

Abstract
Two novel reductive dehalogenases (RDases) that are highly similar to each other but catalyse distinct dechlorination reactions were identified from Dehalobacter-containing mixed cultures. These two RDases were partially purified from crude protein extracts of anaerobic dechlorinating enrichment cultures using blue native polyacrylamide gel electrophoresis. Gel slices were assayed for dechlorinating activity, and associated proteins were identified using liquid chromatography tandem mass spectrometry with the metagenome of the parent culture as the reference database. The two RDases identified, annotated as CfrA and DcrA, share an amino acid identity of 95.2 per cent, but use different substrates: CfrA dechlorinates chloroform (CF) and 1,1,1-trichloroethane (1,1,1-TCA), but not 1,1-dichloroethane; DcrA dechlorinates 1,1-dichloroethane, but not CF or 1,1,1-TCA. These two novel RDases share no more than 40 per cent amino acid identity to any other known or putative RDases, but both have a twin-arginine motif and two iron–sulfur binding motifs conserved in most RDases. Peptides specific to two putative membrane anchor proteins, annotated as CfrB and DcrB, were also detected in gel slices.
Keywords: reductive dehalogenation, reductive dehalogenases, Dehalobacter, blue native PAGE, chloroform, chlorinated ethanes
1. Introduction
Chloroform (CF) and 1,1,1-trichloroethane (1,1,1-TCA) are persistent groundwater contaminants owing to their historical and widespread industrial use as organic solvents, and improper disposal in the past. In the USA, CF is present at 416 of 1723 National Priorities List (NPL) site, whereas 1,1,1-TCA is present at 393 sites (NPL database search, June 2012, http://cfpub.epa.gov/supercpad/cursites/srchsites.cfm). In addition to having adverse impacts on human health and the environment [1–3], CF and 1,1,1-TCA inhibit many microbial processes, including methanogenesis [4–9] and reductive dechlorination [10–12]. Because many sites are contaminated by multiple chlorinated organics including CF and 1,1,1-TCA, the removal of these two inhibitory compounds is of special importance to bioremediation efforts [13].
We previously reported the discovery of a Dehalobacter-containing mixed culture (herein referred to as ACT-3) capable of dechlorinating CF, 1,1,1-TCA and 1,1-dichloroethane (1,1-DCA) [13,14]. A Dehalobacter strain (16S rRNA gene, DQ663785) from the ACT-3 culture is the first organism shown to respire CF [14]. The important role of Dehalobacter in organohalide-respiration is clear: they can respire chloroethenes [15], chloroethanes [13,16,17], chloromethane [14,18], chlorobenzenes [19] and other halogenated compounds [20,21]. Although Dehalobacter were previously recognized as strictly organohalide-respiring anaerobes, a strain was recently shown to also ferment dichloromethane (DCM) [18,22].
Organohalide-respiring bacteria such as Dehalococcoides and Dehalobacter catalyse reductive dechlorination using reductive dehalogenaseas (RDases) [23]. Genome sequencing has revealed that these organisms tend to harbour multiple distinct putative reductive dehalogenase genes within their genomes: some have more than 30 [24,25]. In total, hundreds of putative reductive dehalogenase genes have been identified; however, only a handful have been functionally characterized because of difficulties inherent to working with slow-growing strict anaerobes, the lack of genetic systems to manipulate these organisms and the inability to express functional reductive dehalogenases heterologously [3]. In this study, the identification of two RDases that catalyse dechlorination reactions in the ACT-3 culture was reported. The genes of these two RDases were among the 19 Dehalobacter RDase genes determined by metagenomic sequencing of the ACT-3 culture. The RDase-identification process, similar to previous reports [26,27], featured the combination of blue native polyacrylamide gel electrophoresis (BN-PAGE), dechlorination activity assays of gel slices and liquid chromatography tandem mass spectrometry (LC–MS/MS) for protein identification.
2. Material and methods
(a). Cultures and culture history
The ACT-3 enrichment culture was originally derived from microcosms prepared with contaminated aquifer material. It has been maintained for more than 10 years in a mineral medium [14] amended with 1,1,1-TCA as an electron acceptor and a mixture of methanol, ethanol, acetate and lactate (MEAL) as electron donors. This culture sequentially dechlorinates 1,1,1-TCA via 1,1-DCA to monochloroethane (CA). It also dechlorinates CF to DCM. Dehalobacter was shown to be responsible for these dechlorination reactions [13,14]. Two subcultures of the ACT-3 culture were created over 6 years ago with different chlorinated substrates: the DCA subculture has been maintained with 1,1-DCA as an electron acceptor and MEAL as electron donor mixture; the CF subculture has been maintained with CF as an electron acceptor and a mixture of methanol, ethanol and lactate (MEL) as electron donors. Over time, these two subcultures have lost specific dechlorinating activities compared with the parent culture: the CF subculture dechlorinates 1,1,1-TCA and CF, but no longer dechlorinates 1,1-DCA, whereas the DCA subculture dechlorinates 1,1-DCA, but no longer 1,1,1-TCA or CF.
(b). Metagenome sequencing and assembly
DNA from the ACT-3 parent culture was extracted using a CTAB protocol recommended by the US Department of Energy Joint Genome Institute (JGI, Walnut Creek, CA, USA). The protocol is available online: http://my.jgi.doe.gov/general/protocols/DNA_Isolation_Bacterial_CTAB_Protocol.doc. The sequencing was performed by JGI using 454 pyrosequencing. A total of 444 Mb of sequence was generated, including paired-end 454 reads from an 8 kb insert library. Initial assembly was performed with Newbler v. 2.5. The collection of resulting contigs is referred to as the ACT-3 metagenome. The initial assembly and annotation were performed by the JGI and can be accessed by the JGI taxon object ID of 2100351010 (http://img.jgi.doe.gov/cgi-bin/m/main.cgi). DNA samples from the CF and DCA subcultures were extracted with UltraClean soil DNA isolation kit (MOBIO). To determine the community structure, DNA samples from these three mixed cultures were sequenced by 16S rRNA gene pyrotag sequencing. This sequencing and the phylogenetic assignment of sequenced reads were also performed by the JGI.
(c). Identification of putative rdhA and rdhB genes
From the ACT-3 metagenome, contigs encoding putative rdhA genes were identified using a BLASTX search with a query database consisting of hundreds of known or putative RDases from public databases. Fragmentation in some rdhA genes (sequences that belong to one rdhA gene were not assembled into one contig) owing to strain variations of the dominant organism, Dehalobacter, was observed; the re-assembly and curation of these fragmented rdhA genes were achieved by performing sequence alignments using Geneious Pro v. 5.4.2 [28] as illustrated in the electronic supplementary material, figure S2. Two re-assembled rdhA genes were found expressed in the cultures in subsequent LC–MS/MS analysis; therefore, the DNA sequences of these two genes were further confirmed and polished with read mapping using Geneious Pro and additional Sanger sequencing directed by specific PCR primers (see the electronic supplementary material, figure S2). Putative rdhB genes were identified by analysing the gene neighbourhoods of rdhA genes. Two rdhB genes were also re-constructed from fragmented sequences (see the electronic supplementary material, figure S3).
