

Abstract
Herein we report the first attempt of coupling multidimensional separations to matrix-assisted laser desorption/ionization (MALDI) mass spectrometric imaging detection. Complex neuropeptide mixtures extracted from crustaceans were first fractionated by reversed-phase liquid chromatography (RPLC), and then subjected to a capillary electrophoresis-mass spectrometric imaging platform. With a specific focus on orcokinin family neuropeptides, we demonstrated that these trace-level analytes from complex neural tissue samples can be fully separated from chemical noise and interfering components and visualized as mass spectrometric imaging signals. A total of 19 putative orcokinins were detected, with highly efficient separations within the family being achieved for the first time. The results indicate that two-dimensional separation coupling to mass spectrometric imaging can serve as a novel and powerful tool in proteomics and peptidomics studies.
Keywords: Capillary electrophoresis, Liquid chromatography, Mass spectrometric imaging, Multidimensional separation, Neuropeptide
1 Introduction
Since its introduction by Caprioli and co-workers in 1997 [1], mass spectrometric imaging (MS imaging, MSI) has been widely adopted to create molecular ion images without the use of radioactive or fluorescent labeling tags [2, 3]. Being introduced into several ionization methods, including but not limited to matrix-assisted laser desorption/ionization (MALDI) [4–6], desorption electrospray ionization (DESI) [7], secondary ion mass spectrometry (SIMS) [8, 9], and laser ablation electrospray ionization (LAESI) [10], numerous MSI studies have shown capabilities to create distribution maps for compounds from small molecules to large proteins with a variety of MS platforms [11, 12]. Although separations, most commonly including liquid chromatography (LC) and capillary electrophoresis (CE), have been widely employed and coupled to a variety of MS instruments to achieve highly efficient separation and enhanced MS signal for the analysis of complex samples such as trace-level peptide mixtures [13–17], no separation tools, until very recently, have ever been coupled to MS imaging since separation tends to cause loss of spatial distribution information. On the other hand, however, MS imaging offers unique advantages when it is coupled with separation tools such as LC and CE. The separation dimensions are able to provide highly efficient fractionation to reduce the inherent complexity and to enable more accurate quantitation for MSI. Furthermore, by converting discrete mass spectra to a continuous ion trace with enhanced MS imaging signals, analytes can be studied from a new perspective as individual colored regions with MSI, offering enriched information for complex sample analysis.
In order to add separation dimensions to MS imaging, a well-designed interface is the key to enable smooth sample collection and to form homogeneous trace on the sampling plate. Recently, studies have been reported attempting to couple both LC and CE to MS imaging. Weidner and Falkenhagen reported an interface with an electrospray desorption device to spray LC elutes, which was premixed with matrix, to the MALDI plate. Two polymers could be separated and visualized via MS imaging [18]. We have shown the first example of coupling CE with MALDI MS imaging [19]. By using a MALDI plate with grooves etched on its surface, we were able to limit the width of CE trace to submillimeter scale with highly efficient separation. Quantitation could be achieved simultaneously by incorporating isotopic labeling. This interface was further developed by coupling a pressure-assisted CE (PACE) with MS imaging using a commercially available ground stainless steel MALDI plate, which demonstrated significantly enhanced robustness and limited diffusion (Z. Zhang, et al., unpublished data). More than 100 putative neuropeptides extracted from crustaceans were detected and accurately quantified after a single run, suggesting great potential for the CE-MSI platform as a novel tool for proteomics and peptidomics studies. With the established interface to MS imaging, more separation dimensions can be added for enhanced separations with orthogonal modalities; however, no multidimensional separations have ever been tested so far.
