

Abstract
The genome sequence of psychrophilic Shewanella sediminis revealed the presence of five putative reductive dehalogenases (Rdhs). We found that cell extracts of pyruvate/fumarate-grown S. sediminis cells catalysed reduced methyl viologen-dependent reductive dechlorination of tetrachloroethene (PCE) to trichloroethene (TCE) at a specific activity of approximately 1 nmol TCE min−1 (mg protein)−1. Dechlorination of PCE followed Michaelis–Menten kinetics with an apparent Km of 120 μM PCE. No PCE dechlorination was observed with heat-denatured extract or when cyanocobalamin was omitted from the growth medium; however, the presence of PCE in the growth medium increased PCE transformation rates. Analysis of mutants carrying in-frame deletions of all five Rdhs encoding genes showed that only deletion of Ssed_3769 resulted in the loss of PCE dechlorination activity suggesting that Ssed_3769 is a functional Rdh. This is the first study to show reductive dechlorination activity of PCE in a sediment-dwelling Shewanella species that may be important for linking the flux of organohalogens to organic carbon via reductive dehalogenation in marine sediments.
Keywords: Shewanella, tetrachloroethene, trichloroethene, reductive dehalogenase, marine sediment
1. Introduction
Microbial reductive dehalogenation is a biochemically and metabolically intriguing activity found as co-metabolic process in a variety of micro-organisms such as methanogens or homoacetogens and more importantly as a catabolic process in phylogenetically diverse groups of bacteria including Desulfitobacterium, Sulfurospirillum, Desulfomonile, Desulfovibrio, Anaeromyxobacter, Desulfuromonas, Dehalobacter, Dehalogenimonas and Dehalococcoides [1,2]. The latter two genera have been of special interest as their catabolism is restricted to organohalide respiration [3,4]. Most of the so far isolated organohalide-respiring bacteria have been isolated based on their catabolic activity to dechlorinate anthropogenic chloroaliphatic or chloroaromatic compounds [5,6]. In all cases, the material source for the isolation of microbes was derived from terrestrial environments, such as chloroethene-contaminated aquifers, rivers or soil sediments as well as sewage sludge [2,7]. However, putative reductive dehalogenase (Rdh) genes (rdh) have been also found in uncontaminated sediments [8], including marine deep sea sediments [9]. These observations, in conjunction with the discovery of site-specific mobilization of rdh genes as an important mode for migration of these genes within Dehalococcoides populations [10], raise the question about the role of rdh genes and Rdh activities in these environments, especially in marine deep sea sediments, as well as their evolutionary history.
Reductive dehalogenation activity is associated with a monomeric RdhA enzyme encoded by rdhA. RdhA contains two iron–sulfur clusters, and many studies have shown the presence of a corrinoid cofactor involved in its activity. It is speculated that RdhA is associated in vivo to the outside of the cyptoplasmic membrane via a putative small integrated membrane protein RdhB [11–13]. Reductive dechlorination of PCE and other chloroethenes requires in vitro a low-redox potential electron donor such as reduced methyl viologen. The in vivo electron donor is typically molecular hydrogen for most of the catabolic organohalide-respiring bacteria, especially Dehalococcoides [13]. The cellular electron transfer pathway from hydrogen via membrane-bound hydrogenase(s) to the Rdh is unknown. In this overall catabolic process, the exergonic oxidation of hydrogen with organohalogens is coupled to energy conservation most likely via a chemiosmotic mechanism [12,14].
Analysis of rdh genes from many different micro-organisms revealed common and unique characteristics [15,16]. Both rdh genes are often linked, and experimental evidence has shown that, if tested, they are co-transcribed [13]. Whole genome sequence analyses of several Dehalococcoides mccartyi strains revealed that some genomes can carry as many as 36 (strain VS) full-length, non-identical Rdh homologous genes [4,16,17]. Despite the presence of this unusually high number of Rdhs in some organohalide-respiring bacteria, only a few Rdhs have been characterized biochemically. These biochemical studies collectively have shown that each Rdh seems to be substrate-specific and structurally related halogenated compounds have been observed to be transformed at rates that are orders of magnitudes lower than the primary halogenated substrate [13,18,19].
Interestingly, the genome sequence of the marine sediment-dwelling Shewanella species strain HAW-EB3, named Shewanella sediminis (S. sediminis) [20], revealed the presence of five Rdh homologue genes. Bacteria of this genus are known for their versatile electron-accepting capabilities using a complex electron transfer network composed mainly of c-type cytochromes as well as iron–sulfur proteins [21]. The goal of this study was to gain insights into the function of the putative Rdhs in S. sediminis and to shed light on the function and evolution of these genes in marine sediment environments.
2. Material and methods
(a). Growth conditions and media (growth curves)
All strains and plasmids used in this study are described in table 1. Escherichia coli strains were grown in Luria–Bertani (LB) broth medium at 37°C and S. sedimins strains were grown at 10°C in LB with addition of 1% (wt/vol) NaCl or minimal media (4 M) with the following composition (per litre): 1 mM CaCl2 · 2H2O, 5 μM CoCl2, 0.2 μM CuSO4 · 5H2O, 5.4 μM FeCl2 · 2H2O, 57 μM H3BO3, 5.7 mM K2HPO4, 3.3 mM KH2PO4, 1.0 mM MgSO4 · 7H2O, 1.3 μM MnSO4, 67.2 μM Na2EDTA, 3.9 μM Na2MoO4 · 2H2O, 1.5 μM Na2SeO4, 342 mM NaCl, 2 mM NaHCO3, 5 μM NiCl2 · 6H2O, 1 μM ZnSO4 and 9 mM (NH4)2SO4 and 0.1% (wt/vol) casamino acids, pH 7.4). After autoclaving and before inoculation, a filter-sterilized vitamin solution was added to the medium to reach final concentrations of: 4.5 nM folic acid, 13.5 nM riboflavin, 24 nM dl-6,8-thioctic acid, 41 nM biotin, 365 nM 4-aminobenzoic acid, 46 nM pantothenate, 1.5 μM pyridoxamine, 812 nM nicotinic acid, 66 nM thiamine, 18 nM cyanocobalamin. Where necessary, medium was solidified by 1.5% (wt/vol) agar and supplemented with 10 μg ml−1 gentamycin or 10 μg ml−1 chloramphenicol. For growth under anoxic conditions, 30 mM fumarate was added as terminal electron acceptor and 40 mM pyruvate as electron donor. Anaerobic cultures were either prepared in 150 ml serum vials or 1 l Schott bottles sealed with butyl rubber stoppers. Oxygen was removed from the medium by repeatedly flushing the headspace of each vial for 1 min with nitrogen (99.9% purity; Praxair, Santa Clara, CA) followed by a 1 min application of vacuum for at least 20 cycles. Alternatively, the medium was autoclaved and subsequently cooled down under a nitrogen stream while rigorous stirring. Mineral medium was inoculated (1% inoculum) to a starting OD of 0.02 from S. sediminis stationary phase LB + 1% NaCl cultures.
Table 1.