(d). Sample preparation for blue native polyacrylamide gel electrophoresis
Inside an anaerobic chamber (Coy, MI, USA), culture samples (40 ml) were transferred to a 50 ml Falcon tube sealed with anaerobic tape. The tube was centrifuged at 10 000g for 20 min and returned to the anaerobic chamber. The cell pellet was resuspended in 1 ml remaining supernatant and transferred to a 2 ml O-ring capped microcentrifuge tube, and centrifuged again at 10 000g for 10 min. Inside the anaerobic chamber, with the supernatant discarded, the cell pellet was resuspended in 200 µl BN-PAGE sample buffer (1% digitonin, 50 mM Bis-Tris, 50 mM NaCl, 10% w/v glycerol, pH 7.2 with HCl). Glass beads (approx. 50 mg) were added to the tube, and cells were lysed during three rounds of vortexing in a horizontal vortex (Scientific Industries Inc., Bohemia, NY, USA) at the maximum amplitude; each round consisted of 2 min vortex followed by 1 min cooling in an ice bath. The sample was centrifuged at 10 000g for 10 min, and the supernatant (crude protein extract) was transferred to a new Eppendorf tube. Before being subjected to electrophoresis, 200 µl crude protein extract was supplemented with 10 µl of a 5 per cent Coomassie blue G solution.
(e). BN-PAGE and staining
BN-PAGE and staining were performed outside the anaerobic chamber. Precast 4–16% gradient Bis-Tris gels (NativePAGE Novex, Invitrogen) and an electrophoresis device (XCell SureLock, Invitrogen) were used. The preparation of running buffers and the set-up of the device were performed according to the manufacturer's manual. Typically, a protein ladder (NativeMark, Invitrogen) was loaded into the first lane of the gel, and 20 µl of the crude protein extract was loaded into each of the nine remaining lanes. Electrophoresis was run at 150 V for 60 min and then at 200 V for another 45 min, with the entire chamber cooling in an ice bath. After electrophoresis, the first two lanes (the protein ladder and one of the culture samples) were cut for staining, and the remaining gel lanes were saved for dechlorinating enzyme assays or for further separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). For the staining of the first two lanes, we used the ‘fast Coomassie G-250 staining’ procedure from the manufacturer's manual of the precast gels (page 23).
(f). SDS-PAGE
For SDS-PAGE analysis, BN-PAGE gel slices were cut from one unstained lane, chopped into less than 1 mm pieces and transferred to Eppendorf tubes containing 250 µl elution buffer (100 mM Tris–HCl, pH 7.0 and 0.1% SDS). Protein elution was performed by shaking the tubes at 1000 rpm for 4 h at room temperature. After elution, the 250 µl protein sample was concentrated to a volume of approximately 20 µl by ultrafiltration with 3 kDa cut-off membranes (Pall Co., Canada), before being loaded on a SDS-PAGE gel. The SDS-PAGE gels were stained by silver staining following established protocols.
(g). Assaying dechlorinating activity in gel slices
For assaying dechlorinating activity, an unstained gel lane was aligned with the stained gel lane and was cut into gel slices—this was carried out outside the glove box. The gel-slicing pattern, as shown in figure 1, was designed to cover the regions that were known to have dechlorination activity. In preliminary assays, the other regions of a gel lane were tested and were found to have no dechlorination activity. The same pattern was kept when we prepared gel slices for subsequent separation with SDS-PAGE or LC–MS/MS analysis for protein identification. The same pattern was used for the protein extracts from all three mixed cultures. These gel slices were then transferred to 2 ml screw-top glass vials and were brought into the anaerobic chamber. For each glass vial, 1 ml assay buffer was added, which contained 100 mM Tris–HCl (pH 7.4), 2 mM titanium citrate, 2 mM methyl viologen, and approximately 0.5 mM chlorinated substrate (1,1,1-TCA, 1,1-DCA or CF). These vials were incubated in the anaerobic chamber for 24 h, and then 0.3 ml headspace samples were taken for gas chromatography (GC) analysis to evaluate the extent of dechlorination of the substrate. As positive controls, 20 µl of the original crude cell extract (equal to the volume of sample loaded into one well of the BN-PAGE gel) was assayed. As negative controls, 20 µl heat-killed (incubating at 80°C for 15 min) crude protein extracts were assayed in preliminary tests; no dechlorination activity of 1,1,1-TCA, CF and 1,1-DCA was observed in such negative controls.
Figure 1.

Image of left two lanes of the BN-PAGE gel, showing molecular weight ladder and the first stained sample lane. Remaining identical sample lanes (unstained) from the same gel were sliced at specific positions consistently by aligning to the stained lane as indicated. Each gel slice was then used in dechlorination assays. BP, band position. (Online version in colour.)
(h). LC–MS/MS analysis of gel slices
To identify the proteins contained in the gel slices, the slices were sent for LC–MS/MS analysis at the Advanced Protein Technology Center at SickKids' Hospital (Toronto, Canada). The proteins in the excised gel slices were reduced with 100 mM dithiothreitol (in 50 mM ammonium bicarbonate), alkylated with 10 mM iodoacetamide (in 50 mM ammonium bicarbonate) and digested by overnight incubation with porcine trypsin (13 µg µl−1). The resulting peptides were extracted with 25 mM ammonium bicarbonate buffer and 100 per cent acetonitrile. The peptides thus produced were loaded onto a 150 μm ID pre-column (Magic C18, Michrom Biosciences) at 4 μl min−1 and separated over a 75 μm ID analytical column packed into an emitter tip containing the same packing material. The peptides were eluted over 60 min at 300 nl min−1 using a 0–40% acetonitrile gradient in 0.1 per cent formic acid using an EASY n-LC nano-chromatography pump (Proxeon Biosystems, Odense, Denmark). The peptides were eluted into an LTQ linear ion trap mass spectrometer (Thermo-Fisher, San Jose, CA, USA) operated in a data-dependent mode. Six MS/MS scans were obtained per MS cycle.
The raw data files were searched with X!Tandem (Beavis Informatics Ltd., Canada) using a parent ion accuracy of 1.8 Da, a fragment accuracy of 0.4 Da, no semi-enzymatic cleavage, the maximum missed cleavage sites of 1 and the maximum expectation value for recorded peptides of 0.01. A fixed modification of carbamidomethyl cysteine and a variable modification of oxidized methionine were included in the search and the refinement, which also included variable modifications of the deamidation of asparagine and glutamine. The peptides were searched against two protein databases. The first consists of all proteins and protein fragments (from the ACT-3 metagenome) predicted and annotated through JGI's IMG/m annotation pipelines (accessed by IMG taxon object ID of 2100351010). The second database is a custom database, consisting of only curated rdhA and rdhB genes found in the ACT-3 metagenome (see the electronic supplementary material, table S3). In a separate study, we were able to close the genomes of two Dehalobacter strains present in the ACT-3 culture [29]. The complete sequences of the two genomes were used to pull out proteins that are certain to belong to Dehalobacter (with amino acid (AA) identity >95%) and help to assign taxonomy. The remaining non-Dehalobacter proteins were then searched against the NCBI RefSeq_protein database and assigned taxonomy based on the best hit.