Herein, we report the first attempt on coupling two-dimensional (2-D) separations to MS imaging by linking orthogonal separation modalities including LC and CE (LC-CE-MSI). Neuropeptides extracted from the sinus gland (SG) of blue crab (Callinectes sapidus) with extremely low amounts and wide dynamic ranges were selected for analysis. The samples were first fractionated by reversed-phase LC (RPLC), and then analyzed with PACE-MSI platform. Multiple CE traces were initially collected on the same MALDI plate and then subjected to MS imaging simultaneously. Compared with unseparated control and LC-MALDI-time of flight (TOF)/TOF analysis, LC-CE-MSI exhibited high separation efficiency and enhanced MS sensitivity. With a specific focus on the orcokinin family neuropeptides, we were able to achieve exceptional separations not only among different neuropeptide families and interfering species, but also among numerous peptide isoforms with very similar sequences and structures within the orcokinin neuropeptide family. This platform is robust and easy to operate, indicating its potential applications to more complex analyte analysis.
2 Materials and methods
2.1 Reagents and sample preparation
All reagents were of analytical grade and purchased from Fisher Scientific (Pittsburgh, PA, USA) unless otherwise stated. The blue crabs (C. sapidus) were purchased from local grocery store and kept in tank with artificial seawater at 10–12°C. Detailed dissection procedure can be found in our previous reports [20]; briefly, the crabs were anesthetized with ice, and the SGs from eyestalk were dissected in physiological saline. Neuropeptides from SG were extracted with cold acidified methanol (methanol/water/acetic acid = 90:9:1 v/v/v) and centrifuged to collect the resulting supernatant. The extraction was repeated for three times with all supernatants combined, dried, and reconstituted in 0.1% formic acid (v/v). The samples were stored at −80°C until analyzed.
2.2 Off-line HPLC fractionation
The re-suspended extracts were first vortexed and briefly centrifuged, and the resulting supernatants were subsequently fractionated on a Waters Alliance high-performance liquid chromatography (HPLC) system (Milford, MA, USA). The mobile phases included solution A (deionized water containing 0.1% v/v formic acid) and solution B (acetonitrile containing 0.1% v/v formic acid). Approximately 50 μL of extract was injected into a Gemini C18 column (2.1 mm id × 150 mm length, 5 μm particle size; Phenomenex; Torrance, CA, USA). The separations consisted of a 130 min gradient of 5–95% solution B with a flow rate of 0.2 mL/min and column temperature 40°C. Fractions were automatically collected every 4 min with a Rainin Dynamax FC-4 fraction collector (Rainin Dynamax, Woburn, MA, USA).
2.3 PACE and its interface to MS imaging
Ten HPLC fractions from 0 to 40 min were collected for the second-dimensional CE separation and MS imaging detection. A recently established PACE-MSI platform was employed (Z. Zhang, et al., unpublished data). Briefly, a fracture was opened and covered with cellulose acetate membrane near the outlet end of CE capillary (75 μm id/190 μm od from Polymicro Technologies, Phoenix, AZ, USA) to enable sheathless CE separation, and a height difference between inlet and outlet ends were created to provide CE an additional elution force. We have demonstrated that this design was highly efficient in separating trace amount of peptide mixtures with wide dynamic ranges. In addition to the CE capillary, another capillary was added to deliver 10 mg/mL CHCA (α-cyano-4-hydroxycinnamic acid, from Sigma-Aldrich, St. Louis, MO, USA), in 1:1 acetonitrile/water (v/v) as matrix. CE eluent and matrix solution were mixed on the surface of a ground stainless steel MALDI plate from Bruker Daltonics (Bremen, Germany), which was mechanically controlled to move at 4.5 mm/min (Fig. 1). Approximately 0.8-mm-wide, continuous, and homogenous traces could be deposited on the MALDI plate surface with limited sample diffusion when the trace was dried quickly under the heat from a lamp placed above the plate. CE traces from 2 to 25 min fractionation were collected on the MALDI plate for each HPLC fraction, and multiple HPLC fractions were collected as individual traces on the MALDI plate and analyzed with MS imaging as multiple “regions of interest” in one experiment, thus increasing the analytical throughput.
Figure 1.
