

Abstract
Variation in the social environment can have profound effects on survival and reproduction in wild social mammals. However, we know little about the degree to which these effects are influenced by genetic differences among individuals, and conversely, the degree to which social environmental variation mediates genetic reaction norms. To better understand these relationships, we investigated the potential for dominance rank, social connectedness and group size to modify the effects of genetic variation on gene expression in the wild baboons of the Amboseli basin. We found evidence for a number of gene–environment interactions (GEIs) associated with variation in the social environment, encompassing social environments experienced in adulthood as well as persistent effects of early life social environment. Social connectedness, maternal dominance rank and group size all interacted with genotype to influence gene expression in at least one sex, and either in early life or in adulthood. These results suggest that social and behavioural variation, akin to other factors such as age and sex, can impact the genotype–phenotype relationship. We conclude that GEIs mediated by the social environment are important in the evolution and maintenance of individual differences in wild social mammals, including individual differences in responses to social stressors.
Keywords: gene–environment interaction, dominance rank, Amboseli baboons, social connectedness, allele-specific gene expression
1. Introduction
The social interactions and social structures that characterize group-living mammals are not only products of adaptive change, but can themselves influence the evolutionary process. For example, behavioural patterns that govern mating and dispersal are directly reflected in patterns of population genetic structure [1–5]. Social behaviour thus affects the distribution of genetic variation upon which selection can act. Social behaviour also shapes the environment experienced by individual animals within a social group. Low-status versus high-status individuals, or socially integrated versus socially isolated animals may differ in adaptively important aspects of steroid hormone physiology ([6,7]; reviewed in Cavigelli & Chaudhry [8]), immune function [9–14] and access to mates or other resources [15–20]. Such differences provide scope for social behaviour to create new sources of environmental selective pressure. Indeed, this observation led West-Eberhard [21] to argue that evolutionary transitions to obligate sociality often alter the set of traits shaped by strong selection.
Much of what we know about the evolutionary impact of social behaviour in social mammals has arisen from field studies, which have produced detailed illustrations of the relationship between social structure and genetic structure [22,23] and the impact of social interactions on fitness [24,25]. By contrast, we know far less about a third potential effect of social behaviour on the genetics of these species: the role of the social environment in shaping genetic reaction norms. Specifically, we know little about whether and to what extent the social environment, similarly to other environmental effects, can produce norms of reaction that differ for individuals of different genotypes (i.e. gene–environment interactions, GEIs [26]). Viewed from a complementary perspective, we also do not know the degree to which physiological changes in response to the social environment are contingent on genotype. GEIs involving the social environment are thus important for two reasons. First, GEIs may help us understand how genetic differences among individuals affect susceptibility to selectively relevant social environmental conditions. Second, by altering how genetic variation is translated into trait variation, such GEIs may alter the strength of selection on the genetic variants themselves.
At least two lines of evidence suggest that GEIs involving social environmental effects are likely to arise in natural animal populations. First, data from a range of species indicate that GEIs often involve environmental variation that has large direct effects on fitness. Low-quality or quantity of food resources, for example, alters genetic effects on sexually selected traits in collared flycatchers [27], body size in blue tits [28] and lifespan in Drosophila [29–31]. Similarly, in plants, classical ecological stressors such as drought and leaf damage influence genetic effects on flowering time [32]. Because the social environment impacts fitness for many social mammals, it might also participate in GEIs. Second, studies of captive rhesus macaques have indicated the potential for socially mediated GEIs to take place [33]. For instance, in male rhesus macaques, early social environment (rearing of male infants with their mothers versus rearing of male infants with same-age peers) appears to mediate genetic effects on adult aggression [34], alcohol consumption [35] and stress hormone physiology [36]. Whether similar effects occur in the context of natural variation in the social environment, however, remains unknown.
We set out to test this possibility by taking advantage of a long-term study of a well-characterized population of social mammals: the baboons of the Amboseli basin of Kenya. The Amboseli baboons have been under continuous study for over 41 years [37], providing an opportunity to focus on aspects of the social environment of known importance to these animals [7,9,15,19]. We combined detailed observational data on dominance rank, social connectedness and group size with new data on genetic variation in the same set of individuals.
We also gathered data on gene expression variation as the phenotype of interest for testing for GEIs. We chose gene expression levels, because they represent accessible quantitative traits that can be readily measured at multiple loci, are responsive to social environmental variation [11,38], and are influenced by GEIs. For example, for 47 per cent of genes in the yeast genome, the effects of genetic variation on gene expression levels depend on feeding substrate (glucose or ethanol). That is, differences in gene expression between yeast strains were either present in only one feeding condition, or were larger in one condition than the other [39]. We also took advantage of the fact that genetic variants that affect gene expression often lie close to the genes they regulate, on the same physical chromosome. Hence, the maternally inherited allele ‘controls’ gene expression of the maternally inherited copy of the gene, and the paternally inherited allele ‘controls’ gene expression of the paternally inherited copy. Genetic effects on gene expression that behave in this manner (often referred to as cis-regulatory variants) can therefore be detected by measuring allele-specific gene expression (ASGE), which measures differences in gene expression between the two alleles of a gene, within each individual [40,41]. ASGE assays therefore capture the ratio of gene expression between two alleles in the same environmental and genetic background (because the two alleles are contained within the same individual). Importantly for this study, ASGE levels can also indicate the presence of GEIs when differences in ASGE across individuals are associated with environmental variation. Specifically, GEIs are implicated in cases in which ASGE values for a given genotype vary across environments, implying that environmental exposure changes the relative amounts of gene expression driven by the two alleles present in a study subject [42–44].
Here, we took advantage of ASGE measurements to identify genes for which gene expression is affected by cis-regulatory variation and to detect GEIs. We asked two sets of questions. First, we tested for evidence that several aspects of the social environment are involved in naturally occurring GEIs, focusing on dominance rank, social connectedness and group size as the key social environments of interest. To place these analyses in context, we also tested for interactions between genotype and age, and between genotype and sex, two interactions that are distinct from (although potentially related to) social environmental GEIs. Second, we investigated whether GEIs were more likely to be associated with early life social environmental variables, in aggregate, rather than with adult social environments, and whether one sex was more likely to experience rank- or social connectedness-related GEIs than the other.
2. Methods
(a). Study subjects
Study subjects were 96 members (50 females and 46 males) of a natural population of baboons monitored by the Amboseli Baboon Research Project in the Amboseli basin, Kenya. This population consists primarily of yellow baboons (Papio cynocephalus) with some hybrid admixture from immigration of anubis baboons (Papio anubis) from outside the basin [45,46]. Ninety-one of the 96 individuals included in this study were members of one of five intensively studied social groups (i.e. ‘study groups’) sampled between 2005 and 2009. The other five individuals were born into study groups but emigrated to non-study group as adults and were sampled in those non-study groups. All study subjects were recognized on sight by observers based on unique physical characteristics.
For most individuals (n = 78), social environmental information was available for both early life and adult life (close to the time of darting), as a consequence of near-daily behavioural and demographic monitoring. For a subset of males who immigrated into the study population as adults (n = 15), data were missing on early life social environment, and for three females, maternal rank and social connectedness data were missing because of sparse data collection during their early lives. Birthdates and ages for the majority of individuals were known within several days' error (n = 81); for immigrant males, birthdates were estimated based on known patterns of age-related change in physical characteristics [47].
(b). Collection of blood samples
To obtain blood samples for gene expression analysis and genotyping, study subjects were anaesthetized with a Telazol-loaded dart using a handheld blowpipe. Adult animals were darted opportunistically, resulting in an overall sample that was randomized with respect to age, sex and the environmental characteristics we analysed here, except that in addition to only darting adult (post-pubertal) animals, we also avoided females with dependent infants and pregnant females beyond the first trimester of pregnancy. Following anaesthetization, study subjects were quickly transferred to a processing site distant from the rest of the group. Blood samples for gene expression analysis were collected by drawing whole blood into PaxGene Vacutainer tubes (BD Vacutainer), and blood samples for sequencing and genotyping were collected into BD Vacutainer EDTA tubes. Following sample collection, study subjects were allowed to regain consciousness in a covered holding cage until fully recovered from the effects of the anaesthetic. They were then released within view of their social group; all subjects promptly rejoined their respective groups upon release, without incident.