(i). PCRs
Four primers were designed to specifically amplify each of the two similar rdhA genes identified in this study. They were cfrA-413f (CCCGAACCTCTAGCACTTGTAG), cfrA-531r (ACGGCAAAGCTTGCACGA), dcrA-424f (AGCACTCAGAGAGCGTTTTGC) and dcrA-533r (CAACGGCCCAGCTTGCAT). For PCRs, a Taq DNA polymerase (Fermentas, Canada) was used. The thermocycling programme was as follows: initial denaturation of 10 min at 94°C; 40 cycles of 30 s denaturation at 94°C, 30 s annealing at 60°C and 30 s extension at 72°C; and final extension of 10 min at 72°C.
(j). Sequence analysis
The multiple sequence alignment and phylogenetic analysis of the two identified RdhA proteins and 18 additional characterized RdhA proteins were performed using MUSCLE [30] and PHYML [31] accessed through Geneious Pro. Potential promoter sites were predicted using NNPP v. 2.2 [32]. Potential ribosome binding sites were predicted using SIGSCAN v. 4.05 [33]. The twin-arginine signal peptide was predicted by tatP [34]. Transmembrane helixes were predicted by TMHMM v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/). The theoretical molecular weights of proteins were calculated by the Compute PI/Mw tool of ExPASy (http://expasy.org/tools/pi_tool.html).
(k). Nucleotide sequence accession numbers
The DNA sequences of cfrA, dcrA, cfrB and dcrB have been deposited in GenBank with the following accession numbers: JX282329 for cfrA, JX282330 for dcrA, JX282334 for cfrB and JX282335 for dcrB.
3. Results
(a). Metagenome sequencing
Dehalobacter was previously shown to be responsible for the reductive dechlorination of 1,1,1-TCA, 1,1-DCA and CF in ACT-3 [13,14]. This corresponds well with the dominance of Dehalobacter in the three ACT-3-related cultures as determined by pyrotag sequencing of the 16S rRNA gene (see the electronic supplementary material, figure S1). Shotgun and paired-end 454 pyrosequencing of the ACT-3 culture metagenomic DNA produced 13 479 contigs (greater than 500 bp) with N50 of 1708 bp and the largest contig of 169 374 bp. In a separate study [29], we successfully assembled all Dehalobacter contigs into a closed circular assembly. However, certain locations in this assembly consist of alternative contigs that belong to two highly similar but different Dehalobacter genomes; by comparing this assembly with additional sequencing data from the CF subculture, which happens to have only one Dehalobacter strain, we managed to assemble and separate two complete Dehalobacter genomes. The coexistence of two Dehalobacter strains/genomes in ACT-3 resulted in fragmentation of some Dehalobacter contigs in the initial assembly produced by Newbler. The read depth of the contigs that were shared by both strains was approximately 80, and the two strains were found in similar abundance as indicated by read depth analysis. With homology search and manual curation, 19 putative intact rdhA genes and one truncated rdhA homologous gene (786 bp) were retrieved from the ACT-3 metagenome. The genome context investigation of these putative rdhA genes revealed 17 putative rdhB genes. Read depth analysis of these rdhA and rdhB genes (data not shown) showed that they belong to the dominant genus, Dehalobacter, which was further confirmed after the closure of the two Dehalobacter genomes [29].
(b). Functional differentiation of the three mixed cultures
The ACT-3 parent culture dechlorinates CF, 1,1,1-TCA and 1,1-DCA, whereas the CF subculture dechlorinates only CF and 1,1,1-TCA, and the DCA subculture dechlorinates only 1,1-DCA [14]. These observations were confirmed in dechlorination assays performed using the crude protein extracts from the three cultures (table 1). The crude protein extract from the CF subculture did not dechlorinate 1,1-DCA, the one from the DCA subculture did not dechlorinate 1,1,1-TCA or CF, and the one from the parent ACT-3 culture dechlorinated all three substrates (table 1).
Table 1.
Results of 24 h enzyme assays of the crude protein extracts from the three mixed cultures (data are averages (± s.d.) of triplicate extracts). Values in bold show the highest activities.
| samples | dechlorination products (nmol) |
||
|---|---|---|---|
| CF assay: DCM | 1,1,1-TCA assay: 1,1-DCA (CA)a | 1,1-DCA assay: CA | |
| buffer controlb | BDc | BD (BD)a | BD |
| ACT-3 | 228 ± 3 | 98 ± 1 (6.4 ± 0.1)a | 56 ± 2 |
| CF subculture | 139 ± 2 | 64 ± 2 (BD)a | 0.1 ± 0.0 |
| DCA subculture | 2.5 ± 0.1 | 0.9 ± 0.1 (BD)a | 47 ± 1 |
aIn the assays of 1,1,1-TCA, CA can also be produced. The amount of CA produced is shown in parentheses.
bThe negative control using the reaction buffer without protein addition.
cBD means below detection limit.
(c). RDase expression in the chloroform subculture
The crude protein extract from the CF subculture was shown to have negligible dechlorinating activity on 1,1-DCA (table 1 and the positive control of figure 2a); therefore, the gel slices from this sample were assayed only with 1,1,1-TCA and CF. Testing for dechlorination activity in gel slices revealed two regions of enriched dechlorination activity on the gel (figure 2a): one was around band position 3 or 4 (BP-3 or BP-4), slightly below the 242 kDa marker; the other was around BP-7, slightly below the 146 kDa marker. Although the dechlorination profile with CF as substrate appears slightly different from that with 1,1,1-TCA (figure 2a), this difference was probably caused by slight variations in the position of the gel slice in adjacent lanes (one individual lane was used for one specific assay). Nevertheless, we found these two regions of enriched dechlorination activity consistently in over three preliminary trials. In the gel slices of enriched activity, proteins with molecular weight similar to that of an RDase (approx. 45 kDa) were identified by two-dimensional separation using SDS-PAGE (see the electronic supplementary material, figure S4). The presence of RDases was further confirmed by LC–MS/MS analysis of the proteins in gel slices from BP-2, BP-3, BP-4, BP-6, BP-7 and BP-8. Matching the MS spectra against the IMG-predicted proteins of the ACT-3 metagenome identified two protein hits related to RdhA proteins (DHTCA2_00197470 and DHTCA2_00327390, table 2). Subsequent analysis showed that they were two fragmented sequences of one RdhA protein, and the fragmentation was caused by strain variations in the initial assembly with Newbler. When the MS spectra were matched to the custom RDase database consisting of only curated putative rdhA and rdhB genes curated from the ACT-3 metagenome as described earlier, we found only one RdhA and only one RdhB were expressed in the CF subculture (figure 2b). These two proteins were named CfrA and CfrB, with corresponding genes cfrA and cfrB. The assembly and separation of two Dehalobacter genomes from the ACT-3 metagenome [29] confirmed that the cfrA and cfrB genes were adjacent and located in one gene operon in one genome. Because only CfrA was identified in the CF subculture, we can conclude that CfrA dechlorinates both CF and 1,1,1-TCA, but not 1,1-DCA. The fact that CfrA dechlorinates both CF and 1,1,1-TCA is actually not surprising because these two substrates are similar in structure: 1,1,1-TCA is methyl chloroform.