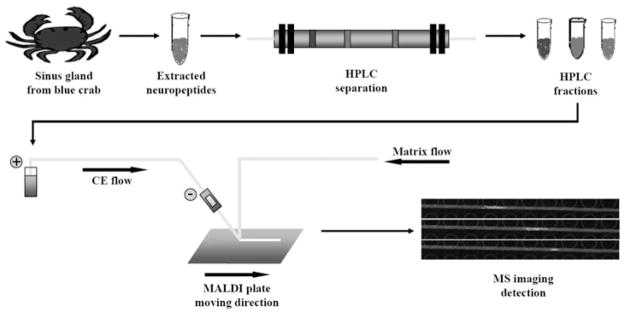
Schematic diagram of neuropeptide analysis with HPLC-CE-MSI platform. The sinus glands dissected from blue crabs were extracted with cold acidified methanol. Extracted neuropeptide mixtures were first separated with C18 RPLC with fractions collected every 4 min. Each of the fractions was then subjected to pressure-assisted capillary electrophoresis (PACE) separation platform and collected on a ground stainless steel MALDI plate as a straight and homogenous trace. These traces were subsequently analyzed with MALDI MS imaging to produce molecular ion images, in which peaks were separated as individual colored regions.
2.4 MS imaging and data processing
MALDI MS imaging was performed with Autoflex III MALDI-TOF/TOF (Bruker Daltonics) equipped with 200 Hz Smartbeam II laser beam. The mass range from m/z 600 to 4000 was recorded with the following parameters under positive reflectron mode: ion source 1 voltage 19.00 kV, ion source 2 voltage 16.62 kV, reflector 1 voltage 20.90 kV, reflector 2 voltage 9.64 kV, and lens voltage 8.70 kV. For MS imaging, the step size for each pixel was set at 150 μm on the x-axis × 150 μm on the y-axis, and 200 shots were accumulated in a random mode for each pixel. Instrument-specific software FlexImaging from Bruker Daltonics was used for data analysis.
3 Results and discussions
MALDI MS has been extensively employed in the study of neuropeptides due to its flexibility for reanalysis and tolerance to impurities. Because of the complexity of biological samples extracted from animals, separation and purification techniques were frequently involved in neuropeptide analysis [21,22]. Considering the low sample consumption and convenience for assembling the platform, CE [23] and capillary iso-electric focusing (CIEF) [24–26] have been used as preferred microscale separation techniques for coupling to MALDI-Fourier transform ion cyclotron resonance (MALDI-FTICR) or MALDI-TOF/TOF MS detections. Adding a separation dimension, either pressure-driven or electrically driven, usually resulted in significantly enhanced MS signal with several folds of increase in the number of analytes detected due to alleviation of analyte suppression and dynamic range issues [27–30]. Although in most cases multiple neuropeptide families could be identified after separation, neuropeptides with similar characteristics or within the same family were difficult to separate even when CE or CIEF technique was employed. An effective approach is to employ MALDI MS imaging for detection. Rather than collecting discrete CE fractions as individual spots on MALDI plate, continuous CE trace can be collected that provides higher separation sensitivity and resolution. In addition, the imaging signal converts peptide peaks to separated colored regions, which has been demonstrated to be extremely beneficial to the analysis of overlapping peaks in mass spectra [19] (Z. Zhang, et al., unpublished data).
