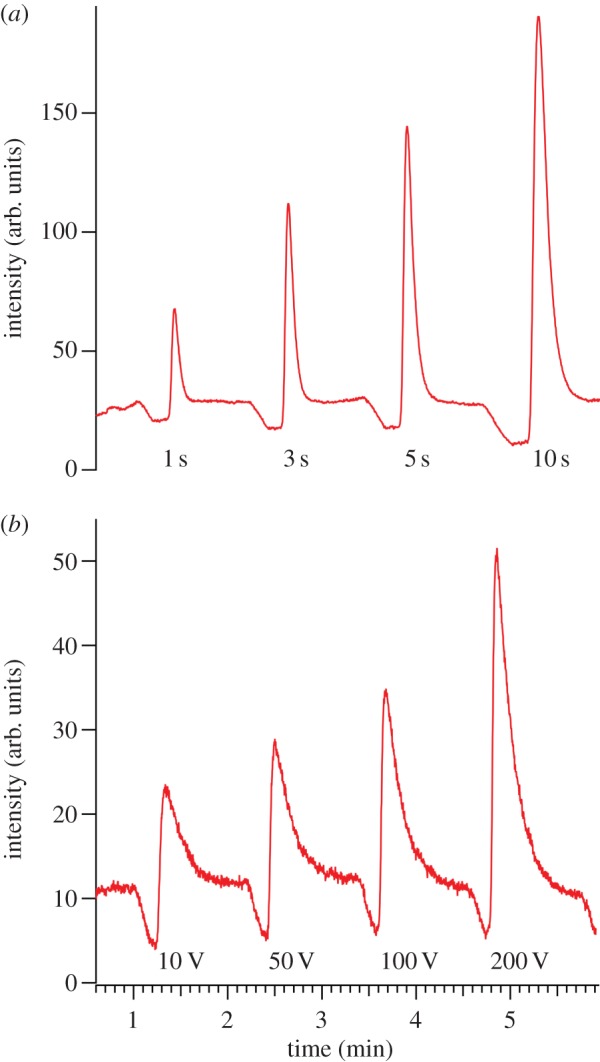
Electropherograms depicting how the duration, Δtinj, and voltage magnitude, ΔVinj, of gated injection influence the lipid (10 μM NBD-PA in 10 mM TBA-TPhB/NMF) band observed 400 μm downstream in the separation channel. (a) Series of NBD-PA bands injected at 50 V for 1, 3, 5 and 10 s. (b) Series of NBD-PA bands injected for 1 s at 10, 50, 100 and 200 V. In both experiments, Esep = 212 V cm−1. (Online version in colour.)
3.2. Lipid diffusion coefficient
The diffusion coefficient of the injected lipid molecules (Dm) determines the longitudinal dispersion of injected bands and thus can be employed to assess separation quality. Here, Dm was calculated using the ‘on-the-fly-by-electrophoresis’ method [37,38], according to
| 3.3 |
where
| 3.4 |
where σ represents the lipid bandwidth (μm) and t represents the time for the injected lipid packet to migrate from the injection to the observation point (400 μm). Although equation (3.4) accounts for the finite width of the injected band, it does not account for the tailing (see figure 3), which is caused by electrical limitations of the high-voltage supply. Currently, the minimum applied voltage (10 V) and application time (1 s) for electrokinetic injections are too high, resulting in significant injection on both sides of the NCAM, producing an apparent band tail. Improvements to incorporate a power supply that permits millivolt potentials and millisecond application times are being implemented. The observed band tailing also influences interpretation of diffusion coefficients calculated based on Gaussian peak shapes, where based on analysis of repeated injections identical to those depicted in figure 3 (where t varies by ±7%), Dm,NBD−PA = 4.48 × 10−7 cm2 s−1.
Band broadening, Δσ, increases with the square root of both Dm and t. Although this undesirable effect, which ultimately reduces resolution, is inevitable, the width of the injected band (στ = 0) is limited by the cross-sectional area (100 × 100 μm) of the overlapping microchannels. Since separations are performed well above the concentration limit of detection (LOD), much smaller channels could be used which would result in further improvements in resolution. However, the need for a PDMS adhesion layer dictates that the microchannel width be sufficiently large to prevent blockage by uncured PDMS during assembly.