Figure 2.
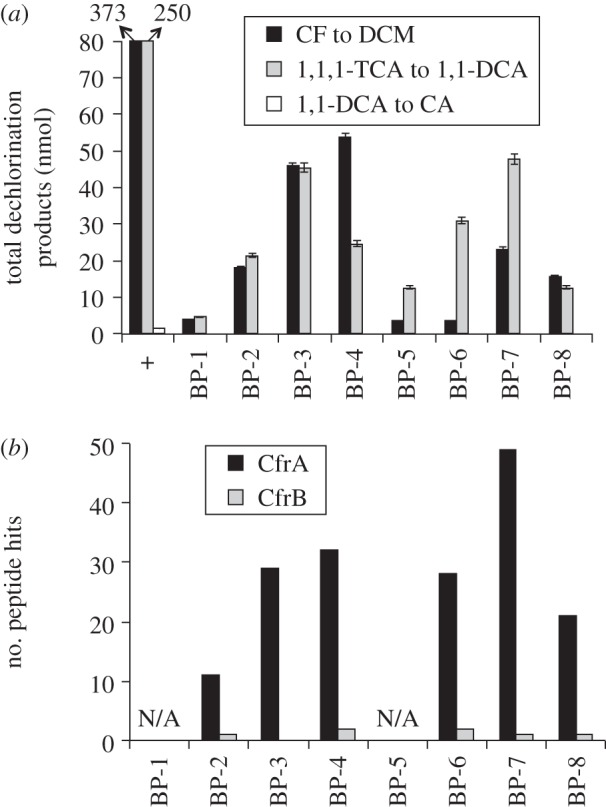
Results of BN-PAGE with protein samples from the CF subculture. (a) Quantification of dechlorination products in enzyme assays with gel slices. The arrow bars indicate the regular technical error in headspace GC measurements. Plus symbol indicates the positive control, where activity was measured using 20 µl of protein extracts equal to the volume loaded onto each BN-PAGE well. Black bars, DCM detected from CF; grey bars, 1,1-DCA detected from 1,1,1-TCA; white bars, CA detected from 1,1-DCA. (b) Counts of LC–MS/MS-detected peptide hits in gel slices when searched against curated RdhA and RdhB proteins derived from the ACT-3 metagenome. ‘N/A’ means the gel slice was not analysed.
Table 2.
Proteins identified in the gel slices of enriched activity from BN-PAGE gels using the reference database of all proteins identified in the ACT-3 metagenome (only the top seven hits were listed for each gel slice). Bold indicates reductive dehalogenases.
| culture | BN-PAGE slice | IMG gene locus tag | peptide hits | annotation | putative organism |
|---|---|---|---|---|---|
| CF subculture | BP-3 | DHTCA2_00302240 | 25 | OAH/OAS sulfhydrylase | Dehalobacter |
| DHTCA2_00199880 | 20 | chaperone protein DnaK | Dehalobacter | ||
| DHTCA2_00295340 | 14 | chaperonin GroEL | Dehalobacter | ||
| DHTCA2_00242140 | 13 | citrate lyase beta subunit | Dehalobacter | ||
| DHTCA2_00272470 | 10 | dihydroxy-acid dehydratase | Dehalobacter | ||
| DHTCA2_00327390 | 8 | reductive dehalogenase CfrA fragment | Dehalobacter | ||
| DHTCA2_00269400 | 7 | formate dehydrogenase alpha subunit | Dehalobacter | ||
| BP-7 | DHTCA2_00197470 | 23 | reductive dehalogenase CfrA fragment | Dehalobacter | |
| DHTCA2_00327390 | 18 | reductive dehalogenase CfrA fragment | Dehalobacter | ||
| DHTCA2_00217090 | 12 | uroporphyrin-III C-methyltransferase | Dehalobacter | ||
| DHTCA2_00199880 | 7 | chaperone protein DnaK | Dehalobacter | ||
| DHTCA2_00306260 | 7 | Hup-type Ni,Fe-hydrogenase large subunit | Dehalobacter | ||
| DHTCA2_00474830 | 5 | alcohol dehydrogenase, class IV | Desulfovibrio | ||
| DHTCA2_00941900 | 5 | translation elongation factor TU | Desulfovibrio | ||
| DCA subculture | BP-2 | DHTCA2_00532520 | 27 | carbon monoxide dehydrogenase large subunit | Desulfovibrio |
| DHTCA2_00486690 | 11 | methanol–cobalamin methyltransferase subunit B | Methanosarcina | ||
| DHTCA2_00114900 | 10 | carbon monoxide dehydrogenase large subunit | Desulfovibrio | ||
| DHTCA2_00239010 | 10 | formyltetrahydrofolate synthetase | Dehalobacter | ||
| DHTCA2_00199880 | 9 | chaperone protein DnaK | Dehalobacter | ||
| DHTCA2_00269400 | 8 | formate dehydrogenase alpha subunit | Dehalobacter | ||
| DHTCA2_00474830 | 5 | alcohol dehydrogenase, class IV | Desulfovibrio | ||
| BP-6 | DHTCA2_00474830 | 17 | alcohol dehydrogenase, class IV | Desulfovibrio | |
| DHTCA2_00199880 | 14 | chaperone protein DnaK | Dehalobacter | ||
| DHTCA2_00502190 | 9 | alcohol dehydrogenase, class IV | Desulfovibrio | ||
| DHTCA2_00603650 | 6 | reductive dehalogenase DcrA fragment | Dehalobacter | ||
| DHTCA2_00306260 | 5 | Hup-type Ni,Fe-hydrogenase large subunit | Dehalobacter | ||
| DHTCA2_00201920 | 4 | branched-chain amino acid transporter | Dehalobacter | ||
| DHTCA2_00609600 | 4 | pyridoxamine 5’-phosphate oxidase | Dehalobacter | ||
| ACT-3 parent culture | BP-4 | DHTCA2_00202930 | 9 | putative cell wall-binding domain | Dehalobacter |
| DHTCA2_00199880 | 8 | chaperone protein DnaK | Dehalobacter | ||
| DHTCA2_00269400 | 5 | formate dehydrogenase alpha subunit | Dehalobacter | ||
| DHTCA2_00755650 | 4 | DNA polymerase sliding clamp subunit | Methanohalobium | ||
| DHTCA2_00474830 | 3 | alcohol dehydrogenase, class IV | Desulfovibrio | ||
| DHTCA2_00941900 | 3 | translation elongation factor TU | Desulfovibrio | ||
| DHTCA2_00331370 | 3 | acyl CoA:acetate/3-ketoacid CoA transferase | Clostridium | ||
| BP-7 | DHTCA2_00637940 | 11 | methanol–cobalamin methyltransferase subunit B | Methanohalophilus | |
| DHTCA2_01115960 | 11 | methanol–cobalamin methyltransferase subunit B | Methanosalsum | ||
| DHTCA2_00197470 | 10 | reductive dehalogenase CfrA fragment | Dehalobacter | ||
| DHTCA2_00474830 | 9 | alcohol dehydrogenase, class IV | Desulfovibrio | ||
| DHTCA2_00941900 | 8 | translation elongation factor TU | Desulfovibrio | ||
| DHTCA2_00327390 | 7 | reductive dehalogenase CfrA fragment | Dehalobacter | ||
| DHTCA2_00306770 | 6 | translation elongation factor TU | Dehalobacter |
(d). RDase expression in the dichloroethane subculture
The crude protein extract of the DCA subculture had negligible dechlorinating activity on either 1,1,1-TCA or CF (table 1 and the positive control of figure 3a); therefore, the gel slices were assayed only with 1,1-DCA (figure 3a). Dechlorination assays on gel slices revealed the presence of one region of highly enriched activity (centred on BP-6, figure 3a). LC–MS/MS analysis revealed the presence of only one RdhA protein and one RdhB protein (figure 3b), which were named DcrA and DcrB with the corresponding genes dcrA and dcrB. The dcrA and dcrB genes were adjacent and located in one operon in one genome in the newly assembled genomes [29]. Because DcrA was the only RDase identified, we concluded that DcrA dechlorinates 1,1-DCA, but not 1,1,1-TCA or CF.
