

Abstract
We describe the presentation, management, and clinical outcome of a massive acinic cell carcinoma of the parotid gland. The primary tumor and blood underwent exome sequencing which revealed deletions in CDKN2A as well as PPP1R13B, which induces p53. A damaging nonsynonymous mutation was noted in EP300, a histone acetylase which plays a role in cellular proliferation. This study provides the first insights into the genetic underpinnings of this cancer. Future large-scale efforts will be necessary to define the mutational landscape of salivary gland malignancies to identify therapeutic targets and biomarkers of treatment failure.
1. Introduction
Salivary gland cancers account for 0.3–0.9% of all cancers [1, 2], and acinic cell carcinoma (AciCC) accounts for 5–11% of these [3, 4]. AciCC most commonly arises in the parotid gland and typically presents at an early stage allowing surgical treatment with favorable five-year survival rates in excess of 90% [4]. However, approximately 19% of cases present with advanced stage disease, which is associated with a higher rate of distant metastases and poorer survival [4]. To date, this tumor type has not been genetically characterized.
2. Case Report
Our patient is a morbidly obese (375 lbs) 58-year-old woman who presented to the head and neck surgery clinic with a right parotid mass and intact facial nerve function. She underwent a fine needle aspiration which was consistent with a Warthin's tumor. CT imaging demonstrated a 5.8 cm right parotid mass. She was scheduled for surgery, but was lost to followup. She presented again to the head and neck surgery clinic one year later with significant interval growth of the mass and a partial right facial paresis (Figures 1(a) and 1(b)). A CT scan demonstrated a 11.2 × 9.4 × 10.7 cm mass centered in the right parotid effacing the jugular vein and abutting the mandible and skull base with extension along the facial nerve to the geniculate ganglion (Figures 1(c) and 1(d)). She was taken to the operating room and underwent a radical parotidectomy with facial nerve sacrifice, radical neck dissection, parapharyngeal space resection, and lateral temporal bone resection (Figures 2(a), 2(b), and 2(c)). The tumor was found to be invading the jugular foramen requiring occlusion of the sigmoid sinus and packing of the jugular foramen for vascular control. The tumor was also noted to be extending medially to the geniculate ganglion of the facial nerve. Gross tumor removal was accomplished (Figure 2(d)). We were not able to obtain a negative margin on the proximal facial nerve; thus, the ipsilateral masseter nerve was grafted to the buccal and marginal mandibular branches of the distal facial nerve. Her face was further rehabilitated with a static palmaris longus sling and temporary tarsorraphy, which was later replaced with a gold weight. The defect was reconstructed with a large cervicofacial rotation flap and a radial forearm free flap (Figure 2(e)). Pathologic examination revealed a 14 cm low-grade acinic cell carcinoma with extensive perineural and lymphovascular spread. The lymph node dissection yielded 29 lymph nodes, all of which were negative for malignancy. Postoperatively, she received 6000 cGy in 30 fractions using intensity modulated radiation therapy (IMRT) to the primary site and neck. During and after radiation, the patient experienced massive weight loss, losing approximately 200 lbs. Follow-up imaging one year after treatment revealed no evidence of local or regional recurrence; however, there was interval development of multiple new bilateral lung nodules up to 0.9 cm highly suspicious for metastases (Figures 3(a) and 3(b)). They were deemed too small to be biopsied percutaneously. The patient was referred for consideration of palliative chemotherapy; however, as she was asymptomatic the decision was made to follow her with serial imaging.
Figure 1.
