

Abstract
Dehalobacter restrictus strain PER-K23 is an obligate organohalide respiring bacterium, which displays extremely narrow metabolic capabilities. It grows only via coupling energy conservation to anaerobic respiration of tetra- and trichloroethene with hydrogen as sole electron donor. Dehalobacter restrictus represents the paradigmatic member of the genus Dehalobacter, which in recent years has turned out to be a major player in the bioremediation of an increasing number of organohalides, both in situ and in laboratory studies. The recent elucidation of the D. restrictus genome revealed a rather elaborate genome with predicted pathways that were not suspected from its restricted metabolism, such as a complete corrinoid biosynthetic pathway, the Wood–Ljungdahl (WL) pathway for CO2 fixation, abundant transcriptional regulators and several types of hydrogenases. However, one important feature of the genome is the presence of 25 reductive dehalogenase genes, from which so far only one, pceA, has been characterized on genetic and biochemical levels. This study describes a multi-level functional genomics approach on D. restrictus across three different growth phases. A global proteomic analysis allowed consideration of general metabolic pathways relevant to organohalide respiration, whereas the dedicated genomic and transcriptomic analysis focused on the diversity, composition and expression of genes associated with reductive dehalogenases.
Keywords: Dehalobacter, organohalide respiration, metabolism, genome, reductive dehalogenase
1. Introduction
Dehalobacter restrictus strain PER-K23 has been isolated from a tetrachloroethene (PCE) dechlorinating enrichment culture originally obtained from sediment of the Rhine River mixed with anaerobic granular sludge [1]. Dehalobacter restrictus is a Gram-positive member of the Firmicutes growing exclusively via organohalide respiration (OHR) with H2 as electron donor, PCE or trichloroethene (TCE) as sole electron acceptor and acetate as carbon source. The key catalytic enzyme in OHR with PCE, the reductive dehalogenase PceA, has been purified and shown to harbour a corrinoid and two 4Fe/4S clusters [2]. In D. restrictus, the PceA enzyme is encoded by a gene that is part of the pceABCT gene cluster that has been shown to be highly conserved in several other OHR strains belonging to the genus Desulfitobacterium [3,4]. The newly available genome sequence of D. restrictus has revealed a high number of 25 predicted reductive dehalogenase homologue (rdhA) encoding genes (no Genbank accession number has yet been attributed to the genome of D. restrictus, however it is possible to access it via http://img.jgi.doe.gov/cgi-bin/w/main.cgi) [5], though only PCE and TCE have been recognized as physiological substrates. This observation clearly raises the question of the true bioremediation potential of D. restrictus.
Two other Dehalobacter isolates have been reported: Dehalobacter sp. TEA able to dechlorinate PCE and TCE [6], and Dehalobacter sp. TCA1 dechlorinating 1,1,1-trichloroethane to chloroethane [7], both strains however not yet having been characterized in detail on biochemical and genetic levels. Many studies have described cocultures or enrichment cultures where Dehalobacter spp. have been considered as the key player in the dechlorination of several other organohalides. A coculture containing Dehalobacter sp. E1 and Sedimentibacter sp. B4 has been obtained for the dechlorination of β-hexachlorocyclohexane (β-HCH) to benzene and chlorobenzene [8]. The draft genome of Dehalobacter sp. E1 has been recently reconstituted and was shown to harbour 10 putative rdhA genes, including a gene cluster with a high level of similarity to pceABCT present in D. restrictus, although strain E1 has not been shown to grow on PCE [9,10].
Further organohalides such as dichloroethane, chloroform, dichlorobenzenes or 4,5,6,7-tetrachlorophthalide were shown to be dechlorinated by enrichment cultures dominated by Dehalobacter spp. [11–14], suggesting that the degradation potential of the genus Dehalobacter is largely beyond PCE and TCE. Finally, fermentation of dichloromethane by members of Dehalobacter has been shown [15,16], suggesting that not necessarily all members of this genus are obligate OHR bacteria (OHRB).
The apparent redundancy in rdhA genes can be rather considered as a genuine property of OHRB that are otherwise restricted in their metabolism. For example, genomes of members of the OHR-obligate Dehalococcoides genus, for which five different genomes are already available (and three more pending), display between 10 and 36 rdhA genes [17], most of which have unknown substrate range. By contrast, completed genomes of members of the metabolically versatile Desulfitobacterium genus reveal the presence of only a limited number of rdhA genes, with Desulfitobacterium hafniense DCB-2 harbouring a maximum of seven copies [18,19]. While the composition of the genes associated with rdhA genes is strongly varying in the genomes of OHRB, rdhA subunits are almost invariably accompanied by a short open reading frame, rdhB, with the exception of the recently sequenced genome of Dehalogenimonas lykanthroporepellens [20]. Despite a very low level of sequence similarity, RdhB proteins display consensually two or three transmembrane helices strongly indicating a role in anchoring the catalytic subunit in the membrane.
Recently, proteomics and transcriptomics studies were used to study the metabolism of two OHRB, Desulfitobacterium hafniense strain TCE1 [21] and Y51 [22], respectively, under different growth conditions, both confirming the apparent lack of regulation of the pceA gene that was postulated earlier [4]. Most omics studies involving OHRB have however focused on members of the Dehalococcoides genus. This genus, although phylogenetically distant to Dehalobacter, inhabits similar ecological niches, and is exclusively dependent on OHR metabolism with H2 as electron donor. These studies have used both transcriptomics using full genome microarrays and proteomics to identify key components of the metabolism of OHRB under different growth conditions or growth phases [23–29].
In addition to genes directly linked to reductive dehalogenation, the genome of D. restrictus (http://img.jgi.doe.gov/cgi-bin/w/main.cgi) furthermore encodes one formate dehydrogenase (Fdh), and eight hydrogenase complexes, among which are three uptake hydrogenases (Hup-type), one energy-conservation hydrogenase (Ech-type) and one hydrogenase-3 (Hyc-type; [5]), similar to what has been described for Dehalococcoides. No data are yet available, however, concerning the role of these enzymes in the metabolism of D. restrictus.
Detailed studies of the metabolism of members of the Dehalobacter genus have so far been hampered by the lack of full genome information. Hence, the recently elucidated genome sequence of D. restrictus now provides the necessary basis for detailed studies of the metabolism of this obligate OHR bacterium using a tiered functional genomics approach.
2. Material and methods
(a). Bacteria and growth conditions
Dehalobacter restrictus strain PER-K23 (DSM 9455) was cultivated as described earlier [1,2]. Anaerobic serum flasks were supplemented with hydrogen as electron donor, inoculated with 2 per cent (v/v) inoculum, and finally 1 per cent (v/v) of 2 M PCE solution in hexadecane was added as electron acceptor. Nine batch cultures of D. restrictus were cultivated in 300 ml medium at 30°C under agitation (100 r.p.m.), and their growth was monitored by chloride production and not optical density as it is biased by precipitation of medium component. The true nature of OHR (i.e. the link between dechlorination and growth) was already demonstrated for D. restrictus [1]. Triplicate cultures were each harvested at three different growth stages of chloride release (20, 30 and 40 mM) that we have defined as the exponential (E), late-exponential (LE) and stationary (S) phases (see the electronic supplementary material, figure S1). Aliquots of 50 ml culture were collected for transcriptomic analysis, whereas the rest of each culture was harvested for proteomic analysis. For RNA extraction, 50 ml was collected by 2 min centrifugation at 4600g at 15°C, the pellet was readily resuspended in 1 ml of LifeGuard (MO-BIO, Carlsbad, CA, USA), incubated for 1 min and flash-frozen in liquid nitrogen. The remaining 250 ml of culture was centrifuged for 10 min as above for proteomic analysis. The pellet was washed in 10 mM Tris–HCl (pH 7.5) containing 1 mM EDTA, and then flash-frozen in liquid nitrogen. All biomass samples were stored at −80°C until use. Escherichia coli DH5α was cultivated on standard liquid or solid LB medium containing 100 μg l−1 ampicillin when transformed with derivatives of the pGEM-T easy vector (Promega, Duebendorf, Switzerland).
(b). Sequence analysis
All sequences mentioned in this study are taken from the recently published genome of D. restrictus strain PER-K23 [5]. The annotation of specific genes was verified, using a manual search with BLAST [30]. Rho-independent transcription terminators were identified with TransTerm from the Nano+Bio-Center of Kaiserslautern Technical University (http://www.cs.jhu.edu/~genomics/TransTerm/transterm.html) using default parameters. Protein sequences were aligned using ClustalX v. 2.0 [31]. The RdhA tree was built with MEGA5 [32].
(c). RNA extraction
RNA was extracted using the TRIzol method according to Prat et al. [33] with the following modification. The DNaseI treatment was stopped by adding 1× DNase stop solution and incubating for 10 min at 65°C. RNA concentration was estimated using the Nanodrop ND-1000 spectrophotometer (Thermo Scientific, Ecublens, Switzerland).
(d). Reverse transcription
Two micrograms of RNA was added to 4.5 μg of random hexamer (Microsynth GmbH, Balgach, Switzerland) in a volume of 85 μl. This mixture was incubated at 70°C for 5 min and then placed on ice. A 75 μl reverse transcription (RT) mix contained 32 μl 5× buffer, 8 μl 10 mM dNTPs, 19.2 μl 25 mM MgCl2, 4 μl RNasin (40 U μl−1) and 8 μl ImProm-II reverse transcriptase (Promega). The RT was performed as follows: 25°C for 5 min, 42°C for 60 min and 70°C for 15 min in a T3 Thermocycler (Biometra, Goettingen, Germany).
(e). Primer design
Specific primers were designed for each rdhA gene present in the D. restrictus genome by targeting unique regions. The primers were chosen such that the amplified products would fall in a size range suitable for quantitative PCR (qPCR; see below). Primer sequences and expected amplicon sizes are given in the electronic supplementary material, table S1.
(f). Endpoint PCR approaches
Different PCR strategies were applied in this study: standard endpoint PCR, multiplex endpoint PCR (mPCR) and qPCR (see below). Standard PCRs were carried out in 10 μl containing 4.25 μl ddH2O, 1 μl 10 × buffer, 0.3 μl dNTPs at 10 mM each, 0.4 μl 25 mM MgCl2, 1 μl each primer at 10 μM and 0.05 μl Taq polymerase at 5 U μl−1 (Peqlab, Erlangen, Germany). Two microlitres of genomic DNA or cDNA was added as template. For mPCR, a solution with eight different primers (four targets) was prepared containing 10 μM of each primer. Two microlitres of that solution was added in the standard reaction mix. Standard PCR and mPCR were performed in a Thermocycler (Biometra) using the following conditions: 5 min of initial denaturation at 95°C, followed by 30 cycles of 1 min denaturation at 95°C, one primer annealing at 52°C and 1 min elongation at 72°C. A final extension step of 10 min at 72°C was added at the end. The PCR products were routinely analysed in 1.5 per cent (w/v) agarose gels stained with GelRed (Biotium, Hayward, CA, USA). DNA was visualized using the Syngene gel imaging system (Syngene, Cambridge, UK).
