

Abstract
Acute lung injury due to influenza infection is associated with high mortality, an increase in neutrophils in the airspace, and increases in tissue myeloperoxidase (MPO). Because IL-17A and IL-17F, ligands for IL-17 receptor antagonist (IL-17RA), have been shown to mediate neutrophil migration into the lung in response to LPS or Gram-negative bacterial pneumonia, we hypothesized that IL-17RA signaling was critical for acute lung injury in response to pulmonary influenza infection. IL-17RA was critical for weight loss and both neutrophil migration and increases in tissue myeloperoxidase (MPO) after influenza infection. However, IL-17RA was dispensable for the recruitment of CD8+ T cells specific for influenza hemagglutinin or nucleocapsid protein. Consistent with this, IL-17RA was not required for viral clearance. However, in the setting of influenza infection, IL-17RA−/− mice showed significantly reduced levels of oxidized phospholipids, which have previously been shown to be an important mediator in several models of acute lung injury, including influenza infection and gastric acid aspiration. Taken together, these data support targeting IL-17 or IL-17RA in acute lung injury due to acute viral infection.
A cute lung injury (ALI)2 is a severe clinical state, characterized by noncardiogenic pulmonary edema, capillary leak, and hypoxemia. It can be triggered by both infectious and noninfectious stimuli. The most severe form of ALI is acute respiratory distress syndrome (ARDS) (1). Most patients who died in the severe acute respiratory syndrome virus outbreak in 2003 developed ARDS (2). Likewise, the Spanish influenza pandemic of 1918 and current outbreaks of H5N1 influenza have both been documented to cause ARDS in mice and primates (3– 6). Neutrophils play an important part in the pathogenesis of ALI, in that they rapidly infiltrate the lung and are an important source of proinflammatory cytokines such as IL-1β and TNF-α as well as reactive oxygen intermediates (ROI; Refs. 7–10).
The role of neutrophils in severe influenza infections has been debated. Studies using the reconstructed 1918 virus show that highly virulent strains of influenza result in an overwhelming neutrophilic infiltrate that is associated with worsening lung injury (5, 6, 11). However, neutrophils have also been shown to play a role in control and clearance of influenza virus in experimental models (12–16), and neutrophil depletion resulted in an uncontrolled and ultimately lethal viremia in the reconstructed 1918 influenza model (11). However, neutrophil depletion using the mAb RB6-8C5 is complicated by the concurrent depletion of other monocytes, dendritic cells, and even some CD8+ T cells (17–19).
Recent studies have shown that oxidized phospholipids have many proinflammatory properties (20) and are an important signal in the development of ALI. Upon oxidation, airway phospholipids are capable of signaling through TLR4. This signal is MyD88 independent and involves the Toll/IL-1R domain-containing adapter-inducing IFN-β (TRIF)-TNFR-associated factor-6 signaling pathway, and blocking this signal can prevent ALI. This role for oxidized phospholipids in the pathogenesis of ALI has been shown for both infectious (influenza) and noninfectious (acid aspiration) models (21). Given the ability of neutrophils to generate ROIs, it is possible that these cells are responsible for the oxidation of airway phospholipids.
IL-17 is a potent regulator of the neutrophil response. Both IL-17A and IL-17F are capable of signaling through IL-17RA to induce granulopoietic factors such as G-CSF and stem cell factor which leads to expansion of neutrophil progenitors in the bone marrow and spleen, as well as expansion of mature neutrophils in peripheral blood (22, 23). Further, IL-17RA signaling up-regulates production of neutrophilic chemokines such as CXCL1 (KC), CXCL2, CXCL5, CXCL6, and CXCL8, which in turn regulate neutrophil emigration to mucosal sites (24 –26). Due to this role in neutrophil expansion and recruitment, IL-17RA signaling is an important part of the host response to extracellular bacterial infections. It has previously been demonstrated that defects in the IL-17 axis result in a decreased neutrophil response associated with higher bacterial burdens and decreased survival when mice are challenged with the Gram-negative bacterium Klebsiella pneumoniae (24, 27).
Given the critical role of IL-17RA in neutrophil recruitment and the ability of neutrophils to generate ROIs, along with the important role oxidized phospholipids play in the pathogenesis of ALI, we wanted to determine whether blocking IL-17RA signaling conferred protection against ALI. Using an acute infection model, influenza A/PR/8/34 (H1N1)(PR/8), we sought to identify a pathway by which the neutrophilic infiltrate is able to be regulated to reduce lung injury.