Strains and plasmids used in this study.
| strain | relevant genotype and phenotype | reference |
|---|---|---|
| Escherichia coli | ||
| DH5-λpir | host used for mating with MR-1. ϕ80dlacZΔM15 Δ(lacZYA-argF)U196 recA1 hsdR17 deoR thi-1 supE44 gyrA96 relA1/λpir | [22] |
| WM3064-λpir | host used for mating with MR-1. thrB1004 pro thi rpsL hsdS lacZΔM15 RP4-1360 Δ(araBAD) 567ΔdapA 1341::[erm pir(wt)] | [23] |
| S17-λpir | host used for mating with MR-1. thi pro recA hsdR [RP4-2Tc::Mu-Km::Tn7]λpir Tpr Smr | [24] |
| Shewanella sediminis | ||
| AS1028 | Shewanella sediminis strain HAW-EB3, wild-type (WT) | [20] |
| AS1029 | in-frame deletion of Ssed_4120 in AS (WT), ΔSsed_4120 | this study |
| AS1030 | in-frame deletion of Ssed_3769 in AS (WT), ΔSsed_3769 | this study |
| AS1031 | in-frame deletion of Ssed_2100 in AS (WT), ΔSsed_2100 | this study |
| AS1032 | in-frame deletion of Ssed_2103 in AS (WT), ΔSsed_2103 | this study |
| AS1033 | in-frame deletion of Ssed_1729 in AS (WT), ΔSsed_1729 | this study |
| AS1034 | AS1030 complemented with Ssed_3769 by knock-in, Ssed_3769+ | this study |
| plasmid | description | reference |
| pDS3.0 | suicide plasmid for constructing in-frame deletions; Gmr | [25] |
| pDS132 | suicide plasmid for constructing in-frame deletions; Cmr | [26] |
| pDS132_Δssed_4120 | Ssed_4120 in-frame deletion fragment in pDS132; Cmr | this study |
| pDS3.0_ΔSsed_3769 | Ssed_3769 in-frame deletion fragment in pDS3.0; Gmr | this study |
| pDS3.0_ΔSsed_2100 | Ssed_2100 in-frame deletion fragment in pDS3.0; Gmr | this study |
| pDS3.0_ΔSsed_2103 | Ssed_2103 in-frame deletion fragment in pDS3.0; Gmr | this study |
| pDS3.0_ΔSsed_1729 | Ssed_1729 in-frame deletion fragment in pDS3.0; Gmr | this study |
| pDS132_Ssed_3769 | Plasmid for replacing WT Ssed_3769 allele into ΔSsed_3769; Cmr | this study |
(b). Mutant construction and cloning in Shewanella sediminis
All genetic work was carried out according to standard protocols. Kits for isolation and/or purification of DNA were obtained from Promega (Madison, WI, USA), and enzymes were purchased from New England Biolabs (NEB, Ipswich, MA, USA). Ssed_1729, Ssed_2100, Ssed_2103, Ssed_3769 and Ssed_4120 knockout mutants were constructed via homologous recombination, resulting in a mutant lacking the entire open reading frame except for the start and stop codon. Briefly, approximately 750 bp upstream and downstream fragments of the target gene were PCR-amplified from wild-type (AS1028) genomic DNA and subsequently joined via a complementary tag that was added to the 5′-end of each inner primer (table 2). The fusion products of Ssed_1729, Ssed_2100, Ssed_2103 and Ssed_3769 were ligated into pDS3.0 via the SmaI restriction site, whereas for Ssed_4120, the fused fragment was ligated into the SacI site of pDS132 after digestion of an introduced SacI restriction site. Escherichia coli DH5α-λpir or E. coli S17-λpir strains were transformed with the ligation mixture and plated on LB containing 10 μg ml−1 gentamycin (pDS3.0) or 10 μg ml−1 chloramphenicol (pDS132). The resulting plasmids pDS132_ΔSsed_4120, pDS3.0_ΔSsed_3769, pDS3.0_ΔSsed_2100, pDS3.0_ΔSsed_2103 and pDS3.0_ΔSsed_1729 (table 1) were verified by sequencing and transformed into the WT (AS1028) through bi-parental mating using E. coli WM3064 as conjugal donor. After 24 h incubation at room temperature, the mating mix was resuspended in LB + 1%NaCl and subsequently plated on LB + 1%NaCl agar plates containing 10 μg ml−1 gentamycin or 10 μg ml−1 chloramphenicol. Colonies were screened for single crossover events using PCR primers flanking the recombination region. Resolution of the integrated vector by a second crossover event was performed with a confirmed first crossover strain. This strain was grown in LB + 1%NaCl medium without selection and plated onto solid LB + 1%NaCl medium containing 10 per cent sucrose. Deletion events were verified by PCR using primer rdhX-F and primer rdhX-R and DNA sequencing of the resolved mutant strain.
Table 2.
Sequences of primers used in this study.
| primer name | primer sequence |
|---|---|
| knockout primers | |
| rdh1_5o_SacI | GACTACTTGCGAGCTCGATGAGCAGAAAGTCAGTTATGG |
| rdh1_5i | TGAAGTTCATGTCACGGATCTCATAATGCTTAATTCTCTTCA |
| rdh1_3o_SacI | GACTACTTGCGAGCTCAAAGTACGGCATGAACTAAAT |
| rdh1_3i | AGATCCGTGACATGAACTTCATAACATATAGGAGAAAGAGTG |
| rdh1_Fo | GCAGCTTGCATACATTTACAT |
| rdh1_Ro | AAGCGAAGTTGAAGCACCTCT |
| rdh2_5o | TTACTGCAAACTATCGACAGC |
| rdh2_5i | AGATAGCTCCTTCAGATCCTTCATAGGGTTACCTTAATTAAC |
| rdh2_3o | GAGCCCCTTGGTGCAAGGCC |
| rdh2_3i | AAGGATCTGAAGGAGCTATCTTAGCAGGCAATGTCGGAGGCG |
| rdh2_Fo | TGCAGCACAGTCTGCAACTTAT |
| rdh2_Ro | AGGGCGACCATCAGGGCTTCA |
| rdh3_5o | TATAAACTTGACCTGTAACGA |
| rdh3_5i | CGTCATGGGTTTACAGCCGTTCATTTTCACCCTTATTACGTT |
| rdh3_3o | ATCGTGCAATTCAGCTTGGTT |
| rdh3_3i | AACGGCTGTAAACCCATGACGTAAATCTTTCTCTCCTTGCTC |
| rdh3_Fo | CTCACCGATGCAGGAATAGAG |
| rdh3_Ro | AGTTTAGTTGTTAACGTTTTC |
| rdh4_5o | TAAATGCTTTACAGAATGTAC |
| rdh4_5i | AGCATACTAGCTGTCATCACTCATTGATTACCTCTGACTATT |
| rdh4_3o | GGATCCCCATTGTCGATGATG |
| rdh4_3i | AGTGATGACAGCTAGTATGCTTAACGCAATATCCCCCGGTGT |
| rdh4_Fo | TAGCCTTCTATAACGCCATCA |
| rdh4_Ro | GGGACAGAGAGTATTGCAATA |
| rdh5_5o | CATTATTATCAGTCAGGGTAT |
| rdh5_5i | AGTAATCACTCGTTCCCAGTTCATACTTATCCCTACATCTTT |
| rdh5_3o | CCAAGACCTGCTTTAGCAGGC |
| rdh5_3i | AACTGGGAACGAGTGATTACTTGATCTCGTAAGATTGCGGCT |
| rdh5_Fo | GACGTTATAGCCCCAGTTTGC |
| rdh5_Ro | GCCTTGGACGATAACGCTTCG |
| qPCR primers | |
| rdh1_F | GCAAGCCATCTTACCCATGT |
| rdh1_R | CCGTGTCGGTATCGCTAAAT |
| rdh2_F | GTTCCAGCGTGGATCGTTAT |
| rdh2_R | CCCGGTTACCAGCAGATAGA |
| rdh3_F | GTCCCAGCGTTTATCACGTT |
| rdh3_R | TTGCTGGCATGGAATGAATA |
| rdh4_F | CGCGGTTGGTTATTCAACTT |
| rdh4_R | CACTTCTGGCGTCATCAAGA |
| rdh5_F | GGGTCCGATTAAGCCGGGCG |
| rdh5_R | CATCGGCGCCATCCGGTTCA |
| gyrA_F | CACGCAGCCCACTGATGCCT |
| gyrA_R | GTGGCCGAGCTGTCATGCGT |
To complement the mutant of Ssed_3769 (AS1030), the wild-type gene was reintroduced into the Ssed_3769 locus by gene replacement. This was carried out similar to the earlier mentioned method, except that the Ssed_3769 wild-type gene, and its flanking regions were cloned into pDS132 resulting in pDS132_ Ssed_3769. The mating was performed using E. coli WM3064-λpir and the ΔSsed_3769 (AS1030) strain instead of the wild-type strain of S. sediminis.