(g). Cloning and sequencing of PCR products
PCR products were purified with the QIAquick PCR purification kit (Qiagen, Hombrechtikon, Switzerland) according to the manufacturer's instructions. The products were then A-tailed following instructions from the pGEM T-Easy vector manual (Promega), and finally ligated into pGEM T-Easy overnight at 16°C. The ligated products were cloned by heat shock transformation of CaCl2-competent E. coli DH5α. Transformants were screened using colony PCR with primers T7 and SP6, and positive clones were cultivated overnight at 37°C followed by plasmid preparation with the QIAprep Spin Miniprep kit (Qiagen). Plasmid inserts were verified by sequencing using the BigDye Terminator 3.1 kit on the ABI Prism 3130 Genetic Analyzer according to the manufacturer's instructions (Applied Biosystems).
(h). Quantitative PCR
Standards for qPCR were prepared from plasmids containing the gene targets as follows. One microgram of plasmid DNA was digested with five units of ScaI restriction enzyme (Promega) for 2 h at 37°C. The linearized plasmid was dephosphorylated during 1 h at 37°C by adding 1 μl shrimp alkaline phosphatase (Takara, Clontech Laboratories, Mountain View, CA, USA), followed by purification with the QIAquick PCR purification kit (Qiagen). The DNA concentration was measured with the Nanodrop ND-1000 spectrophotometer (Thermo Scientific). Serial dilutions (from 10−1 to 10−8 copies μl−1) of the purified sample were finally prepared and used as standards. A typical 10 μl qPCR contained 5 μl KAPA SYBR FAST universal 2× qPCR master mix (KAPA Biosystems, Woburn, MA, USA), 0.2 μl of each primer at 10 μM, 2.1 μl ddH2O and 2.5 μl template DNA (standards or samples). The reactions were performed in the Rotor Gene qPCR machine (RG-3000, Corbett Research, Qiagen) using the following programme: 2 min of initial denaturation at 95°C, then 40 cycles of 30 s denaturation at 95°C, 30 s primer annealing at 58°C and 20 s elongation at 72°C. Fluorescence was measured at the end of each elongation step. Each run consisted of triplicate reactions for both the standards and the samples. Run performances are given in the electronic supplementary material, table S2 for each considered gene target. The obtained data were expressed as transcript copy number per μl of initial cDNA samples.
(i). Protein extraction and SDS-PAGE
Cell pellets were transferred to 2 ml low binding microcentrifuge tubes (Eppendorf, Nijmegen, The Netherlands) prior to protein extraction. Protein extraction was conducted in 500 µl SDT-lysis buffer (100 mM Tris–HCl pH 7.6, 4% sodium dodecyl sulfate (SDS), 0.1 M dithiothreitol). Cells were lysed by sonication, using a Branson sonifier equipped with a 3 mm tip (six pulses of 30 s with 30 s rest on ice in-between each pulse, strength of the pulse was increased stepwise from setting 2 to 4). Proteins were denatured by boiling for 5 min, followed by 10 min centrifugation at 15 700g. Protein concentrations were determined using the Bradford method [34]. Finally, SDS-polyacrylamide gel electrophoresis (PAGE) was performed with gels containing 10 per cent acrylamide using a MiniProtean III system (Bio-Rad, Veenendaal, The Netherlands). Samples containing 10 µg protein were mixed with 2× loading buffer (100 mM Tris–HCl pH 6.8, 200 mM dithiothreitol, 4% SDS, 0.2% bromophenol blue and 20% glycerol) and briefly heated to 95°C before loading on gels. Gels were stained with Coomassie brilliant blue.
(j). In-gel trypsin digestion
For the growth phase experiment, each lane was cut in five slices of approximately equal size. Each slice was cut into approximately 1 mm3 pieces and transferred to independent 500 µl low binding microcentrifuge tubes (Eppendorf). All solutions were prepared using 50 mM NH4HCO3 unless otherwise stated. Tubes were briefly centrifuged, and the liquid phase removed between each step. Proteins were reduced by incubating in 50 mM dithiothreitol for 1 h at 60°C while slowly shaking, and alkylated by incubation in 100 mM iodoacetamide for 1 h in the dark at room temperature, washed once and incubated with 20 ng trypsin (sequencing grade, Roche Diagnostics, Almere, The Netherlands) over night at room temperature. Samples were sonicated in a water bath for 30 min before the supernatant was transferred to fresh 500 µl low binding microcentrifuge tubes. To increase the yield, the gel pieces were covered with 10 per cent trifluoroacetic acid in H2O and sonicated for another 30 min. Then, an equal volume of a solution containing 15 per cent acetonitrile and 1 per cent trifluoroacetic acid in H2O were added. The samples were sonicated for 1 min, before supernatants were combined in the low binding microcentrifuge tubes mentioned earlier. Peptides were concentrated using StageTip C18 columns essentially as described by Rappsilber et al. [35]. Finally, the volume was reduced to 10 µl using a SpeedVac vacuum centrifuge, and increased to 25 μl with 0.1 per cent (v/v) formic acid. Samples were measured by nLC–MS/MS with a Proxeon nLC and a LTQ-Orbitrap mass spectrometer as described by Lu et al. [36].
(k). LC–MS data analysis
LC–MS runs with all MS/MS spectra obtained were analysed with MaxQuant v. 1.2.2.5 [37] using default settings for the Andromeda search engine [38], except that extra variable modifications were set for de-amidation of N and Q.
A protein database was generated based on the genomes of D. restrictus and Dehalobacter sp. E1 [10], using the Artemis genome browser, and combined with a database that contains sequences of common contaminants such as, for instance, BSA (P02769, bovine serum albumin precursor), trypsin (P00760, bovine), trypsin (P00761, porcine), keratins K22E (P35908, human), K1C9 (P35527, human), K2C1 (P04264, human) and K1CI (P35527, human) [39]. The label-free quantification (LFQ) as well as the match between runs options (with ± 2 min retention time deviation) were enabled. De-amidated peptides were allowed to be used for protein quantification, and all other quantification settings were kept default.
Filtering and further bioinformatic analysis of the MaxQuant/Andromeda workflow output and the analysis of the abundances of the identified proteins were performed with the Perseus v. 1.2.0.16 module (available at the MaxQuant suite). Accepted were peptides and proteins with a false discovery rate (FDR) of less than 1 per cent and proteins with at least two identified peptides of which one should be unique. Also, quantification was carried out by the MaxQuant software for which MS data of at least three isotopes per peptide are used [37], and at least two quantified peptides per protein. This method makes label-free relative quantification reliable and therefore possible [40,41].
Reversed hits were deleted from the MaxQuant result table as well as all results showing a LFQ value of 0 for both sample and control. Zero values for one of the two LFQ columns were replaced by a value of 5 to make sensible ratio calculations possible. Relative protein quantification of sample to control was conducted with Perseus v. 1.2.0.16 by applying a two sample t-test using the ‘LFQ intensity’ columns obtained with threshold 0.10 and S0 = 1.
3. Results
(a). Proteomic analysis of Dehalobacter restrictus along growth phases
The genome of D. restrictus strain PER-K23 was predicted to encode 2826 proteins [5]. Using a combined protein database generated from the genomes of D. restrictus and Dehalobacter sp. E1, we identified 1055 proteins by proteome analysis (see the electronic supplementary material, table S3 and figure S2), of which 15 have been previously annotated as pseudogenes in D. restrictus, and one was newly discovered (see the electronic supplementary material, table S4). Data obtained from biological triplicates taken at the designated exponential (E), late-exponential (LE) and stationary (S) phases (see §2) were used to calculate the relative abundance ratio of proteins at stationary versus exponential phase (S/E), late-exponential versus exponential phase (LE/E) and stationary versus late-exponential phase (S/LE). The S/E protein abundance ratios of only 38 proteins were considered as statistically different (with FDR < 0.1), and corresponded to ratios between 25- to 3000-fold (table 1). However, in a mere qualitative approach, we considered a threefold increase/decrease in relative protein abundance as cut-off to define the proteins that differed between growth phases. This selection allowed investigation of general trends in protein changes across the different growth phases. The largest differences were seen between stationary and exponential phases, where the production of 29 per cent of all identified proteins seemed to be regulated. Comparing late-exponential and exponential phase, or stationary and late-exponential phase, only 16 per cent and 18 per cent of all identified proteins were produced at different levels, respectively. In the following, we focused on selected proteins and metabolic pathways most directly linked to the organohalide respiratory lifestyle of D. restrictus (table 2), but the complete dataset is given in the electronic supplementary material, table S5. The housekeeping enzyme RNA polymerase (RpoB, Dehre_0495) was detected at stable levels throughout all growth phases.
Table 1.
Detected proteins showing a significant increase/decrease in abundance (expressed as S/E ratio) during the transition from exponential (E) to stationary (S) phases. The S/LE and LE/E ratios are also indicated.
| locus tag (Dehre_#) | annotated function | protein abundance ratioa |
||
|---|---|---|---|---|
| S/E | S/LE | LE/E | ||
| proteins displaying significant increase in S/E ratio | ||||
| 1215 | late competence development protein (ComFB) | 3.0 × 103 | 1.8 × 10 | 1.7 × 102 |
| 0983 | cupin-domain protein | 2.0 × 103 | 1.5 × 10 | 1.3 × 102 |
| 0318 | uncharacterized protein conserved in bacteria | 8.4 × 102 | 4.2 | 2.0 × 102 |
| 2151 | aspartyl/glutamyl-tRNA (Asn/Gln) amidotransferase | 6.6 × 102 | 7.1 | 9.4 × 10 |
| 0568 | similar to acyl-coenzyme A synthetase/AMP-fatty acid ligase | 4.4 × 102 | 8.7 | 5.1 × 10 |
| 0109 | predicted transcriptional regulator | 3.9 × 102 | 1.4 × 10 | 2.8 × 10 |
| 1963 | uncharacterized protein conserved in bacteria | 3.7 × 102 | 1.1 × 10 | 3.3 × 10 |
| 0668 | RelE-type toxin (TA system) | 2.6 × 102 | 3.6 × 10 | 7.3 |
| 0856 | response regulator with CheY-like and AraC-type domains | 2.6 × 102 | 2.4 × 10 | 1.1 × 10 |
| 2645 | uncharacterized domain 1 protein | 2.3 × 102 | 1.6 × 10 | 1.5 × 10 |
| 2325 | hypothetical protein | 2.3 × 102 | 9.0 × 10 | 2.5 |
| 1400 | nitrogen regulatory protein PII | 2.3 × 102 | 1.7 × 10 | 1.4 × 10 |
| 0147 | CODH/ACS, maturation factor | 1.8 × 102 | 3.1 | 5.7 × 10 |
| 1786 | hypothetical protein | 1.2 × 102 | 1.5 × 10 | 8.0 |
| 0146 | CODH/ACS, maturation factor | 1.2 × 102 | 4.5 | 2.7 × 10 |
| 1237 | acetate-CoA ligase | 7.3 × 10 | 2.6 | 2.8 × 10 |
| 0264 | hypothetical protein | 7.1 × 10 | 5.3 | 1.3 × 10 |
| 2205 | YGGT protein family | 5.1 × 10 | 1.1 × 10 | 4.8 |
| 2544 | hypothetical protein | 5.0 × 10 | 7.8 | 6.5 |
| 0651 | predicted transcriptional regulator | 4.9 × 10 | 2.2 | 2.2 × 10 |
| 1310 | transcription antitermination factor NusB | 3.7 × 10 | 1.0 × 10 | 3.5 |
| 0143 | pterin-binding enzyme | 3.3 × 10 | 2.5 | 1.3 × 10 |
| 2265 | response regulator containing CheY-like receiver | 3.0 × 10 | 6.5 | 4.6 |
| 2560 | transcriptional regulator | 2.7 × 10 | 8.8 | 3.1 |
| 0144 | CODH/ACS, γ-subunit (CFeSP) | 2.5 × 10 | 1.6 | 1.5 × 10 |
| 0198 | bacterial nucleoid DNA-binding protein | 2.5 × 10 | 7.3 | 3.4 |
| proteins displaying significant decrease in S/E ratio | ||||
| 2864 | Cobalt transport protein | 3.4 × 10−2 | 1.5 × 10−1 | 2.3 × 10−1 |
| 1795 | predicted transcriptional regulator containing CBS domains | 1.5 × 10−2 | 2.9 × 10−2 | 5.0 × 10−1 |
| 2895 | uncharacterized protein conserved in bacteria | 2.0 × 10−3 | 1.6 × 10−1 | 1.3 × 10−2 |
| 0194 | transcription-repair coupling factor | 2.0 × 10−3 | 5.0 × 10−3 | 5.2 × 10−1 |
| * | LSU ribosomal protein L34p | 3.0 × 10−3 | 4.0 × 10−3 | 6.6 × 10−1 |
| 0258 | excisionase-like DNA-binding domain | 3.0 × 10−3 | 9.0× 10−3 | 3.6 × 10−1 |
| 2307 | uncharacterized protein conserved in bacteria | 5.0 × 10−3 | 1.0 | 5.0 × 10−3 |
| 2254 | chemotaxis protein stimulating methylation of MCP proteins | 6.0 × 10−3 | 8.0 × 10−3 | 7.6 × 10−1 |
| 2873 | trypsin-like serine protease | 6.0 × 10−3 | 9.0 × 10−3 | 7.2 × 10−1 |
| 1962 | predicted Fe-S oxidoreductase | 9.0 × 10−3 | 3.8 × 10−2 | 2.3 × 10−1 |
| 2146 | rRNA (uracil-5-)-methyltransferase (RumA) | 9.0 × 10−3 | 1.0 | 9.0 × 10−3 |
| 2280 | hypothetical protein | 1.0 × 10−2 | 6.7 | 1.0 × 10−3 |
aRatios above 1 mean increase in protein abundance, ratios below 1 mean decrease.