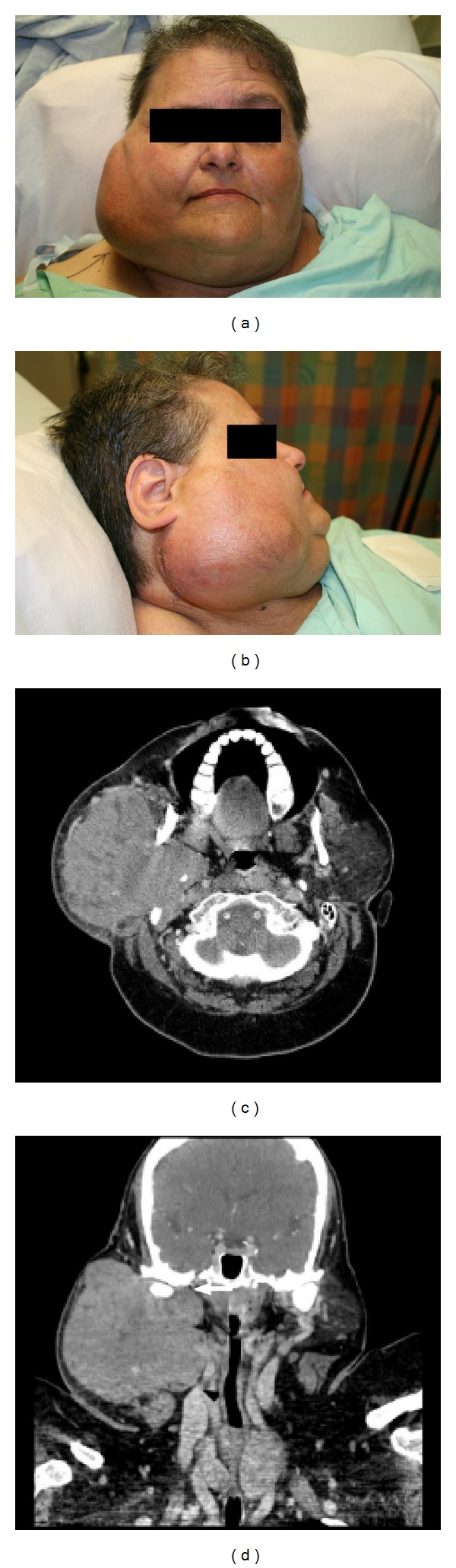
Anterior (a) and lateral (b) pictures of the patient at presentation. Axial (c) and coronal (d) CT scan images of the primary tumor. Note extension of the tumor into the parapharyngeal space with occlusion of the jugular vein (black arrowhead) and skull base involvement (white arrowhead).
Figure 2.
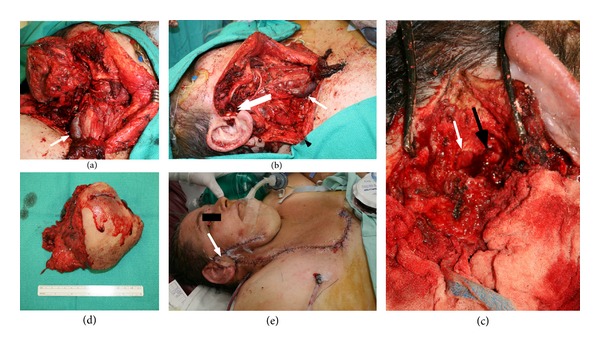
Intraoperative photographs demonstrating the radical parotidectomy with facial nerve sacrifice (a and b). (a) The large arrowhead indicates the divided jugular vein. (b) The single arrowhead (white) again marks the divided jugular vein, the double arrowhead (white) indicates residual tumor at the jugular foramen, and the black arrowhead indicates the divided end of the accessory nerve that was grafted to a cervical rootlet. (c) Temporal bone resection with surgicel occluding the sigmoid sinus (white arrowhead) and a fascia and muscle plug occluding the eustachian tube (black arrowhead). Demonstration of the primary tumor (d) and reconstruction with a large cervicofacial advancement flap and radial forearm free flap ((e) arrow marks forearm flap skin paddle).
Figure 3.