Here, in addition to coupling CE with MS imaging, we further attempted to add another separation dimension prior to CE-MSI in order to achieve enhanced separation of complex neuropeptides with a specific focus on the orcokinin family neuropeptides based on both polarity and charge state of analytes prior to MS imaging detection. Modified from 2-D LC separations commonly adopted for large-scale proteomics studies [31–33], an off-line method of coupling LC to CE-MSI has been developed. Considering the extremely low quantities of neuropeptides, we collected LC fractions for every 4 min, and ten LC fractions from 0 to 40 min were collected, which included all of the targeted peptides. These LC fractions were further separated with PACE-MSI, and as comparison they were also directly spotted on the MALDI plate and analyzed with MALDI-TOF/TOF as controls to the 2-D LC-CE-MSI analysis. In order to obtain better MS signals, we accumulated and averaged 1500 laser shots for each spot of control samples. As shown in Table 1, a total of 14 orcokinins were separated by HPLC from fractions 5–8. These orcokinins were all previously identified with recognizable CE migration behaviors. Followed by CE-MSI detection of the same sample, we were able to observe a total of 19 putative orcokinins, with only 200 laser shots being accumulated for each selected pixels. Of the five additional orcokinins detected, three were previously identified. The other two peptide peaks at m/z 1542.6 and 1585.6 were not observed before, but they shared the same migration patterns with all the other orcokinins. Thus, these two new peptide peaks have been assigned as putative novel orcokinin peptides whose identities need to be further confirmed with additional investigations.
Table 1.
List of orcokinin family neuropeptides detected with LC-MALDI MS and LC-CE-MSI
| Theoretical mass | Sequence | Fraction # with LC-MALDI MS | Fraction # with LC-CE-MSI |
|---|---|---|---|
| 1186.54 | FDEIDRSSFA | 6 | 6 |
| 1256.55 | NFDEIDRSGFG | 5 | 5 |
| 1270.57 | NFDEIDRSGFA | 6 | 6 |
| 1271.55 | DFDEIDRSGFA | 7 | 7 |
| 1286.56 | NFDEIDRSSFG | 5 | 5 |
| 1300.58 | NFDEIDRSSFA | 6 | 6 |
| 1301.56 | DFDEIDRSSFA | – | 7 |
| 1403.62 | NFDEIDRSGFGF | – | 7 |
| 1433.63 | NFDEIDRSSFGF | – | 7 |
| 1474.66 | NFDEIDRSGFGFA | 7 | 7 |
| 1475.64 | DFDEIDRSGFGFA | 7 | 7 |
| 1502.69 | NFDEIDRSGFGFV | 8 | 8 |
| 1504.67 | NFDEIDRSSFGFA | 7 | 7 |
| 1532.70 | NFDEIDRSSFGFV | 8 | 8 |
| 1533.69 | DFDEIDRSSFGFV | 7 | 7 |
| 1542.60 | – | – | 7 |
| 1547.68 | NFDEIDRSSFGFN | 7 | 7 |
| 1548.66 | DFDEIDRSSFGFN | 8 | 8 |
| 1585.60 | – | – | 7 |
–: Unknown or undetected
We attribute the increased number of detected orcokinins to the continuous collection of CE elutes on the MALDI plate, but the enhanced separation is also a factor. Figure 2 shows LC fraction 5 with direct MALDI-TOF/TOF analysis in the mass range of m/z 800–1400. Although separated by HPLC, the orcokinins were still mixed with other peptides and noise peaks from a variety of sources. However, with the help of PACE-MSI, these coeluting peaks can be more readily distinguished from each other. As shown in Fig. 2, peak at m/z 909.0 is a chemical background noise with high intensity. It was difficult to determine whether this mass spectral peak was a chemical noise simply by looking at the mass spectrum of LC fraction 5. In contrast, the uniformly high abundance of the signal at m/z 909.0 throughout the CE trace in MS imaging spectra (reflected as false color images) strongly suggested that this peak was a chemical noise peak, rather than analyte of interest. Another peak at m/z 1203.7 is probably an unidentified peptide, separated from m/z 1256.6 (orcokinin, NFDEIDRSGFG) in the molecular imaging traces. The distinct image patterns can be attributed to different CE migration behaviors as this putative peptide is not highly negatively charged in CE buffer such as orcokinins. Compared with “regular” mass spectra, peaks in MSI images are shown as colored regions that are separated from each other. By simply clicking on the colored regions, separated peaks can be extracted from the image and shown as “regular” mass spectra. This unique characteristic is extremely beneficial for the study of complex samples with low concentrations.
Figure 2.