(c). RNA purification and cDNA synthesis
Total RNA was isolated from triplicate samples using the Trizol reagent protocol according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). RNA samples were treated with DNase I amplification grade (Invitrogen) according to the manufacturer's instructions to remove genomic DNA, with subsequent purification performed with an RNeasy minikit (Qiagen, Valencia, CA, USA). Electrophoretic analysis was performed with an Agilent 2100 bioanalyzer (Agilent Technologies Inc., Palo Alto, CA, USA), to assess RNA integrity. Absence of PCR amplification of a genomic region of 100 bp using primers and Phire polymerase (NEB) was determined for verification of the lack of genomic contamination. cDNA synthesis from total RNA was carried out using SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Reverse transcription control reactions were performed with and without reverse transcriptase enzyme.
(d). Quantitative PCR
Quantitative PCR (qPCR) was performed with the IQ SYBR green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) and iCycler iQ RT-PCR detection system (Bio-Rad Laboratories), using equal amounts of cDNA (added in 10 μl volume to a 25 μl reaction) for amplification of each gene region (ca 200 bp). Triplicate reactions were run for each sample with the following programme: 95.0°C for 3 min, 50 cycles of 95.0°C for 30 s, 59.0°C for 30 s, and 72.0°C, and a melt curve to determine primer specificity: 95.0 for 1 min, 55.0°C for 1 min, and 80 cycles of a stepwise increase by 0.5°C starting at 55.0°C for 10 s. Each RT-PCR was performed in triplicate based on three independent RNA extractions. A 200 bp fragment from the gyrA gene was amplified and used for derivation of the standard curve. Expression ratios are given as the log2-fold difference in the quantity of product from the experimental sample (WT+PCE) versus that from the control sample (WT−PCE). Data analysis was performed as previously described [27], and normalization of all expression ratios was conducted using gyrA.
(e). PCR amplification of putative rdh targets from DNA and cDNA
Specific primers were used to amplify an approximately 200 bp fragment of Ssed_4120, SSed_3768, Ssed_2100, Ssed_2103 and Ssed_1729 from S. sediminis. The PCR was performed with Dreamtaq Mastermix (Fermentas, Glen Burnie, MD, USA) using the following conditions: an initial incubation at 95°C for 5 min, followed by 30 cycles of 30 s at 95°C, 45 s at 59°C, 10 s at 72°C and a final extension of 10 min at 72°C. Genomic DNA extracted from S. sediminis served as positive control.
(f). Cell suspension assays
Shewanella sediminis wild-type was grown anaerobically in 30 mM fumarate and 40 mM pyruvate 4 M mineral medium as described earlier and harvested in early stationary phase (approx. 72 h). Cells were washed in phosphate buffer (5.7 mM K2HPO4, 3.3 mM KH2PO4, 2 mM NaHCO3, 342 mM NaCl, pH 7.4) and resuspended in 100 mM Tris–HCl (pH 8) to reach an OD of approximately 1. Pyruvate (2.5 mM), hydrogen (0.6 bar) and reduced methyl viologen (2.5 mM) were tested as electron donors for dechlorination of PCE (2.5 mM were added to reach a saturated solution). In additional vials, fumarate (2.5 mM) was added as competitive electron acceptor. Headspace volume (500 μl) was withdrawn at different time points to monitor TCE formation via gas chromatography. Each cell suspension experiment was carried out twice with biological duplicates each time.
(g). Preparation of cell extracts
Preparation of cell extracts was performed in an anaerobic glovebox (Coy Laboratory Products, Inc., MI, USA) supplied with a gas mix of nitrogen and hydrogen (95 : 5). Anaerobically grown S. sediminis cells were harvested in early stationary phase (ca 72 h) by centrifugation for 10 min at 5000g at 4°C. The supernatant was removed, and the cell pellet was washed with phosphate buffer (5.7 mM K2HPO4, 3.3 mM KH2PO4, 2 mM NaHCO3, 342 mM NaCl, pH 7.4) before being resuspended in 0.5 or 1 ml of anaerobic resuspension buffer (100 mM Tris–HCl, pH 8). Cells were lysed by bead beating on a vortex 10 times for 3 s with 30 s incubation periods on ice in between, and the lysis mixture was clarified by centrifugation at 122 500g for 50 min at 4°C. The supernatant was designated as cell-free extract and stored under nitrogen at −80°C in a butyl rubber stoppered serum vial until used.
(h). Reductive dechlorination assays
All cell extract assays were carried out in an anaerobic glove box with a N2–H2 (95 : 5) atmosphere using anoxic stock solutions. Dehalogenation assays were performed in 5 ml vials sealed with polytetrafluoroethylene Mininert valves. The standard assay mixture contained in a total volume of 1 ml, 100 mM Tris HCl, pH 8, 5 mM Ti(III)–NTA, 2.5 mM methyl viologen and 2.5 mM PCE were added to reach saturation concentrations. Ti(III)–NTA solution was prepared according to Moench & Zeikus [28]. The reaction was started by adding 100 μl cell extract corresponding to 300–500 μg protein. The reaction vials were incubated at room temperature in the glove box and stirred with a magnetic stirrer. Chlorinated compounds were monitored over time by taking 500 μl headspace samples with a gas-tight syringe for analysis via gas chromatography. All data represent the mean of at least two independent experiments with biological duplicates.
(i). Analytical methods
Analysis of chloroethenes was from a 500 μl headspace sample using a gas chromatograph from Hewlett–Packard (series II 5890) equipped with flame ionization detector (FID). Separation was accomplished in a DB-624 capillary column (30 m length, 0.530 μM ID, 3 μM film thickness). Helium was used as carrier gas at a flow rate of 5.1 ml min−1. The injector and detector temperatures were 200°C and 280°C (FID), respectively, and analyses were isothermal at 100°C. Compounds were identified by comparison of their retention times with the retention times of external standards. Quantification of PCE and TCE was based on 16-point external calibration curves. Standards were prepared by adding appropriate ethanol stock solutions to sterilized assay buffer in vials having the same headspace : liquid ratio as the vials used in the assays.