Blood samples were stored for no more than 3 days in an evaporatively cooled charcoal structure at Amboseli, which maintains a daily maximum temperature of 20–25°C. They were then shipped to Nairobi, where they were frozen at −20°C until transport to the USA. Our previous work has demonstrated stability of ASGE measurements under these conditions [44,48]. For pyrosequencing assays, RNA was extracted using the PaxGene RNA blood kit (Qiagen) and reverse transcribed into cDNA (High Capacity cDNA Archive Kit; Applied Biosystems). For genotyping and sequencing, DNA samples were extracted using the DNeasy DNA extraction kit (Qiagen).
(c). Social environmental effects
(i). Dominance rank
In baboons, dominance rank of mature individuals is linear within sexes and is measured by the ability of dominant individuals to consistently win agonistic encounters with their subordinates. Adult dominance ranks in Amboseli are assigned on a monthly basis, separately for each sex, based on the outcomes of all pairwise encounters during that month. Baboons do not achieve adult dominance ranks until they are 2–4 years old (for females) or 6–8 years old (for males) [49,50]. Hence, an individual's status during early life largely reflects the status of its mother, which we term its maternal dominance rank. For our measure of rank effects during early life, we therefore considered maternal dominance rank, measured as the rank of each focal individual's mother during the month that individual was conceived [51]. As our measure of rank in adulthood, we used the sex-specific dominance rank for each individual, assigned in the month that individual was darted.
(ii). Social connectedness
Social connectedness measures (SCI-M for males and SCI-F for females) capture the degree to which individuals are socially integrated with other individuals in their groups. We calculated one SCI value per individual per year of age, as a composite index of the frequency the individual was groomed and groomed others (for males) at that age; for females, we also included whether the individual was in close proximity to others [19]. Specifically, we identified the number of times the focal individual was groomed by another adult, the number of times the focal individual groomed another adult and (in the case of females) the number of times the focal animal was the nearest neighbour of an adult female (within 5 m) [52]. These counts were not directly comparable across groups of different sizes because the number of observations per animal was reduced in larger groups relative to smaller groups. Hence, to control for these differences in observer intensity across groups, we collated the same data for all other same-sex adults alive in the population during the same interval. We then regressed each measure (grooming, being groomed and proximity data) separately against the number of point samples per adult female per day the study subject was in a given group. Finally, we calculated social connectedness as the mean value of the residuals of the respective models for grooming, being groomed and proximity for the focal individual. Note that while SCI-F was a consequence of interactions between focal females and both adult males and adult females, SCI-M reflects interactions between focal males and adult females.
For measures of social connectedness during early life, we used the focal individual's mother's social connectedness value (SCI-F), during the year the focal individual was conceived (i.e. if the focal individual was conceived when its mother was 4.5 years old, then we used the year interval from age 4 to age 5 for the mother). In 15 cases for natal individuals, maternal SCI measures were not available for the time interval surrounding conception. We then used the closest available measure of SCI for that individual's mother, provided it overlapped the calendar year before or after the year of the focal individual's conception (n = 6; otherwise, the SCI was considered missing data: n = 9). For measures of social connectedness in adulthood, we considered the SCI-M or SCI-F value for the age–year overlapping the date each individual was darted (n = 83), or the closest available measure of SCI to the dart date, within 1 year (n = 3 for females and n = 2 for males).
(iii). Group demography
To capture social competition for resources and availability of mates, we measured the number of adults present in an individual's social group either at the month of birth (to measure the influence of early life group demography) or at the month of darting (to measure the influence of group demography during adulthood).
(d). Pyrosequencing assay development
We measured ASGE using pyrosequencing on a PyroMark Q96 MD instrument. This approach depends on the presence of at least one single nucleotide polymorphism (SNP) in the transcribed region of a target gene (the ‘assay SNP’), which allows a PCR-based assay to discriminate between the two variants of the target gene (these variants need not be functionally different in the protein they produce). In heterozygotes for the assay SNP, ASGE can then be measured as the log2-transformed ratio of the signal derived from one variant of the target gene versus the signal derived from the alternative variant of the target gene [40]. The advantage of this approach is that correlations between ASGE and environmental variation indicate GEIs, not simply changes in total gene expression; in other words, they indicate differences in the proportional expression of two different alleles as a function of changes in the social environment. A limitation is that ASGE measurements can only be taken for those genes that harbour a common SNP in a transcribed region; if such a SNP is not available, then this method will not work.
Because of this limitation, we began our assay development efforts using a large initial set of 166 loci (figure 1). This set was chosen because they were likely to be expressed in our samples (i.e. in blood) and because they scored highly on a predictive algorithm for common ASGE [53]. We also added several loci because they had previously been studied in association with gene expression variation in humans or other primates. This set was then filtered to those genes (i) for which we could generate high-quality Sanger sequence from putative transcribed regions, based on primers derived from the then-current draft baboon genome sequence (Pham1.0; 18 genes failed this filter); (ii) that harboured one or more common SNPs in these regions, based on Sanger sequencing runs from 10 to 12 unrelated Amboseli individuals (33 genes failed this filter; note that we did not sequence all transcribed regions for each gene); and (iii) for which both variants of a potential assay SNP were detectable in ASGE assays, based on successful amplification of the region surrounding a target assay SNP from cDNA and good pyrosequencing signal strength (22 genes failed this filter). An additional four genes were filtered due to unacceptably high variance across technical replicates. After filtering, we measured ASGE for 89 genes (figure 1). This gene set was enriched for genes involved in immunity, as cell types found in blood play important roles in the immune response.
Figure 1.

Overall workflow. (a) An initial set of genes was screened for those loci for which we could perform ASGE measurements and then for which we detected common ASGE within an initial test set of Amboseli individuals. (b) Genes that exhibited common ASGE (n = 34) were subjected to ASGE measurements for all ASGE assay SNP heterozygotes and approximately 7–9 kb of the putative cis-regulatory region was resequenced to identify genetic effects on gene expression (see Table S2 for exact numbers). (c) These genes were then analysed jointly to test for evidence of GEIs involving each environment of interest. The number of genes that survived each progressive screening set is shown in bold at each transition between steps.
(e). Allele-specific gene expression measurements
To test for GEIs, we further restricted our analysis to genes that exhibited common ASGE, which signified the presence of common segregating genetic variation that affects gene expression levels. This filter was necessary because our interest lay in testing whether the social environment modifies the magnitude of ASGE, and we did not have power to detect such effects for genes that rarely exhibited non-zero ASGE. To identify cases of common ASGE, we genotyped the assay SNPs identified for each gene to identify heterozygous individuals. We then tested for common ASGE in six to eight individuals, based on four replicate cDNA PCRs (to measure gene expression) and two replicate genomic DNA (gDNA) PCRs (to control for technical bias in the relative signal strength for the two alleles) for each individual. We log2-transformed the ratio of the signal strength from the two alleles for each reaction and tested whether the distribution of ASGE values obtained from cDNA differed from the corresponding distribution obtained from gDNA (two-tailed, non-parametric Wilcoxon-summed ranks test, with significance assessed via permutation, following Tung et al. [44]). This procedure allowed us to exclude cases in which ASGE measurements from cDNA were largely indistinguishable from the same assay run on gDNA, indicating absent or rare ASGE in the Amboseli baboons.
Based on this procedure, we identified 35 genes that putatively showed common ASGE. Upon further evaluation (see §3), we believe one of these genes (CYP17A1) was a false positive; hence, 34 of these genes formed the core of the remainder of our study. For each of these genes, we measured ASGE in all individuals in the study sample that were heterozygous for the assay SNP. For each individual–gene combination, we ran four replicate cDNA measurements and two replicate gDNA measurements on two separately prepared pyrosequencing plates. After log2-transforming the relative intensities of the two alternative alleles at the assay SNP, we performed three levels of quality filtering. First, we removed measurements in which one of the two alleles was detected at low intensity (less than 20 units; assay variance is higher for low-intensity measurements because ASGE measurements are ratios). Second, we removed outlier gDNA measurements (approx. 10% of all measurements), conservatively identified as those deviating by more than 0.5 log2 units from the plate-specific median for all gDNA measurements. This approach corrected for potential assay failures. If all gDNA measurements for an individual–gene combination were outliers, then the individual was removed from the analysis for that gene. Finally, we averaged the log2-transformed cDNA measurements and gDNA measurements for each individual–gene combination, and corrected the cDNA measurements by subtracting the mean gDNA log2-transformed ratio. This is standard practice for assessing ASGE using pyrosequencing [40,41,48]: the idea is that some level of technical bias may be inherent to an ASGE assay itself, and this bias can be corrected based on estimating the magnitude of the bias from gDNA samples (which have a known ratio; 1 : 1 in most cases). This procedure also corrects for plate effects (i.e. systematically higher or lower signal from one of the two alternative bases on a specific plate) because they affect both cDNA and gDNA measurements. After correcting cDNA measurements with gDNA measurements on the same plate, these plate effects are removed. Following these three quality-filtering steps, we obtained a single measure of corrected ASGE for each individual–gene combination (see the electronic supplementary material, table S1 for a summary of numbers of individuals assayed for each gene).