Figure 3.
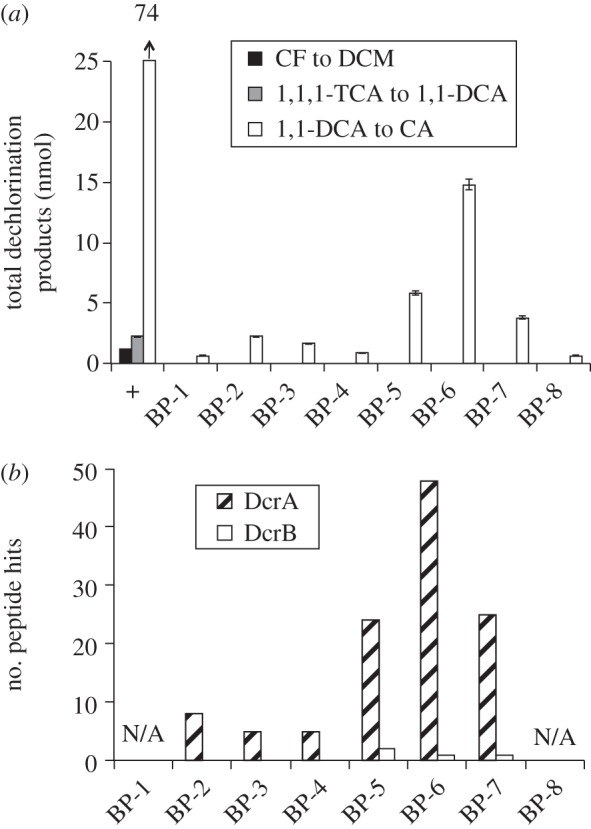
Results of BN-PAGE with protein samples from the DCA subculture. (a) Quantification of dechlorination products in enzyme assays with gel slices; legend as in figure 2. (b) Counts of LC–MS/MS peptide hits in gel slices when searched against curated RdhA and RdhB proteins derived from the ACT-3 metagenome. ‘N/A’ means the gel slice was not analysed.
(e). RDase expression in the ACT-3 parent culture
The dechlorination profiles (figure 4a) and RDase peptide hit profiles (figure 4b) in gel slices from the ACT-3 parent culture are just like a combination of the results we found for each of the two subcultures. Again, two similar gel regions of enriched dechlorination activity were found, and the number of RDase peptide hits is higher in the second region. With the functions assigned to CfrA and DcrA in the earlier analyses, the coexistence of CfrA and DcrA explains perfectly why the ACT-3 culture dechlorinates all three substrates. Notably, both CfrA and DcrA were found in all gel slices analysed except BP-2 (figure 4b) and both RdhB proteins, CfrB and DcrB, were also found expressed in the ACT-3 culture.
Figure 4.
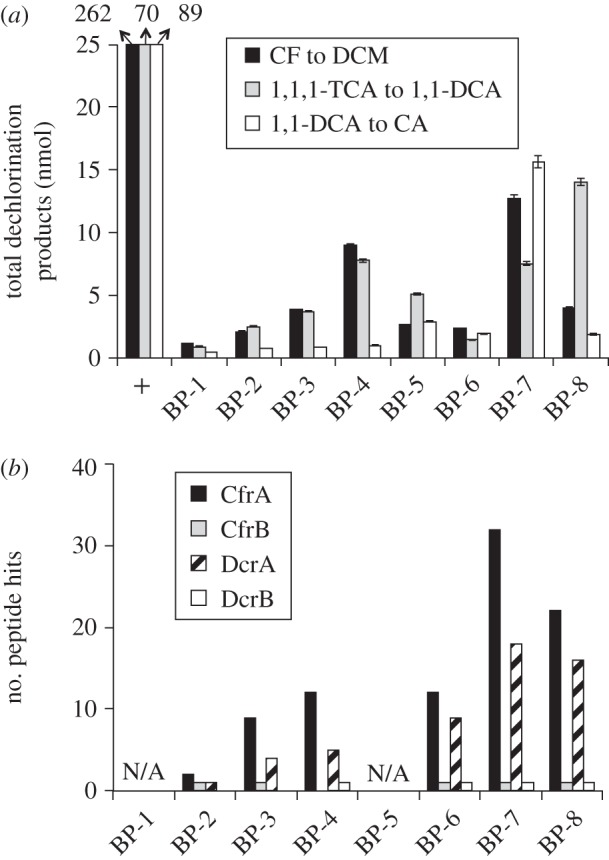
Results of BN-PAGE with protein samples from the ACT-3 culture. (a) Quantification of dechlorination products in enzyme assays with gel slices; legend as in figure 2. (b) Counts of LC–MS/MS peptide hits in gel slices when searched against curated RdhA and RdhB proteins derived from the ACT-3 metagenome. ‘N/A’ means the gel slice was not analysed.
(f). Distribution of cfrA and dcrA genes
With PCRs using the primers (cfrA-413f, cfrA-531r, dcrA-424f and dcrA-533r) that specifically target cfrA and dcrA genes, we investigated the distribution of these two genes in the three mixed cultures (see the electronic supplementary material, figure S5). The distribution of these two genes is identical to that of the encoded RDases as determined by LC–MS/MS analysis. Therefore, we can further confirm the absence of DcrA in the CF subculture and the absence of CfrA in the DCA subculture.