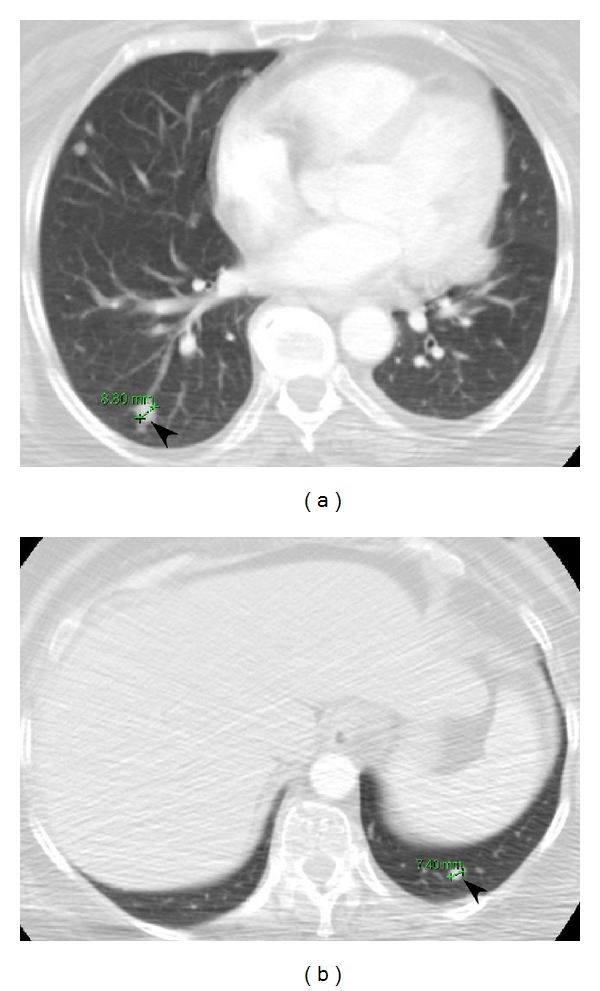
Axial CT scan images of the thorax (panels (a) and (b)) demonstrating the interval development of pulmonary metastases (arrowheads).
3. DNA Extraction, Exome Sequencing, and Bioinformatics Methods
Ethical approval was obtained from the University of Western Ontario Health Sciences Research Ethics and informed consent was obtained from the patient. DNA extraction from blood and tumor samples was carried out as previously described [5]. Exome sequencing was performed by Otogenetics (Tucker, Georgia) using the Agilent Human All Exon 50 Mbp exome capture kit with 30-fold coverage with 100 base-pair paired-end reads. The reference blood and primary tumor samples were aligned to the human hg19 reference sequence using Novoalign (v2.07.14). A maximum of five repeat alignments, defined as having a score difference of zero, were reported in the final output. SAM formatted output was specified with appropriate read group information provided. The remaining parameters were set to default values. Low-quality alignments, defined as alignments with low confidence in the reported position due to multiple alignment hits or poor base quality, were removed from the BAM files using SAMtools (v0.1.18) [6] by specifying the -q 30 quality filter. Additionally, unaligned and nonprimary reads (only 1 alignment, called the primary alignment, was retained in cases of multiple alignments) were removed by again using SAMtools (v0.1.18) and specifying the -F4 and –F 256 flags, respectively [6]. PCR artifacts were removed using MarkDuplicates tool from Picard (v1.66) with default settings. Samples were then processed as a matched set through the GATK (v1.3-16) pipeline [7, 8]. Samples were initially locally realigned using the IndelRealigner walker from the GATK package with known insertions and deletions found in dbSNP build 135. This was followed by base quality recalibration using GATK. Finally, variants were called and filtered using the GATK UnifiedGenotyper and VariantFiltration walkers again with default settings. Somatic variants within the targeted regions were identified using an in-house Perl library. To be classified as a somatic variant, the following conditions had to be met: (1) a tumor variant was identified by GATK and had a minimum 20x coverage and (2) the variant base was not seen at that position in the corresponding normal sample (20x coverage). The genes were annotated using RefGene and the consequences of the variations were identified using ANNOVAR dated 2012-03-08 [9]. As a final filtration step, any somatic variants, found in genes identified by Fuentes et al. as possible problematic genes for sequencing data, were removed [10]. Next, copy-number variants in the target regions were predicted with contra (v2.0.3) using default parameters [11]. DNAcopy (v1.32.0) was used to segment the copy numbers for visualization. This was done using default settings. Visualizations were generated using R (v2.15.2) and the lattice (v0.20-11) and latticeExtra (v0.6-24) packages. Of the target bases, approximately 60% were covered at a minimum of 20x collapsed coverage in the blood sample and 63% in the tumor.