(f). Cis-regulatory genotyping
Sequence variants that influence gene expression differences in cis tend to be clustered near transcription start sites (TSSs). We therefore focused on these regions to search for sequence variants that might help explain ASGE in the Amboseli population. To identify and genotype these putative regulatory variants, we used a target enrichment approach (Agilent SureSelect) [54], followed by high-throughput sequencing on the Illumina HiSeq 2000 platform. For each of the 34 genes with common ASGE, we identified an approximately 7–9 kb target (see the electronic supplementary material, table S2) covering the region upstream of the gene TSS and the region between the TSS and its downstream translation start site. To do so, we used the rhesus macaque genome (rhemac2), because the draft baboon genome available at the time (Pham1.0) contained too many gaps and missing regions to adequately cover these regions. Importantly, rhesus macaque and baboon have highly similar sequence in these regions, and cross-species sequence capture has been validated in primates, including more distant pairs than macaque–baboon [55,56]. We then designed 120 bp biotinylated RNA probes tiled to cover our target regions at a mean 2× coverage. We used these probes to capture our regions of interest. We added a unique 6 bp barcode (Agilent) to each library, which enabled us to pool captured DNA from all 96 individuals and sequence a single, multiplexed sample on a single lane of the HiSeq 2000.
We generated 182 million, 50 bp reads from this sample (see the electronic supplementary material, table S3). Reads were generally evenly distributed across the 96 individual subjects (median = 1.85 million reads±0.060 million reads s.d.), with the exception of two outlier individuals for whom we obtained very few reads (‘Face’ and ‘Morris’: electronic supplementary material, table S3). Reads were mapped to the baboon genome (Panu2.0, released after the probes were designed) using the default settings in NovoAlign (NovoCraft). Across individuals, a median of 82 per cent (median range: 60–85%) of reads mapped to the genome with Phred-scaled mapping quality greater than or equal to 20. To translate between the regions we targeted using rhesus macaque genome sequence and the baboon genome assembly, we used lastz [57] and axtChain [58] to find the corresponding target regions in baboon. For 89 per cent of genes, we could clearly identify a single region that corresponded to the size expected based on the rhesus macaque sequence from which the probes were designed. For four genes (CLC, GBP1, APOBE3G and RNASE2), our probes mapped to a larger region than we had anticipated in the original probe design; we therefore analysed genotype data from all baboon regions that were on the same chromosome as the ASGE assay SNP and that matched the target macaque sequence, as SNPs that were farther than expected from a gene transcription start site could still plausibly be functionally relevant. Note that for these four genes, we also performed SNP and genotype calls based only on reads from the captured sequences that mapped uniquely to the baboon genome; we simply captured a larger region than we had originally intended. In total, a median of 72 per cent of high-quality-mapped reads fell within the targeted regions in baboon (range: 24–80%).
We conducted variant discovery and genotyping on the mapped, quality-filtered reads, using the Genome Analysis Toolkit (GATK) ([59,60]; see electronic supplementary material for additional details). We then used the program Beagle [61] to impute missing genotypes in the resulting dataset. As input, we used the genotype likelihoods produced by GATK; we then filtered the Beagle results to include only genotypes with a posterior probability greater than 0.98.
(g). Identification of gene–environment interactions
To identify GEIs in our dataset, we reasoned that an environment that has an unconditional effect on gene expression (i.e. is independent of cis-regulatory variation that might be associated with the gene) should influence the expression levels of both alleles of a gene similarly. An environmental effect involved in a GEI, on the other hand, should influence the expression levels of the two alleles of a gene differently, depending on the identity of the cis-regulatory variant(s) linked to that allele [42–44]. Under this model, individuals heterozygous for a functional cis variant will exhibit different levels of ASGE depending on the environment (provided that this variant is, at least to some degree, linked to the transcribed SNP used in the ASGE assay and the environment is not confounded by genetic background effects: the social environments we considered are unlikely to be confounded by genetic background in our sample, as they are poorly correlated with measures of both admixture and kinship; see electronic supplementary material, figures S1 and S2). We took advantage of this property to test whether models of ASGE that included GEIs involving the social environments of interest were favoured over models that did not include GEIs.
ASGE itself signifies the presence of functional cis-regulatory variation, where the responsible functional variant(s) is linked, to some degree, to the ASGE assay SNP. The a priori expectation is therefore that heterozygotes for the assay SNP (i.e. the individuals we were able to assay) are probably heterozygotes for the functional site(s) as well, which is likely to be the case if the assay SNP is closely linked to this site. Such a pattern would be reflected by non-zero ASGE levels for all assayed individuals. Alternatively, some individuals might not be heterozygous for the functional cis-regulatory site if the assay SNP and the functional variant were not tightly linked (e.g. in the cases when the assay SNP is far from the putative promoter region). This possibility, in turn, would be reflected by a pattern in which some assayed individuals (those heterozygous for the functional SNP) exhibited non-zero ASGE, but others (homozygotes for the functional SNP) exhibited ASGE levels close to zero. In these cases, heterozygosity/homozygosity at a site more closely linked to the functional SNP would better explain variance in ASGE levels. Because ASGE levels in homozygotes would not correlate with environmental variation even in the presence of GEIs, including these individuals would dilute any signal of GEIs in the population. For each gene, we therefore compared models reflecting these two alternative possibilities: either non-zero ASGE in all assayed individuals (reflecting close linkage disequilibrium between the assay SNP and the functional site), or non-zero ASGE only in heterozygotes for a putative regulatory SNP (reflecting closer linkage between the functional site and this SNP instead of the assay SNP). We chose the best model as that which yielded the highest model r2-value. This model identified probable heterozygotes at the (unknown) functional regulatory site responsible for ASGE as either heterozygotes at a regulatory region SNP or heterozygotes for the assay SNP itself (see the electronic supplementary material, figure S3). We used only these heterozygous individuals in subsequent GEI analyses. In all these analyses, we excluded putative regulatory SNPs that were in apparently perfect linkage disequilibrium with other sites for the same gene (we retained a single SNP for each correlated set) and putative regulatory SNPs with missing or little data (fewer than five heterozygous individuals and five homozygous individuals). Note that this process was completely blind to data on environmental variation.
We then asked whether, for each environmental variable of interest, our dataset supported the potential for GEIs involving the social environment. We did so using the following nested set of models (including heterozygotes or inferred heterozygotes at a putative functional regulatory SNP only):
and
where yij is the log2-transformed, normalized ASGE measure for gene i in individual j; gi fits an intercept for each gene; gi × vj fits a separate regression slope for each gene, relating the focal environmental variable to ASGE values for that gene; and eij is a residual, which we assumed to be independent and normally distributed with E[eij] = 0 and variance 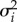 (different for each gene). For each environment, except dominance rank and social connectedness (which are calculated differently for males and females and have different biological interpretations), we tested males and females both separately and jointly.
(different for each gene). For each environment, except dominance rank and social connectedness (which are calculated differently for males and females and have different biological interpretations), we tested males and females both separately and jointly.
Models were fit using a maximum-likelihood criterion with the function gls in R [62], after excluding genes with fewer than five heterozygous individuals. If M1 was a better fit to the data than M0, as assessed by a likelihood ratio test, we interpreted the data as supportive of GEI(s) arising as a consequence of the social environment tested in those models. Importantly, this approach specifically tests the hypothesis that a given social environment participates in GEIs in Amboseli, rather than testing each gene–environment combination for each gene separately (which would incur an unacceptably high multiple testing burden, given our sample size). We adjusted for multiple testing using the false discovery rate method of Benjamini & Hochberg [63] in the function p.adjust in R.