3.3. System limit of detection
Robust biomarker detection in mammalian biofluids requires low LODs. To determine the LOD for the NAME experiment, discrete volumes of NBD-PA were injected into the separation channel at varying analyte concentrations. The excitation source was set to maximum power (30 mW), and the resulting signal-to-noise (S/N) ratio of the fluorescence peak was measured downstream (see electronic supplementary material for more information). Table 1 depicts the S/N ratio for lipid concentrations in the range 100 pM < C < 1 μM. Conservatively interpreting these data indicates that the LOD of the current system is approximately 1–10 pM, a range acceptable for fluorescence-based assay development and easily competent for the determination of circulating plasma biomarkers [39–41]. For example, millimolar lipid and protein levels and micromolar vitamin concentrations in plasma were used by Karaouzene et al. [42] to determine that obesity induces both oxidative stress and lipid composition changes in men. Conversely, lipid biomarkers for some conditions are present at much lower femtomolar to picomolar concentrations in human biofluids. Picomolar concentrations of isoprostanes in plasma, for example, have been identified as early indicators of Rett's syndrome [43]. Addressing these more challenging biomarker assays in the NAME system described here will require improvements in LOD, physically concentrating the sample [44–46] or both.
Table 1.
S/N ratio as a function of concentration for NBD-PA bands.
| NBD-PA concentration | S/Na |
|---|---|
| 100 pM | 31 ± 7 |
| 1 nM | 98 ± 33 |
| 10 nM | 175 ± 57 |
| 100 nM | 202 ± 83 |
| 1 μM | 358 ± 135 |
aMeasured 400 μm downstream in 10 mM TBA-TPhB/NMF.
3.4. Separation performance metrics
3.4.1. Plate number (N)
The number of theoretical plates is an indirect measure of the microchannel separation efficiency, indicating how well the system performs in the face of longitudinal diffusion and subsequent band broadening. N is defined by
| 3.5 |
where Vapp is the voltage applied across the separation channel, l is the effective channel length and L is the total channel length over which the voltage is applied. Plate numbers ranging from 104 to 105 are common in high-quality electrophoretic separations [47].
3.4.2. Background electrolyte concentration
Table 2 shows the dependence of N on the ionic strength of the TBA-TPhB/NMF solution. Using a single device, TBA-TPhB/NMF solutions (100 μM, 1 mM and 10 mM, respectively) were introduced into both the analyte reservoir and the separation microchannel. Migrating peaks were then observed 400 μm downstream from the injection point, and the number of theoretical plates at 400 μm was determined by averaging the observed mobility values from several peaks produced at each electrolyte concentration. Qualitatively, EOF was observed to become less reproducible at the highest electrolyte concentrations. The thickness of the electrical double layer (κ−1) for the TBA-TPhB/NMF separation media at the wall–solution interface is given by
 |
3.6 |
where ɛr, ɛ0, R, T, F and Celectrolyte represent the dielectric constant of the NMF solvent, permittivity of free space, gas constant, temperature, Faraday constant and the TBA-TPhB concentration, respectively.
Table 2.
Number of theoretical plates (N) versus ionic strength of the BGE.
| TBA-TPhB concentration | Na |
|---|---|
| 100 μM | 538 ± 21 |
| 1 mM | 508 ± 61 |
| 10 mM | 428 ± 40 |
aLipid injections performed at ΔVinj = 10 V, Δtinj = 1 s, Esep = 424 V cm−1, for each concentration. Measured 400 μm downstream.
Based on equation (3.6), κ−1 for the concentrations investigated spans the range 5–50 nm. The dimensions of microchannels accommodating electrokinetic flow (of the order of 100 μm) are considerably larger than κ−1, which suggests that the electrolyte concentration should not have a significant impact on the EOF, and subsequent separation quality. However, table 2 clearly shows a modest but statistically significant improvement in the quality of separation with decreasing BGE concentration and is best when 100 μM TBA-TPhB/NMF is used. Furthermore, preliminary work has shown that the presence of TBA-TPhB at this concentration does not obviate analysis of NBD-PA using ambient ionization mass spectrometry.