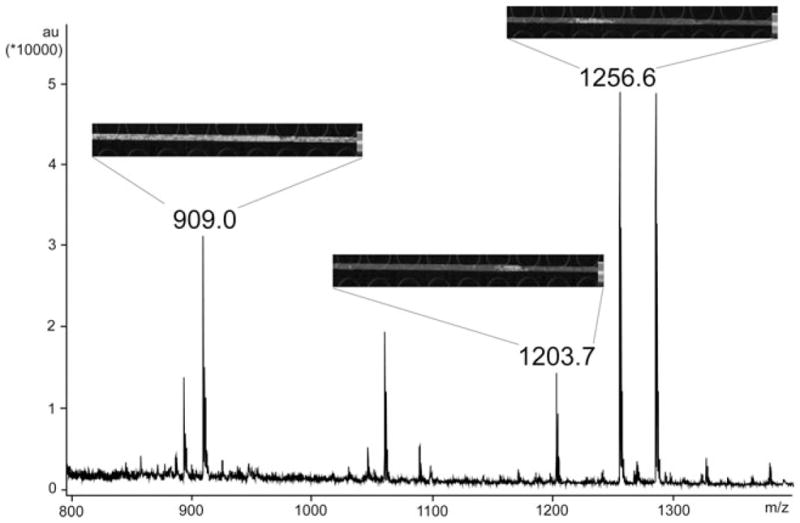
Separation of orcokinin neuropeptides from other peptides and noise peaks in an LC fraction with CE-MSI. The mass spectrum of LC fraction #5 with direct MALDI-TOF/TOF analysis is shown. The CE-MSI imaging signals were inserted as insets for three individual peaks. Upper left panel shows images of m/z 909.0: a noise peak with abundant image signal all over the trace. Middle panel shows images of m/z 1203.7: unidentified putative neuropeptide that does not belong to the orcokinin family. It was coeluted with other orcokinins in the LC fraction, but was separated by subsequent CE-MSI platform. Upper right panel shows ion images of m/z 1256.6: Orcokinin family neuropeptide NFDEIDRSGFG.
The orcokinin family neuropeptides share very similar sequences and characteristics. It was difficult to separate them from each other in our previous experiments [24, 25]. However, via 2-D LC-CE coupling to MS imaging, we separated orcokinins into four different LC fractions, and followed by efficient separation via CE-MSI analysis within each LC fractions. Figure 3 shows an example where two closely related orcokinin sequences are unambiguously distinguished by multidimensional separation coupled to MSI platform. Here, m/z 1474.7 (NFDEIDRSGFGFA) and m/z 1475.7 (DFDEIDRSGFGFA) are a pair of orcokinins previously identified with N-terminal deamidation (asparagine–aspartic acid) modification [25]. They share the same retention time in LC and are overlapped with each other in the mass spectrum (Fig. 3A). So we separated this LC fraction with CE and analyzed with MS imaging. In order to display MSI image, mass filters in the software FlexImaging were centered at m/z 1474.7, m/z 1475.7, and m/z 1476.7, respectively, with 1 Da width, and we were able to acquire three MSI images as shown in Fig. 3B. For mass filter m/z 1474.7, we only observed one colored region. When we clicked on this region, we extracted the m/z 1474.7 peak, separated from m/z 1475.7, as shown in Fig. 3C. By moving the mass filter to m/z 1475.7, we observed less-intense signal from the m/z 1474.7 region, but found a newly separated colored region to the right indicating different CE migration time. After we clicked this region, we observed a peak of m/z 1475.7 (Fig. 3D). If we move the mass filter further to m/z 1476.7, we only observed the colored region for m/z 1475.7. By employing this new LC-CE-MSI platform, we were able to eliminate the ambiguity of peptide assignment by visualizing each peak separated into distinct MSI images.
Figure 3.