Finally, we tested two hypotheses about how GEIs differ between sexes and in relationship to the timing of environmental effects. First, we tested whether GEIs were more strongly associated with early life social environments (maternal dominance rank, maternal social connectedness and social group size in early life) than with adult social environments (the individual's own rank, social connectedness index and group size at the time of darting), or vice versa. Because rank and social connectedness were calculated separately for males and females, we tested for early versus late life GEIs separately for each sex. Second, we tested whether, among sex-specific effects (dominance rank and social connectedness), GEIs were more evident in male or female subjects.
For both tests, we compared the variance in ASGE explained for each gene by (i) early versus late environmental effects, and (ii) male-specific versus female-specific environmental effects, using a two-sample paired t-test. Genes were removed from these analyses if fewer than nine individuals for the early versus adult life comparison, or eight individuals for the male versus female comparison (which involved fitting fewer parameters), were testable for each of the two competing models.
3. Results
(a). Genetic effects on gene expression
We identified common ASGE in 34 blood-expressed genes in the Amboseli baboon population, out of 166 genes we originally surveyed and 89 genes for which we could reliably measure ASGE. We therefore estimate that commonly segregating functional cis-regulatory variants influence gene expression in approximately 38 per cent of genes in the Amboseli baboon population. This frequency agrees with a previous, smaller-scale analysis of this population, which yielded an estimate of 36.4 per cent [44]. In addition, the putative cis-regulatory regions near gene TSSs that we surveyed exhibited substantial segregating genetic variation. Overall, we identified 3527 high confidence segregating sites (among 250 333 total base pairs for which at least 10 individuals were sequenced at a coverage of at least 30×), indicating a frequency of variants identified (including rare variants) of about one per 70 bp. Diversity levels based on these SNPs were in excellent concordance with estimated diversity levels from prior, Sanger sequencing-based estimates of genetic diversity for the same population (see the electronic supplementary material).
For 13 of the 34 genes that exhibited common ASGE, the presence or absence of ASGE in an individual baboon was best explained by heterozygosity versus homozygosity at a SNP in the captured, resequenced regulatory region (figure 2a). In these cases, genetic variation responsible for the observed ASGE was likely to be more closely linked to variants in the upstream regulatory region than to the assay SNP itself (consistent with the expectation that these regions are enriched for functional cis-regulatory variants). For these genes, genotype at the putative regulatory site explained a large proportion of overall variance in ASGE (median r2 = 55%; range = 31–81%: note that in subsequent GEI analysis, we were concerned only with variance among heterozygotes, not the variance explained by the contrast between heterozygotes and homozygotes reported here). For 21 genes, heterozygosity at the assay SNP best explained the patterns of ASGE in our sample, and thus we included all assayed individuals (all of whom were heterozygotes at the assay SNP) in downstream GEI analyses. That is, for these 21 genes, the assay SNP was likely to be more closely linked to the functional variant driving ASGE than any of the resequenced upstream SNPs in the sample. Thus, the resequenced variants explained variance in ASGE levels more poorly than a model in which all assayed individuals were probably heterozygous for the (unknown) functional site (figure 2b: the best SNP identified in the resequenced regulatory region is shown against the assay SNP for comparison). Finally, for one gene, CYP17A1, we identified no best SNP: neither the assay SNP nor any of the resequenced, putative regulatory SNPs explained ASGE variation well (no SNP explained variance in ASGE levels with a p-value below a nominal threshold of 0.05). This gene (CYP17A1) probably represents a false positive result from our earlier, more restricted test for common ASGE and was excluded from further analysis.
Figure 2.
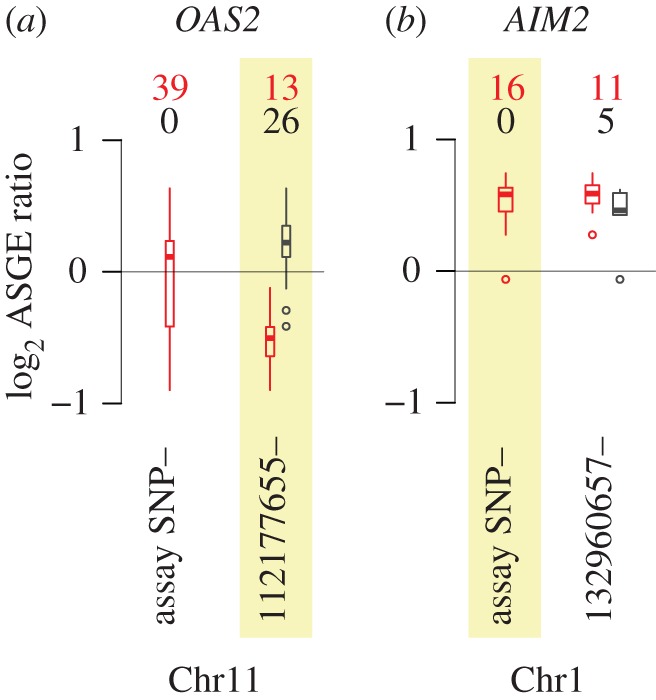
Example genetic associations with ASGE. (a) Variation in the ASGE data for OAS2 is best explained by heterozygosity/homozygosity at a SNP contained in the captured, resequenced region upstream of the OAS2 transcription start site (r2 for a model including the best site in the resequencing data = 0.54 versus r2 for a model including the assay SNP = 0.01). (b) Variation in the ASGE data for AIM2 is best explained by heterozygosity/homozygosity at the assay SNP, and not at any SNP in the captured, resequenced region (r2 for a model including the assay SNP = 0.88 versus r2 for a model including the best site in the resequencing data = 0.73). (a,b) In both panels, boxplots show the range of ASGE variation across all assayed individuals (left: ‘assay SNP’) and ASGE variation subdivided by a SNP in the captured, resequenced region (right: base pair coordinates for these SNPs are provided as labels). Numbers above each set of boxplots provide the number of heterozygotes (red) and homozygotes (black) for each site, and the site associated with the best ASGE partition is highlighted in yellow. For the assay SNP, all assayed individuals are heterozygous because assay SNP heterozygosity is a requirement for the assay to be performed. Boxplots represent the data distributions as follows: heavy bars show the sample median; boxes cover the interquartile range of the data and whiskers extend to the most extreme data point (excluding outliers that were more than 1.5 times the interquartile range from the box; small open circles mark outliers beyond this range).
(b). Environmental modification of allele-specific gene expression levels
We asked whether any of the social environmental variables we tested had the capacity to influence the genotype–gene expression relationship identified through our ASGE assays (see table 1 and electronic supplementary material, tables S4 and S5). Of all the social environments we analysed, we observed the most consistent GEIs in relationship to group size. The number of adults in an individual's social group at the time of darting, which indexes competition for mates and resources, was associated with GEIs in males (p = 2.9 × 10−3), females (p = 9.2 × 10−3) and also jointly when males and females were tested together (p = 0.012; figure 3a,c). Similarly, we found evidence that group size at birth, the early life analogue of number of adults in the group at darting, was also involved in GEIs in females (p = 4.72 × 10−4), but no evidence for such GEIs in males (p = 0.433). Support for this early life effect was weaker but still evident when males and females were modelled together (p = 0.024; see also electronic supplementary material, table S4 for a summary of all models).
Table 1.
Evidence for GEIs involving tested interaction effects.
| interaction effect | p-value (females)a | p-value (males) | p-value (both sexes) |
|---|---|---|---|
| social environments | |||
| dominance rank | |||
| early (maternal) | 0.464 (31) | 0.020 (24) | 0.304 (34) |
| adult | 0.133 (31) | 0.285 (30) | n.a.b |
| social connectedness | |||
| early (maternal) | 0.122 (30) | 0.237 (24) | 0.108 (34) |
| adult | 0.014 (31) | 1.13 × 10−3 (28) | n.a. |
| group size | |||
| early | 4.7 × 10−4 (31) | 0.433 (24) | 0.024 (34) |
| adult | 9.20 × 10−3 (31) | 2.88 × 10−3 (30) | 0.012 (34) |
| non-social effects | |||
| age | 0.0023 (31) | 0.542 (30) | 0.098 (34) |
| sex | n.a. | n.a. | 6.50 × 10−3 (34) |
ap-values from likelihood ratio tests comparing a model with GEIs to a model without GEIs are provided in each cell. Bold values are those that survive multiple hypothesis test correction using the method of Benjamini & Hochberg [63], at a false discovery rate of 10%. Values in parentheses provide the number of genes included in each test.
bSocial connectedness and dominance ranks in adulthood could not be evaluated for both sexes combined, as both statistics are calculated separately for males and females.