3.4.3. Separation electric field
Figure 4 shows the effect of electric field magnitude on band broadening in 100 μM TBA-TPhB. As shown, band broadening decreases with increasing electric field strength, and hence the efficiency of the lipid separation, as measured by N, improves at higher fields up to 500 V cm−1. Although these findings suggest that optimal separations are achieved using the highest applied voltage the equipment permits, the maximum electric field is limited by Joule heating, which can decrease the viscosity of the separation media, promoting molecular diffusion and subsequent band broadening. In addition, fields in the 400–500 V cm−1 range were sufficient to accomplish the separations of model compounds in these studies.
Figure 4.
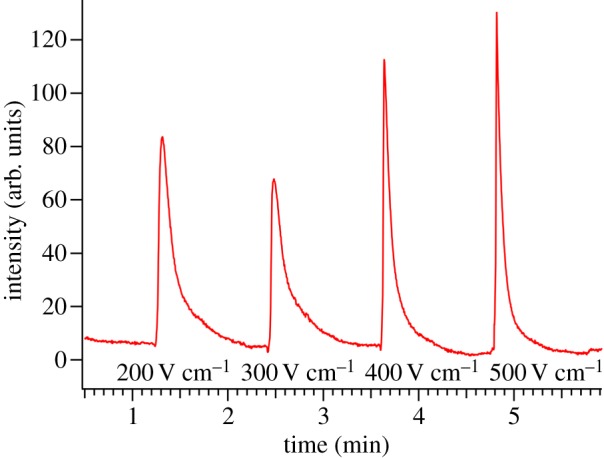
Electropherograms illustrating the relationship between dispersion of injected lipid (10 μM NBD-PA in 100 μM TBA-TPhB/NMF solution) bands and the magnitude of the electric field driving separation. ΔVinj = 10 V and Δtinj = 1 s. (Online version in colour.)
3.4.4. Cation hydrophobicity
Owing to its large dielectric constant (ɛr = 182), NMF exhibits excellent solvation properties for the TBA-TPhB BGE chosen for these experiments. The association of the TBA cation with the negatively charged PDMS surface drives EOF in the presence of an applied field. In order to investigate how EOF affects performance of the NAME system, tetraphenylphosphonium, a more hydrophobic cation, was selected for comparison. Using NBD-PA, peak shapes in TBA-TPhB/NMF and TPhP-TPhB/NMF solutions were compared, and it was determined that the tetraphenylphosphonium cation improves μobs of NBD-PA by approximately 8 per cent, a small, but statistically significant, effect.
3.5. Non-aqueous microchip electrophoresis of binary and ternary lipid mixtures
As shown in figure 5, fully resolved electrophoretic separations of both binary and ternary lipid mixtures are obtained 3.5 cm downstream of the injection point. In the case of cation-driven EOF, the electrophoretic driving force, determined by lipid charge-to-size ratio, opposes the bulk fluid motion. The order of migration of the three species agrees with predictions based on the molecular weights and charges of NBD-PA (MW = 563.5, net charge = −1), NBD-PG (MW = 637.6, net charge = −1) and NBD-CoA (MW = 1234.4, net charge = −3), since NBD-CoA has the largest electrophoretic mobility (largest charge-to-size ratio), and NBD-PG has the smallest.
Figure 5.