(g). Expression of non-RDase proteins
A large number of other non-RDase proteins were identified from BN-PAGE gel slices using LC–MS/MS analysis (table 2 and electronic supplementary material, table S1). As expected, a large proportion of identified proteins belong to Dehalobacter (see the electronic supplementary material, table S1), corresponding to the abundance of this organism in these three cultures (see the electronic supplementary material, figure S1). Looking for potential protein–protein interactions with CfrA and DcrA, we focused on the Dehalobacter proteins identified from the two regions of enriched activity on the BN-PAGE gels (table 2). Besides CfrA or DcrA, there were another two proteins that were repeatedly identified in all three cultures and in relatively high peptide counts: formate dehydrogenase alpha subunit (DHTCA2_00269400) in the first region of enriched activity and hup-type Ni,Fe-hydrogenase large subunit (DHTCA2_00306260) in the second region of enriched activity. Other subunits to these proteins, the formate dehydrogenase beta subunit (DHTCA2_00269390) and hup-type Ni,Fe-hydrogenase small subunit (DHTCA2_00306250), were also found in corresponding gel slices (see the electronic supplementary material, table S1).
(h). Sequence analysis of cfrA and dcrA
DNA sequence analysis identified potential promoter sites and ribosome binding sites upstream of cfrA and dcrA genes. Based on a start codon 1 bp downstream of the nearest ribosome binding site, cfrA and dcrA genes with a full length of 1521 bp were predicted. However, when the corresponding proteins translated from these two DNA sequences were aligned with other characterized RdhA proteins, the twin-arginine motif, RRQFLK, was found located 66 AAs from the N-terminal. By contrast, this motif is located only several residues from the N-terminal in other known RDases. Therefore, we chose an alternative start codon for these two rdhA genes so that the twin-arginine motif is only 16 AAs from the N-terminal. With this adjustment, the gene length shortens to 1371 bp and the protein length to 456 AAs.
CfrA and DcrA differ from each other in 22 of 456 AAs (figure 5). Despite such high similarity, they were well discriminated by LC–MS/MS analysis (figure 5), highlighting the sensitivity of this technique. A signal peptide (39 AAs long), which contains the twin-arginine motif, was predicted in both RDases. No peptide from the region of the predicted signal peptide was detected by LC–MS/MS analysis, indicating the cleavage of the signal peptide in the mature forms of CfrA and DcrA. The molecular weights of CfrA before and after the cleavage of the signal peptide were 50 and 46 kDa; those of DcrA were similar. The molecular weight of 46 kDa agreed well with bands seen by SDS-PAGE (see the electronic supplementary material, figure S4). In addition to the twin-arginine signal peptides, CfrA and DcrA also share the two iron–sulfur binding motifs that are common to RdhA proteins (figure 5).
Figure 5.

Amino acid sequence alignment for CfrA versus DcrA, and CfrB versus DcrB. Sequences shown are for CfrA and CfrB. Residues that differ in DcrA and DcrB are in bold on next line. The twin-arginine motif is boxed and the two iron–sulfur binding motifs are shaded. The dashed underline indicates the predicted signal peptide. The peptides detected by LC–MS/MS are underlined.
CfrA and DcrA are not closely related to other known or putative RdhA proteins. Their best hit in BLASTP search (5 October 2012) against NCBI non-redundant database was a putative reductive dehalogenase (YP_004971810) with AA identity of approximately 40 per cent. Their novelty was further confirmed in the phylogenetic analysis with 12 other RDases that have some functional characterizations (figure 6). CfrA and DcrA were also not closely related to other putative RdhA proteins recovered from their source metagenome (see the electronic supplementary material, figure S6).
Figure 6.

Maximum-likelihood phylogenetic tree of the RDases that have functional characterization. The alignment was generated using the MUSCLE algorithm, and the tree generated using the PhyML plugin in Geneious under the WAG model of evolution. Bootstrap support values (from 100 bootstrap iterations) are indicated where greater than 50 per cent. The scale bar represents the average number of substitutions per site. For a complete tree of curated RDase sequences see the introductory chapter to this issue [35].
4. Discussion
(a). CfrA and DcrA
CfrA and DcrA are the first RDases described to specifically dechlorinate CF and 1,1-DCA. Maillard et al. [36] described the purification and characterization of a PceA RDase, which was reported to dechlorinate 1,1,1-TCA, but at a rate that is 1.4 per cent of the rate for tetrachloroethene (PCE) dechlorination. The novelty of CfrA and DcrA is also evident from their lack of homology to other putative or known RDases in public databases or even to other putative RDases recovered from the same metagenome (figure 6 and electronic supplementary material, figure S6). Another distinctive feature of these two RDases is that they are highly similar to each other (95.2% identical in AA sequence, 97.9% identical in DNA sequence), but have exclusively different substrates. Perhaps one of the RDases evolved from the other or they evolved from a common ancestor; either way, the discovery of these two RDases shows how new functions can evolve in RDases. In a separate study [29], we showed that there were two Dehalobacter strains, and hence genomes, in the ACT-3 culture, and that CfrA belongs to one strain and DcrA belongs to the other strain. The existence of these two strains appears to be an example of recent strain divergence with niche selection.
(b). CfrB and DcrB
Possessing typically three transmembrane α-helixes, RdhB proteins have been predicted as the membrane anchors for the catalytic units (RdhA proteins) [37]. This is the first time that RdhB proteins have been detected by mass spectrometry. There are multiple reasons why RdhB proteins have not been detected in previous proteomic studies [27,38,39]. Membrane proteins such as RdhB proteins are poor substrates for trypsin digestion, a common sample preparation step in typical LC–MS/MS analysis. Trypsin cleaves proteins after lysine and arginine residues, which are rare in membrane proteins such as RdhBs. Small peptides (less than 6 AAs) and large hydrophobic peptides resulting from trypsin digestion are difficult to identify by LC–MS/MS owing to different reasons [40]. This explains why only one peptide for CfrB (KDHTGGISQST) and one peptide for DcrB (KDHIGGISQST) were detected in the LC–MS/MS analyses (figure 5). We examined some other putative RdhB sequences from other organisms and found that they would be very difficult to detect by LC–MS/MS analysis using trypsin digestion (see the electronic supplementary material, figure S7).
(c). Non-RDase proteins
We repeatedly observed two regions of enriched activity (one below 242 kDa and the other one below 146 kDa) in the BN-PAGE gels in the studies of the three mixed cultures. Interestingly, when we applied the same analysis to a Dehalococcoides-containing mixed culture [26], we found only one region of enriched activity, which was close to the upper region of enriched activity (242 kDa) described in this study. In the work done by Adrian et al. [27] using clear native PAGE, only one region of dechlorination activity was reported when protein extracts from a Dehalococcoides pure culture were used. Therefore, the observation of two distinct regions of enriched activity might be specific to Dehalobacter cultures.