Protein concentrations in the cell extract were determined with the Bradford assay using the Bio-Rad reagent (Bio-Rad Laboratories). Bovine serum albumin was used as the standard.
(j). Sequence analysis
Rdh gene sequences were obtained from GenBank (http://www.ncbi.nlm.nih.gov). All sequence analysis including translation of nucleotide sequences, alignments of amino acid sequences (using the ClustalW algorithm, default settings) and generation of neighbour-joining phylogenetic trees (Jukes–Cantor model, default settings) was performed with the software Geneious Pro v. 5.6.3.
3. Results
(a). Evolutionary relationship of reductive dehalogenases in Shewanella sediminis
Five putative Rdhs genes, Ssed_1729, Ssed_2100, Ssed_2103, Ssed_3769 and Ssed_4120 were identified in the genome of Shewanella sediminis, a cold temperature-adapted γ-proteobacterium isolated from marine sediments near Halifax Harbour (NCIMB 14036T, DSM 17055T, GenBank accession no. CP000821; [20]). The predicted amino acid sequences encoded by these genes were aligned with each other as well as with several putative dehalogenases identified in genomes of organohalide-respiring micro-organisms and compared. Although the predicted amino acid sequence identity among the five putative S. sediminis Rdhs (except for Ssed_2103) varied between 36 per cent and 47 per cent, identities to characterized Rdhs from Dehalococcoides sp., Sulfurospirillum multivorans and Desulfitobacterium were 12.5–17.5%, 19.5–22.5% and 23.5–26.5%, respectively (figure 1a). Similar to characterized Rdhs, the encoded amino acid sequences of Ssed_1729, Ssed_2100, Ssed_2103, Ssed_3769 and Ssed_4120 contained two iron sulfur binding motifs in the C-terminal region (figure 1b). Both motifs are equivalent to the conserved ferredoxin-type 4Fe4S consensus sequence characteristic for bacterial ferredoxins [29], with the first one being CX2CX2CX3XP, whereas the second one exhibits a region of 10 residues between the first two cysteines (Ssed_1729, Ssed_2100, Ssed_3769, Ssed_4120Ssed) and 14 for Ssed_2103. The presence of an N-terminal twin-arginine signal peptide sequence as previously described in Rdhs was also observed in all predicted S. sediminis Rdhs, indicating that the mature enzymes are most likely periplasmatic and translocated as assembled holoenzymes from the cytoplasm via the TAT pathway [30,31]. A conserved amino acid sequence motif for corrinoid cofactor binding as previously derived from other corrinoid-dependent enzymes (DXHX2G) could not be identified [32]. Other highly conserved amino acid residues include proline, histidine and tyrosine (position 480, 579 and 608 of the consensus sequence), whose functions are unknown so far [33,34]. Another feature common to all S. sediminis putative Rdhs, with the exception of Ssed_1729, is the location of a second open reading frame (putative rdhB) 0–15 bp downstream of the rdhA gene. Collectively, this sequence analysis strongly suggests that the identified putative Rdhs in S. sediminis represent true homologues of previously biochemically characterized Rdhs.
Figure 1.

(a) Neighbour-Joining consensus tree of the deduced amino acid sequence from S. sediminis and a selection of organohalide-respiring bacteria. RdhA are labelled with the following abbreviations: Dhc, Dehalococcoides; Desulfitob, Desulfitobacterium; Sulfurosp, Sulfurospirillum and Ssed, Shewanella. (b) Multiple alignment of the C-terminal region of deduced amino acid sequences of reductive dehalogenase genes from S. sediminis and a range of known organohalide-respiring organisms. Consensus motifs for the iron sulfur clusters are highlighted in grey, other shared residues are marked with boxes.
(b). Reductive tetrachloroethene dechlorination activity in whole cells and cell-free extract of Shewanella sediminis
Most of the putative Rdhs in S. sediminis (Ssed_1729, Ssed_2100, Ssed_3769 and Ssed_4120) were annotated as putative tetrachloroethene (PCE) Rdhs, which motivated us to initiate experiments in cell suspensions of wild-type cells to test for PCE dechlorination activity. Pyruvate, hydrogen or reduced methyl viologen was used as low-redox potential electron donors in these experiments. When reduced methyl viologen was provided as electron donor, the formation of TCE was observed that increased linearly over time, whereas neither hydrogen nor pyruvate could serve as electron donor for PCE dechlorination in whole cell suspensions. When fumarate was added in addition to PCE as competitive electron acceptor, no significant TCE was produced as the electrons from reduced methyl viologen were apparently directed towards fumarate reduction to succinate (data not shown).
In a subsequent batch culture screen, we tested whether anaerobically grown cells (10 mM fumarate, 10 mM pyruvate, 0.1% casamino acids) carry reductive dehalogenation activity towards organohalogen compounds other than PCE. Mineral medium batch cultures of S. sediminis (4 M) were amended with the halogenated compounds tetrachloroethene, trichloroethene, cis-dichloroethene, 1,1-dichloroethene, 1,1,2-trichloroethane, 1,1-dichloroethane, 1,2-dichloroethane, bromoethane, 1,2-dichloropropane, 2,2-dichloropropane, 1,2,3-trichloropropane, 2-bromo-1-chloropropane, 1,2-dibromo-3-chloropropane, chloroform, 2-chlorobutane, 2,2-dichloroethanol at concentrations ranging between 0.2 and 2.3 mM, and hydrogen as electron donor. Under these conditions, except for an abiotic 2,2-dichloropropane transformation, no catabolic dechlorination activity was observed nor was growth detected for all substrates over a period of six months (data not shown). No significant TCE formation from PCE was observed with hydrogen and pyruvate as in vivo electron donor as it was the case in the cell suspension experiments.
As we had identified PCE Rdh activity in whole cells, we assayed cell-free extracts of S. sediminis for PCE dehalogenation activity. Extracts of early stationary phase (OD approx. 0.45) S. sediminis cells that were grown on 30 mM fumarate and 40 mM pyruvate catalysed the reductive dechlorination of PCE to TCE with TCE being the only metabolite at a constant rate (figure 2). PCE dechlorination activity followed Michaelis–Menten kinetics as shown in figure 3. Fitting the Michaelis–Menten kinetic model to the data revealed an apparent Km of 120 μM PCE and a vmax of approximately 1 nmol TCE produced min−1 (mg protein)−1. In control assays without cell extract or with growth medium alone, no dechlorination activity was observed. To test whether the observed dehalogenation activity might have been due to dehalogenase-independent dechlorination by cellular corrinoid(s) alone and not to a corrinoid-containing Rdh, we tested heat-treated cell extract (10 min at 95°C) for Rdh activity. Heat-treated cell extract resulted in an at least 10-fold lower apparent activity than the non-heat-treated cell extract (figure 2). These data collectively show that the observed PCE dechlorination activity is due to a cellular Rdh activity and not to an abiotic reductive dehalogenation of PCE.
Figure 2.