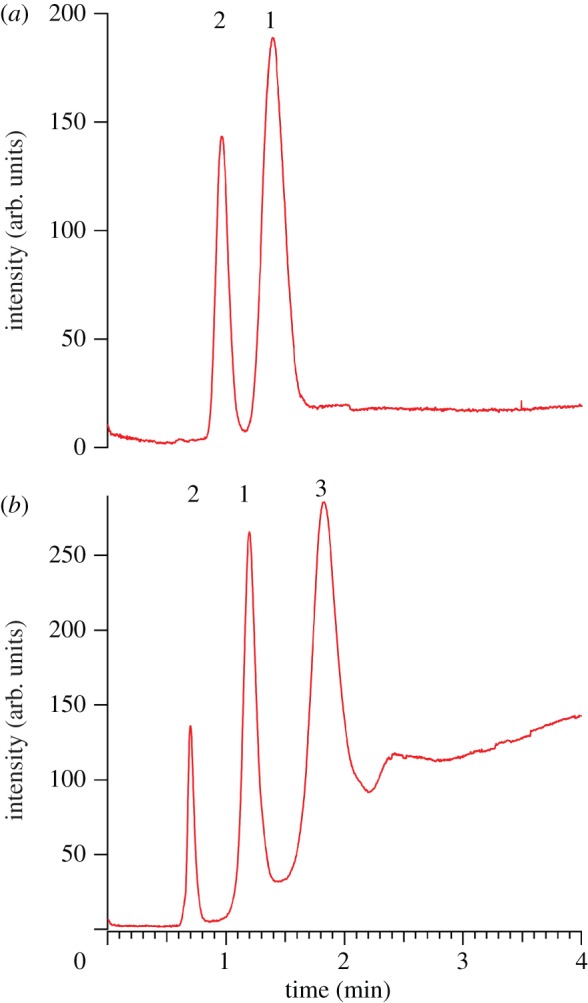
Electropherogram demonstrating high-resolution lipid separation via NAME. Peaks are observed 3.5 cm downstream of the injection point. (a) Electrophoretic separation of a binary analyte mixture: 10 μM NBD-PA (1) and NBD-PG (2) in 100 μM TBA-TPhB/NMF. (b) Electrophoretic separation of a ternary analyte mixture: 10 μM NBD-PA (1), NBD-PG (2) and CoA (3) in 100 μM TBA-TPhB/NMF. ΔVinj = 50 V, Δtinj = 1 s and Esep = 424 V cm−1. (Online version in colour.)
Another useful separation metric is the resolution, Rs, defined by equation (3.7),
| 3.7 |
where w represents the peak widths of the observed species. Acceptable baseline separation is achieved when Rs > 1.5. The resolution in figure 5 for the binary separation is Rs,21 = 2.16, and in the case of the ternary separation, Rs,21 = 5.02 and Rs,13 = 3.48. The plate numbers for each species in the binary (N1 = 1.65 × 105, N2 = 2.38 × 105) and ternary (N1 = 1.91 × 105, N2 = 3.26 × 105, N3 = 1.26 × 105) separations are notable. However, peak capacity is relatively low, since it depends on the quantity of injected analyte, which is governed by the injection parameters (see above). Regardless, the combination of the initial separation and EOF results is sufficiently promising to vigorously pursue NAME as a viable on-site separation strategy for in vivo monitoring of lipid biomarkers.
4. Conclusions
Micromolar tetraalkylammonium salts in NMF constitute effective media for electrophoretic separations of intact lipids and their oxidation products. Furthermore, these solutions are chemically compatible with low-cost microchips that offer superb fluid control for precise handling and manipulation of analyte mixtures. Discrete lipid packets are controllably injected from a sample reservoir microchannel, across an NCAM, using low injection voltages (ΔVinj < 100 V) for Δtinj = 1–10 s. The sample voxels are introduced into a separation channel containing low ionic strength BGE, and, in the presence of sufficiently high applied electric fields, yield high-resolution molecular separations of a quality comparable to those of commercial CE and NACE systems. In addition, relatively short separation channel lengths (roughly one-tenth the column length used in bench-top separation instruments) featured in this three-dimensional architecture afford very rapid fluidic processing of lipid mixtures (less than 3 min, typically). Unlike time-consuming immunoassays, when NAME is coupled to an appropriate pre-processing strategy, such as microdialysis, it can rapidly separate and monitor lipid biomarkers obtained directly from patient biofluids. In addition to monitoring the levels of known biomarkers to track disease progression, NAME can be implemented in programmes of biomarker discovery. Although separation performance in this work is assessed using fluorescence detection, the three-dimensional NAME microchip can be coupled directly to a mass spectrometer (MS) for universal label-free detection. Softer ionization strategies promise to better maintain lipid integrity during analyte introduction into the MS, so current work in this laboratory is addressing the interfacing of NAME microchips to ambient ionization MS in order to combine this promising new approach to lipid separations and biomarker detection with highly sensitive label-free detection.