4. Interpretation of Identified Mutations
Our bioinformatics pipeline identified 14 nonsynonymous mutations (Table 1) and significant copy-number variations (CNVs) in 35 genes including 31 amplifications and 4 deletions (Table 2). CNVs were classified as significant if they met an adjusted P value threshold value of 0.05. A comprehensive listing of the single-nucleotide variants, (SNVs) identified including noncoding regions is provided in the Supplementary Table S1 available online at http://dx.doi.org/10.1155/2013/270362. The amplifications occurred preferentially in chromosome 2 (P = 2.91 × 10−21) and chromosome 9 (P = 1.59 × 10−18), with single-amplification loci in chromosomes 14 and 22 (Table 2). The coding SNVs and all CNVs are illustrated in Figure 4. In addition, there were two deletions on chromosome 9 (CDKN2A and MTAP), one on chromosome 14 (PPP1R13B) and one on chromosome 3 (ETV5). All of the identified single-nucleotide variant mutations were heterozygous. Thirteen exhibited nonsynonymous changes in the respective proteins and one was a nonsense mutation which led to premature termination of the protein (GRIK3).
Table 1.
Somatic non-synonymous single nucleotide variations.
| Gene | Chr | Position | Reference allele | Tumor allele | Zygosity | Region | Type | Transcript | Exon | CDS position | Protein change |
|---|---|---|---|---|---|---|---|---|---|---|---|
| APBA1 | 9 | 72067091 | C | T | Hetero | Exonic | Nonsynonymous | NM_001163 | 9 | c.G1915A | p.D639N |
| ARHGAP5 | 14 | 32560713 | G | T | Hetero | Exonic | Nonsynonymous | NM_001030055 | 2 | c.G838T | p.V280L |
| CCDC74A | 2 | 132288362 | T | C | Hetero | Exonic | Nonsynonymous | NM_138770 | 3 | c.T506C | p.M169T |
| EP300 | 22 | 41523603 | A | T | Hetero | Exonic | Nonsynonymous | NM_001429 | 4 | c.A1019T | p.Q340L |
| GRIK3 | 1 | 37324744 | G | A | Hetero | Exonic | Nonsynonymous | NM_000831 | 7 | c.C1069T | p.R357C |
| KIAA0319L | 1 | 35972628 | G | T | Hetero | Exonic | Stop-gain | NM_024874 | 3 | c.C251A | p.S84X |
| KIAA1109 | 4 | 123207860 | G | C | Hetero | Exonic | Nonsynonymous | NM_015312 | 51 | c.G9202C | p.D3068H |
| KRT18 | 12 | 53343059 | C | A | Hetero | Exonic | Nonsynonymous | NM_199187 | 2 | c.C102A | p.S34R |
| LRRC1 | 6 | 53778709 | A | G | Hetero | Exonic | Nonsynonymous | NM_018214 | 11 | c.A1048G | p.I350V |
| LRRC8E | 19 | 7963808 | G | A | Hetero | Exonic | Nonsynonymous | NM_025061 | 3 | c.G401A | p.S134N |
| MYO10 | 5 | 16694556 | G | A | Hetero | Exonic | Nonsynonymous | NM_012334 | 27 | c.C3724T | p.R1242C |
| SAMD8 | 10 | 76910566 | A | G | Hetero | Exonic | Nonsynonymous | NM_144660 | 2 | c.A280G | p.M94V |
| SLC25A36 | 3 | 140675512 | C | A | Hetero | Exonic | Nonsynonymous | NM_018155 | 2 | c.C185A | p.P62H |
| WARS2 | 1 | 119575781 | T | C | Hetero | Exonic | Nonsynonymous | NM_015836 | 6 | c.A836G | p.H279R |
Chr: chromosome, Bolded genes are in cosmic v61.