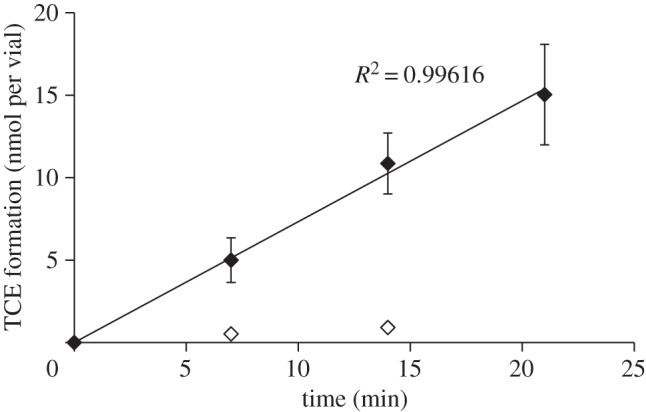
Product (TCE) formation from PCE by cell extracts of Shewanella sediminis in a standard dechlorination assay using (filled diamonds) 100 μl cell extract and (open diamonds) 100 μl heat-treated cell extract. The reaction mixture contained 100 mM Tris HCl buffer at pH 8, 2.5 mM methyl viologen, 5 mM Ti(III)–NTA and 2.5 mM PCE were added to reach saturation concentrations.
Figure 3.
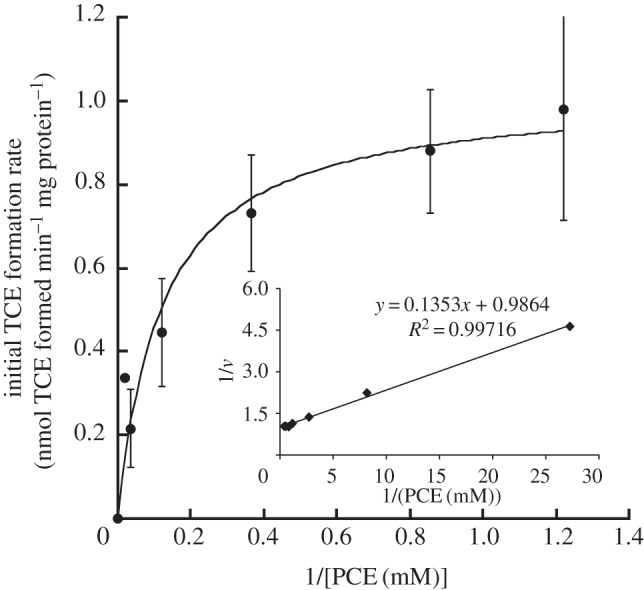
Kinetics of PCE dechlorination in cell extracts of Shewanella sediminis. Points represent initial dechlorination rates determined from individual dechlorination assays with PCE as substrate. The line represents the best fit of the data to the kinetic Michaelis–Menten model. The inset shows the Lineweaver–Burk plot. Values are means of results of at least duplicate biological experiments±s.d.
Dechlorination activity of S. sediminis required that vitamin B12 was present in the growth medium of the cells used for cell-free extract experiments as cell extracts from cells grown without cyanocobalamin in the growth medium did not show any TCE formation (data not shown). In addition, dechlorination activity occurred only in cell extracts from anaerobically grown cultures (data not shown).
(c). Induction of putative reductive dehalogenases gene expression
Although we showed above that PCE is not a catabolic substrate for S. sediminis, we tested whether PCE can induce Rdh activity by assaying for PCE dehalogenase activity in vitro in cell extracts of cells grown in a pyruvate/fumarate minimal medium that was amended with PCE. Extracts of cells grown in the presence of 0.1 mM PCE carried a 65 per cent higher specific activity than cells recovered from unamended media (1.82±0.18 versus 1.19±0.1 nmol min−1 (mg protein)−1, respectively; figure 4).
Figure 4.
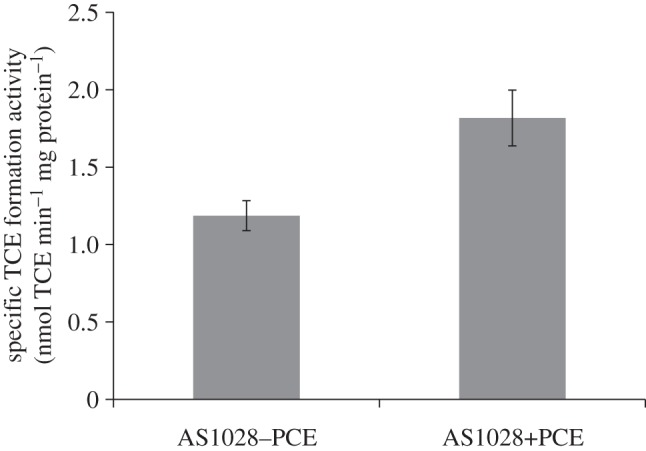
Specific PCE dechlorination activities in wild-type Shewanella sediminis cell extracts from cultures grown on 30 mM fumarate, 40 mM pyruvate and with or without addition of PCE (ca 0.1 mM). The assay mixtures contained 100 mM Tris–HCl buffer at pH 8, 2.5 mM methyl viologen and 5 mM Ti(III)–NTA and 2.5 mM PCE were added to reach saturation concentrations. Values are means of results of at least duplicate biological experiments±s.d.
In order to examine whether this difference in specific reductive dehalogenation activity was due to increased rdh gene transcription or to some post-transcriptional regulation, we prepared RNA from both PCE-amended and non-amended cultures, and tested for the presence of transcripts of Ssed_4120, Ssed_3769, Ssed_2100, Ssed_2103 and Ssed_1729 by qRT-PCR using gyrA as control gene for normalization. No differential regulation was seen for any of the putative Rdh genes expressed under both conditions (figure 5). Using more sensitive RT-PCR assays, we found that the transcript numbers in general were too low to obtain conclusive data from the qPCR assays. This apparent low level of rdh gene transcription is consistent with the low dechlorination activity we observed for PCE.
Figure 5.
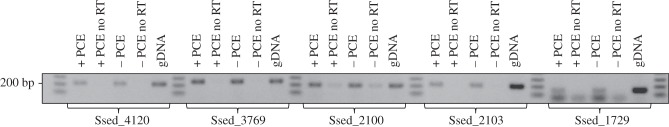
Agarose gel picture of RT-PCR products obtained with Ssed_4120, Ssed_3769, Ssed_2100, Ssed_2103 and Ssed_1729 specific primers from Shewanella sediminis cultures with and without supplementation of PCE to the growth medium. The approximate size of the respective amplicons according to a marker is given on the left. The same amount of cDNA was added to each reaction. Genomic DNA (gDNA) of S sediminis served as positive control.
(d). Identification of the PCE reductive dehalogenase gene
In order to determine which, if any, of the five putative rdh genes in S. sediminis are associated with the PCE dehalogenation activity, we constructed markerless in-frame deletion mutations in each putative rdh in S. sediminis wild-type. The resulting mutants, designated AS1029–AS1033, were tested in vitro for PCE dechlorination activity. Growth rate and final optical density of all mutant strains when grown on pyruvate and fumarate was indistinguishable from wild-type (figure 6a). Figure 6b shows that of all strains tested, only deletion of Ssed_3769 resulted in a significantly reduced TCE formation rate, which was about 10 times less than wild-type activity. Interestingly, although all other mutant strains catalysed PCE reduction at rates similar to wild-type, ΔSsed_2100 showed a slightly increased PCE transformation rate (1.4±0.2 nmol min−1 (mg protein)−1). To show unambiguously that the loss of activity in this mutant was not due to a secondary mutation, we complemented strain AS1030 (ΔSsed_3769) by introducing the wild-type Ssed_3769 allele at the chromosomal locus by homologous recombination (‘knock-in’). The in vitro PCE reduction rate of this complemented mutant (AS1034) was restored to wild-type level (1.1±0.03 nmol min−1 (mg protein)−1; figure 6b). From these results, we concluded that the gene product of Ssed_3769 is necessary and sufficient for the in vivo PCE dehalogenation activity, and constitutes a functional Rdh.