Acknowledgements
We thank Kayla Shaw, who programmed a user-friendly LabView application for the high-voltage equipment operation. This work is supported by the National Science Foundation Instrument Development for Biological Research Program grant no. NSF 0852741.
References
- 1.Migliore L, Fontana I, Colognato R, Coppede F, Siciliano G, Murri L. 2005. Searching for the role and the most suitable biomarkers of oxidative stress in Alzheimer's disease and in other neurodegenerative diseases. Neurobiol. Aging. 26, 587–595 10.1016/J.Neurobiolaging.2004.10.002 (doi:10.1016/J.Neurobiolaging.2004.10.002) [DOI] [PubMed] [Google Scholar]
- 2.Ogino K, Wang DH. 2007. Biomarkers of oxidative/nitrosative stress: an approach to disease prevention. Acta Med. Okayama 61, 181–189 [DOI] [PubMed] [Google Scholar]
- 3.Pulfer M, Murphy RC. 2003. Electrospray mass spectrometry of phospholipids. Mass Spectrom. Rev. 22, 332–364 10.1002/Mas.10061 (doi:10.1002/Mas.10061) [DOI] [PubMed] [Google Scholar]
- 4.Tyurin VA, et al. 2000. Oxidative stress following traumatic brain injury in rats: quantitation of biomarkers and detection of free radical intermediates. J. Neurochem. 75, 2178–2189 (doi:10.1046/J.1471–4159.2000.0752178.X) [DOI] [PubMed] [Google Scholar]
- 5.Cracowski JL, Durand T, Bessard G. 2002. Isoprostanes as a biomarker of lipid peroxidation in humans: physiology, pharmacology and clinical implications. Trends Pharmacol. Sci. 23, 360–366 10.1016/S0165-6147(02)02053-9 (doi:10.1016/S0165-6147(02)02053-9) [DOI] [PubMed] [Google Scholar]
- 6.Mattsson N, Haghighi S, Andersen O, Yao YM, Rosengren L, Blennow K, Pratico D, Zetterberg H. 2007. Elevated cerebrospinal fluid F2-isoprostane levels indicating oxidative stress in healthy siblings of multiple sclerosis patients. Neurosci. Lett. 414, 233–236 10.1016/J.Neulet.2006.12.044 (doi:10.1016/J.Neulet.2006.12.044) [DOI] [PubMed] [Google Scholar]
- 7.Riccobene R, Arsena R, Mule G, Vaccaro F, Altieri C, Tornese F, Ocello A, Cerasola G, Cottone S. 2010. The relationship between an oxidative stress biomarker and plasma haemoglobin in patients with chronic kidney disease. High Blood Press. Cardiovasc. Prev. 17, 227–233 10.2165/11311980-000000000-00000 (doi:10.2165/11311980-000000000-00000) [DOI] [Google Scholar]
- 8.Sicilia T, Mally A, Schauer U, Pahler A, Volkel W. 2008. LC-MS/MS methods for the detection of isoprostanes (iPF(2 alpha)-III and 8,12-iso-iPF(2 alpha)-VI) as biomarkers of CCl4-induced oxidative damage to hepatic tissue. J. Chromatogr. B 861, 48–55 10.1016/J.Jchromb.2007.11.021 (doi:10.1016/J.Jchromb.2007.11.021) [DOI] [PubMed] [Google Scholar]
- 9.Proudfoot J, Barden A, Mori TA, Burke V, Croft KD, Beilin LJ, Puddey IB. 1999. Measurement of urinary F2-isoprostanes as markers of in vivo lipid peroxidation: a comparison of enzyme immunoassay with gas chromatography/mass spectrometry. Anal. Biochem. 272, 209–215 10.1006/Abio.1999.4187 (doi:10.1006/Abio.1999.4187) [DOI] [PubMed] [Google Scholar]
- 10.Ghosal S. 2006. Electrokinetic flow and dispersion in capillary electrophoresis. Annu. Rev. Fluid Mech. 38, 309–338 10.1146/Annurev.Fluid.38.050304.092053 (doi:10.1146/Annurev.Fluid.38.050304.092053) [DOI] [Google Scholar]