Table 2.
Proteomic analysis of selected metabolic pathways of D. restrictus.
| locus tag (Dehre_#) | protein | annotated function | protein abundance ratio |
||
|---|---|---|---|---|---|
| S/E | S/LE | LE/E | |||
| proteins associated with organohalide respiration | |||||
| 2022 | RdhA14 | reductive dehalogenase | 3.4 × 10−1 | 5.4 × 10−1 | 6.3 × 10−1 |
| 2025 | RdhK15 | CPR/Fnr-type regulator | 1.2 | 3.1 | 3.8 × 10−1 |
| 2048 | RdhK20 | CPR/Fnr-type regulator | 4.7 × 10−1 | 4.0 × 10−1 | 1.2 |
| 2395 | PceT | chaperone (trigger factor) | 1.2 × 10−1 | 2.8 × 10−1 | 4.3 × 10−1 |
| 2396 | PceC | FMN-binding domain | 6.9 × 10−1 | 8.0 × 10−1 | 8.6 × 10−1 |
| 2397 | PceB | membrane anchor | 5.3 × 10−1 | 6.8 × 10−1 | 7.8 × 10−1 |
| 2398 | PceA | PCE reductive dehalogenase | 8.0 × 10−1 | 1.2 | 6.9 × 10−1 |
| proteins associated with corrinoid synthesis and uptake | |||||
| 0286 | ABC-type iron transporter, substrate-binding | 6.6 × 10−1 | 1.3 | 8.3 × 10−1 | |
| 0289 | Mg/Co protoporphyrin IX chelatase | 6.6 × 10−2 | 1.0 | 6.6 × 10−2 | |
| 0291 | NodI | ABC-type Nod export system, ATP-binding | 5.2 × 10−1 | 2.9 | 1.5 |
| 1488 | CobT | nicotinate-nt-DMB phosphoribosyltransferase | 2.0 × 10−1 | 3.2 | 6.4 × 10−1 |
| 1606 | CobA | cob(I)yrinic acid a,c-diamide adenosyltransferase | 1.2 | 1.0 | 1.2 |
| 1607 | CbiP | cobyric acid synthase | 6.8 × 10−1 | 1.1 | 7.5 × 10−1 |
| 1608 | phosphoglycerate mutase | 8.3 × 10−1 | 1.1 | 9.3 × 10−1 | |
| 1609 | CobD | l-Thr-O-3-phosphate decarboxylase | 9.9 × 10−1 | 8.4 × 10−1 | 8.3 × 10−1 |
| 1610 | CbiB | adenosylcobinamide-phosphate synthase | 3.3 | 3.8 × 10−1 | 1.2 |
| 1611 | CobC | α-ribazole-5′-phosphate phosphatase | 1.3 | 8.1 × 10−1 | 1.0 |
| 1612 | CobU/CobP | cobinamide kinase/phosphate guanylyltransferase | 7.4 × 10−1 | 1.7 | 1.3 |
| 1614 | CobU/CobP | cobinamide kinase/phosphate guanylyltransferase | 1.1 | 1.1 | 1.3 |
| 1615 | CbiA | cobyrinic acid a,c-diamide synthase | 6.9 × 10−1 | 1.0 | 7.0 × 10−1 |
| 2535 | BtuF | ABC-type Cbl/Fe3+ transporter, substrate-binding | 1.5 | 8.5 × 10−1 | 1.2 |
| 2537 | BtuD | ABC-type Cbl/Fe3+ transporter, ATPase | 5.8 × 10−1 | 1.2 | 7.2 × 10−1 |
| 2538 | CbiZ | adenosylcobinamide amidohydrolase | 2.5 | 2.3 | 5.6 |
| 2848 | CbiC | precorrin-8x methylmutase | 8.3 × 10−1 | 9.4 × 10−1 | 8.9 × 10−1 |
| 2850 | CbiX | sirohydrochlorin cobalt chelatase | 1.7 | 2.1 | 8.1 × 10−1 |
| 2851 | HemL | glutamate-1-semialdehyde 2,1-aminomutase | 3.2 × 10−1 | 5.0 × 10−1 | 6.4 × 10−1 |
| 2852 | HemB | d-aminolevulinic acid dehydratase | 5.9 × 10−1 | 7.6 × 10−1 | 7.8 × 10−1 |
| 2853 | CysG/HemD | uroporphyrinogen-III synthase/C-methyltransferase | 8.6 × 10−1 | 8.7 × 10−1 | 9.9 × 10−1 |
| 2854 | HemC | porphobilinogen deaminase | 6.5 × 10−1 | 7.2 × 10−1 | 9.0 × 10−1 |
| 2857 | HemA | glutamyl-tRNA reductase | 6.9 × 10−2 | 8.2 | 5.7 × 10−1 |
| 2859 | CbiF | precorrin-4 C11-methyltransferase | 1.1 | 9.0 × 10−1 | 9.6 × 10−1 |
| 2860 | CbiL | precorrin-2 C20-methyltransferase | 1.1 | 1.0 | 1.1 |
| 2862 | CbiO | ECF-type cobalt transporter, ATPase | 4.0 × 10−2 | 9.6 × 10−1 | 3.8 × 10−2 |
| 2864 | CbiN | ECF-type cobalt transporter, bipartite component | 3.4 × 10−2 | 6.7 | 2.3 × 10−1 |
| proteins belonging to the Wood–Ljundahl pathway | |||||
| 0140 | predicted RNA-binding protein | 3.8 × 10 | 1.1 | 3.5 × 10 | |
| 0142 | CODH/ACS, α-subunit | 1.0 × 10 | 2.0 | 5.2 | |
| 0143 | pterin-binding enzyme | 3.3 × 10 | 2.5 | 1.3 × 10 | |
| 0144 | CODH/ACS, γ-subunit | 2.5 × 10 | 1.6 | 1.5 × 10 | |
| 0145 | CODH/ACS, δ-subunit | 1.1 × 10 | 1.3 | 8.0 | |
| 0146 | CODH/ACS, maturation factor | 1.2 × 102 | 4.5 | 2.7 × 10 | |
| 0147 | CODH/ACS, maturation factor | 1.8 × 102 | 3.1 | 5.7 × 10 | |
| 0148 | CODH/ACS, β-subunit | 5.4 | 1.5 | 3.5 | |
| 0150 | pterin-binding enzyme | 1.3 | 1.0 | 1.2 | |
| 0151 | methylene-H4F-DH/methenyl-H4F cyclohydrolase | 2.3 | 1.1 | 2.1 | |
| 0152 | methenyl-H4F cyclohydrolase | 6.3 × 10−1 | 5.3 × 10−1 | 1.2 | |
| 0153 | formyl-H4F synthetase | 9.3 × 10−1 | 8.3 × 10−1 | 1.1 | |
| 2348 | formate dehydrogenase, α-subunit | 6.5 × 10−1 | 1.1 | 6.2 × 10−1 | |
| 2349 | NADH : ubiquinone oxidoreductase | 1.8 × 10−1 | 2.1 × 10−1 | 8.9 × 10−1 | |
(i). Reductive dehalogenases
The genome of D. restrictus contains 25 genes predicted to encode rdhA (see below). Overall, a total of 86 genes are potentially associated with reductive dehalogenase expression and maturation, including genes that are predicted to encode putative membrane anchors, transcriptional regulators, chaperones and other rdh associated genes. Two of the reductive dehalogenase catalytic subunits (RdhA) were detected in the proteome: RdhA14 (Dehre_2022) and RdhA24 (PceA, Dehre_2398). The former shows a very high amino acid sequence identity (89%) with RdhA2 from Desulfitobacterium hafniense DCB-2 (Dhaf_0693) [18], whereas the latter is the biochemically characterized PceA [2] (see the electronic supplementary material, table S6). All four proteins encoded by the pceABCT gene cluster (Dehre_2398 to Dehre_2395) were also identified in the proteome (table 2). PceA was among the most abundant proteins at all growth stages (data not shown). The protein abundance ratio of PceA, PceB and PceC remained within the threefold cut-off value when comparing any of the three growth phases considered. The absence of a regulatory component in the direct vicinity of the pce gene cluster (see below) suggests that PceA is constitutively expressed, although it needs to be further investigated. PceT, however, was the only member of the gene cluster that seemed to be regulated as the relative protein abundance ratios were 0.12, 0.43 and 0.28 for S/E, LE/E and S/LE, respectively. Although the value for LE/E did not exceed the cut-off value, the data suggest that PceT was most abundant at the exponential phase and then became slightly less abundant at later growth stages (figure 1 and table 2).
Figure 1.

Metabolic map showing selected pathways of D. restrictus. Metabolic pathways, both predicted from D. restrictus genome and analysed by functional genomics, are presented in a simplified bacterial cell. The cytoplasmic membrane is depicted in grey, the cytoplasm in blue shading. Dehalobacter restrictus genomic loci (Dehre_#) are indicated in parentheses, with black label showing proteins detected in the proteomic analysis and those not detected indicated in red. Three important pathways are given in detail: the Wood–Ljungdahl pathway (WL), the menaquinone biosynthesis pathway (MK) and the corrinoid biosynthesis pathway (Cbl).