Materials and Methods
Animal infections and HCl challenge
IL-17ra−/− on a BALB/CJ background (28) and C57BL/6 (B6) background (24), p55/p75−/− on a B6.129 background (29), and γ/σ−/− mice on the B6 background (30) have been previously described. Wild-type controls included BALB/c, B6.129, and C57BL6 mice, respectively. Mice were challenged intranasally with an LD90 of influenza A/PR/8/34 (H1N1). Following infection, mice were monitored daily for weight loss and signs of clinical illness. Mice were sacrificed at the indicated time points. For HCl challenge, mice were given 100 μl of HCl (pH 1.5) intratracheally and sacrificed at 24 h postchallenge. Control mice received equivalent volumes of PBS.
Lung harvest and bronchoalveolar lavage
At indicated time points, mice were euthanized with an overdose of isofluorane. The trachea was exposed and cannulated. One milliliter of sterile PBS was injected into the airway and then collected. Animals were then perfused with PBS via injection in the right ventricle to flush blood vessels in the lung. Left lungs were collected in TRIzol reagent and homogenized using an electric tissue grinder. RNA was purified according to the manufacturer’s instructions. Right lungs were collected and homogenized in PBS for use in plaque assays and ELISA. Lavage fluid was spun down at 500 × g for 5 min to pellet cells. Bronchoalveolar lavage (BAL) cells were counted using a Coulter Counter (BD Biosciences), and 105 cells were spun onto a glass slide, then fixed and stained for counting.
IL-6 neutralization studies
A mAb capable of neutralizing IL-6 (clone MP5-20F3; BD Pharmingen) was administered to mice via iv injection at a concentration of 3 μg/g of body weight at the time of influenza challenge. A second dose was administered at 3 days postinfection (dpi). Mice were monitored daily and sacrificed at 6 dpi. To ensure that sufficient neutralizing Ab was delivered, lung homogenates were used in a direct ELISA. Briefly, 96-well ELISA plates were coated with 100 ng of rIL-6 at room temperature for 4 h. Plates were washed four times, blocked overnight with 1% BSA at 4°C, and then washed four times again. Lung homogenates were added at 100 μl/well, incubated for 2 h at room temperature, and then washed four times, followed by detection with anti-IgG1-HRP. Plates were developed using tetramethylbenzidine substrate reagents (BD Biosciences).
Real-time PCR
RNA collected from the left lung was used as template to generate cDNA using iScript reagents and protocol (Bio-Rad). cDNA was then used in fast real-time PCR using the Applied Biosystems 7900HT. Reaction conditions were 95°C for 1 s and 60°C for 20 s, repeated for 40 cycles, with a 20-s hot start at 95°C. Primer and probe sequences were as follows: IL-17A forward, 5′-GCTCCAGAAGGCCCTCAG A-3′, IL-17A reverse, 5′-TCCC TCCGCATTGACA-3′; IL-17A probe, 5′-TCAACCGTTCCACGTCAC CCTG-3′, IL-17F forward, 5′-AGGGCATTTCTGTCCCACGTGAAT-3′, IL-17F reverse, 5′-GCATTGATGCAGCCTGAGTGTCT-3′, IL-17R probe, 5′-CATGGGATTACAACATCACTCGAGACCC-3′; influenza M protein forward, 5′-GGACTGCAGCGTAGACGCTT-3′, influenza M protein reverse, 5′-CATCCTGTTGTATATGAGGCCCAT-3′, influenza M protein probe, 5′-CTCAGTTATTCTGCTGGTGCACTTGCCA-3′. 18s RNA was used as a housekeeping gene with the following primer/probe sequences: forward, 5′-GATCCATTGGAGGGCAAGTCT-3′; reverse, 5′-GCAGCAACTTTAATATACGCTATTGC-3′; probe, 5′-TGCCAGCAGC CGCGGTAATTC-3′. Fold-change in mRNA was quantified using the ΔΔCt method.
Measurement of cytokines
IL-17A and IL-17F in lung homogenate were measured by ELISA (R&D DuoSet) according to the manufacturer’s instructions. All other cytokines were measured by multiplex analysis using Luminex (Millipore) on the Bioplex reader (Bio-Rad).
IL-17A ELISpot
For ELISpot, lungs were collected in PBS and dissected into 1-mm pieces under sterile conditions. They were then digested with collagenase and DNase for 45 min at 37°C. The digest was passed through a 40-μm pore size filter, RBC were lysed, and cells were washed three times with PBS. Cells were plated at 1 × 105 cells/well on an IL-17A ELISpot plate (eBioscience) in DMEM and stimulated with 50 ng/ml PMA and 750 ng/ml ionomycin overnight. Plate was developed according to the manufacturer’s instructions.