As described earlier, subunits of two interesting protein complexes were found in the two regions of enriched activity in all three cultures: formate dehydrogenase in the upper region and hup-type hydrogenase in the lower region. The identified formate dehydrogenase alpha subunit (DHTCA2_00269400) is homologous (34.5% AA identity) to a protein (CBDB1A195) in Dehalococcoides mccartyi strain CBDB1, annotated as formate dehydrogenase major unit. This Dehalococcoides protein was found in high expression level in different Dehalococcoides-containing cultures [27,38,39]. Unlike Dehalococcoides, which cannot use formate as an electron donor, one Dehalobacter strain can [17]. The hup-type Ni,Fe-hydrogenase found in the lower region of enriched activity (146 kDa) is potentially involved in hydrogen uptake in Dehalobacter. These two protein complexes could be parts of the electron transfer chain directed to RDases. Based on the patterns we found in the BN-PAGE gels, it is tempting to speculate that they are physically associated with RDases, although we do not have conclusive evidence.
Acknowledgements
The authors are very grateful to Dr Lorenz Adrian (UFZ, Leipzig) and Winnie Chan for teaching us native PAGE techniques. We thank Ariel Grostern for establishing the three mixed cultures and Paul Taylor for help with the analysis of LC–MS/MS data. Support was provided by the Government of Canada through Genome Canada and the Ontario Genomics Institute (2009-OGI-ABC-1405). Support was also provided by the Government of Ontario through the ORF-GL2 programme and the United States Department of Defense through the Strategic Environmental Research and Development Program (SERDP) under contract no. W912HQ-07-C-0036 (project no. ER-1586). Metagenome sequencing was conducted by the US Department of Energy Joint Genome Institute supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231. S.T. received awards from the Government of Ontario through the Ontario Graduate Scholarships in Science and Technology and the Natural Sciences and Engineering Research Council of Canada (NSERC PGS B).
References
- 1.Doherty RE. 2000. A history of the production and use of carbon tetrachloride, tetrachloroethylene, trichloroethylene and 1,1,1-trichloroethane in the United States. I. Historical background; carbon tetrachloride and tetrachloroethylene. Environ. Forensics 1, 69–81 10.1006/enfo.2000.0010 (doi:10.1006/enfo.2000.0010) [DOI] [Google Scholar]
- 2.Meek ME, Beauchamp R, Long G, Moir D, Turner L, Walker M. 2002. Chloroform: exposure estimation, hazard characterization, and exposure-response analysis. J. Toxicol. Environ. Health B Crit. Rev. 5, 283–334 10.1080/10937400290070080 (doi:10.1080/10937400290070080) [DOI] [PubMed] [Google Scholar]
- 3.Sjuts H, Fisher K, Dunstan MS, Rigby SE, Leys D. 2012. Heterologous expression, purification and cofactor reconstitution of the reductive dehalogenase PceA from Dehalobacter restrictus. Protein Express. Purif. 85, 224–229 10.1016/j.pep.2012.08.007 (doi:10.1016/j.pep.2012.08.007) [DOI] [PubMed] [Google Scholar]
- 4.Adamson DT, Parkin GF. 2000. Impact of mixtures of chlorinated aliphatic hydrocarbons on a high-rate, tetraehloroethene-dechlorinating enrichment culture. Environ. Sci. Technol. 34, 1959–1965 10.1021/es990809f (doi:10.1021/es990809f) [DOI] [Google Scholar]
- 5.de Best JH, Hage A, Doddema HJ, Janssen DB, Harder W. 1999. Complete transformation of 1,1,1-trichloroethane to chloroethane by a methanogenic mixed population. Appl. Microbiol. Biotechnol. 51, 277–283 10.1007/s002530051393 (doi:10.1007/s002530051393) [DOI] [Google Scholar]
- 6.Suidan MT, Wuellner AM, Boyer TK. 1991. Anaerobic treatment of a high-strength industrial waste bearing inhibitory concentrations of 1,1,1-trichloroethane. Water Sci. Technol. 23, 1385–1393 [Google Scholar]
- 7.Yang C-HJ. 1981. The effects of cyanide and chloroform toxicity on methane fermentation, p. xiii, 230 leaves Philadelphia, PA: Drexel University [Google Scholar]
- 8.Weathers LJ, Parkin GF. 1995. Metallic iron-enhanced biotransformation of carbon tetrachloride and chloroform under methanogenic conditions. Bioremediation Chlorinated Solvents 3, 117–122 [Google Scholar]
- 9.Yu ZT, Smith GB. 2000. Inhibition of methanogenesis by C-1- and C-2-polychlorinated aliphatic hydrocarbons. Environ. Toxicol. Chem. 19, 2212–2217 [Google Scholar]
- 10.Bagley DM, Lalonde M, Kaseros V, Stasiuk KE, Sleep BE. 2000. Acclimation of anaerobic systems to biodegrade tetrachloroethene in the presence of carbon tetrachloride and chloroform. Water Res. 34, 171–178 10.1016/S0043-1354(99)00121-9 (doi:10.1016/S0043-1354(99)00121-9) [DOI] [Google Scholar]
- 11.Duhamel M, Wehr SD, Yu L, Rizvi H, Seepersad D, Dworatzek S, Cox EE, Edwards EA. 2002. Comparison of anaerobic dechlorinating enrichment cultures maintained on tetrachloroethene, trichloroethene, cis-dichloroethene and vinyl chloride. Water Res. 36, 4193–4202 10.1016/S0043-1354(02)00151-3 (doi:10.1016/S0043-1354(02)00151-3) [DOI] [PubMed] [Google Scholar]
- 12.Futagami T, Goto M, Furukawa K. 2008. Biochemical and genetic bases of dehalorespiration. Chem. Record 8, 1–12 10.1002/Tcr.20134 (doi:10.1002/Tcr.20134) [DOI] [PubMed] [Google Scholar]
- 13.Grostern A, Edwards EA. 2006. A 1,1,1-trichloroethane-degrading anaerobic mixed microbial culture enhances biotransformation of mixtures of chlorinated ethenes and ethanes. Appl. Environ. Microbiol. 72, 7849–7856 10.1128/Aem.01269-06 (doi:10.1128/Aem.01269-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grostern A, Duhamel M, Dworatzek S, Edwards EA. 2010. Chloroform respiration to dichloromethane by a Dehalobacter population. Environ. Microbiol. 12, 1053–1060 10.1111/J.1462-2920.2009.02150.X (doi:10.1111/J.1462-2920.2009.02150.X) [DOI] [PubMed] [Google Scholar]
- 15.Holliger C, Hahn D, Harmsen H, Ludwig W, Schumacher W, Tindall B, Vazquez F, Weiss N, Zehnder AJ. 1998. Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration. Arch. Microbiol. 169, 313–321 10.1007/s002030050577 (doi:10.1007/s002030050577) [DOI] [PubMed] [Google Scholar]