Figure 6.

Effect of ΔSsed_4120, ΔSsed_3769, ΔSsed_2100, ΔSsed_2103 and ΔSsed_1729 deletions on (a) growth in 4 M minimal medium with 30 mM fumarate as electron acceptor and 40 mM pyruvate as electron donor and (b) PCE dechlorination activity in cell extracts from cells harvested at early stationary phase. The assay mixtures contained 100 mM Tris HCl buffer at pH 8, 2.5 mM methyl viologen and 5 mM Ti(III)–NTA and 2.5 mM PCE were added to reach saturation concentrations. Growth was measured by optical density at 600 nm (abs. (600 nm)). Values are means of results of at least duplicate biological experiments±s.d.
4. Discussion
Previous work on reductively dehalogenating bacteria showed that a phylogenetically heterogeneous group of micro-organisms can catalyse reductive dechlorination of PCE catabolically or co-metabolically under anaerobic conditions [2]. In this study, we report the identification of an Rdh in S. sediminis, a marine psychrophilic γ-proteobacteria, that was isolated from an unexploded-ordnance-dumping site in the Baltic Sea, based on its ability to degrade hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) [20]. Shewanella sediminis clusters phylogenetically to other psychrophilic marine Shewanella strains that were all retrieved from deeper parts of marine ecosystems. Costal sediments, especially those close to the shore, are often a sink for anthropogenic pollutants, and Halifax Harbour has been shown to exhibit high levels of hydrocarbon and metal contamination [35]. All putative S. sediminis rdh sequences revealed the presence of features functionally similar to those of characterized Rdhs, and grouped into cluster 2 of the two major phylogenetic clades of RdhA sequences, which contains mainly non-Dehalococcoides rdhA genes [16]. Based on their amino acid sequence, they were most similar (46–58%) to a putative Rdh gene of Vibrio sp. RC586, another marine sediment micro-organism, isolated from Chesapeake Bay, MD, about 800 miles south of Halifax Harbour [36].
Using cell-free extract assays, we demonstrated, for the first time, reductive dechlorination of PCE to TCE in a Shewanella species, and were able to link this dechlorination activity to Ssed_3769, one of the putative Rdhs in S. sediminis. Similar to previous observations, transformation of PCE in S. sediminis was B12-dependent based on growth experiments, which is of particular interest as most Rdhs characterized so far are corrinoid-dependent enzymes [12,13,18]. Moreover, many organohalide-respiring microbes require the addition of cyanocobalamin or B12 in the medium for successful growth [37]. Although there are many genes present in S. sediminis that are involved in the anaerobic and aerobic de novo B12 biosynthesis, according to BioCyc (v. 16.1) some of them are missing or not yet identified, such as cbiK, cbiX, cbiD, cbiT or cobF, cobI, cobG, cobK and cobNST, which would explain the necessity of cyanocobalamin supplementation to the growth medium for PCE dechlorination. In contrast to many catabolic organohalide-respiring bacteria, molecular hydrogen could not serve as electron donor to support in vivo dechlorination, in catabolic organohalide-respiring Dehalococcides, the HupL hydrogenase has been postulated to be the key hydrogenase coupling reductive dehalogenation of hydrogen consumption. Shewanella sediminis however does not contain a HupL-type hydrogenase but rather a Ni–Fe hydrogenase (Ssed_1907) that, although containing matching PROSITE patterns to an uptake hydrogenase, is only 37 per cent identical to Dehalococcoides HupL at the amino acid level. Moreover, under the conditions tested, PCE reduction could not support growth suggesting that PCE dechlorination is a co-metabolic process in S. sediminis. Co-metabolic dechlorination is mostly mediated by protein-bound tetrapyrrole cofactors such as iron(II)porphyrins, corrinoids or factor F430 [38]. Compared with metabolic dehalogenation, co-metabolic dechlorination processes have been shown to be ubiquitous in anaerobic bacteria such as homoacetogens or methanogens, and to proceed at much lower rates [2,39,40]. Anaerobic microbes such as Methanosarcina sp., and Acetobacterium woodii were shown to dechlorinate PCE co-metabolically at rates of 5.8 × 10−4 and 6 × 10−2 nmol min−1 (mg protein)−1, respectively [41]. Metabolic PCE dechlorination rates in cell extracts of organohalide respirers range from 0.05 (PceA, Desulfitobacterium, strain Y51) to 1.5 μmol min−1 (mg protein)−1 (PceA, S. multivorans) [12,14,42]. Interestingly, another Shewanella strain, Shewanella oneidensis MR1, was shown to co-metabolically dechlorinate tetrachloromethane (CT) to trichloromethane (CF) using c-type cytochromes produced during microaerophilic growth [43,44]. However, PCE could not be degraded by this strain, and the gene required for CT dehalogenation was not identified. Although for S. sediminis the vmax for PCE dehalogenation of about 1 nmol min−1 (mg protein)−1 was about two to three orders of magnitude lower than previously reported PCE dechlorination activities in Dehalococoides or Desulfitobacterium, it was still significantly higher than the observed rates for co-metabolic PCE transformation in methanogens or homoacetogens. The relatively high apparent Km for PCE suggests that PCE may not be the natural substrate for Ssed_3769. Thus, other yet unidentified organohalogens in the sediment might be the primary substrate for Ssed_3769, and perhaps the other putative Rdhs, although we have not shown yet that they are functional.
While the organohalogens tested here as substrates for reductive dehalogenation in S. sediminis structurally resemble the most common anthropogenic halogenated compounds, there are many naturally occurring organohalogens that are not commercially available and therefore difficult to test as substrates for these Rdhs. This explains partially the so far limited spectrum of substrates identified for the catabolic organohalogen-respiring bacteria such as Dehalococcoides mccartyi or other Dehalococcoides-like Chloroflexi present in pristine environments [8,9]. According to Gribble [45,46], marine life is the largest source of naturally occurring organohalogen compounds. However, many organohalogens characterized from pristine marine environments are more complex, aromatic compounds, including brominated compounds such as indoles, phenols or pyrroles [45–47], which we did not include in our dehalogenation screen due to a lack in availability. On the other hand, PCE and TCE have been shown to be produced in various organisms such as algae, plants, bacteria or mammals [47,48]. In a recent study, it has been demonstrated that a cocktail of enzymatically generated chlorinated compounds from organic soil matter supported growth of Dehalococcoides-like Chloroflexi [8]; these undefined chlorinated organic compounds may function as terminal electron acceptors for these micro-organisms.