Figure 3.

Socially mediated GEIs. ASGE levels that are modified by an individual's environment imply GEIs, because the relative gene expression levels of the two alleles of a gene change depending on the environment. Each panel depicts, for one exemplar gene, a change in ASGE with a social environment (note that our core questions focused on analyses for each environment, across genes, however). We identified consistent socially mediated GEIs related to group size and social connectedness in adulthood for both (a,b) males and (c,d) females. (a) PHF11, (b) RNASE2, (c) CD8A and (d) SLAMF7.
In contrast to group size, for which both early life and adult exposures were associated with GEIs, adult social connectedness was associated with GEIs for both males (p = 1.13 × 10−3) and females (p = 1.41 × 10−2; figure 3b,d), but maternal social connectedness was not associated with GEIs for either sex. Conversely, for dominance rank, we observed no signal of an individual's own rank near the time of sampling. The only evidence for a rank effect was also sex-specific: maternal dominance rank was detected as a contributor to GEIs for males (p = 1.97 × 10−2) but not for females (p = 0.464).
(c). Sex- and timing-related differences in the environmental components of gene–environment interactions
Finally, we took advantage of our analysis of multiple social environmental effects to ask whether, in aggregate, social environments relevant to early versus adult life stages (i.e. all three early life environments versus all three adult environments) tended to explain more variance in ASGE across all measurable genes. That is, we asked whether early or adult social environmental variation more consistently contributed to variance in gene expression via GEIs. For females, early life social environmental characteristics (maternal dominance rank, maternal social connectedness and group size) and social environmental characteristics in adulthood (group size, adult dominance rank and social connectedness at darting) were indistinguishable with respect to explaining ASGE (p = 0.96; figure 4a). In males, despite our reduced power to detect such differences compared with females (for whom we had a larger sample size for early life effects, and thus retained more genes for this analysis), we observed a trend towards a greater impact of adult social environments than early life environments on ASGE (p = 0.085; figure 4b).
Figure 4.

Testing for biases in the variance explained by GEIs. In each panel, the difference in percentage variance in ASGE explained by early life versus adult environments (a,b) or male-specific versus female-specific environments (c) is plotted for each gene. (a) The distribution of variance in ASGE explained by early life effects versus effects during adulthood does not differ in females (paired t-test: p = 0.960). (b) The distribution of variance in ASGE explained by early life effects versus effects during adulthood exhibits a weak trend suggesting bias towards adult social environmental exposures in males (paired t-test: p = 0.085). (c) For sex-specific environmental effects of dominance rank and social connectedness, females and males do not differ in the degree of variance accounted for by GEIs (paired t-test: p = 0.687).
We performed a similar analysis to test whether female-specific social environments (SCI-F and adult dominance rank) tended to be more or less important than male-specific social environments (SCI-M and adult dominance rank). We observed no evidence for sex differences in the impact of these effects (p = 0.687; figure 4c).
4. Discussion
Genetic differences make important contributions to variation in behavioural phenotypes. However, behavioural traits also feed back to influence population genetic structure and evolutionary genetic change. Understanding flexibility and constraint in the evolution of behaviour therefore demands that we develop a better understanding of the reciprocal mechanisms through which genes and behaviour are linked. In this study, we tested whether GEIs involving the social environment act as one such mechanism. To do so, we combined a long-term dataset on the demography and behaviour of wild baboons with novel data on ASGE variation and genotype, focusing on individually identified individuals that had been tracked over the course of their lives.
Our results serve as proof of principle that, like age and sex [64,65], social environmental characteristics have the capacity to influence the relationship between genotype and gene expression in wild social mammals. Although not all of the individual variables that we tested revealed GEIs, we identified significant support for GEIs for each of the broad categories of social environments we investigated: dominance rank, social connectedness and group demography. Our data thus suggest that the importance of these predictors to social competition, survival and reproductive success extends, to some degree, to genetic reaction norms operating at the molecular level. Viewed from a complementary perspective, gene regulatory responses to the social environment in social mammals [11] are therefore likely to be mediated in part by genetic differences among individuals. Given that social status and the quality of social interactions are powerful predictors of survival and longevity [20], untangling these interactions will be important for understanding how individual animals differ in susceptibility to these effects.
By investigating the potential for GEIs at multiple loci, we were also able to ask whether social environment-mediated GEIs tended to be biased by sex or by the timing of environmental exposures. We found no evidence for an overall sex bias related to the variance in ASGE accounted for by GEIs. However, we did observe support for GEIs involving maternal rank for males, but not females. This result is somewhat surprising, as maternal rank is a pervasive maternal effect in female baboons that influences growth during development [66,67], the timing of reproductive and social maturation in females [50,66], and the rank they achieve in adulthood [68–70]. By contrast, while maternal rank affects male growth as it does female growth [66,67], it seems to have less of an impact on a male's life history than in females [50,68], and maternal rank appears to have little or no influence on the dominance rank that sons eventually achieve [68]. However, long-term effects of maternal rank on male physiology have been previously demonstrated in the Amboseli baboons: subadult sons of higher-ranking females have lower glucocorticoid levels than sons of lower-ranking mothers [51]. In the light of these previous findings, our results suggest that maternal rank-mediated GEIs in males may fit into a broader pattern of persistent maternal effects on physiology that, as yet, is poorly understood.
Possible differences in the timing of the social environmental exposures involved in GEIs are also suggested by our analysis of early life versus adult life social environments. Early life, especially the gestational and periparturitional periods, has been proposed as a ‘sensitive period’ in phenotypic development, with potentially profound effects on later life traits [71,72]. This idea has garnered support from empirical evidence tying early life exposures to disease susceptibility and stress reactivity in adulthood, raising the possibility that early life environmental effects might be important for social environment-mediated GEIs. Among the females we studied, however, the amount of variance in ASGE explained by early life social environments was indistinguishable from that explained by adult social environments. By contrast, in males, we observed weak evidence that adult social environments might exert stronger effects. Although follow-up work will be necessary to confirm these results, they suggest that early life social environments, while important, are not necessarily privileged over social exposures in adulthood with respect to their effects on gene expression reaction norms.
Interestingly, male baboons arguably experience increased social environmental variability over their lifetimes relative to females: unlike females, males often change social groups multiple times as adults, thus also changing their rank, social bonds and demographic context. Male dominance rank is more dynamic within groups than female dominance rank as well [68,69,73]. It therefore seems quite possible that the environment that a male currently experiences, which affects immediate access to mates [15,17,74], tenure length in a group [47] and testosterone and glucocorticoid levels [7], may be more physiologically relevant than their experiences during early life. We conjecture that this pattern may hold especially true for blood-expressed genes such as those we studied, as many of these genes are related to immune function that itself can vary depending on social environmental conditions. In Amboseli, for example, higher-ranking males heal faster from wounds and injuries than low-ranking males [9]. Tissues that have less of a role in sensing or responding to the external environment may be less likely to exhibit social environment-mediated GEIs. Additionally, because immune-related genes sometimes harbour unusually high levels of genetic variation, GEIs may also be more important in blood. Testing these hypotheses—as well as whether specific classes of genes are susceptible to GEIs and whether GEIs are plastic in the face of environmental change—will require gathering considerably more data on a wider variety of samples.
Such work will be facilitated by new methods for measuring ASGE on a genome-wide scale. Genome-wide approaches could extend the approach we used here to full transcriptomes [75], although they would likely use different methods for both measuring and analysing ASGE levels. In an alternative strategy, genes that are highly responsive to a given social environment could be tested for environment–contingent genetic effects. Indeed, recent work has successfully discovered genetic modifiers of the gene expression response to physical stressors such as tuberculosis infection [76], radiation exposure [77] and synthetic glucocorticoid treatment [78]. Intriguingly, these studies suggest that genetic differences contribute only weakly to the effects of the environment when environmental effects are strong (i.e. explain a large fraction of variance in gene expression levels). In comparison, our observations here suggest that environmental variation only weakly modifies the effects of genotype on gene expression levels when genotype effects are strong. Applying these complementary approaches together might therefore yield a more complete understanding of the nature and effect size of social environment-mediated GEIs.