- 11.Sickman A, Dormeyer W, Wortelkamp S, Woitalla D, Kuhn W, Meyer HE. 2002. Towards a high resolution separation of human cerebrospinal fluid. J. Chromatogr. B 771, 167–196 10.1016/S1570-0232(01)00626-2 (doi:10.1016/S1570-0232(01)00626-2) [DOI] [PubMed] [Google Scholar]
- 12.Zhao ZW, Xu Y. 2010. An extremely simple method for extraction of lysophospholipids and phospholipids from blood samples. J. Lipid Res. 51, 652–659 10.1194/Jlr.D001503 (doi:10.1194/Jlr.D001503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terabe S. 2009. Capillary separation: micellar electrokinetic chromatography. Annu. Rev. Anal. Chem. 2, 99–120 10.1146/Annurev.Anchem.1.031207.113005 (doi:10.1146/Annurev.Anchem.1.031207.113005) [DOI] [PubMed] [Google Scholar]
- 14.Collier A, Wang J, Diamond D, Dempsey E. 2005. Microchip micellar electrokinetic chromatography coupled with electrochemical detection for analysis of synthetic oestrogen mimicking compounds. Anal. Chim. Acta. 550, 107–115 10.1016/J.Aca.2005.06.053 (doi:10.1016/J.Aca.2005.06.053) [DOI] [Google Scholar]
- 15.Zhang L, Hu S, Cook L, Dovichi NJ. 2002. Analysis of aminophospholipid molecular species by methyl-beta-cyclodextrin modified micellar electrokinetic capillary chromatography with laser-induced fluorescence detection. Electrophoresis 23, 3071–3077 (doi:10.1002/1522-2683(200209)23:17<3071::Aid-Elps3071>3.0.Co;2-K) [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Krylov SN, Hu S, Dovichi NJ. 2000. Methyl-beta-cyclodextrin modified micellar electrokinetic capillary chromatography with laser-induced fluorescence for separation and detection of phospholipids. J. Chromatogr. A 894, 129–134 10.1016/S0021-9673(00)00706-8 (doi:10.1016/S0021-9673(00)00706-8) [DOI] [PubMed] [Google Scholar]
- 17.Yang LY, Lee CS. 1997. Micellar electrokinetic chromatography mass spectrometry. J. Chromatogr. A 780, 207–218 10.1016/S0021-9673(97)00295-1 (doi:10.1016/S0021-9673(97)00295-1) [DOI] [PubMed] [Google Scholar]
- 18.Steiner F, Hassel M. 2000. Nonaqueous capillary electrophoresis: a versatile completion of electrophoretic separation techniques. Electrophoresis 21, 3994–4016 (doi:10.1002/1522-2683(200012)21:18<3994::Aid-Elps3994>3.0.Co;2-T) [DOI] [PubMed] [Google Scholar]
- 19.Jussila M, Sundberg S, Hopia A, Makinen M, Riekkola ML. 1999. Separation of linoleic acid oxidation products by micellar electrokinetic capillary chromatography and nonaqueous capillary electrophoresis. Electrophoresis 20, 111–117 (doi:10.1002/(Sici)1522–2683(19990101)20:1<111::Aid-Elps111>3.3.Co;2-L) [DOI] [PubMed] [Google Scholar]
- 20.Varga A, Nilsson S. 2008. Nonaqueous capillary electrophoresis for analysis of the ethanol consumption biomarker phosphatidylethanol. Electrophoresis 29, 1667–1671 10.1002/Elps.200700548 (doi:10.1002/Elps.200700548) [DOI] [PubMed] [Google Scholar]
- 21.Hernandez M, Borrull F, Calull M. 2002. Using nonaqueous capillary electrophoresis to analyze several quinolones in pig kidney samples. Electrophoresis 23, 506–511 (doi:10.1002/1522-2683(200202)23:3<506::Aid-Elps506>3.0.Co;2-C) [DOI] [PubMed] [Google Scholar]
- 22.Tivesten A, Folestad S, Schonbacher V, Svensson K. 1999. Nonaqueous capillary electrophoresis for the analysis of labile pharmaceutical compounds. Chromatographia 49, S7–S11 10.1007/Bf02468970 (doi:10.1007/Bf02468970) [DOI] [Google Scholar]