Dechlorination processes in sediments, especially in deep sea sediments, not only play an important role in the natural chlorine cycle but also have implications for the organic carbon flux in these environments. Organohalogens are organic carbon compounds that are typically inaccessible for most known heterotrophic micro-organisms because they are unable of reductive dechlorination. Yet, the vast majority of the members of deep sea microbial communities most likely depend on catabolizing organic compounds. Microbes such as S. sediminis may represent a group of micro-organisms that can uniquely access organic carbon resources locked as organohalogens in sediments that may not be available to non-dehalogenating heterotrophic microbes. Thus, S. sediminis might use the dehalogenated organic compounds eventually as catabolic electron donor rather than the organohalogens as catabolic electron acceptor. A variety of simple and complex organic compound degradation pathways such as for ethanol, glycerol, aldehyde or phenylacetate degradation present in its genome, might be involved in the utilization of these dehalogenated organic compounds anaerobically, fermentative or under metal reducing conditions.
Acknowledgements
This work was supported by the Strategic Environmental Research Defense Project (SERDP) grant no. ER-1588 and an ENI grant to A.M.S. S.T.L. was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft), fellowship no. Lo 1597/1-1. We thank Frank E. Löffler for helpful discussions.
References
- 1.Smidt H. 2004. Anaerobic microbial dehalogenation. Annu. Rev. Microbiol. 58, 43–73 10.1146/annurev.micro.58.030603.123600 (doi:10.1146/annurev.micro.58.030603.123600) [DOI] [PubMed] [Google Scholar]
- 2.Damborský J. 1999. Tetrachloroethene-dehalogenating bacteria. Folia Microbiol. 144, 247–262 10.1007/BF02818543 (doi:10.1007/BF02818543) [DOI] [PubMed] [Google Scholar]
- 3.Shivakumara S, et al. 2012. Complete genome sequence of Dehalogenimonas lykanthroporepellens type strain (BL-DC-9T) and comparison to ‘Dehalococcoides’ strains. Standards Genomic Sci. 6, 251–264 10.4056/sigs.2806097 (doi:10.4056/sigs.2806097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seshadri R, Adrian L, Fouts D, Eisen J. 2005. Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes. Science 307, 105–108 10.1126/science.1102226 (doi:10.1126/science.1102226) [DOI] [PubMed] [Google Scholar]
- 5.Mohn W, Tiedje J. 1992. Microbial reductive dehalogenation. Microbiol. Rev. 56, 482–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatt P, Kumar MS, Mudliar S, Chakrabarti T. 2007. Biodegradation of chlorinated compounds: a review. Crit. Rev. Environ. Sci. Technol. 37, 165–198 10.1080/10643380600776130 (doi:10.1080/10643380600776130) [DOI] [Google Scholar]
- 7.Tas N, van Eekert MHA, de Vos WM, Smidt H. 2010. The little bacteria that can—diversity, genomics and ecophysiology of ‘Dehalococcoides’ spp. in contaminated environments. Microbial Biotechnol. 3, 389–402 10.1111/j.1751-7915.2009.00147.x (doi:10.1111/j.1751-7915.2009.00147.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krzmarzick MJ, Crary BB, Harding JJ, Oyerinde OO, Leri AC, Myneni SCB, Novak PJ. 2012. Natural niche for organohalide-respiring Chloroflexi. Appl. Environ. Microbiol. 78, 393–401 10.1128/AEM.06510-11 (doi:10.1128/AEM.06510-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Futagami T, Morono Y, Terada T, Kaksonen AH, Inagaki F. 2009. Dehalogenation activities and distribution of reductive dehalogenase homologous genes in marine subsurface sediments. Appl. Environ. Microbiol. 75, 6905–6909 10.1128/AEM.01124-09 (doi:10.1128/AEM.01124-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McMurdie PJ, Hug LA, Edwards EA, Holmes S, Spormann AM. 2011. Site-specific mobilization of vinyl chloride respiration islands by a mechanism common in Dehalococcoides. BMC Genomics 12, 287. 10.1186/1471-2164-12-287 (doi:10.1186/1471-2164-12-287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smidt H, Akkermans A, Van Der Oost J, de Vos W. 2001. Molecular characterisation of key enzymes in halorespiration. In Biotechnology for the environment: strategy and fundamentals (eds Agathos S, Reineke W.), pp. 23–46 Dordrecht, The Netherlands: Kluwer Academic Publishers [Google Scholar]
- 12.Wohlfarth G, Diekert G. 1999. Reductive dehalogenases. In Chemistry and biochemistry of B12 (ed. Banerjee R.), pp. 871–893 New York, NY: John Wiley & Sons Inc [Google Scholar]
- 13.Müller JA, Rosner BM, Abendroth von G, Meshulam-Simon G, Mccarty PL, Spormann AM. 2004. Molecular identification of the catabolic vinyl chloride reductase from Dehalococcoides sp. strain VS and its environmental distribution. Appl. Environ. Microbiol. 70, 4880–4888 10.1128/AEM.70.8.4880-4888.2004 (doi:10.1128/AEM.70.8.4880-4888.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holliger C, Wohlfarth G, Diekert G. 1998. Reductive dechlorination in the energy metabolism of anaerobic bacteria. FEMS Microbiol. Rev. 22, 383–398 10.1111/j.1574-6976.1998.tb00377.x (doi:10.1111/j.1574-6976.1998.tb00377.x) [DOI] [Google Scholar]
- 15.Smidt H, Akkermans ADL, Van Der Oost J, de Vos WM. 2007. Halorespiring bacteria—molecular characterization and detection. Enzyme Microbial Technol. 27, 812–820 10.1016/S0141-0229(00)00316-1 (doi:10.1016/S0141-0229(00)00316-1) [DOI] [PubMed] [Google Scholar]
- 16.McMurdie PJ, et al. 2009. Localized plasticity in the streamlined genomes of vinyl chloride respiring Dehalococcoides. PLoS Genet. 5, e1000714. 10.1371/journal.pgen.1000714 (doi:10.1371/journal.pgen.1000714) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kube M, Beck A, Zinder S, Kuhl H, Reinhardt R, Adrian L. 2005. Genome sequence of the chlorinated compound respiring bacterium Dehalococcoides species strain CBDB1. Nat. Biotechnol. 23, 1269–1273 10.1038/nbt1131 (doi:10.1038/nbt1131) [DOI] [PubMed] [Google Scholar]
- 18.Adrian L, Rahnenfuhrer J, Gobom J, Hölscher T. 2007. Identification of a chlorobenzene reductive dehalogenase in Dehalococcoides sp. strain CBDB1. Appl. Environ. Microbiol. 73, 7717–7724 10.1128/AEM.01649-07 (doi:10.1128/AEM.01649-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waller AS, Krajmalnik-Brown R, Löffler FE, Edwards EA. 2005. Multiple reductive-dehalogenase-homologous genes are simultaneously transcribed during dechlorination by Dehalococcoides-containing cultures. Appl. Environ. Microbiol. 71, 8257–8264 10.1128/AEM.71.12.8257-8264.2005 (doi:10.1128/AEM.71.12.8257-8264.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao J-S, Manno D, Beaulieu C, Paquet L, Hawari J. 2005. Shewanella sediminis sp. nov., a novel Na+-requiring and hexahydro-1,3,5-trinitro-1,3,5-triazine-degrading bacterium from marine sediment. Int. J. Syst. Evol. Microbiol. 55, 1511–1520 10.1099/ijs.0.63604-0 (doi:10.1099/ijs.0.63604-0) [DOI] [PubMed] [Google Scholar]