Our study suggests that behaviour and social structure influence the function of genetic variation in wild social mammals on a proximate (mechanistic) timescale by changing its effects on gene regulation. However, like other behavioural phenomena, social environment-mediated GEIs can also be investigated on an evolutionary timescale [79], and our findings raise new questions about the degree to which socially mediated GEIs might shape evolution over the long term. GEIs have long been proposed, albeit with mixed support from theoretical analyses, as a mechanism through which selectively relevant genetic variation might be maintained in natural populations [80–83]. Such analyses have generally assumed that the relevant environmental players in GEIs are structured either across space (across populations that experience different ecological conditions) or time (as ecological conditions change within a population). By contrast, social environmental variation tends to be structured among individuals, within populations or social groups. Unlike many other types of environmental variation, it is also frequently omnipresent, as a direct consequence of species social structure: for example, dominance rank-mediated environmental variation always occurs within baboon social groups because no two individuals can occupy the same rank. These differences raise an intriguing possibility that the evolution of complex social structures among social mammals has also had an effect on the ways in which genetic variation is maintained and expressed—strong motivation for investigating social environment-mediated GEIs in the context of long-term genetic evolution as well.
Acknowledgements
This work was supported by the National Science Foundation (IOS-0919200 to S.C.A., DEB-0846286 to S.C.A and G.A.W., and BCS-0846532 to J.A.) and the National Institute of Aging (NIA R01-AG034513-01 and NIA P01-AG031719 to S.C.A.). We thank the Office of the President, Republic of Kenya; the Kenya Wildlife Service and its Amboseli staff and wardens; our local sponsor, the Institute of Primate Research; the National Museums of Kenya; and the members of the Amboseli–Longido pastoralist communities. Particular thanks go to the Amboseli fieldworkers who contribute to genetic and environmental sampling, especially R. Mututua, S. Sayialel and K. Warutere, and to T. Wango and V. Atieno, who provided assistance with the samples and logistical support in Nairobi. We are also grateful for the contributions of J. Stroud, J. Gordon, and S. Morrow to the pyrosequencing and genotyping data, S. Mukherjee for statistical advice, T. Brown-Brandl and J. Horvath for reading an early draft of the manuscript, two anonymous reviewers for their helpful comments and Baylor College of Medicine (Human Genome Sequencing Center) for the preliminary draft assembly of the baboon genome. Data included in this study were obtained in accordance with the Institutional Animal Care and Use Committee protocols approved by Duke University and Princeton University. Data included in this study are deposited in Dryad (doi:10.5061/dryad.s5j81) and in the NCBI Short Read Archive (SRP018756). Mention of trade names or commercial products in this publication is solely for providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture (USDA). USDA is an equal opportunity provider and employer.
References
- 1.Sugg DW, Chesser RK, Stephen Dobson F, Hoogland JL. 1996. Population genetics meets behavioral ecology. Trends Ecol. Evol. 11, 338–342 10.1016/0169-5347(96)20050-3 (doi:10.1016/0169-5347(96)20050-3) [DOI] [PubMed] [Google Scholar]
- 2.Altmann J, et al. 1996. Behavior predicts genetic structure in a wild primate group. Proc. Natl Acad. Sci. USA 93, 5797–5801 10.1073/pnas.93.12.5797 (doi:10.1073/pnas.93.12.5797) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guschanski K, Caillaud D, Robbins MM, Vigilant L. 2008. Females shape the genetic structure of a gorilla population. Curr. Biol. 18, 1809–1814 10.1016/j.cub.2008.10.031 (doi:10.1016/j.cub.2008.10.031) [DOI] [PubMed] [Google Scholar]
- 4.Nussey DH, Coltman DW, Coulson T, Kruuk LE, Donald A, Morris SJ, Clutton-Brock TH, Pemberton J. 2005. Rapidly declining fine-scale spatial genetic structure in female red deer. Mol. Ecol. 14, 3395–3405 10.1111/j.1365-294X.2005.02692.x (doi:10.1111/j.1365-294X.2005.02692.x) [DOI] [PubMed] [Google Scholar]
- 5.Reich D, Thangaraj K, Patterson N, Price AL, Singh L. 2009. Reconstructing Indian population history. Nature 461, 489–494 10.1038/nature08365 (doi:10.1038/nature08365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbott DH, et al. 2003. Are subordinates always stressed? A comparative analysis of rank differences in cortisol levels among primates. Horm. Behav. 43, 67–82 10.1016/S0018-506X(02)00037-5 (doi:10.1016/S0018-506X(02)00037-5) [DOI] [PubMed] [Google Scholar]
- 7.Gesquiere LR, Learn NH, Simao MCM, Onyango PO, Alberts SC, Altmann J. 2011. Life at the top: rank and stress in wild male baboons. Science 333, 357–360 10.1126/science.1207120 (doi:10.1126/science.1207120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavigelli SA, Chaudhry HS. 2012. Social status, glucocorticoids, immune function, and health: can animal studies help us understand human socioeconomic-status-related health disparities? Horm. Behav. 62, 295–313 10.1016/j.yhbeh.2012.07.006 (doi:10.1016/j.yhbeh.2012.07.006) [DOI] [PubMed] [Google Scholar]
- 9.Archie EA, Altmann J, Alberts SC. 2012. Social status predicts wound healing in wild baboons. Proc. Natl Acad. Sci. USA 109, 9017–9022 10.1073/pnas.1206391109 (doi:10.1073/pnas.1206391109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sapolsky RM. 2004. Social status and health in humans and other animals. Annu. Rev. Anthropol. 33, 393–418 10.1146/annurev.anthro.33.070203.144000 (doi:10.1146/annurev.anthro.33.070203.144000) [DOI] [Google Scholar]
- 11.Tung J, Barreiro LB, Johnson ZP, Hansen KD, Michopoulos V, Toufexis D, Michelini K, Wilson ME, Gilad Y. 2012. Social environment is associated with gene regulatory variation in the rhesus macaque immune system. Proc. Natl Acad. Sci. USA 109, 6490–6495 10.1073/pnas.1202734109 (doi:10.1073/pnas.1202734109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hawley DM, Jennelle CS, Sydenstricker KV, Dhondt AA. 2007. Pathogen resistance and immunocompetence covary with social status in house finches (Carpodacus mexicanus). Funct. Ecol. 21, 520–527 10.1111/j.1365-2435.2007.01254.x (doi:10.1111/j.1365-2435.2007.01254.x) [DOI] [Google Scholar]
- 13.Lindstrom KM. 2004. Social status in relation to Sindbis virus infection clearance in greenfinches. Behav. Ecol. Sociobiol. 55, 236–241 10.1007/s00265-003-0703-3 (doi:10.1007/s00265-003-0703-3) [DOI] [Google Scholar]
- 14.Rantala MJ, Kortet R. 2004. Male dominance and immunocompetence in a field cricket. Behav. Ecol. 15, 187–191 10.1093/beheco/arg103 (doi:10.1093/beheco/arg103) [DOI] [Google Scholar]
- 15.Alberts SC, Buchan JC, Altmann J. 2006. Sexual selection in wild baboons: from mating opportunities to paternity success. Anim. Behav. 72, 1177–1196 10.1016/j.anbehav.2006.05.001 (doi:10.1016/j.anbehav.2006.05.001) [DOI] [Google Scholar]
- 16.Cameron EZ, Setsaas TH, Linklater WL. 2009. Social bonds between unrelated females increase reproductive success in feral horses. Proc. Natl Acad. Sci. USA 106, 13 850–13 853 10.1073/pnas.0900639106 (doi:10.1073/pnas.0900639106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Packer C. 1979. Male dominance and reproductive activity in Papio anubis. Anim. Behav. 27, 37–45 10.1016/0003-3472(79)90127-1 (doi:10.1016/0003-3472(79)90127-1) [DOI] [PubMed] [Google Scholar]