Table 2.
Somatic copy-number aberrations.
| Gene | Chr | Start coordinate | End coordinate | Adjusted mean log ratio | P value | Adjusted P value | Gain/loss |
|---|---|---|---|---|---|---|---|
| AAK1 | 2 | 69752135 | 69752264 | 1.719 | 1.76E − 006 | 1.54E − 002 | Gain |
| ABCA1 | 9 | 107586742 | 107586862 | 1.908 | 1.50E − 007 | 2.40E − 003 | Gain |
| ABCA12 | 2 | 215890389 | 215890509 | 1.846 | 7.60E − 006 | 3.83E − 002 | Gain |
| AGAP1 | 2 | 236791980 | 236792100 | 2.084 | 6.34E − 009 | 1.35E − 004 | Gain |
| ALDH1A1 | 9 | 75533626 | 75533746 | 1.595 | 5.94E − 006 | 3.25E − 002 | Gain |
| ASAP2 | 2 | 9508540 | 9508660 | 1.931 | 1.08E − 006 | 1.13E − 002 | Gain |
| CDKN2A | 9 | 21974440 | 21974830 | −4.943 | 1.48E − 031 | 1.42E − 026 | Loss |
| CERKL | 2 | 182430138 | 182430258 | 1.672 | 1.12E − 006 | 1.13E − 002 | Gain |
| CLASP1 | 2 | 122098419 | 122098539 | 1.990 | 2.29E − 008 | 4.38E − 004 | Gain |
| ETV5 | 3 | 185823397 | 185823517 | −1.623 | 4.52E − 006 | 2.71E − 002 | Loss |
| EXD3 | 9 | 140250649 | 140250877 | 1.883 | 1.22E − 005 | 4.65E − 002 | Gain |
| GABBR2 | 9 | 101156436 | 101156556 | 1.864 | 9.79E − 008 | 1.70E − 003 | Gain |
| GSN | 9 | 124076166 | 124076316 | 1.636 | 8.28E − 006 | 3.96E − 002 | Gain |
| IL18RAP | 2 | 103040817 | 103040937 | 1.602 | 6.85E − 006 | 3.64E − 002 | Gain |
| INPP4A | 2 | 99136444 | 99136684 | 1.894 | 1.99E − 006 | 1.65E − 002 | Gain |
| ITGA4 | 2 | 182388877 | 182388997 | 1.965 | 1.44E − 006 | 1.38E − 002 | Gain |
| LOC375190 | 2 | 24390449 | 24390569 | 2.155 | 1.82E − 007 | 2.68E − 003 | Gain |
| LOC96610 | 22 | 22657565 | 22657685 | 1.907 | 1.01E − 005 | 4.29E − 002 | Gain |
| MERTK | 2 | 112765945 | 112766169 | 1.473 | 7.37E − 006 | 3.81E − 002 | Gain |
| MERTK | 2 | 112778145 | 112778265 | 1.707 | 9.88E − 006 | 4.29E − 002 | Gain |
| MFSD6 | 2 | 191354482 | 191354602 | 1.980 | 5.64E − 007 | 6.35E − 003 | Gain |
| MTAP | 9 | 21837897 | 21838017 | −4.841 | 4.56E − 045 | 8.72E − 040 | Loss |
| NGEF | 2 | 233745847 | 233745967 | 1.612 | 8.93E − 006 | 4.17E − 002 | Gain |
| NUP188 | 9 | 131719210 | 131719330 | 1.627 | 2.65E − 006 | 2.03E − 002 | Gain |
| POLR1E | 9 | 37489294 | 37489414 | 2.029 | 2.27E − 007 | 3.10E − 003 | Gain |
| POMT2 | 14 | 77753065 | 77753185 | 1.796 | 3.35E − 006 | 2.46E − 002 | Gain |
| PPIG | 2 | 170460673 | 170460793 | 1.944 | 2.96E − 007 | 3.77E − 003 | Gain |
| PPP1R13B | 14 | 104201451 | 104201571 | −1.855 | 3.88E − 006 | 2.48E − 002 | Loss |
| PSMD5 | 9 | 123591371 | 123591491 | 1.845 | 9.39E − 006 | 4.28E − 002 | Gain |
| RNASEH1 | 2 | 3596211 | 3596331 | 1.911 | 5.03E − 006 | 2.83E − 002 | Gain |
| SH3RF3 | 2 | 109988029 | 109988149 | 2.909 | 2.23E − 013 | 7.12E − 009 | Gain |
| SLC46A2 | 9 | 115642010 | 115642130 | 1.621 | 1.14E − 005 | 4.45E − 002 | Gain |
| SULT6B1 | 2 | 37415566 | 37415686 | 1.530 | 1.04E − 005 | 4.32E − 002 | Gain |
| TESK1 | 9 | 35607906 | 35608026 | 1.688 | 4.54E − 006 | 2.71E − 002 | Gain |
| TTC27 | 2 | 32929895 | 32930015 | 1.466 | 1.09E − 005 | 4.43E − 002 | Gain |
Chr: chromosome, bolded genes are in cosmic v61.