- 16.Grostern A, Edwards EA. 2009. Characterization of a Dehalobacter coculture that dechlorinates 1,2-dichloroethane to ethene and identification of the putative reductive dehalogenase gene. Appl. Environ. Microbiol. 75, 2684–2693 10.1128/AEM.02037-08 (doi:10.1128/AEM.02037-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun B, Griffin BM, Ayala-del-Río HL, Hashsham SA, Tiedje JM. 2002. Microbial dehalorespiration with 1,1,1-trichloroethane. Science 298, 1023–1025 10.1126/science.1074675 (doi:10.1126/science.1074675) [DOI] [PubMed] [Google Scholar]
- 18.Lee M, Low A, Zemb O, Koenig J, Michaelsen A, Manefield M. 2012. Complete chloroform dechlorination by organochlorine respiration and fermentation. Environ. Microbiol. 14, 883–894 10.1111/j.1462-2920.2011.02656.x (doi:10.1111/j.1462-2920.2011.02656.x) [DOI] [PubMed] [Google Scholar]
- 19.Nelson JL, Fung JM, Cadillo-Quiroz H, Cheng X, Zinder SH. 2011. A role for Dehalobacter spp. in the reductive dehalogenation of dichlorobenzenes and monochlorobenzene. Environ. Sci. Technol. 45, 6806–6813 10.1021/es200480k (doi:10.1021/es200480k) [DOI] [PubMed] [Google Scholar]
- 20.van Doesburg W, van Eekert MH, Middeldorp PJ, Balk M, Schraa G, Stams AJ. 2005. Reductive dechlorination of beta-hexachlorocyclohexane (β-HCH) by a Dehalobacter species in coculture with a Sedimentibacter sp. FEMS Microbiol. Ecol. 54, 87–95 10.1016/j.femsec.2005.03.003 (doi:10.1016/j.femsec.2005.03.003) [DOI] [PubMed] [Google Scholar]
- 21.Yoshida N, Ye L, Baba D, Katayama A. 2009. A novel Dehalobacter species is involved in extensive 4,5,6,7-tetrachlorophthalide dechlorination. Appl. Environ. Microbiol. 75, 2400–2405 10.1128/AEM.02112-08 (doi:10.1128/AEM.02112-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Justicia-Leon SD, Ritalahti KM, Mack EE, Löffler FE. 2012. Dichloromethane fermentation by a Dehalobacter sp. in an enrichment culture derived from pristine river sediment. Appl. Environ. Microbiol. 78, 1288–1291 10.1128/AEM.07325-11 (doi:10.1128/AEM.07325-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smidt H, de Vos WM. 2004. Anaerobic microbial dehalogenation. Annu. Rev. Microbiol. 58, 43–73 10.1146/annurev.micro.58.030603.123600 (doi:10.1146/annurev.micro.58.030603.123600) [DOI] [PubMed] [Google Scholar]
- 24.McMurdie PJ, et al. 2009. Localized plasticity in the streamlined genomes of vinyl chloride respiring Dehalococcoides. PLoS Genet. 5, e1000714. 10.1371/journal.pgen.1000714 (doi:10.1371/journal.pgen.1000714) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kube M, Beck A, Zinder SH, Kuhl H, Reinhardt R, Adrian L. 2005. Genome sequence of the chlorinated compound-respiring bacterium Dehalococcoides species strain CBDB1. Nat. Biotechnol. 23, 1269–1273 10.1038/nbt1131 (doi:10.1038/nbt1131) [DOI] [PubMed] [Google Scholar]
- 26.Tang S, Chan WW, Fletcher KE, Liang X, Seifert J, Löffler FE, Edwards EA, Adrian L. 2013. Functional characterization of Dehalococcoides reductive dehalogenases using blue native polyacrylamide gel electrophoresis. Appl. Environ. Microbiol. 79, 974–981 10.1128/AEM.01873-12 (doi:10.1128/AEM.01873-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adrian L, Rahnenfuhrer J, Gobom J, Hölscher T. 2007. Identification of a chlorobenzene reductive dehalogenase in Dehalococcoides sp. strain CBDB1. Appl. Environ. Microbiol. 73, 7717–7724 10.1128/AEM.01649-07 (doi:10.1128/AEM.01649-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drummond AJ, et al. 2011. Geneious v. 5.4.2. Auckland, New Zealand: Biomatters Ltd [Google Scholar]
- 29.Tang S, Gong Y, Edwards EA. 2012. Semi-automatic in silico gap closure enabled de novo assembly of two Dehalobacter genomes from metagenomic data. PLoS ONE 7, e52038. 10.1371/journal.pone.0052038 (doi:10.1371/journal.pone.0052038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 (doi:10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 [DOI] [PubMed] [Google Scholar]
- 32.Reese MG. 2001. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 26, 51–56 10.1016/S0097-8485(01)00099-7 (doi:10.1016/S0097-8485(01)00099-7) [DOI] [PubMed] [Google Scholar]
- 33.Prestridge DS. 1991. SIGNAL SCAN: a computer program that scans DNA sequences for eukaryotic transcriptional elements. Comput. Appl. Biosci. 7, 203–206 [DOI] [PubMed] [Google Scholar]
- 34.Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. 2005. Prediction of twin-arginine signal peptides. BMC Bioinformatics 6, 167. 10.1186/1471-2105-6-167 (doi:10.1186/1471-2105-6-167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hug LA, Maphosa F, Leys D, Löffler FE, Smidt H, Edwards EA, Adrian L. 2013. Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Phil. Trans. R. Soc. B 368, 20120322. 10.1098/rstb.2012.0322 (doi:10.1098/rstb.2012.0322) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maillard J, Schumacher W, Vazquez F, Regeard C, Hagen WR, Holliger C. 2003. Characterization of the corrinoid iron–sulfur protein tetrachloroethene reductive dehalogenase of Dehalobacter restrictus. Appl. Environ. Microbiol. 69, 4628–4638 10.1128/AEM.69.8.4628-4638.2003 (doi:10.1128/AEM.69.8.4628-4638.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neumann A, Wohlfarth G, Diekert G. 1998. Tetrachloroethene dehalogenase from Dehalospirillum multivorans: cloning, sequencing of the encoding genes, and expression of the pceA gene in Escherichia coli. J. Bacteriol. 180, 4140–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris RM, Sowell S, Barofsky D, Zinder S, Richardson R. 2006. Transcription and mass-spectroscopic proteomic studies of electron transport oxidoreductases in Dehalococcoides ethenogenes. Environ. Microbiol. 8, 1499–1509 10.1111/j.1462-2920.2006.01090.x (doi:10.1111/j.1462-2920.2006.01090.x) [DOI] [PubMed] [Google Scholar]
- 39.Morris RM, Fung JM, Rahm BG, Zhang S, Freedman DL, Zinder SH, Richardson RE. 2007. Comparative proteomics of Dehalococcoides spp. reveals strain-specific peptides associated with activity. Appl. Environ. Microbiol. 73, 320–326 10.1128/AEM.02129-06 (doi:10.1128/AEM.02129-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran BQ, Hernandez C, Waridel P, Potts A, Barblan J, Lisacek F, Quadroni M. 2011. Addressing trypsin bias in large scale (phospho)proteome analysis by size exclusion chromatography and secondary digestion of large post-trypsin peptides. J. Proteome Res. 10, 800–811 10.1021/pr100951t (doi:10.1021/pr100951t) [DOI] [PubMed] [Google Scholar]