- 18.Schulke O, Bhagavatula J, Vigilant L, Ostner J. 2010. Social bonds enhance reproductive success in male macaques. Curr. Biol. 20, 2207–2210 10.1016/j.cub.2010.10.058 (doi:10.1016/j.cub.2010.10.058) [DOI] [PubMed] [Google Scholar]
- 19.Silk JB, Alberts SC, Altmann J. 2003. Social bonds of female baboons enhance infant survival. Science 302, 1231–1234 10.1126/science.1088580 (doi:10.1126/science.1088580) [DOI] [PubMed] [Google Scholar]
- 20.Silk JB, Beehner JC, Bergman TJ, Crockford C, Engh AL, Moscovice LR, Wittig RM, Seyfarth RM, Cheney DL. 2010. Strong and consistent social bonds enhance the longevity of female baboons. Curr. Biol. 20, 1359–1361 10.1016/j.cub.2010.05.067 (doi:10.1016/j.cub.2010.05.067) [DOI] [PubMed] [Google Scholar]
- 21.West-Eberhard MJ. 1979. Sexual selection, social competition, and evolution. Proc. Am. Philos. Soc. 123, 222–234 [Google Scholar]
- 22.Perrin N, Petit EJ, Menard N. 2012. Social systems: demographic and genetic issues. Mol. Ecol. 21, 443–446 10.1111/j.1365-294X.2011.05404.x (doi:10.1111/j.1365-294X.2011.05404.x) [DOI] [PubMed] [Google Scholar]
- 23.Tung J, Alberts SC, Wray GA. 2010. Evolutionary genetics in wild primates: combining genetic approaches with field studies of natural populations. Trends Genet. 26, 353–362 10.1016/j.tig.2010.05.005 (doi:10.1016/j.tig.2010.05.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sapolsky RM. 2005. The influence of social hierarchy on primate health. Science 308, 648–652 10.1126/science.1106477 (doi:10.1126/science.1106477) [DOI] [PubMed] [Google Scholar]
- 25.Silk JB. 2007. Social components of fitness in primate groups. Science 317, 1347–1351 10.1126/science.1140734 (doi:10.1126/science.1140734) [DOI] [PubMed] [Google Scholar]
- 26.Falconer DS, Mackay TFC. 1996. Introduction to quantitative genetics. Essex, UK: Longman [Google Scholar]
- 27.Qvarnstrom A. 1999. Genotype-by-environment interactions in the determination of the size of a secondary sexual character in the collared flycatcher (Ficedula albicollis). Evolution 53, 1564–1572 10.2307/2640901 (doi:10.2307/2640901) [DOI] [PubMed] [Google Scholar]
- 28.Merila J, Fry JD. 1998. Genetic variation and causes of genotype-environment interaction in the body size of blue tit (Parus caeruleus). Genetics 148, 1233–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leips J, Mackay TFC. 2000. Quantitative trait loci for life span in Drosophila melanogaster: interactions with genetic background and larval density. Genetics 155, 1773–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leroi AM, Chippindale AK, Rose MR. 1994. Long-term laboratory evolution of a genetic life-history trade-off in Drosophila melanogaster 1. The role of genotype-by-environment interaction. Evolution 48, 1244–1257 10.2307/2410382 (doi:10.2307/2410382) [DOI] [PubMed] [Google Scholar]
- 31.Vieira C, Pasyukova EG, Zeng ZB, Hackett JB, Lyman RF, Mackay TFC. 2000. Genotype–environment interaction for quantitative trait loci affecting life span in Drosophila melanogaster. Genetics 154, 213–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juenger TE, Sen S, Stowe KA, Simms EL. 2005. Epistasis and genotype–environment interaction for quantitative trait loci affecting flowering time in Arabidopsis thaliana. Genetica 123, 87–105 10.1007/s10709-003-2717-1 (doi:10.1007/s10709-003-2717-1) [DOI] [PubMed] [Google Scholar]
- 33.Barr CS, Newman TK, Becker ML, Parker CC, Champoux M, Lesch KP, Goldman D, Suomi SJ, Higley JD. 2003. The utility of the non-human primate model for studying gene by environment interactions in behavioral research. Genes Brain Behav. 2, 336–340 10.1046/j.1601-1848.2003.00051.x (doi:10.1046/j.1601-1848.2003.00051.x) [DOI] [PubMed] [Google Scholar]
- 34.Newman TK, Syagailo YV, Barr CS, Wendland JR, Champoux M, Graessle M, Suomi SJ, Higley JD, Lesch K-P. 2005. Monoamine oxidase A gene promoter variation and rearing experience influences aggressive behavior in rhesus monkeys. Biol. Psychiatry 57, 167–172 10.1016/j.biopsych.2004.10.012 (doi:10.1016/j.biopsych.2004.10.012) [DOI] [PubMed] [Google Scholar]
- 35.Barr CS, Newman TK, Lindell S, Shannon C, Champoux M, Lesch KP, Suomi SJ, Goldman D, Higley JD. 2004. Interaction between serotonin transporter gene variation and rearing condition in alcohol preference and consumption in female primates. Arch. Gen. Psychiatry 61, 1146–1152 10.1001/archpsyc.61.11.1146 (doi:10.1001/archpsyc.61.11.1146) [DOI] [PubMed] [Google Scholar]
- 36.Barr CS, et al. 2004. Rearing condition and rh5-HTTLPR interact to influence limbic-hypothalamic-pituitary-adrenal axis response to stress in infant macaques. Biol. Psychiatry 55, 733–738 10.1016/j.biopsych.2003.12.008 (doi:10.1016/j.biopsych.2003.12.008) [DOI] [PubMed] [Google Scholar]
- 37.Alberts SC, Altmann J. 2012. The Amboseli baboon research project: forty years of continuity and change. In Long-term field studies of primates (eds Kappeler PM, Watts DP.). Berlin, Germany: Springer [Google Scholar]
- 38.Cole SW, Hawkley LC, Arevalo JM, Sung CY, Rose RM, Cacioppo JT. 2007. Social regulation of gene expression in human leukocytes. Genome Biol. 8, R189. 10.1186/gb-2007-8-9-r189 (doi:10.1186/gb-2007-8-9-r189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith EN, Kruglyak L. 2008. Gene–environment interaction in yeast gene expression. PLoS Biol. 6, e83. 10.1371/journal.pbio.0060083 (doi:10.1371/journal.pbio.0060083) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wittkopp P, Haerum B, Clark A. 2004. Evolutionary changes in cis and trans gene regulation. Nature 430, 85–88 10.1038/nature02698 (doi:10.1038/nature02698) [DOI] [PubMed] [Google Scholar]
- 41.Wittkopp PJ, Haerum BK, Clark AG. 2008. Regulatory changes underlying expression differences within and between Drosophila species. Nat. Genet. 40, 346–350 10.1038/ng.77 (doi:10.1038/ng.77) [DOI] [PubMed] [Google Scholar]
- 42.de Meaux J, Goebel U, Pop A, Mitchell-Olds T. 2005. Allele-specific assay reveals functional variation in the chalcone synthase promoter of Arabidiopsis thaliana that is compatible with neutral evolution. Plant Cell 17, 676–690 10.1105/tpc.104.027839 (doi:10.1105/tpc.104.027839) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Korff M, et al. 2009. Asymmetric allele-specific expression in relation to developmental variation and drought stress in barley hybrids. Plant J. 59, 14–26 10.1111/j.1365-313X.2009.03848.x (doi:10.1111/j.1365-313X.2009.03848.x) [DOI] [PubMed] [Google Scholar]
- 44.Tung J, Akinyi MY, Mutura S, Altmann J, Wray GA, Alberts SC. 2011. Allele-specific gene expression in a wild nonhuman primate population. Mol. Ecol. 20, 725–739 10.1111/j.1365-294X.2010.04970.x (doi:10.1111/j.1365-294X.2010.04970.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alberts SC, Altmann J. 2001. Immigration and hybridization patterns of yellow and anubis baboons in and around Amboseli, Kenya. Am. J. Primatol. 53, 139–154 10.1002/ajp.1 (doi:10.1002/ajp.1) [DOI] [PubMed] [Google Scholar]
- 46.Tung J, Charpentier MJ, Garfield DA, Altmann J, Alberts SC. 2008. Genetic evidence reveals temporal change in hybridization patterns in a wild baboon population. Mol. Ecol. 17, 1998–2011 10.1111/j.1365-294X.2008.03723.x (doi:10.1111/j.1365-294X.2008.03723.x) [DOI] [PubMed] [Google Scholar]
- 47.Alberts SC, Altmann J. 1995. Balancing costs and opportunities: dispersal in male baboons. Am. Nat. 145, 279–306 10.1086/285740 (doi:10.1086/285740) [DOI] [Google Scholar]
- 48.Tung J, Primus A, Bouley AJ, Severson TF, Alberts SC, Wray GA. 2009. Evolution of a malaria resistance gene in wild primates. Nature 460, 388–391 [DOI] [PubMed] [Google Scholar]