Figure 4.
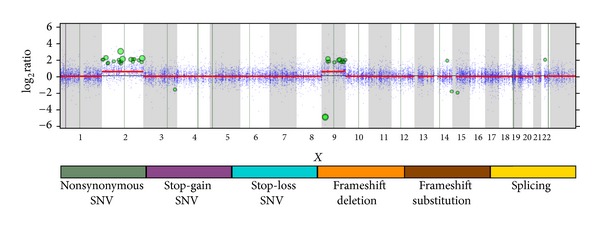
Somatic whole exome sequencing results across the genome. Vertical lines represent the single-nucleotide variants, while the points represent copy-number variant calls. Significant CNVs are represented by the green dots with its size reflecting its significance. Segmented copy-number variants are depicted in red and show clear gains in chromosome 2 and chromosome 9.
Several of these aberrations are plausibly associated to tumor formation and growth. Most critically, somatic deletion of the potent tumor-suppressor CDKN2A was identified. CDKN2A is one of the most widely mutated genes in human malignancies. According to the ICGC data-coordinating centre, it is mutated in up to half of glioblastoma multiforme, 14% of squamous cell carcinomas of the lung, and a third of all pancreatic adenocarcinomas [12]. It functions by stabilizing TP53 by sequestering the MDM2 ubiquitin ligase and by inhibiting CDK4-mediated G1 progression through the cellcycle. In addition, a deletion of 120 nucleotides of methylthioadenosine phosphorylase (MTAP) was identified, which is an important protein for salvaging adenine and methionine [13]. MTAP is located upstream of CDKN2A, frequently deficient in cancers, and often codeleted with p16 [13].
Also of great note, a nonsynonymous mutation in the E1A binding protein p300 (EP300) was identified. This protein functions as a histone acetyltransferase that regulates transcription via chromatin remodeling and is important in cell proliferation and differentiation [14]. EP300 interacts with hundreds of cellular transcriptional regulators [15] and is a key regulator of p53 function [16]. The observed mutation converts a glutamine to leucine at position 340 within the TAZ1 (CH1) zinc finger domain, which interacts with numerous transcription factors [17] and viral oncoproteins including human papillomavirus E7 [18]. The glutamine that is mutated is known to be involved in the interaction with STAT2, a key component of the interferon response, and potentially many other targets of the TAZ1 domain [19]. EP300 is frequently mutated in several tumour types [20–22] and its inactivation is thought to play a major role in the development of small cell lung cancer [21]. Loss of EP300 function would result in unopposed histone deacetylation, potentially creating an opportunity for targeted therapy with histone deacetylase (HDAC) inhibitors [23].
There was also a deletion identified in PPP1R13B which encodes the apoptosis stimulating of p53 protein 1 (ASPP1) [24]. Specifically, ASPP1 binds to p53 and enhances its ability to specifically stimulate expression of proapoptotic target genes, but not genes involved in cell cycle arrest [25]. Thus, ASPP1 functions as a tumor suppressor gene and has been shown to be downregulated in breast cancer [25] and leukemia cell lines [26], suggesting that the loss of this gene may play an important role in cancer progression. The role of the mutated genes in initiation and progression of AciCC identified in our study will require further investigation.