(ii). Hydrogenases
Hydrogen is the only electron donor that D. restrictus has been shown to use. The key role of hydrogenases is underscored by the fact that the genome of D. restrictus is predicted to encode eight multi-subunit hydrogenase complexes. Three of these (Dehre_0551–0553, 1061–1063 and 2405–2407) belong to the group of periplasmic membrane-bound Ni/Fe uptake hydrogenases (Hup) consisting of three subunits, a membrane-bound b-type cytochrome, a Fe/S cluster protein and the catalytic subunit (electronic supplementary material, table S5 and figure 1). Two membrane-bound energy-conserving Ni/Fe hydrogenases (Dehre_1568–1573 and 1645–1650) resemble the Hyc and Ech clusters found in Dehalococcoides mccartyi 195 [42]. These two hydrogenase complexes each consist of six subunits, a large and small subunit, and four subunits resembling elements of the proton-translocating respiration complex I (see the electronic supplementary material, table S5 and figure 1). The three Fe-only hydrogenases (Hym) consist of the catalytic unit and two or three subunits predicted to be involved in electron transfer. Unlike what was observed in Dehalococcoides mccartyi 195 [42], none of the Fe-only complexes contains any predicted transmembrane region, which suggests that they are either located in the cytoplasm or form a complex with other membrane-bound proteins (see the electronic supplementary material, table S5 and figure 1).
A constant amount of the large and small subunits from one of the Hup-type hydrogenases (Dehre_0552–0553) was detected throughout all growth phases. We did not detect the b-type cytochrome subunit (Dehre_0551), possibly as a consequence of its strong association with the membrane. Both putative energy-conserving hydrogenase complexes were detected in the cells. We detected the large and small subunit of the Hyc-type hydrogenase (Dehre_1568–1569), but none of the four subunits (Dehre_1570–1573) predicted to be involved in electron transfer and proton transport across the cell membrane. Interestingly, the small subunit (Dehre_1568) was most abundant at late-exponential phase and least abundant in stationary phase with S/E, S/LE and LE/E ratios of 0.24, 0.03 and 9.74, respectively, whereas the abundance of the large subunit did not differ between growth phases (electronic supplementary material, table S5 and figure 1). All but one (Dehre_1649) component of the Ech complex (Dehre_1645–1650) were detected. The only protein that differed in abundance between growth phases was Dehre_1647, predicted to encode an NADH-ubiquinone oxidoreductase. This protein became gradually more abundant at later growth stages with S/E, S/LE and LE/E ratios of 8.4, 1.8 and 4.8, respectively. We detected both three subunit Fe-only hydrogenases (Dehre_1739–1741 and Dehre_2372–2374) in the proteome, but none of the components of the four subunit complex (Dehre_2317–2320). The abundance of Dehre_1739–1741 did not change with the growth phases, whereas Dehre_2372–2374 showed a weak trend of decreasing abundance at later growth phases, most pronounced for Dehre_2373 with S/E, S/LE and LE/E ratios of 0.30, 0.32 and 0.96, respectively (see the electronic supplementary material, table S5).
(iii). Corrinoid synthesis and uptake
The genome of D. restrictus encodes a seemingly complete de novo corrinoid biosynthesis pathway starting from glutamyl-tRNA (figure 1). This pathway is encoded by two distinct gene clusters in D. restrictus: cluster I (Dehre_2848–2865), the upper pathway, and cluster II (Dehre_1606–1615), corresponding to the lower pathway. One additional gene (Dehre_1488) belonging to the lower pathway is located elsewhere in the genome (table 2).
Cluster I contains all genes necessary for the synthesis of cobyrinic acid starting from glutamyl-tRNA. This pathway, however, appears to be incomplete because cbiH (Dehre_2856) encoding precorrin-3B C17-methyltransferase displays a frame-shift mutation, and consequently is annotated as a pseudogene. We identified several proteins of the corrinoid synthesis pathway until cobyrinic acid, except CbiH and all enzymes responsible for the conversion of cobalt-precorrin-5A to cobalt-precorrin-8. From the upper pathway, only HemA (Dehre_2857) and HemL (Dehre_2851) showed a decreasing relative abundance from exponential to stationary phases with S/E ratios of 0.07 and 0.32, respectively. Most enzymes of the lower corrinoid synthesis pathway encoded by cluster II were found in stable amounts throughout the growth phases with exception of CbiB and CobS. CbiB (Dehre_1610) is responsible for the conversion of adenosylcobyric acid to adenosylcobinamide and was found in slightly increasing amounts at stationary phase (S/E: 3.26), whereas CobS (Dehre_1613) which is responsible for the conversion of adenosylcobinamide-GDP to adenosylcobalamin was not detected at all.
The genome of D. restrictus contains several gene clusters predicted to be involved in cobalt and corrinoid uptake. One predicted ABC-type cobalt transporter (Dehre_0850–0852) and two ECF-type cobalt transporters (Dehre_0278–0280 and Dehre_2862–2865) are present in D. restrictus. While none of Dehre_0850–0852 or Dehre_0278–0280 was detected in the proteome, we identified both CbiO (Dehre_2862) and CbiN (Dehre_2864) proteins from the transport system encoded in corrinoid synthesis gene cluster I. Both showed a decreasing trend when going from exponential to stationary phase with S/E ratio of 0.04 and 0.03, respectively (table 2). Two gene clusters (Dehre_0281–0292 and Dehre_2535–2538) are predicted to encode proteins possibly involved in uptake of various corrinoid precursors as part of salvaging pathways. From the first cluster, three proteins (Dehre_0286, 0289 and 0291) were detected. Their protein abundance ratio did not change over time, except for Dehre_0289, which was detected only during exponential phase (table 2), whereas from the second cluster, all proteins except the membrane-associated Dehre_2536 were detected. Only Dehre_2538 showed an LE/E ratio exceeding the threefold cut-off (5.63). Interestingly, this protein is predicted to encode a CbiZ homologue that salvages cobinamides and converts it back to cobyric acid [43].
(iv). Additional elements of the general energy metabolism
Constant amounts of six proteins (Dehre_2797–2802) of the 10 subunits of the ATP synthase (Dehre_2797–2806) were detected in the proteome. The proton-translocating respiration complex I encoded in the genome of D. restrictus consists of 11 subunits (Dehre_0889–899) instead of the canonical 14 [5], lacking the components NuoEFG that usually receive electrons from NADH. Three subunits (NuoBCD, Dehre_890–892) were clearly detected in the proteome, whereas none of the membrane components could be seen.
The genome encodes enzymes of a putative WL pathway for CO2 fixation (Dehre_0130–0155 and Dehre_2348–2351). Most proteins belonging to this pathway were detected in the proteome (see the electronic supplementary material, table S5). They were observed at constant level throughout the growth phases with the exception of proteins representing the carbonyl branch of the WL pathway and the acetyl-CoA synthase/CO dehydrogenase (ACS/CODH) complex. Generally, these proteins showed a gradual and significant increase in relative abundance towards later growth stages with S/E ratios between 25 and 175 (table 1).
We also identified a putative three component Fdh, consisting of a membrane-bound b-type cytochrome, a Fe/S cluster protein and the catalytic subunit (Dehre_1730–1734), which were detected at all growth phases (see the electronic supplementary material, table S5 and figure 1). The catalytic unit contains probably a selenocysteine as it is encoded by two in-frame genes (Dehre_1733–1734) separated by a UGA stop codon.
The genomic loci Dehre_2245–2284 and 2297–2314 contain large numbers of genes involved in the synthesis of flagella, motor proteins and chemotaxis (figure 1). In the proteome, we identified 32 of 62 proteins encoded in these genomic regions. Sixteen of them were less abundant in stationary than in exponential phase, only three increased in abundance, and the remaining 13 were equally abundant during stationary and exponential phase (see the electronic supplementary material, table S5), indicating that the cells are reducing their motility when entering the stationary phase.
Proteins showing significant changes in abundance between stationary and exponential phase are displayed in table 1. Generally, many proteins associated with regulation of transcription, chemotaxis and sensing were among the proteins displaying significant changes in their abundance. The protein showing the greatest change in abundance, with an S/E value of 2954, is Dehre_1215, annotated as ComFB, an uncharacterized protein possibly involved in development of late competence [44,45]. The gene cluster containing the comFB gene (Dehre_1214–1220) in D. restrictus contains genes predicted to encode an RNA helicase, an ABC transporter, and two genes encoding proteins of unknown function. We detected the periplasmic component of the ABC transporter and one of the hypothetical proteins in the proteome, the latter increasing in abundance at later growth phases (see the electronic supplementary material, table S5). The genome of D. restrictus encodes other competence factors such as ComEA and ComEC (Dehre_0586–0587), and ComFA (Dehre_2784), suggesting that it is capable of natural competence. None of these additional proteins, however, were detected in the proteomic analysis. Two gene clusters encoding pili (Dehre_1166–1175 and Dehre_1272–1289) possibly involved in DNA uptake are also present.
Another protein (Dehre_0668) that was among those with the strongest increase in abundance in stationary phase (table 1) has a high similarity with RelE toxin and builds with Dehre_0667 a toxin/antitoxin addiction module system that could be involved in modulating the persistence of cell growth in unfavourable growth conditions [46]. The antitoxin component (Dehre_0667) was however never detected in the proteome. The direct vicinity of Dehre_0668 displays several phage- or plasmid-related genes, suggesting that Dehre_0667-0668 could have been acquired by horizontal gene transfer and represent a phage-like defence mechanism [47].
(b). Diversity and composition of reductive dehalogenase gene clusters in Dehalobacter restrictus
(i). Multiple reductive dehalogenase homologue gene clusters in Dehalobacter restrictus
A thorough analysis of the D. restrictus genome [5] has revealed the presence of 25 rdhA genes, among which 20 are full-length, four harbour one or several frame-shifts (rdhA04, 05, 13 and 21), and one is a partial gene (rdhA25; see electronic supplementary material, table S6). The biochemically characterized reductive dehalogenase PceA [2] is encoded by rdhA24. While most rdhA genes are grouped in two genomic regions (rdhA01–10 and rdhA13–23), a detailed analysis of the genetic structure around them allowed definition of 13 clusters consisting of one to six rdhA surrounded by genes encoded on the same strand. It is however rather unlikely that these clusters represent actual operons as several rho-independent transcription terminators were predicted within the clusters (figure 2). Three general rdh genetic organizations can be considered here. Together with the well-characterized pceABCT cluster (rdhA24), two other rdhA genes are embedded in a similar configuration (rdhA20 and -22), albeit harbouring an additional rdhK subunit at the 3′-end. Seven rdhA genes are accompanied by rdhB and rdhC subunits, five of them in the orientation rdhABC (rdhA02, -05, -06, -13 and -17) and two as rdhBAC (rdhA14 and -21). Finally, the remaining rdhA subunits are accompanied only by their respective B subunit exclusively in the orientation rdhBA. Most of rdh gene clusters are also associated with one rdhK subunit in various orientations. The rdhK-encoded proteins clearly belong to the large family of CRP/Fnr regulatory proteins from which CprK members of Desulfitobacterium dehalogenans and Desulfitobacterium hafniense DCB-2 were extensively studied and represent the paradigmatic DNA-binding regulatory protein for the respective chlorophenol reductive dehalogenase (cpr) operons [48–56]. Screening of the genome of D. restrictus for RdhK protein-encoding genes revealed 25 paralogues from which 22 are located within the 13 rdh gene clusters, and the remaining three in their direct vicinity. This strongly suggests that RdhK are regulatory proteins dedicated to OHR metabolism.