- 23.Wang F, Khaledi MG. 1996. Chiral separations by nonaqueous capillary electrophoresis. Anal. Chem. 68, 3460–3467 10.1021/Ac960537o (doi:10.1021/Ac960537o) [DOI] [PubMed] [Google Scholar]
- 24.Valko IE, Siren H, Riekkola ML. 1996. Chiral separation of dansyl-amino acids in a nonaqueous medium by capillary electrophoresis. J. Chromatogr. A 737, 263–272 10.1016/0021-9673(96)00003-9 (doi:10.1016/0021-9673(96)00003-9) [DOI] [Google Scholar]
- 25.Nandi P, Desaiasa DP, Lunte SM. 2010. Development of a PDMS-based microchip electrophoresis device for continuous online in vivo monitoring of microdialysis samples. Electrophoresis 31, 1414–1422 10.1002/Elps.200900612 (doi:10.1002/Elps.200900612) [DOI] [PubMed] [Google Scholar]
- 26.Schulze P, Ludwig M, Kohler F, Belder D. 2005. Deep UV laser-induced fluorescence detection of unlabeled drugs and proteins in microchip electrophoresis. Anal. Chem. 77, 1325–1329 10.1021/Ac048596m (doi:10.1021/Ac048596m) [DOI] [PubMed] [Google Scholar]
- 27.Cannon DM, Kuo TC, Bohn PW, Sweedler JV. 2003. Nanocapillary array interconnects for gated analyte injections and electrophoretic separations in multilayer microfluidic architectures. Anal. Chem. 75, 2224–2230 10.1021/Ac020629f (doi:10.1021/Ac020629f) [DOI] [PubMed] [Google Scholar]
- 28.Kim BY, Yang J, Gong MJ, Flachsbart BR, Shannon MA, Bohn PW, Sweedler JV. 2009. Multidimensional separation of chiral amino acid mixtures in a multilayered three-dimensional hybrid microfluidic/nanofluidic device. Anal. Chem. 81, 2715–2722 10.1021/Ac802630p (doi:10.1021/Ac802630p) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo TC, Cannon DM, Chen YN, Tulock JJ, Shannon MA, Sweedler JV, Bohn PW. 2003. Gateable nanofluidic interconnects for multilayered microfluidic separation systems. Anal. Chem. 75, 1861–1867 10.1021/Ac025958m (doi:10.1021/Ac025958m) [DOI] [PubMed] [Google Scholar]
- 30.Kuo TC, Cannon DM, Shannon MA, Bohn PW, Sweedler JV. 2003. Hybrid three-dimensional nanofluidic/microfluidic devices using molecular gates. Sens. Actuators A 102, 223–233 10.1016/S0924-4247(02)00394-1 (doi:10.1016/S0924-4247(02)00394-1) [DOI] [Google Scholar]
- 31.Iannacone JM, Jakubowski JA, Bohn PW, Sweedler JV. 2005. A multilayer poly(dimethylsiloxane) electrospray ionization emitter for sample injection and online mass spectrometric detection. Electrophoresis 26, 4684–4690 10.1002/Elps.200500498 (doi:10.1002/Elps.200500498) [DOI] [PubMed] [Google Scholar]
- 32.Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. 1998. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane). Anal. Chem. 70, 4974–4984 10.1021/Ac980656z (doi:10.1021/Ac980656z) [DOI] [PubMed] [Google Scholar]
- 33.Chueh BH, Huh D, Kyrtsos CR, Houssin T, Futai N, Takayama S. 2007. Leakage-free bonding of porous membranes into layered microfluidic array systems. Anal. Chem. 79, 3504–3508 10.1021/Ac062118p (doi:10.1021/Ac062118p) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JN, Park C, Whitesides GM. 2003. Solvent compatibility of poly(dimethylsiloxane)-based microfluidic devices. Anal. Chem. 75, 6544–6554 10.1021/Ac0346712 (doi:10.1021/Ac0346712) [DOI] [PubMed] [Google Scholar]
- 35.Fa K, Tulock JJ, Sweedler JV, Bohn PW. 2005. Profiling pH gradients across nanocapillary array membranes connecting microfluidic channels. J. Am. Chem. Soc. 127, 13 928–13 933 10.1021/Ja052708p (doi:10.1021/Ja052708p) [DOI] [PubMed] [Google Scholar]