- 49.Alberts SC, Altmann J. 1995. Preparation and activation: determinants of age at reproductive maturity in male baboons. Behav. Ecol. Sociobiol. 36, 397–406 10.1007/BF00177335 (doi:10.1007/BF00177335) [DOI] [Google Scholar]
- 50.Charpentier MJE, Tung J, Altmann J, Alberts SC. 2008. Age at maturity in wild baboons: genetic, environmental and demographic influences. Mol. Ecol. 17, 2026–2040 10.1111/j.1365-294X.2008.03724.x (doi:10.1111/j.1365-294X.2008.03724.x) [DOI] [PubMed] [Google Scholar]
- 51.Onyango PO, Gesquiere LR, Wango EO, Alberts SC, Altmann J. 2008. Persistence of maternal effects in baboons: mother's dominance rank at son's conception predicts stress hormone levels in subadult males. Horm. Behav. 54, 319–324 10.1016/j.yhbeh.2008.03.002 (doi:10.1016/j.yhbeh.2008.03.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altmann J. 1974. Observational study of behavior: sampling methods. Behaviour 49, 227–267 10.1163/156853974X00534 (doi:10.1163/156853974X00534) [DOI] [PubMed] [Google Scholar]
- 53.Tung J, Fedrigo O, Haygood R, Mukherjee S, Wray GA. 2009. Genomic features that predict allelic imbalance in humans suggest patterns of constraint on gene expression variation. Mol. Biol. Evol. 26, 2047–2059 10.1093/molbev/msp113 (doi:10.1093/molbev/msp113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gnirke A, et al. 2009. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189 10.1038/nbt.1523 (doi:10.1038/nbt.1523) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.George RD, McVicker G, Diederich R, Ng SB, MacKenzie AP, Swanson WJ, Shendure J, Thomas JH. 2011. Trans genomic capture and sequencing of primate exomes reveals new targets of positive selection. Genome Res. 21, 1686–1694 10.1101/gr.121327.111 (doi:10.1101/gr.121327.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vallender EJ. 2011. Expanding whole exome resequencing into non-human primates. Genome Biol. 12, R87. 10.1186/gb-2011-12-9-r87 (doi:10.1186/gb-2011-12-9-r87) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harris RS. PhD thesis, Pennsylvania State University: 2007. Improved pairwise alignment of genomic DNA. [Google Scholar]
- 58.Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. 2003. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl Acad. Sci. USA 100, 11 484–11 489 10.1073/pnas.1932072100 (doi:10.1073/pnas.1932072100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 10.1038/ng.806 (doi:10.1038/ng.806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKenna A, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 10.1101/gr.107524.110 (doi:10.1101/gr.107524.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Browning BL, Browning SR. 2009. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 84, 210–223 10.1016/j.ajhg.2009.01.005 (doi:10.1016/j.ajhg.2009.01.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.R Development Core Team 2011. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing [Google Scholar]
- 63.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 [Google Scholar]
- 64.Ober C, Loisel DA, Gilad Y. 2008. Sex-specific genetic architecture of human disease. Nat. Rev. Genet. 9, 911–922 10.1038/nrg2415 (doi:10.1038/nrg2415) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G. 2001. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat. Genet. 29, 389–395 10.1038/ng766 (doi:10.1038/ng766) [DOI] [PubMed] [Google Scholar]
- 66.Altmann J, Alberts SC. 2005. Growth rates in a wild primate population: ecological influences and maternal effects. Behav. Ecol. Sociobiol. 57, 490–501 10.1007/s00265-004-0870-x (doi:10.1007/s00265-004-0870-x) [DOI] [Google Scholar]
- 67.Johnson SE. 2003. Life history and the competitive environment: trajectories of growth, maturation, and reproductive output among Chacma baboons. Am. J. Phys. Anthropol. 120, 83–98 10.1002/ajpa.10139 (doi:10.1002/ajpa.10139) [DOI] [PubMed] [Google Scholar]
- 68.Packer C, Collins DA, Eberly LE. 2000. Problems with primate sex ratios. Phil. Trans. R. Soc. Lond. B 355, 1627–1635 10.1098/rstb.2000.0725 (doi:10.1098/rstb.2000.0725) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheney DL, Seyfarth RM. 2007. Baboon metaphysics. Chicago, IL: University of Chicago Press [Google Scholar]
- 70.Hausfater G, Altmann J, Altmann SA. 1982. Long-term consistency of dominance relations among female baboons (Papio cynocephalus). Science 217, 752–755 10.1126/science.217.4561.752 (doi:10.1126/science.217.4561.752) [DOI] [PubMed] [Google Scholar]
- 71.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. 2008. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 359, 61–73 10.1056/NEJMra0708473 (doi:10.1056/NEJMra0708473) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lindstrom J. 1999. Early development and fitness in birds and mammals. Trends Ecol. Evol. 14, 343–348 10.1016/S0169-5347(99)01639-0 (doi:10.1016/S0169-5347(99)01639-0) [DOI] [PubMed] [Google Scholar]
- 73.Altmann J, Gesquiere L, Galbany J, Onyango PO, Alberts SC. 2010. Life history context of reproductive aging in a wild primate model. Ann. NY Acad. Sci. 1204, 127–138 10.1111/j.1749-6632.2010.05531.x (doi:10.1111/j.1749-6632.2010.05531.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alberts SC, Watts HE, Altmann J. 2003. Queuing and queue-jumping: long-term patterns of reproductive skew in male savannah baboons, Papio cynocephalus. Anim. Behav. 65, 821–840 10.1006/anbe.2003.2106 (doi:10.1006/anbe.2003.2106) [DOI] [Google Scholar]
- 75.Pastinen T. 2010. Genome-wide allele-specific analysis: insights into regulatory variation. Nat. Rev. Genet. 11, 533–538 10.1038/nrg2815 (doi:10.1038/nrg2815) [DOI] [PubMed] [Google Scholar]
- 76.Barreiro LB, Tailleux L, Pai AA, Gicquel B, Marioni JC, Gilad Y. 2012. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proc. Natl Acad. Sci. USA 109, 1204–1209 10.1073/pnas.1115761109 (doi:10.1073/pnas.1115761109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smirnov DA, Morley M, Shin E, Spielman RS, Cheung VG. 2009. Genetic analysis of radiation-induced changes in human gene expression. Nature 459, 587–591 10.1038/nature07940 (doi:10.1038/nature07940) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maranville JC, Luca F, Richards AL, Wen X, Witonsky DB, Baxter S, Stephens M, Di Rienzo A, Gibson G. 2011. Interactions between glucocorticoid treatment and cis-regulatory polymorphisms contribute to cellular response phenotypes. PLoS Genet. 7, e1002162. 10.1371/journal.pgen.1002162 (doi:10.1371/journal.pgen.1002162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tinbergen N. 1963. On aims and methods of ethology. Zeitschrift Tierpsychol. 20, 410–433 10.1111/j.1439-0310.1963.tb01161.x (doi:10.1111/j.1439-0310.1963.tb01161.x) [DOI] [Google Scholar]
- 80.Gillespie JH, Turelli M. 1989. Genotype–environment interactions and the maintenance of polygenic variation. Genetics 121, 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gimelfarb A. 1990. How much genetic variation can be maintained by genotype–environment interactions? Genetics 124, 443–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Turelli M, Barton NH. 2004. Polygenic variation maintained by balancing selection: pleiotropy, sex-dependent allelic effects and G × E interactions. Genetics 166, 1053–1079 10.1534/genetics.166.2.1053 (doi:10.1534/genetics.166.2.1053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Via S, Lande R. 1985. Genotype–environment interaction and the evolution of phenotypic plasticity. Evolution 39, 505–522 10.2307/2408649 (doi:10.2307/2408649) [DOI] [PubMed] [Google Scholar]