Figure 2.

Genetic map of D. restrictus gene clusters containing reductive dehalogenase genes (rdhA, red numbered arrows). For each rdh cluster, all the genes present on the same DNA strand were considered together with the direct flanking genes in opposite orientation. The numbers indicated above each cluster are the corresponding loci in the D. restrictus genome (Dehre_#).
(ii). Diversity of Dehalobacter restrictus RdhA proteins
Protein sequence alignment of RdhA subunits of D. restrictus with selected sequences from other OHRB revealed several interesting features (figure 3). First, a strong correlation could be established between the level of sequence identity (see also electronic supplementary material, table S7) and the genetic organization of the predicted rdh operons. Indeed, the dominating group of 14 RdhA proteins encoded by minimal rdhBA operons forms a separate branch, which also contains the well-characterized cpr (CprA) of Desulfitobacterium dehalogenans. All three rdhABCT predicted operons in D. restrictus also cluster together, however with homology to enzymes with different substrate specificities. PceA (RdhA24) is highly similar to other PceA enzymes from members of the closely related genus Desulfitobacterium, but also highly similar (88% sequence identity) to DcaA of Desulfitobacterium dichloroeliminans, as already reported [3,57]. By contrast, both RdhA20 and -22 of D. restrictus have a rather strong sequence identity with CprA5 and RdhA3 of Desulfitobacterium hafniense strain PCP-1 and strain DCB-2, respectively, which have been shown to use 3,5-dichlorophenol [18,58], these two latter enzymes being encoded in a similar genetic structure (rdhABCT). Interestingly, two pairs of RdhA proteins (RdhA03 with -04; RdhA16 with -19) show a very high level of sequence identity (see the electronic supplementary material, table S7). Another striking feature is the high conservation degree of RdhA proteins between D. restrictus and the newly available RdhA sequences identified in the metagenome of the β-HCH dechlorinating co-culture containing Dehalobacter sp. E1 (DhbE1 in figure 3) [10]. Indeed, five of nine DhbE1 proteins have identical counterparts in D. restrictus (99–100% sequence identity), whereas three RdhA have highly similar homologues (70–92% identity) in D. restrictus. One last sequence (DhbE1_1222) is partial.
Figure 3.

Diversity analysis of D. restrictus RdhA proteins. All D. restrictus RdhA proteins are indicated in red together with their genetic structure: rdhA (red box), rdhB (orange), rdhC (yellow), rdhT (green). Protein sequences were aligned with selected RdhA proteins from other OHRB. When known, the corresponding substrates are also indicated. Dre, Dehalobacter restrictus; Dhb, Dehalobacter spp. (strains E1, MS and WL); Dha, Desulfitobacterium hafniense (strains DCB-2, PCP-1, TCE1 and Y51); Dde, Desulfitobacterium dehalogenans; Ddi, Desulfitobacterium dichloroeliminans; Desor, Desulfosporosinus orientis; Dmc, Dehalococcoides mccartyi (strains 195 and VS); Smu, Sulfurospirillum multivorans.
(c). Transcriptomic analysis of Dehalobacter restrictus reductive dehalogenase genes
(i). Screening of rdhA gene transcription by RT-multiplex PCR
From the global proteomic analysis, only two RdhA proteins were clearly detected: the main PCE reductive dehalogenase (PceA) and RdhA14, albeit at a much lower abundance. A specific approach was then conducted in order to evaluate the transcriptional level of the 24 full-length rdhA genes in D. restrictus along the growth phases. First, a reverse transcription (RT)-multiplex PCR method was developed allowing screening of groups of rdhA genes at the mRNA level in the triplicate cultures collected at the exponential (E), late-exponential (LE) and stationary (S) growth phases. Figure 4 illustrates the qualitative data obtained for a combination of four rdhA genes using that method (the complete set of data is presented in the electronic supplementary material, figure S3). Five rdhA gene transcripts (rdhA08, -14, -16, -19 and -24) were strongly amplified, however showing various transcription levels. The pceA gene (rdhA24) was clearly dominant and was still detected in the RNA samples collected in stationary phase (figure 4 and electronic supplementary material, S2). Those five rdhA genes were further analysed by RT-qPCR.
Figure 4.
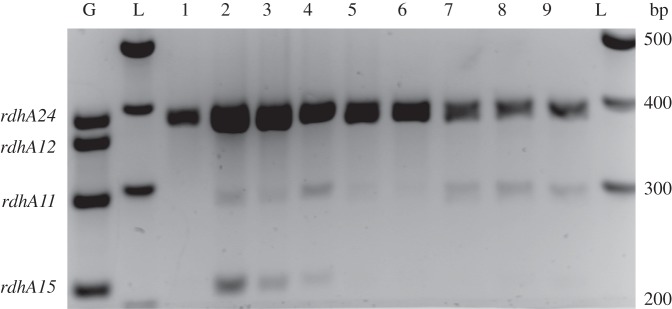
Growth-phase-dependent transcription level of rdhA genes in D. restrictus analysed by RT-multiplex PCR. One multiplex PCR result is depicted as illustration. The targeted genes are indicated on the left of the gel. G, positive control on genomic DNA; L, 100 bp ladder from which the corresponding fragment sizes are indicated on the right. Samples 1–3, 4–6, and 7–9 were taken from triplicate cultures harvested in exponential, late-exponential and stationary phases, respectively.
(ii). Quantitative assessment of selected rdhA gene transcription by RT-qPCR
Based on individual standards for each target gene, transcript copy numbers per μl of cDNA samples were measured for rdhA08, -14, -16, -19 and -24 (pceA) along with rpoB (Dehre_0495), which was chosen as a constitutively expressed housekeeping gene (figure 5 and see electronic supplementary material, table S2 for qPCR parameters). A decrease in transcription level was generally observed for all genes along the growth phases, some of them dropping below the detection limit of the method applied. These data confirmed the trend observed by the qualitative multiplex PCR approach. In the exponential phase, the pceA gene (rdhA24) was highly transcribed in comparison with all other genes considered (between 51- and 3688-fold, depending on the gene, see electronic supplementary material, table S8 for details). Although decreasing, pceA remained strongly transcribed even at stationary phase. The level of transcription of the remaining rdhA genes decreased with the following order: rdhA19 > rdhA14 ≫ rdhA16 > rdhA08.
Figure 5.

Growth-phase-dependent transcription level of selected rdhA genes in D. restrictus analysed by RT-qPCR. The graph depicts the gene copy number of selected rdhA genes along with rpoB as control obtained from one culture replicate harvested in exponential (light grey), late-exponential (dark grey), and stationary (black) phases, respectively. The same trend was observed for all replicates. Standard deviation of qPCR replicates was below 15% of the measured data. The dotted line (50 copies μl−1) displays the lower detection limit that was generally considered for the data obtained.
4. Discussion
Although D. restrictus was among the first OHRB to be isolated, a significant part of its metabolism remained largely unresolved, mainly owing to the lack of the genome sequence, but also owing to the restricted conditions in which this bacterium has been found to grow, namely exclusively by anaerobic respiration with hydrogen as electron acceptor and PCE or TCE as unique terminal electron acceptor. We recently obtained the genome sequence of D. restrictus strain PER-K23 [5], which allowed us in this study to consider general questions about its metabolism and a more specific investigation line focusing on the key players in OHR, the reductive dehalogenases.
The 2.9 Mb genome of D. restrictus can be considered to occupy an intermediate position among OHRB between the reduced genome size of the OHR obligate Dehalococcoides genus (approx. 1.4 Mb) and the largely redundant genomes of the versatile Desulfitobacterium genus (greater than 5 Mb). Metabolically, however, D. restrictus is closer to Dehalococcoides, suggesting that, besides additional genetic information responsible for peptidoglycan synthesis and motility, some parts of the D. restrictus genome may be not functional or encode for yet unsuspected metabolic pathways. A remarkable example is the presence of a complete biosynthetic pathway for cobalamin, an essential cofactor for OHR metabolism. Indeed, based on the anaerobic pathway described by Roessner & Scott [59] and the cobinamide salvaging pathway studied by Escalante-Semerena and co-workers [43,60–62], all genes were clearly identified in D. restrictus, although it cannot grow without a supply of vitamin B12 in the medium ([1]; J. Maillard 2012, unpublished data). The proteomic data obtained here showed that about half of the proteins of the biosynthetic pathway mostly from the upper pathway were not detected, indicating that under the growth conditions applied, D. restrictus used the corrinoid amended and possibly modified according to its needs. On the genetic level, the frame-shift mutation observed in cbiH (Dehre_2856) needs to be confirmed, but could also be a reason why D. restrictus is not able to synthesize cobalamin de novo. Preliminary proteomic data obtained from cells that were partially depleted of vitamin B12 revealed that the production of corrinoid transporters and proteins of the salvaging pathway increased significantly rather than the biosynthetic proteins (J. Maillard & T. Kruse 2012, unpublished data), suggesting that the biosynthetic pathway is not functional in D. restrictus.
Enzymes belonging to the WL pathway for CO2 fixation were clearly detected on the proteomic level in D. restrictus. A significant increase in the CODH/ACS complex (Dehre_0143–8, corresponding roughly to the carbonyl branch of the pathway) was observed in the late-exponential and stationary phases when compared with the exponential phase. This suggested that acetate might be depleted in the medium already during the late-exponential phase, and that D. restrictus could partially assimilate CO2 via the acetyl-CoA synthase. This is however contrasting with a previous dataset where heterotrophic CO2 assimilation (probably via pyruvate : ferredoxin oxidoreductase, PFOR) has been postulated for an enrichment of D. restrictus [63]. Several homologues of the latter enzyme were also detected in the proteome (see the electronic supplementary material, table S5). The role of the WL pathway and more generally the assimilation of CO2 have been questioned for other OHRB such as various isolates of Desulfitobacterium [18,19,21,64] or of Dehalococcoides [65–67]. In Desulfitobacterium hafniense strains, components of the WL pathway have been shown to participate in the use of phenyl methyl ethers as electron donor [64]. There, however, the methyl branch was mainly used together with O-demethylases. For strain TCE1, it has been reported that components of this pathway increased in abundance when H2 and PCE were used as a combination of electron donor and acceptor [21]. The work by Tang et al. [67] suggested that the WL pathway was not involved in CO2 fixation in Dehalococcoides. As illustrated by these examples, the role of the WL pathway in OHRB might be diverse, and further dedicated experiments are required to fully understand why D. restrictus recruits it at late growth phases.