- 21.Cordova CD, Schicklberger MFR, Yu Y, Spormann AM. 2011. Partial functional replacement of CymA by SirCD in Shewanella oneidensis MR-1. J. Bacteriol. 193, 2312–2321 10.1128/JB.01355-10 (doi:10.1128/JB.01355-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170, 2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saltikov CW, Newman DK. 2003. Genetic identification of a respiratory arsenate reductase. Proc. Natl Acad. Sci. USA 100, 10 983–10 988 10.1073/pnas.1834303100 (doi:10.1073/pnas.1834303100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon R, Priefer UA, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Biotechnology 1, 784–791 10.1038/nbt1183-784 (doi:10.1038/nbt1183-784) [DOI] [Google Scholar]
- 25.Wan XF, et al. 2004. Transcriptomic and proteomic characterization of the Fur modulon in the metal-reducing bacterium Shewanella oneidensis. J. Bacteriol. 186, 8385–8400 10.1128/JB.186.24.8385-8400.2004 (doi:10.1128/JB.186.24.8385-8400.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Philippe N, Alcaraz J-P, Coursange E, Geiselmann J, Schneider D. 2004. Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51, 246–255 10.1016/j.plasmid.2004.02.003 (doi:10.1016/j.plasmid.2004.02.003) [DOI] [PubMed] [Google Scholar]
- 27.Livak K, Schmittgen T. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 10.1006/meth.2001.1262 (doi:10.1006/meth.2001.1262) [DOI] [PubMed] [Google Scholar]
- 28.Moench TT, Zeikus J. 1983. An improved preparation method for a titanium (III) media reductant. J. Microbiol. Methods 1, 199–202 10.1016/0167-7012(83)90024-6 (doi:10.1016/0167-7012(83)90024-6) [DOI] [Google Scholar]
- 29.Bruschi M, Guerlesquin F. 1988. Structure, function and evolution of bacterial ferredoxins. FEMS Microbiol. Rev. 54, 155–176 10.1111/j.1574-6968.1988.tb02741.x (doi:10.1111/j.1574-6968.1988.tb02741.x) [DOI] [PubMed] [Google Scholar]
- 30.Palmer T, Sargent F, Berks B. 2005. Export of complex cofactor-containing proteins by the bacterial Tat pathway. Trends Microbiol. 13, 175–180 10.1016/j.tim.2005.02.002 (doi:10.1016/j.tim.2005.02.002) [DOI] [PubMed] [Google Scholar]
- 31.Futagami T, Goto M, Furukawa K. 2008. Biochemical and genetic bases of dehalorespiration. Chem. Rec. 8, 1–12 10.1002/tcr.20134 (doi:10.1002/tcr.20134) [DOI] [PubMed] [Google Scholar]
- 32.Ludwig ML, Matthews RG. 1997. Structure-based perspective on B12-dependent enzymes. Annu. Rev. Biochem. 66, 269–313 10.1146/annurev.biochem.66.1.269 (doi:10.1146/annurev.biochem.66.1.269) [DOI] [PubMed] [Google Scholar]
- 33.Marsh A, Ferguson D. 1997. Knowledge-based modeling of a bacterial dichloromethane dehalogenase. Proteins 28, 217–226 (doi:10.1002/(SICI)1097-0134(199706)28:2<217::AID-PROT10>3.0.CO;2-L) [DOI] [PubMed] [Google Scholar]
- 34.Damborský J, Koča J. 1999. Analysis of the reaction mechanism and substrate specificity of haloalkane dehalogenases by sequential and structural comparisons. Protein Eng. 12, 989–998 10.1093/protein/12.11.989 (doi:10.1093/protein/12.11.989) [DOI] [PubMed] [Google Scholar]
- 35.Gearing J, Buckley D, Smith J. 1991. Hydrocarbon and metal contents in a sediment core from Halifax Harbor: a chronology of contamination. Can. J. Fish Aquat. Sci. 48, 2344–2354 10.1139/f91-275 (doi:10.1139/f91-275) [DOI] [Google Scholar]
- 36.Haley BJ, et al. 2010. Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae. BMC Microbiol. 10, 154. 10.1186/1471-2180-10-154 (doi:10.1186/1471-2180-10-154) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He J, Holmes VF, Lee PKH, Alvarez-Cohen L. 2007. Influence of vitamin B12 and cocultures on the growth of Dehalococcoides isolates in defined medium. Appl. Environ. Microbiol. 73, 2847–2853 10.1128/AEM.02574-06 (doi:10.1128/AEM.02574-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gantzer CJ, Wackett LP. 1991. Reductive dechlorination catalyzed by bacterial transition-metal coenzymes. Environ. Sci. Technol. 25, 715–722 10.1021/es00016a017 (doi:10.1021/es00016a017) [DOI] [Google Scholar]
- 39.Terzenbach DP, Blaut M. 1994. Transformation of tetrachloroethylene to trichloroethylene by homoacetogenic bacteria. FEMS Microbiol. Lett. 123, 213–218 10.1111/j.1574-6968.1994.tb07224.x (doi:10.1111/j.1574-6968.1994.tb07224.x) [DOI] [PubMed] [Google Scholar]
- 40.Fathepure BZ, Nengu J, Boyd SA. 1987. Anaerobic bacteria that dechlorinate perchloroethene. Appl. Environ. Microbiol. 53, 2671–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fetzner S. 1998. Bacterial dehalogenation. Appl. Microbiol. Biotechnol. 50, 633–657 10.1007/s002530051346 (doi:10.1007/s002530051346) [DOI] [PubMed] [Google Scholar]
- 42.Fetzner S, Lingens S. 1994. Bacterial dehalogenases: biochemistry, genetics, and biotechnical applications. Microbiol. Rev. 58, 641–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Picardal F, Arnold R, Couch H, Little A, Smith M. 1993. Involvement of cytochromes in the anaerobic biotransformation of tetrachloromethane by Shewanella putrefaciens 200. Appl. Environ. Microbiol. 59, 3763–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ward MJ, Fu QS, Rhoads KR, Yeung CHJ, Spormann AM, Criddle CS. 2004. A derivative of the menaquinone precursor 1,4-dihydroxy-2-naphthoate is involved in the reductive transformation of carbon tetrachloride by aerobically grown Shewanella oneidensis MR-1. Appl. Microbiol. Biotechnol. 63, 571–577 10.1007/s00253-003-1407-3 (doi:10.1007/s00253-003-1407-3) [DOI] [PubMed] [Google Scholar]
- 45.Gribble GW. 1998. Naturally occurring organohalogen compounds. Acc. Chem. Res. 31, 141–152 10.1021/ar9701777 (doi:10.1021/ar9701777) [DOI] [Google Scholar]
- 46.Gribble GW. 1992. Naturally occurring organohalogen compounds: a survey. J. Nat. Prod. 55, 1353–1395 10.1021/np50088a001 (doi:10.1021/np50088a001) [DOI] [PubMed] [Google Scholar]
- 47.Vetter W, Gribble G. 2007. Anthropogenic persistent organic pollutants: lessons to learn from halogenated natural products. Environ. Toxicol. Chem. 26, 2249–2252 10.1897/06-615R.1 (doi:10.1897/06-615R.1) [DOI] [PubMed] [Google Scholar]
- 48.Cabrita MT, Vale C, Rauter AP. 2010. Halogenated compounds from marine algae. Mar. Drugs 8, 2301–2317 10.3390/md8082301 (doi:10.3390/md8082301) [DOI] [PMC free article] [PubMed] [Google Scholar]