Assessment of lung injury
Total protein in the BAL fluid (BALF) was measured using the BCA Protein Assay Kit (Pierce Chemical). Lactate dehydrogenase (LDH) activity was measured using the LDH assay (Sigma-Aldrich). Both assays were performed in a 96-well plate for 30 min according to manufacturer’s instructions and analyzed using the Benchmark Plus plate reader (Bio-Rad).
Assessment of T cell response and intracellular cytokine staining
For intracellular cytokine staining, lungs were collected in PBS and dissected into 1-mm pieces under sterile conditions. They were then digested with collagenase and DNase for 45 min at 37°C. The digest was passed through a 40-μm pore size filter, RBC were lysed, and cells were washed three times with PBS. Cells were plated at a concentration of 1–2 × 106 cells/well in a sterile 96-well round-bottom plate and stimulated overnight in DMEM with 50 ng/ml PMA and 750 ng/ml ionomycin at 37°C. Brefeldin A was added during the last 6 h of incubation. Cells were then washed and resuspended in PBS plus 2% FBS, and FcRs were blocked. Cells were then stained with fluorochrome-conjugated mAbs for TCR β-chain, TCR δ-chain, CD4, and CD8, along with nucleocapsid protein (NP)-specific (TYQRTRALV) and hemagglutinin (HA)-specific (IYSTVASSL) pentamer in the dark, on ice, for 30 min. After staining, cells were washed before fixation and permeabilization. Intracellular staining was performed with fluorochrome-conjugated mAb directed toward IFN-γ or IL-17A.
Lung fixation and histological examination
Animals were sacrificed at the indicated time points, and the trachea was exposed and cannulated. Lungs were inflated with 10% neutral buffered formalin (Sigma-Aldrich) for 10 –15 min, removed from the animal, and placed in fresh 10% neutral buffered formalin for at least 24 h at 4°C before processing and embedding. Sections were cut (4 μm) and stained with H&E for histopathological evaluation.
Slides were blinded and scored by Dr. D. A. Pociask in a semiquantitative manner according to the relative degree of inflammatory infiltration. Inflammation was scored as follows: 0, no inflammation; 1, perivascular cuff of inflammatory cells; 2, mild inflammation, extending throughout <25% of the lung; 3, moderate inflammation covering 25–50% of the lung; 4, severe inflammation involving over one-half of the lung.
Measurement of oxidized phospholipids
Oxidized phospholipids were detected by direct ELISA. Briefly, BALF was adsorbed onto 96-well ELISA plates, normalized for 100 ng of total protein per well, overnight at 4°C. Plates were washed four times with PBS plus 0.5% Tween 20 before blocking with 1% BSA for 1 h at room temperature. Plates were then washed four times again and probed with 100 μl of the mAb EO6 (a gift from Dr. J. L. Witztum; see also Ref. 31), diluted to 5 μg/ml in PBS. Plates were incubated for 2 h at room temperature and then washed four times. Anti-IgM conjugated to HRP (Southern Biotech) was used as a secondary Ab, diluted 1/2000, for 2 h at room temperature. Plates were washed once more before being developed with tetramethylbenzidine substrate reagents (BD Biosciences). Optical density was read at 450 nm, with a wavelength correction at 540 nm subtracted out.
Plaque assay
Influenza titers were measured by traditional plaque assay. Briefly, lungs were homogenized, and homogenates were diluted before being applied to 95% confluent Mardin-Darby canine kidney cells. Virus was adsorbed onto the cells for 1 h at room temperature before being washed off four times with serum-free DMEM. Cells were then covered with serum-free DMEM containing trypsin and 0.8% agarose and incubated for 48 h at 37°C with 5% carbon dioxide. Agarose was then removed, and the monolayer was stained with 1% gentian violet to allow for the visualization and counting of plaques.
Statistics
All data are presented as the mean ± SEM. Significance was tested using unpaired t tests, ANOVA, or nonparametric tests where appropriate, or, in the case of survival curves, Kaplan-Meier analysis. All statistics were analyzed using GraphPad Prism 4 software.