The presence of eight different hydrogenases underscores the central role of hydrogen in the metabolism of D. restrictus. The genomes of Desulfitobacterium hafniense DCB-2 [18] and Y51 [19] encode, like D. restrictus, three Hup-type hydrogenases. The fact that we detected only one of them (Dehre_0551–0553) at relatively constant abundance across growth phases indicates that (i) the three Hup complexes have different roles in the metabolism and (ii) the detected Hup plays a role in the core metabolism. Concerning the three Fe-only hydrogenases (Hym), two complexes were present in stable abundance at all growth phases, whereas one was not observed at all. Unlike what is predicted for Hym in Dehalococcoides mccartyi 195 [28,42], we did not find any membrane-associated components in the Hym-type hydrogenases in D. restrictus. We therefore suggest that these enzymes are located in the cytoplasm, where they might be involved in generating reducing equivalents (e.g. NADH, FADH) for biosynthetic reactions or maybe directly in generating a proton motive force with respiration complex I, as speculatively indicated in figure 1. The respiration complex I in D. restrictus lacks the NuoEFG subunits that usually receive electrons from NADH. The electron donor for this type of respiration complex I is not yet known, but it has been speculated that they act as a docking station able to receive electrons from various electron donors [68]. It is interesting that we find two large membrane-bound putatively proton-translocating hydrogenase complexes, Hyc and Ech, in D. restrictus like in Dehalococcoides mccartyi 195, whereas the more closely related Desulfitobacterium hafniense Y51 and DCB-2 only contain a Hyc homologue [18,19,42]. The role of these remains unclear; however, disrupting hyc in Desulfitobacterium dehalogenans resulted in loss of ability to use 3-chloro-4-hydroxyphenylacetic acid and nitrate as electron acceptor when formate was used as electron donor, suggesting a role in the electron transport chain [69]. It has been suggested that Ech and Hyc may play a role in generating low potential electrons for OHR by reverse electron flow. It was however observed that the expression of both Hyc and Ech decreased when Dehalococcoides mccartyi 195 was cultivated under lower partial pressure of hydrogen. Because hydrogen is a stronger reductant at higher partial pressure, the opposite would have been expected if they played a role in reverse electron flow [28]. Our findings suggest that different hydrogenases play specific and central roles in the metabolism of D. restrictus, but elucidating the exact role of the individual hydrogenases requires further studies.
Significant changes in the protein content between exponential and stationary phases were observed for various unrelated proteins for which the predicted function was often not clear. For example, ComFB (Dehre_1215) and a cupin-domain containing protein (Dehre_0983) were identified with more than 1000-fold increase in the stationary versus exponential phase (table 1). The former protein is predicted to play a role in the late development of competence, although no other competence protein was detected. Competence represents a general strategy for bacteria to survive in unfavourable conditions such as during stationary phase [70]. The latter protein has no clear predicted function, but might be part of an operon involved in the shikimate pathway responsible for the biosynthesis of aromatic amino acids. The list of proteins that increased/decreased significantly between exponential and stationary phase clearly indicates that the cells are adjusting their metabolism when shifting from one growth phase to another.
The discovery of 25 reductive dehalogenase (rdhA) genes in the genome of D. restrictus was surprising given its currently known substrate range for reductive dehalogenation [5], but is in line with what has been observed in all available genomes of Dehalococcoides mccartyi. The detailed analysis of the rdh gene clusters we present here, together with the transcriptional and proteomic data on the components of these clusters, helped us to consider their diversity, evolution and function in D. restrictus during growth on PCE. Analysis of the sequence similarity of D. restrictus RdhA proteins along with the best-characterized RdhA proteins revealed at least three groups of enzymes. The largest and relatively deep-branching first group contains 16 RdhA proteins which are affiliated to the characterized ortho-chlorophenol dechlorinating enzymes (CprA) of Desulfitobacterium isolates [71,72]. Within this group, two gene duplication events must have occurred recently, as the couples RdhA16/19, and RdhA03/04 show 98 per cent and 81 per cent sequence identity, respectively (see the electronic supplementary material, table S7). Further synteny analysis revealed that the sequence conservation was extended to the corresponding rdhB genes (data not shown). A second deep-branching group of RdhA sequences contains D. restrictus PceA (RdhA24) and two slightly more distant members (RdhA20 and -22). These proteins form a closely related family together with some of the best-characterized enzymes, namely PceA of several Desulfitobacterium hafniense isolates [4,73,74], DcaA of Desulfitobacterium dichloroeliminans [57] and CprA5 (dechlorinating 3,5-dichlorophenol) of Desulfitobacterium hafniense PCP-1 [75]. The last seven RdhA proteins build up a group of highly heterogeneous enzymes for which no characterized counterpart is yet available. Among them however, four D. restrictus RdhA proteins (RdhA02, -05, -06 and -17) show 45 per cent sequence identity with a putative RdhA identified in the genome of Desulfosporosinus orientis (Desor_3837 [76]). The genetic organization around rdhA genes is tightly correlated with the sequence diversity of their encoded proteins. Indeed, both deep-branching groups of D. restrictus RdhA show uniform genetic structures, rdhBA and rdhABCT, respectively. The rather heterogeneous third group is made of either rdhABC or rdhBAC operons. This strongly indicates a possible evolutionary line in which a few individual rdh operons might have been acquired by horizontal gene transfer, followed by several rounds of gene duplication.
Functional investigation of the rdh gene clusters along the growth curve of D. restrictus on PCE clearly revealed that the PCE reductive dehalogenase (PceA, RdhA24) was dominating both at transcriptional and proteomic levels, with only little change along the growth phases. This can explain why only PceA could be purified from D. restrictus in earlier studies. On proteomic level, RdhA14 was the only other reductive dehalogenase detected but at an estimated PceA/RdhA14 ratio of 212 during exponential phase. While all subunits encoded by the pceABCT operon were identified, neither RdhB nor RdhC belonging to the rdhBAC14 operon was detected, possibly as a result of their lower expression and high hydrophobicity. On the transcriptional level, the results are somehow contrasting. While rdhA14 was also detected at a copy number ratio similar to the proteomic data (see the electronic supplementary material, table S8, B), other rdhA genes were also significantly transcribed, and among them rdhA19 at a slightly higher level than rdhA14, although not detected in the proteome. Whether this is due to the sensitivity of the proteomic analysis or to a possible post-transcriptional regulation remains to be investigated. Similar to several omics studies on Dehalococcoides [24,27–29,77,78], a relatively tight regulation seems to operate in D. restrictus for rdhA candidates, among which only a few of them are steadily expressed. In contrast to Dehalococcoides, however, where mostly two-component systems and MarR-type regulators are likely to regulate the expression of rdhA genes [79], in D. restrictus, as well as in the closely related Desulfitobacterium isolates, numerous CprK activating regulators (so-called RdhK) are present in or in the direct vicinity of rdh gene clusters. Only two of them, however, were detected in the proteomic analysis (Dehre_2025 and 2048), suggesting that their expression level remains low in the cell or that they are themselves regulated.
Additional proteins encoded in rdh gene clusters were also detected in the proteome. The TatA and TatB components of two Tat systems (Dehre_0836, 0839 and 1843) were detected. Interestingly, the former system is encoded directly downstream of rdhA10 and surprisingly contains an ApbE homologue (Dehre_0837) involved in thiamine biosynthesis. Also possibly linked to the translocation of RdhA proteins across the cytoplasmic membrane, a SppA homologue (Dehre_0809) was detected. The corresponding gene is located directly downstream of rdhA04 and its product is possibly involved in the degradation of signal peptides (such as the Tat signal peptides of RdhA proteins) after they have been cleaved from the mature proteins [80,81].
Our multi-level study of D. restrictus metabolism revealed rather elaborate genomic and proteomic features despite its restricted physiology recognized so far, suggesting that there is much more to discover especially in the energy metabolism of this bacterium. In addition, the high number of reductive dehalogenase genes raises the question of a wider bioremediation spectrum via OHR for D. restrictus.
Acknowledgements
The Netherlands Genomics Initiative as well as the European Community's Seventh Framework Programme (FP7/2007-2013) are acknowledged for support to T.K. and H.S. through the Ecogenomics and BACSIN project (grant agreement no. 211684). Sequencing and annotation of the D. restrictus genome was performed under the auspices of the US Department of Energy's Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract no. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under contract no. DE-AC52-07NA27344 and Los Alamos National Laboratory under contract no. DE-AC02-06NA25396. The Swiss National Science Foundation (SNSF) is acknowledged for support to A.R., J.M. and C.H. in frame of the SNF project no. 31003A_138114.
References
- 1.Holliger C, Hahn D, Harmsen H, Ludwig W, Schumacher W, Tindall B, Vazquez F, Weiss N, Zehnder AJ. 1998. Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration. Arch. Microbiol. 169, 313–321 [DOI] [PubMed] [Google Scholar]
- 2.Maillard J, Schumacher W, Vazquez F, Regeard C, Hagen WR, Holliger C. 2003. Characterization of the corrinoid iron-sulfur protein tetrachloroethene reductive dehalogenase of Dehalobacter restrictus. Appl. Environ. Microbiol. 69, 4628–4638 10.1128/AEM.69.8.4628-4638.2003 (doi:10.1128/AEM.69.8.4628-4638.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duret A, Holliger C, Maillard J. 2012. The opportunistic physiology of Desulfitobacterium hafniense strain TCE1 towards organohalide respiration with tetrachloroethene. Appl. Environ. Microbiol. 78, 6121–6127 10.1128/AEM.01221-12 (doi:10.1128/AEM.01221-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maillard J, Regeard C, Holliger C. 2005. Isolation and characterization of Tn-Dha1, a transposon containing the tetrachloroethene reductive dehalogenase of Desulfitobacterium hafniense strain TCE1. Environ. Microbiol. 7, 107–117 10.1111/j.1462-2920.2004.00671.x (doi:10.1111/j.1462-2920.2004.00671.x) [DOI] [PubMed] [Google Scholar]