5. Discussion
AciCC is the least aggressive major salivary gland malignancy [27]. Typically these lesions are present at an early stage with low-grade histology and are cured at a high rate solely with surgery [4]. However, a subset presents with higher grade histology and/or advanced local, regional, and distant disease that portends a poorer outcome despite the addition of adjuvant radiation [4]. Currently, there are few treatment options available to be offered these patients when they relapse. For rare tumors such as AciCC, the standard mechanism to identify chemotherapeutic agents through a series of phase I, II, and III trials is not feasible due to limited patient numbers. A focused strategy based on tumor biology is necessary. The advent of massively parallel sequencing has led to incredible advances in the understanding of tumor genetics and biology. Recent exome sequencing of head and neck squamous cell carcinoma (HNSCC) has revealed that the mutational landscape of HNSCC is dominated by mutations in tumor suppressor genes, with only rare targetable mutations in oncogenes [28, 29]. However, studies of other cancers such as melanoma have revealed clearly targetable changes such as activating mutations in BRAF, which have already had a profound impact on clinical care [30]. Our study has provided the first glimpse of the genetic underpinnings of AciCC, highlighting changes in the tumor suppressors CDKN2A and PPP1R13B. There was also a mutation in the histone acetyltransferase EP300 that could reduce the acetylation of various targets. This mutation may make this tumor more susceptible to histone deacetylase inhibitors, which are already showing promise in vitro and in early-stage clinical trials [31, 32]. Future large-scale studies of salivary malignancies utilizing next generation sequencing (as for other cancers [33, 34]) will provide hope for improved patient outcomes.
6. Conclusion
Acinic cell carcinoma is a relatively rare salivary gland malignancy that typically has a favorable prognosis when treated solely with surgery. A small subset of these cancers present with advanced-stage disease which can be associated with poorer survival despite the addition of adjuvant radiation. Further study is necessary to understand the biology of salivary gland malignancies in order to develop adjuvant therapies to improve outcomes for patients with high-risk disease.
Supplementary Material
Supplementary Table: Complete list of somatic mutations identified in an acinic cell carcinoma. Single nucleotide polymorphisms (SNPs) were identified in the manner outlined in the bioinformatics methods of Section 3 of the manuscript. Variants were filtered against the gene exclusion list from Fuentes et al. [10].
Authors' Contribution
A. C. Nichols, M. Chan-Seng-Yue, P. C. Boutros, and J. W. Barrett contributed equally to this work.
Disclosure
The authors have no financial interests in companies or other entities that have an interest in the information in the contribution (e.g., grants, advisory boards, employment, consultancies, contracts, honoraria, royalties, expert testimony, partnerships, or stock ownership in medically-related fields).
Conflict of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
Exome sequencing was performed by Otogenetics (Tucker, GA, USA). This work was performed with the support of the Western University Translational Head and Neck Cancer Program to A. C. Nichols and the Ontario Institute for Cancer Research to P. C. Boutros through funding provided by the Government of Ontario.