Separation of orcokinins within the same LC fraction with CE-MSI. (A) The mass spectrum of LC fraction 7 showing that peptide peaks at m/z 1474.7 and m/z 1475.7 were overlapped with each other, which cannot be resolved. (B) MS imaging signals after LC-CE-MSI analysis. Mass filters were centered at m/z 1474.7, 1475.7, and 1476.7, respectively, with 1 Da width window. (C) Orcokinin peptide at m/z 1474.7 separated from m/z 1475.7 was extracted from the highlighted MS imaging signal shown in the red box on the left in panel B. (D) Another orcokinin peptide at m/z 1475.7 separated from m/z 1474.7 was also extracted from the highlighted MS imaging signal shown in the red box on the right in panel B.
4 Concluding remarks
In summary, we have successfully coupled 2-D separation to MS imaging for the first time. By analyzing trace-level orcokinin family neuropeptides from an extracted neuropeptide mixture, we have demonstrated that LC-CE-MSI is a superior platform with unique advantages in comparison to regular approaches, including enhanced separation efficiency, increased MS signal, and the ability to separate analytes with similar characteristics. This new platform also features robustness, fast separation speed, and the unique “visualized peaks” in the MSI images. Although not described here, isobaric and isotopic labeling can be readily integrated with this established platform that offers great potential for its applications to both small molecule and large protein quantitation analysis.
Acknowledgments
This work was supported by the National Science Foundation grant (CHE-0967784) and National Institutes of Health grants (1R01DK071801, 1R56DK071801). The authors thank Bruker Daltonics for graciously loaning the Autoflex III MALDI TOF/TOF mass spectrometer. L. Li acknowledges a Vilas Associate Award and an H.I. Romnes Faculty Research Fellowship.
Abbreviations
- DESI
desorption electrospray ionization
- LAESI
laser ablation electrospray ionization
- MSI
mass spectrometric imaging
- PACE
pressure-assisted CE
- SIMS
secondary ion mass spectrometry
- SG
sinus gland
Footnotes
The authors have declared no conflict of interest.
References
- 1.Caprioli RM, Farmer TB, Gile J. Anal Chem. 1997;69:4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 2.Heeren RMA, Smith DF, Stauber J, Kükrer-Kaletas B, MacAleese L. J Am Soc Mass Spectrom. 2009;20:1006–1014. doi: 10.1016/j.jasms.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 3.Amstalden van Hove ER, Smith DF, Heeren RMA. J Chromatogr A. 2010;1217:3946–3954. doi: 10.1016/j.chroma.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 4.Chen R, Li L. Anal Bioanal Chem. 2010;397:3185–3193. doi: 10.1007/s00216-010-3723-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubakhin SS, Greenough WT, Sweedler JV. Anal Chem. 2003;75:5374–5380. doi: 10.1021/ac034498+. [DOI] [PubMed] [Google Scholar]
- 6.Franck J, Arafah K, Elayed M, Bonnel D, Vergara D, Jacquet A, Vinatier D, Wisztorski M, Day R, Fournier I, Salzet M. Mol Cell Proteomics. 2009;8:2023–2033. doi: 10.1074/mcp.R800016-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eberlin LS, Ifa DR, Wu C, Cooks RG. Angew Chem Int Ed. 2010;49:873–876. doi: 10.1002/anie.200906283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Touboul D, Kollmer F, Niehuis E, Brunelle A, Laprevote O. J Am Soc Mass Spectrom. 2005;16:1608–1618. doi: 10.1016/j.jasms.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Altelaar A, Klinkert I, Jalink K, de Lange RPJ, Adan R, Heeren R, Piersma SR. Anal Chem. 2006;78:734–742. doi: 10.1021/ac0513111. [DOI] [PubMed] [Google Scholar]