Results
IL-17 is induced during influenza expression
Among IL-17 family members, IL-17A and IL-17F share the greatest homology, and both are capable of signaling through IL-17RA, although IL-17F binds with a much lower affinity (32). BALB/c mice were challenged with 100 PFU of influenza A/PR/8/34 (H1N1), and following challenge we were able to detect an increase in IL-17A (Fig. 1A) and IL-17F (Fig. 1B) mRNA in lung tissue compared with baseline as early as 2 dpi. This increase in mRNA remains detectable through 7 dpi. We sought to confirm this increase in mRNA by measuring protein levels. IL-17A (Fig. 1C) and IL-17F (Fig. 1D) were detectable by ELISA from 2 dpi through 7 dpi. Beyond 7 dpi, there was significant mortality among BALB/c mice. Thus, to avoid survival bias in data analysis, we chose to use an early time point (2 dpi) and a late time point (6 dpi) for our studies. To gauge the number of cells producing IL-17, we performed an ELISpot on lung digests at 2 and 6 dpi. We found that the population of IL-17A-producing cells was relatively small and that the size of this population was not statistically different between the early and later time points (Fig. 1E).
FIGURE 1.

IL-17A and IL-17F are expressed in response to influenza. Time course, in BALB/cJ mice, of IL-17A (A and C) and IL-17F (B and D) following influenza challenge. mRNA was detected by quantitative real-time PCR and is expressed relative to naive levels, with all time points having a p value of <0.05 compared with naive controls. Protein levels were detected by ELISA, and all time points had a p value of <0.05 compared with naive controls. E, The number of IL-17A-producing cells was quantified by ELISpot. *, p = 0.016; **, p = 0.003, n = 4/time point.
Th17 cells, a subset of CD4+ T cells, are a main producer of IL-17A in many models (33–38). However, in the influenza model, Ag-specific CD4+ T cells do not appear in the lung until 4 dpi or later (39). Because we observed IL-17A (and IL-17F) as early as 2 dpi, it is likely that there is another cellular source of these cytokines in the early part of the infection. We hypothesized that γδ T cells might be the early source of IL-17A and IL-17F, because they have also been shown to be a major source of IL-17 (35). To test this hypothesis, we challenged mice with influenza and performed intracellular cytokine staining for IL-17A 2 and 6 days postinfection. We found that following PMA-ionomycin re-stimulation, ~7% of γδ T cells stained positive for IL-17 and that this increased to >15% of the γδ gate by 2 dpi, and >20% by 6 dpi (Fig. 2A). The size of the entire γδ population increases from <105 cells in a naive lung to >106 cells by 6 dpi (Fig. 2B). In contrast, very few αβ T cells from naive lungs stained positive for IL-17 (<0.6%), and this percentage did not change following influenza challenge (Fig. 2C). This leads us to conclude that during the first week of influenza challenge, the primary producers of IL-17 are γδ T cells.
FIGURE 2.

Production of IL-17 by γδ T cells. Intracellular cytokine staining following influenza challenge demonstrates an increasing percentage of IL-17+ γδ T cells (A), even as the total population of γδ T cells expands (B) Percentage of IL-17+ αβ T cells is unchanged compared with naive mice (C) Challenge of γδ knockout mice results in lower levels of IL-17A and IL-17F message, expressed as fold change from naive, at 6 dpi (D). *, p = 0.03; **, p = 0.02, n = 8/time point.
We next sought to confirm this by challenging γδ knockout mice (30), along with C57BL/6 control mice, and measured levels of IL-17 mRNA at 2 and 6 days postinfection. The induction of IL-17A and IL-17F mRNA was not as strong in the C57BL/6 strain as in the BALB/c strain at the earlier time point. However, we observed significantly lower levels of both IL-17A and IL-17F mRNA at 6 dpi (Fig. 2D), providing further evidence that γδ T cells are a critical source of IL-17 throughout the first 6 days on influenza challenge. However, others have reported the presence of IL-17+CD8+ T cells later in the infection (40); therefore, it is possible that as the adaptive immune response expands, other cell types increasingly contribute to the production of IL-17, although this falls beyond the timing of our lethal model.