References
- 1.SEER Cancer Statistics Review, 1975–2009. Bethesda, Md, USA: National Cancer Institute; 2012. [Google Scholar]
- 2.Jessup JM, Menck HR, Winchester DP, Hundahl SA, Murphy GP. The National Cancer Data Base report on patterns of hospital reporting. Cancer. 1996;78:1829–1837. doi: 10.1002/(sici)1097-0142(19961015)78:8<1829::aid-cncr26>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 3.Cesinaro AM, Criscuolo M, Collina G, Galetti R, Migaldi M, Lo Bianco F. Salivary gland tumors: revision of 391 cases according to the new WHO classification. Pathologica. 1994;86(6):602–605. [PubMed] [Google Scholar]
- 4.Hoffman HT, Karnell LH, Robinson RA, Pinkston JA, Menck HR. National Cancer Data Base report on cancer of the head and neck: acinic cell carcinoma. Head & Neck. 1999;21:297–309. doi: 10.1002/(sici)1097-0347(199907)21:4<297::aid-hed2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 5.Nichols AC, Chan-Seng-Yue M, Yoo J, et al. A pilot study comparing HPV-positive and HPV-negative head and neck squamous cell carcinomas by whole exome sequencing. ISRN Oncology. 2012;2012:9 pages. doi: 10.5402/2012/809370.809370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Depristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43(5):491–501. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38(16):p. e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuentes Fajardo KV, Adams D, Mason CE, et al. Detecting false-positive signals in exome sequencing. Human Mutation. 2012;33:609–613. doi: 10.1002/humu.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Lupat R, Amarasinghe KC, et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012;28:1307–1313. doi: 10.1093/bioinformatics/bts146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hudson TJ, Anderson W, Aretz A, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins CC, Volik SV, Lapuk AV, et al. Next generation sequencing of prostate cancer from a patient identifies a deficiency of methylthioadenosine phosphorylase, an exploitable tumor target. Molecular Cancer Therapeutics. 2012;11:775–783. doi: 10.1158/1535-7163.MCT-11-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eckner R. p300 and CBP as transcriptional regulators and targets of oncogenic events. Biological Chemistry. 1996;377(11):685–688. [PubMed] [Google Scholar]
- 15.Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics. 2010;5(1):9–15. doi: 10.4161/epi.5.1.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coutts AS, La Thangue NB. The p53 response: emerging levels of co-factor complexity. Biochemical and Biophysical Research Communications. 2005;331(3):778–785. doi: 10.1016/j.bbrc.2005.03.150. [DOI] [PubMed] [Google Scholar]
- 17.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes and Development. 2000;14(13):1553–1577. [PubMed] [Google Scholar]
- 18.Bernat A, Avvakumov N, Mymryk JS, Banks L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22(39):7871–7881. doi: 10.1038/sj.onc.1206896. [DOI] [PubMed] [Google Scholar]
- 19.Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. The EMBO Journal. 2009;28(7):948–958. doi: 10.1038/emboj.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Gallo M, O'Hara AJ, Rudd ML, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nature Genetics. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peifer M, Fernandez-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature Genetics. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersen. CL, Hasselbalch H, Gronbaek K. Lack of somatic mutations in the catalytic domains of CREBBP and EP300 genes implies a role for histone deacetylase inhibition in myeloproliferative neoplasms. Leukemia Research. 2012;36:485–487. doi: 10.1016/j.leukres.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 24.Liu ZJ, Lu X, Zhong S. ASPP—apoptotic specific regulator of p53. Biochimica et Biophysica Acta. 2005;1756(1):77–80. doi: 10.1016/j.bbcan.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 25.Samuels-Lev Y, O’Connor DJ, Bergamaschi D, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Molecular Cell. 2001;8(4):781–794. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- 26.Liu ZJ, Zhang Y, Zhang XB, Yang X. Abnormal mRNA expression of ASPP members in leukemia cell lines. Leukemia. 2004;18(4):p. 880. doi: 10.1038/sj.leu.2403300. [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharyya N, Fried MP. Nodal metastasis in major salivary gland cancer: predictive factors and effects on survival. Archives of Otolaryngology. 2002;128(8):904–908. doi: 10.1001/archotol.128.8.904. [DOI] [PubMed] [Google Scholar]
- 28.Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1 . Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bollag G, Tsai J, Zhang J, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nature Reviews Drug Discovery. 2012;11:873–886. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- 31.Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diyabalanage HV, Granda ML, Hooker JM. Combination therapy: histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Letters. 2012;329(1):1–8. doi: 10.1016/j.canlet.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table: Complete list of somatic mutations identified in an acinic cell carcinoma. Single nucleotide polymorphisms (SNPs) were identified in the manner outlined in the bioinformatics methods of Section 3 of the manuscript. Variants were filtered against the gene exclusion list from Fuentes et al. [10].