- 10.Nemes P, Barton AA, Vertes A. Anal Chem. 2009;81:6668–6675. doi: 10.1021/ac900745e. [DOI] [PubMed] [Google Scholar]
- 11.Chaurand P, Schwartz SA, Caprioli RM. Curr Opin Chem Biol. 2002;6:676–681. doi: 10.1016/s1367-5931(02)00370-8. [DOI] [PubMed] [Google Scholar]
- 12.McDonnell LA, Heeren RMA. Mass Spectrom Rev. 2007;26:606–643. doi: 10.1002/mas.20124. [DOI] [PubMed] [Google Scholar]
- 13.Sandra K, Moshir M, D’hondt F, Verleysen K, Kas K, Sandra P. J Chromatogr B. 2008;866:48–63. doi: 10.1016/j.jchromb.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 14.Sandra K, Moshir M, D’hondt F, Tuytten R, Verleysen K, Kas K, Francois I, Sandra P. J Chromatogr B. 2009;877:1019–1039. doi: 10.1016/j.jchromb.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 15.van den Broek I, Sparidans RW, Schellens JHM, Beijnen JH. J Chromatogr B. 2008;872:1–22. doi: 10.1016/j.jchromb.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 16.Kasicka V. Electrophoresis. 2010;31:122–146. doi: 10.1002/elps.200900442. [DOI] [PubMed] [Google Scholar]
- 17.Herrero M, Ibanez E, Cifuentes A. Electrophoresis. 2008;29:2148–2160. doi: 10.1002/elps.200700404. [DOI] [PubMed] [Google Scholar]
- 18.Weidner SM, Falkenhagen J. Anal Chem. 2011;83:9153–9158. doi: 10.1021/ac202380n. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Ye H, Zhang Z, Xiang F, Girdaukas G, Li L. Anal Chem. 2011;83:3462–3469. doi: 10.1021/ac200708f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Zhang Y, Xiang F, Zhang Z, Li L. J Chromatogr A. 2010;1217:4463–4470. doi: 10.1016/j.chroma.2010.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lapainis T, Sweedler JV. J Chromatogr A. 2008;1184:144–158. doi: 10.1016/j.chroma.2007.10.098. [DOI] [PubMed] [Google Scholar]
- 22.Perry M, Li Q, Kennedy RT. Anal Chim Acta. 2009;653:1–22. doi: 10.1016/j.aca.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Ma M, Chen R, Li L. Anal Chem. 2008;80:6168–6177. doi: 10.1021/ac800382t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Wang J, Hui L, Li L. J Chromatogr A. 2011;1218:5336–5343. doi: 10.1016/j.chroma.2011.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hui L, Cunningham R, Zhang Z, Cao W, Jia C, Li L. J Proteome Res. 2011;10:4219–4229. doi: 10.1021/pr200391g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Wang J, Hui L, Li L. Electrophoresis. 2012;33(4):661–665. doi: 10.1002/elps.201100447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gatlin CL, Eng JK, Cross ST, Detter JC, Yates JR. Anal Chem. 2000;72:757–763. doi: 10.1021/ac991025n. [DOI] [PubMed] [Google Scholar]
- 28.Wang T, Ma J, Zhu G, Shan Y, Liang Z, Zhang L, Zhang Y. J Sep Sci. 2010;33:3194–3200. doi: 10.1002/jssc.201000324. [DOI] [PubMed] [Google Scholar]
- 29.Wang F, Han G, Yu Z, Jiang X, Sun S, Chen R, Ye M, Zou H. J Sep Sci. 2010;33:1879–1887. doi: 10.1002/jssc.200900718. [DOI] [PubMed] [Google Scholar]
- 30.Fang X, Wang W, Yang L, Chandrasekaran K, Kristian T, Balgle BM, Lee CS. Electrophoresis. 2008;29:2215–2223. doi: 10.1002/elps.200700609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei X, Herbst A, Ma D, Aiken J, Li L. J Proteome Res. 2011;10:2687–2702. doi: 10.1021/pr2000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilar M, Olivova P, Daly AE, Gebler JC. Anal Chem. 2005;77:6426–6434. doi: 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- 33.Pinkse MWH, Uitto PM, Hilhorst MJ, Ooms B, Heck AJR. Anal Chem. 2004;76:3935–3943. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]