- 5.Kruse T, et al. Submitted Complete genome sequence of Dehalobacter restrictus PER-K23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wild A, Hermann R, Leisinger T. 1996. Isolation of an anaerobic bacterium which reductively dechlorinates tetrachloroethene and trichloroethene. Biodegradation 7, 507–511 10.1007/BF00115297 (doi:10.1007/BF00115297) [DOI] [PubMed] [Google Scholar]
- 7.Sun B, Griffin BM, Ayala-del-Rio HL, Hashsham SA, Tiedje JM. 2002. Microbial dehalorespiration with 1,1,1-trichloroethane. Science 298, 1023–1025 10.1126/science.1074675 (doi:10.1126/science.1074675) [DOI] [PubMed] [Google Scholar]
- 8.van Doesburg W, van Eekert MH, Middeldorp PJ, Balk M, Schraa G, Stams AJ. 2005. Reductive dechlorination of beta-hexachlorocyclohexane (β-HCH) by a Dehalobacter species in coculture with a Sedimentibacter sp. FEMS Microbiol. Ecol. 54, 87–95 10.1016/j.femsec.2005.03.003 (doi:10.1016/j.femsec.2005.03.003) [DOI] [PubMed] [Google Scholar]
- 9.Maphosa F. 2010. Chasing organohalide respirers: ecogenomics approaches to assess the bioremediation capacity of soils. Wageningen, The Netherlands: Wageningen University [Google Scholar]
- 10.Maphosa F, van Passel MWJ, de Wos WM, Smidt H. 2012. Metagenome analysis reveals yet unexplored reductive dechlorinating potential of Dehalobacter sp. E1 growing in co-culture with Sedimentibacter sp. Environ. Microbiol. Rep. 4, 604–616 10.1111/j.758-2229.012.00376.x (doi:10.1111/j.758-2229.012.00376.x) [DOI] [PubMed] [Google Scholar]
- 11.Grostern A, Duhamel M, Dworatzek S, Edwards EA. 2010. Chloroform respiration to dichloromethane by a Dehalobacter population. Environ. Microbiol. 12, 1053–1060 10.1111/j.1462-2920.2009.02150.x (doi:10.1111/j.1462-2920.2009.02150.x) [DOI] [PubMed] [Google Scholar]
- 12.Grostern A, Edwards EA. 2009. Characterization of a Dehalobacter coculture that dechlorinates 1,2-dichloroethane to ethene and identification of the putative reductive dehalogenase gene. Appl. Environ. Microbiol. 75, 2684–2693 10.1128/AEM.02037-08 (doi:10.1128/AEM.02037-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson JL, Fung JM, Cadillo-Quiroz H, Cheng X, Zinder SH. 2011. A role for Dehalobacter spp. in the reductive dehalogenation of dichlorobenzenes and monochlorobenzene. Environ. Sci. Technol. 45, 6806–6813 10.1021/es200480k (doi:10.1021/es200480k) [DOI] [PubMed] [Google Scholar]
- 14.Yoshida N, Ye L, Baba D, Katayama A. 2009. A novel Dehalobacter species is involved in extensive 4,5,6,7-tetrachlorophthalide dechlorination. Appl. Environ. Microbiol. 75, 2400–2405 10.1128/AEM.02112-08 (doi:10.1128/AEM.02112-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Justicia-Leon SD, Ritalahti KM, Mack EE, Löffler FE. 2012. Dichloromethane fermentation by a Dehalobacter sp. in an enrichment culture derived from pristine river sediment. Appl. Environ. Microbiol. 78, 1288–1291 10.1128/AEM.07325-11 (doi:10.1128/AEM.07325-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee M, Low A, Zemb O, Koenig J, Michaelsen A, Manefield M. 2012. Complete chloroform dechlorination by organochlorine respiration and fermentation. Environ. Microbiol. 14, 883–894 10.1111/j.1462-2920.2011.02656.x (doi:10.1111/j.1462-2920.2011.02656.x) [DOI] [PubMed] [Google Scholar]
- 17.McMurdie PJ, et al. 2009. Localized plasticity in the streamlined genomes of vinyl chloride respiring Dehalococcoides. PLoS Genet. 5, 10. 10.1371/journal.pgen.1000714 (doi:10.1371/journal.pgen.1000714) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SH, Harzman C, Davis JK, Hutcheson R, Broderick JB, Marsh TL, Tiedje JM. 2012. Genome sequence of Desulfitobacterium hafniense DCB-2, a Gram-positive anaerobe capable of dehalogenation and metal reduction. BMC Microbiol. 12, 21. 10.1186/1471-2180-12-21 (doi:10.1186/1471-2180-12-21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nonaka H, et al. 2006. Complete genome sequence of the dehalorespiring bacterium Desulfitobacterium hafniense Y51 and comparison with Dehalococcoides ethenogenes 195. J. Bacteriol. 188, 2262–2274 10.1128/JB.188.6.2262-2274.2006 (doi:10.1128/JB.188.6.2262-2274.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siddaramappa S, et al. 2012. Complete genome sequence of Dehalogenimonas lykanthroporepellens type strain (BL-DC-9(T)) and comparison to ‘Dehalococcoides’ strains. Stand. Genomic Sci. 6, 251–264 10.4056/sigs.2806097 (doi:10.4056/sigs.2806097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prat L, Maillard J, Grimaud R, Holliger C. 2011. Physiological adaptation of Desulfitobacterium hafniense strain TCE1 to tetrachloroethene respiration. Appl. Environ. Microbiol. 77, 3853–3859 10.1128/AEM.02471-10 (doi:10.1128/AEM.02471-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng X, Yamamoto S, Vertes AA, Keresztes G, Inatomi K, Inui M, Yukawa H. 2012. Global transcriptome analysis of the tetrachloroethene-dechlorinating bacterium Desulfitobacterium hafniense Y51 in the presence of various electron donors and terminal electron acceptors. J. Ind. Microbiol. Biotechnol. 39, 255–268 10.1007/s10295-011-1023-7 (doi:10.1007/s10295-011-1023-7) [DOI] [PubMed] [Google Scholar]
- 23.Fung JM, Morris RM, Adrian L, Zinder SH. 2007. Expression of reductive dehalogenase genes in Dehalococcoides ethenogenes strain 195 growing on tetrachloroethene, trichloroethene, or 2,3-dichlorophenol. Appl. Environ. Microbiol. 73, 4439–4445 10.1128/AEM.00215-07 (doi:10.1128/AEM.00215-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson DR, Brodie EL, Hubbard AE, Andersen GL, Zinder SH, Alvarez-Cohen L. 2008. Temporal transcriptomic microarray analysis of ‘Dehalococcoides ethenogenes‘ strain 195 during the transition into stationary phase. Appl. Environ. Microbiol. 74, 2864–2872 10.1128/AEM.02208-07 (doi:10.1128/AEM.02208-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson DR, Nemir A, Andersen GL, Zinder SH, Alvarez-Cohen L. 2009. Transcriptomic microarray analysis of corrinoid responsive genes in Dehalococcoides ethenogenes strain 195. FEMS Microbiol. Lett. 294, 198–206 10.1111/j.1574-6968.2009.01569.x (doi:10.1111/j.1574-6968.2009.01569.x) [DOI] [PubMed] [Google Scholar]
- 26.Lee PK, Dill BD, Louie TS, Shah M, Verberkmoes NC, Andersen GL, Zinder SH, Alvarez-Cohen L. 2012. Global transcriptomic and proteomic responses of Dehalococcoides ethenogenes strain 195 to fixed nitrogen limitation. Appl. Environ. Microbiol. 78, 1424–1436 10.1128/AEM.06792-11 (doi:10.1128/AEM.06792-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris RM, Fung JM, Rahm BG, Zhang S, Freedman DL, Zinder SH, Richardson RE. 2007. Comparative proteomics of Dehalococcoides spp. reveals strain-specific peptides associated with activity. Appl. Environ. Microbiol. 73, 320–326 10.1128/AEM.02129-06 (doi:10.1128/AEM.02129-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris RM, Sowell S, Barofsky D, Zinder S, Richardson R. 2006. Transcription and mass-spectroscopic proteomic studies of electron transport oxidoreductases in Dehalococcoides ethenogenes. Environ. Microbiol. 8, 1499–1509 10.1111/j.1462-2920.2006.01090.x (doi:10.1111/j.1462-2920.2006.01090.x) [DOI] [PubMed] [Google Scholar]
- 29.Rahm BG, Morris RM, Richardson RE. 2006. Temporal expression of respiratory genes in an enrichment culture containing Dehalococcoides ethenogenes. Appl. Environ. Microbiol. 72, 5486–5491 10.1128/AEM.00855-06 (doi:10.1128/AEM.00855-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul S, Gish W, Miller W, Myers E, Lipman D. 1990. Basic alignment search tools. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 31.Larkin MA, et al. 2007. Clustal W and Clustal X, version 2.0. Bioinformatics 23, 2947–2948 10.1093/bioinformatics/btm404 (doi:10.1093/bioinformatics/btm404) [DOI] [PubMed] [Google Scholar]
- 32.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 10.1093/molbev/msr121 (doi:10.1093/molbev/msr121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prat L, Maillard J, Rohrbach-Brandt E, Holliger C. 2012. An unusual tandem-domain rhodanese harbouring two active sites identified in Desulfitobacterium hafniense. FEBS J. 279, 2754–2767 10.1111/j.1742-4658.2012.08660.x (doi:10.1111/j.1742-4658.2012.08660.x) [DOI] [PubMed] [Google Scholar]
- 34.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 (doi:10.1016/0003-2697(76)90527-3) [DOI] [PubMed] [Google Scholar]
- 35.Rappsilber J, Mann M, Ishihama Y. 2007. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protocols 2, 1896–1906 10.1038/nprot.2007.261 (doi:10.1038/nprot.2007.261) [DOI] [PubMed] [Google Scholar]
- 36.Lu J, Boeren S, de Vries SC, van Valenberg HJ, Vervoort J, Hettinga K. 2011. Filter-aided sample preparation with dimethyl labeling to identify and quantify milk fat globule membrane proteins. J. Proteomics 75, 34–43 10.1016/j.jprot.2011.07.031 (doi:10.1016/j.jprot.2011.07.031) [DOI] [PubMed] [Google Scholar]
- 37.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 10.1038/nbt.1511 (doi:10.1038/nbt.1511) [DOI] [PubMed] [Google Scholar]
- 38.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 10.1021/pr101065j (doi:10.1021/pr101065j) [DOI] [PubMed] [Google Scholar]
- 39.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 10.1093/bioinformatics/16.10.944 (doi:10.1093/bioinformatics/16.10.944) [DOI] [PubMed] [Google Scholar]
- 40.Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M. 2010. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J. Cell Biol. 189, 739–754 10.1083/jcb.200911091 (doi:10.1083/jcb.200911091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nesvizhskii AI. 2012. Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics 12, 1639–1655 10.1002/pmic.201100537 (doi:10.1002/pmic.201100537) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seshadri R, et al. 2005. Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes. Science 307, 105–108 10.1126/science.1102226 (doi:10.1126/science.1102226) [DOI] [PubMed] [Google Scholar]
- 43.Woodson JD, Escalante-Semerena JC. 2004. CbiZ, an amidohydrolase enzyme required for salvaging the coenzyme B12 precursor cobinamide in archaea. Proc. Natl Acad. Sci. USA 101, 3591–3596 10.1073/pnas.0305939101 (doi:10.1073/pnas.0305939101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovacs AT, Smits WK, Mironczuk AM, Kuipers OP. 2009. Ubiquitous late competence genes in Bacillus species indicate the presence of functional DNA uptake machineries. Environ. Microbiol. 11, 1911–1922 10.1111/j.1462-2920.2009.01937.x (doi:10.1111/j.1462-2920.2009.01937.x) [DOI] [PubMed] [Google Scholar]
- 45.Londono-Vallejo JA, Dubnau D. 1993. comF, a Bacillus subtilis late competence locus, encodes a protein similar to ATP-dependent RNA/DNA helicases. Mol. Microbiol. 9, 119–131 10.1111/j.1365-2958.1993.tb01674.x (doi:10.1111/j.1365-2958.1993.tb01674.x) [DOI] [PubMed] [Google Scholar]