- 36.Kuo TC, Sloan LA, Sweedler JV, Bohn PW. 2001. Manipulating molecular transport through nanoporous membranes by control of electrokinetic flow: effect of surface charge density and Debye length. Langmuir 17, 6298–6303 10.1021/La010429j (doi:10.1021/La010429j) [DOI] [Google Scholar]
- 37.Culbertson CT, Jacobson SC, Ramsey JM. 2002. Diffusion coefficient measurements in microfluidic devices. Talanta 56, 365–373 10.1016/S0039-9140(01)00602-6 (doi:10.1016/S0039-9140(01)00602-6) [DOI] [PubMed] [Google Scholar]
- 38.Gatimu EN, King TL, Sweedler JV, Bohna PW. 2007. Three-dimensional integrated microfluidic architectures enabled through electrically switchable nanocapillary array membranes. Biomicrofluidics 1, 021502. 10.1063/1.2732208 (doi:10.1063/1.2732208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brantley MA, Osborn MP, Sanders BJ, Rezaei KA, Lu P, Li C, Milne GL, Cai J, Sternberg P. 2012. Plasma biomarkers of oxidative stress and genetic variants in age-related macular degeneration. Am. J. Ophthalmol. 153, 460–467 10.1016/J.Ajo.2011.08.033 (doi:10.1016/J.Ajo.2011.08.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hohl A, et al. 2012. Plasma levels of oxidative stress biomarkers and hospital mortality in severe head injury: a multivariate analysis. J. Crit. Care 27, 523 e11–e19 10.1016/j.jcrc.2011.06.007 (doi:10.1016/j.jcrc.2011.06.007) [DOI] [PubMed] [Google Scholar]
- 41.Tug T, Karatas F, Terzi SM, Ozdemir N. 2005. Comparison of serum malondialdehyde levels determined by two different methods in patients with COPD: HPLC or TBARS method. Lab. Med. 36, 41–44 10.1309/Wteet9tj2lumb3c3 (doi:10.1309/Wteet9tj2lumb3c3) [DOI] [Google Scholar]
- 42.Karaouzene N, Merzouk H, Aribi M, Merzouk SA, Berrouiguet AY, Tessier C, Narce M. 2011. Effects of the association of aging and obesity on lipids, lipoproteins and oxidative stress biomarkers: a comparison of older with young men. Nutr. Metab. Cardiovasc. Dis. 21, 792–799 10.1016/J.Numecd.2010.02.007 (doi:10.1016/J.Numecd.2010.02.007) [DOI] [PubMed] [Google Scholar]
- 43.De Felice C, et al. 2011. F-2-dihomo-isoprostanes as potential early biomarkers of lipid oxidative damage in Rett syndrome. J. Lipid Res. 52, 2287–2297 10.1194/Jlr.P017798 (doi:10.1194/Jlr.P017798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chun HG, Chung TD, Ramsey JM. 2010. High yield sample preconcentration using a highly ion-conductive charge-selective polymer. Anal. Chem. 82, 6287–6292 10.1021/Ac101297t (doi:10.1021/Ac101297t) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang YC, Stevens AL, Han JY. 2005. Million-fold preconcentration of proteins and peptides by nanofluidic filter. Anal. Chem. 77, 4293–4299 10.1021/Ac050321z (doi:10.1021/Ac050321z) [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Timperman AT. 2003. Integration of nanocapillary arrays into microfluidic devices for use as analyte concentrators. Analyst 128, 537–542 10.1039/B300102d (doi:10.1039/B300102d) [DOI] [PubMed] [Google Scholar]
- 47.Steiner SA, Fritz JS. 2008. Separation of organic cations using novel background electrolytes by capillary electrophoresis. J. Chromatogr. A 1192, 152–156 10.1016/J.Chroma.2008.02.022 (doi:10.1016/J.Chroma.2008.02.022) [DOI] [PubMed] [Google Scholar]