- 46.Engelberg-Kulka H, Glaser G. 1999. Addiction modules and programmed cell death and antideath in bacterial cultures. Annu. Rev. Microbiol. 53, 43–70 10.1146/annurev.micro.53.1.43 (doi:10.1146/annurev.micro.53.1.43) [DOI] [PubMed] [Google Scholar]
- 47.Lioy VS, Pratto F, de la Hoz AB, Ayora S, Alonso JC. 2010. Plasmid pSM19035, a model to study stable maintenance in Firmicutes. Plasmid 64, 1–17 10.1016/j.plasmid.2010.04.002 (doi:10.1016/j.plasmid.2010.04.002) [DOI] [PubMed] [Google Scholar]
- 48.Gabor K, Hailesellasse Sene K, Smidt H, de Vos WM, van der Oost J. 2008. Divergent roles of CprK paralogues from Desulfitobacterium hafniense in activating gene expression. Microbiology 154, 3686–3696 10.1099/mic.0.2008/021584-0 (doi:10.1099/mic.0.2008/021584-0) [DOI] [PubMed] [Google Scholar]
- 49.Gabor K, Verissimo CS, Cyran BC, Ter Horst P, Meijer NP, Smidt H, de Vos WM, van der Oost J. 2006. Characterization of CprK1, a CRP/FNR-type transcriptional regulator of halorespiration from Desulfitobacterium hafniense. J. Bacteriol. 188, 2604–2613 10.1128/JB.188.7.2604-2613.2006 (doi:10.1128/JB.188.7.2604-2613.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupta N, Ragsdale SW. 2008. Dual roles of an essential cysteine residue in activity of a redox-regulated bacterial transcriptional activator. J. Biol. Chem. 283, 28 721–28 728 10.1074/jbc.M800630200 (doi:10.1074/jbc.M800630200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joyce MG, et al. 2006. CprK crystal structures reveal mechanism for transcriptional control of halorespiration. J. Biol. Chem. 281, 28 318–28 325 10.1074/jbc.M602654200 (doi:10.1074/jbc.M602654200) [DOI] [PubMed] [Google Scholar]
- 52.Levy C, Pike K, Heyes DJ, Joyce MG, Gabor K, Smidt H, van der Oost J, Leys D. 2008. Molecular basis of halorespiration control by CprK, a CRP-FNR type transcriptional regulator. Mol. Microbiol. 70, 151–167 10.1111/j.1365-2958.2008.06399.x (doi:10.1111/j.1365-2958.2008.06399.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazon H, Gabor K, Leys D, Heck AJ, van der Oost J, van den Heuvel RH. 2007. Transcriptional activation by CprK1 is regulated by protein structural changes induced by effector binding and redox state. J. Biol. Chem. 282, 11 281–11 290 10.1074/jbc.M611177200 (doi:10.1074/jbc.M611177200) [DOI] [PubMed] [Google Scholar]
- 54.Pop SM, Gupta N, Raza AS, Ragsdale SW. 2006. Transcriptional activation of dehalorespiration. Identification of redox-active cysteines regulating dimerization and DNA binding. J. Biol. Chem. 281, 26 382–26 390 10.1074/jbc.M602158200 (doi:10.1074/jbc.M602158200) [DOI] [PubMed] [Google Scholar]
- 55.Pop SM, Kolarik RJ, Ragsdale SW. 2004. Regulation of anaerobic dehalorespiration by the transcriptional activator CprK. J. Biol. Chem. 279, 49 910–49 918 10.1074/jbc.M409435200 (doi:10.1074/jbc.M409435200) [DOI] [PubMed] [Google Scholar]
- 56.Smidt H, van Leest M, van der Oost J, de Vos WM. 2000. Transcriptional regulation of the cpr gene cluster in ortho-chlorophenol-respiring Desulfitobacterium dehalogenans. J. Bacteriol. 182, 5683–5691 10.1128/JB.182.20.5683-5691.2000 (doi:10.1128/JB.182.20.5683-5691.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marzorati M, et al. 2007. A novel reductive dehalogenase, identified in a contaminated groundwater enrichment culture and in Desulfitobacterium dichloroeliminans strain DCA1, is linked to dehalogenation of 1,2-dichloroethane. Appl. Environ. Microbiol. 73, 2990–2999 10.1128/AEM.02748-06 (doi:10.1128/AEM.02748-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bisaillon A, Beaudet R, Lepine F, Villemur R. 2011. Quantitative analysis of the relative transcript levels of four chlorophenol reductive dehalogenase genes in Desulfitobacterium hafniense PCP-1 exposed to chlorophenols. Appl. Environ. Microbiol. 77, 6261–6264 10.1128/AEM.00390-11 (doi:10.1128/AEM.00390-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roessner CA, Scott AI. 2006. Fine-tuning our knowledge of the anaerobic route to cobalamin (Vitamin B12). J. Bacteriol. 188, 7331–7334 10.1128/JB.00918-06 (doi:10.1128/JB.00918-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gray MJ, Escalante-Semerena JC. 2009. In vivo analysis of cobinamide salvaging in Rhodobacter sphaeroides strain 2.4.1. J. Bacteriol. 191, 3842–3851 10.1128/JB.00230-09 (doi:10.1128/JB.00230-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woodson JD, Zayas CL, Escalante-Semerena JC. 2003. A new pathway for salvaging the coenzyme B12 precursor cobinamide in archaea requires cobinamide-phosphate synthase (CbiB) enzyme activity. J. Bacteriol. 185, 7193–7201 10.1128/JB.185.24.7193-7201.2003 (doi:10.1128/JB.185.24.7193-7201.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zayas CL, Escalante-Semerena JC. 2007. Reassessment of the late steps of coenzyme B12 synthesis in Salmonella enterica: evidence that dephosphorylation of adenosylcobalamin-5′-phosphate by the CobC phosphatase is the last step of the pathway. J. Bacteriol. 189, 2210–2218 10.1128/JB.01665-06 (doi:10.1128/JB.01665-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holliger C, Schraa G, Stams AJ, Zehnder AJ. 1993. A highly purified enrichment culture couples the reductive dechlorination of tetrachloroethene to growth. Appl. Environ. Microbiol. 59, 2991–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kreher S, Schilhabel A, Diekert G. 2008. Enzymes involved in the anoxic utilization of phenyl methyl ethers by Desulfitobacterium hafniense DCB2 and Desulfitobacterium hafniense PCE-S. Arch. Microbiol. 190, 489–495 10.1007/s00203-008-0400-8 (doi:10.1007/s00203-008-0400-8) [DOI] [PubMed] [Google Scholar]
- 65.Ahsanul Islam M, Edwards EA, Mahadevan R. 2010. Characterizing the metabolism of Dehalococcoides with a constraint-based model. PLoS Comput. Biol. 6, e1000887. 10.1371/journal.pcbi.1000887 (doi:10.1371/journal.pcbi.1000887) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marco-Urrea E, Seifert J, von Bergen M, Adrian L. 2012. Stable isotope peptide mass spectrometry to decipher amino acid metabolism in Dehalococcoides strain CBDB1. J. Bacteriol. 194, 4169–4177 10.1128/JB.00049-12 (doi:10.1128/JB.00049-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang YJ, Yi S, Zhuang WQ, Zinder SH, Keasling JD, Alvarez-Cohen L. 2009. Investigation of carbon metabolism in ‘Dehalococcoides ethenogenes’ strain 195 by use of isotopomer and transcriptomic analyses. J. Bacteriol. 191, 5224–5231 10.1128/JB.00085-09 (doi:10.1128/JB.00085-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moparthi VK, Hagerhall C. 2011. The evolution of respiratory chain complex I from a smaller last common ancestor consisting of 11 protein subunits. J. Mol. Evol. 72, 484–497 10.1007/s00239-011-9447-2 (doi:10.1007/s00239-011-9447-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smidt H, Song D, van Der Oost J, de Vos WM. 1999. Random transposition by Tn916 in Desulfitobacterium dehalogenans allows for isolation and characterization of halorespiration-deficient mutants. J. Bacteriol. 181, 6882–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Claverys JP, Prudhomme M, Martin B. 2006. Induction of competence regulons as a general response to stress in Gram-positive bacteria. Annu. Rev. Microbiol. 60, 451–475 10.1146/annurev.micro.60.080805.142139 (doi:10.1146/annurev.micro.60.080805.142139) [DOI] [PubMed] [Google Scholar]
- 71.Krasotkina J, Walters T, Maruya KA, Ragsdale SW. 2001. Characterization of the B12- and iron-sulfur-containing reductive dehalogenase from Desulfitobacterium chlororespirans. J. Biol. Chem. 276, 40 991–40 997 10.1074/jbc.M106217200 (doi:10.1074/jbc.M106217200) [DOI] [PubMed] [Google Scholar]
- 72.van de Pas BA, Smidt H, Hagen WR, van der Oost J, Schraa G, Stams AJ, de Vos WM. 1999. Purification and molecular characterization of ortho-chlorophenol reductive dehalogenase, a key enzyme of halorespiration in Desulfitobacterium dehalogenans. J. Biol. Chem. 274, 20 287–20 292 10.1074/jbc.274.29.20287 (doi:10.1074/jbc.274.29.20287) [DOI] [PubMed] [Google Scholar]
- 73.Miller E, Wohlfarth G, Diekert G. 1997. Comparative studies on tetrachloroethene reductive dechlorination mediated by Desulfitobacterium sp. strain PCE-S. Arch. Microbiol. 168, 513–519 10.1007/s002030050529 (doi:10.1007/s002030050529) [DOI] [PubMed] [Google Scholar]
- 74.Suyama A, Yamashita M, Yoshino S, Furukawa K. 2002. Molecular characterization of the PceA reductive dehalogenase of Desulfitobacterium sp. strain Y51. J. Bacteriol. 184, 3419–3425 10.1128/JB.184.13.3419-3425.2002 (doi:10.1128/JB.184.13.3419-3425.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thibodeau J, Gauthier A, Duguay M, Villemur R, Lepine F, Juteau P, Beaudet R. 2004. Purification, cloning, and sequencing of a 3,5-dichlorophenol reductive dehalogenase from Desulfitobacterium frappieri PCP-1. Appl. Environ. Microbiol. 70, 4532–4537 10.1128/AEM.70.8.4532-4537.2004 (doi:10.1128/AEM.70.8.4532-4537.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stackebrandt E, Sproer C, Rainey FA, Burghardt J, Pauker O, Hippe H. 1997. Phylogenetic analysis of the genus Desulfotomaculum: evidence for the misclassification of Desulfotomaculum guttoideum and description of Desulfotomaculum orientis as Desulfosporosinus orientis gen. nov., comb. nov. Int. J. Syst. Bacteriol. 47, 1134–1139 10.1099/00207713-47-4-1134 (doi:10.1099/00207713-47-4-1134) [DOI] [PubMed] [Google Scholar]
- 77.Wagner A, Adrian L, Kleinsteuber S, Andreesen JR, Lechner U. 2009. Transcription analysis of genes encoding homologues of reductive dehalogenases in ‘Dehalococcoides’ sp. strain CBDB1 by using terminal restriction fragment length polymorphism and quantitative PCR. Appl. Environ. Microbiol. 75, 1876–1884 10.1128/AEM.01042-08 (doi:10.1128/AEM.01042-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.West KA, et al. 2008. Comparative genomics of ‘Dehalococcoides ethenogenes’ 195 and an enrichment culture containing unsequenced ‘Dehalococcoides’ strains. Appl. Environ. Microbiol. 74, 3533–3540 10.1128/AEM.01835-07 (doi:10.1128/AEM.01835-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kube M, Beck A, Zinder SH, Kuhl H, Reinhardt R, Adrian L. 2005. Genome sequence of the chlorinated compound-respiring bacterium Dehalococcoides species strain CBDB1. Nat. Biotechnol. 23, 1269–1273 10.1038/nbt1131 (doi:10.1038/nbt1131) [DOI] [PubMed] [Google Scholar]
- 80.Bolhuis A, et al. 1999. Signal peptide peptidase- and ClpP-like proteins of Bacillus subtilis required for efficient translocation and processing of secretory proteins. J. Biol. Chem. 274, 24 585–24 592 10.1074/jbc.274.35.24585 (doi:10.1074/jbc.274.35.24585) [DOI] [PubMed] [Google Scholar]
- 81.Wang P, Shim E, Cravatt B, Jacobsen R, Schoeniger J, Kim AC, Paetzel M, Dalbey RE. 2008. Escherichia coli signal peptide peptidase A is a serine-lysine protease with a lysine recruited to the nonconserved amino-terminal domain in the S49 protease family. Biochemistry 47, 6361–6369 10.1021/bi800657p (doi:10.1021/bi800657p) [DOI] [PMC free article] [PubMed] [Google Scholar]