Decreased morbidity and mortality among IL-17RA knockout mice
To examine the effects of IL-17 signaling during influenza infection, we challenged IL-17RA knockout mice on a BALB/cJ background (28) and BALB/cJ controls with 100 PFU of PR/8. Following viral challenge, we noted that the IL-17RA-deficient mice experienced less weight loss throughout the course of infection (Fig. 3A). This correlated well with higher survival rates among the knockout mice (Fig. 3B), suggesting that IL-17RA signaling may be detrimental in the response to influenza virus challenge. It is important that the differences in weight loss were significant from days 2 through 8, beyond which the data are subject to survival bias. To rule out the possibility that the morbidity and mortality differences that we observed were due to differences in viral clearance, we measured viral burden by real-time PCR, using primers and probe for the influenza M1 gene. Somewhat surprisingly, we found that despite their decrease in morbidity and mortality, the IL-17RA knockout mice had a trend toward a higher viral burden at 6 dpi (Fig. 3C). This data trend was also supported by plaque assay (supplemental Fig. 1).3 Taken together, these data show that despite a trend toward higher viral burdens, IL-17RA knockout mice are able to better tolerate influenza challenge, because they ultimately clear the virus and recover from the challenge. This suggests that these mice may have decreased immunopathology that would explain the observed phenotype.
FIGURE 3.
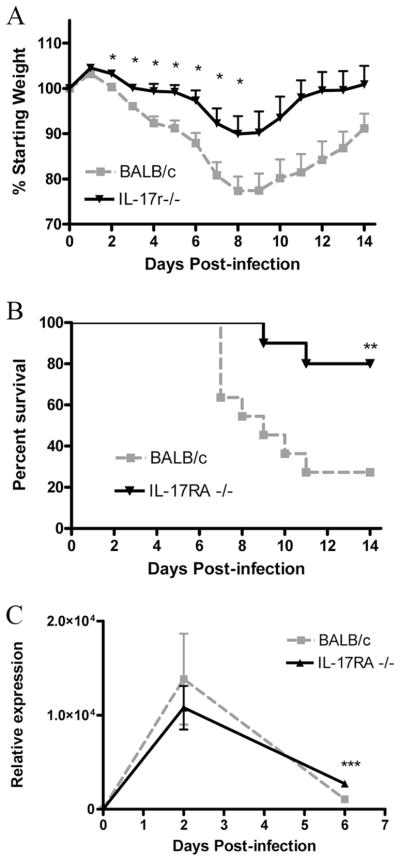
Weight loss and mortality following challenge with influenza A/PR/8. Weight loss (A) and mortality (B) were assessed daily following viral challenge (n = 10/group). Viral burden (C) was determined by quantitative PCR for the influenza M1 protein (n = 4/group/time point). *, p < 0.05; **, p = 0.0067; ***, p = 0.0093.
IL-17RA knockout mice have lower levels of inflammation
Histological examination of lungs at 6 dpi showed substantial decreases in inflammation among the IL-17RA knockout mice (Fig. 4, A and B). Lungs were scored on the basis of the degree and extent of inflammation (41). By day 2, both the wild-type and knockout mice exhibit limited inflammation, mainly restricted to the perivascular areas (Fig. 4C). However, by day 6, wild-type mice showed heavy perivascular inflammatory infiltrates, with inflammation extending out into the parenchyma (Fig. 4, A and C). In contrast, the IL-17RA knockout mice had only mild inflammation mainly limited to the perivascular bed, with the lung parenchyma relatively unaffected.
FIGURE 4.
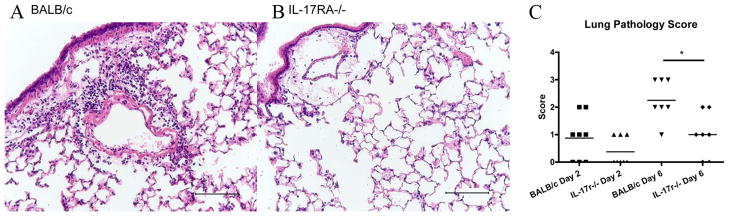
Differences in inflammation following influenza challenge. BALB/c (A) and IL-17RA−/− (B) lungs at 6 dpi, inflated with 10% formalin, paraffin-embedded, and H&E stained. Calibration bar, 100 μm. Pathology scored (C) as in Lung fixation and histological examination. *, p = 0.01.
Given these histological findings, we sought to measure the levels of inflammatory cytokines in lung homogenates. TNF-α (Fig. 5A) and IL-1β (Fig. 5B), both of which can be produced by neutrophils in ALI (9), trended lower in IL-17RA−/− mice. However, IL-6 was greatly reduced in the IL-17RA knockout mice compared with wild-type mice (Fig. 5C). Additionally, levels of the IL-17-responsive chemokines and cytokines, including KC and G-CSF (Fig. 5, D and E) were lower in the IL-17RA knockout mice. Importantly, we also observed a significant decrease in IFN-γ at 6 dpi among the IL-17RA knockout mice (Fig. 5F). These cytokine data support the histological findings of less inflammation in the IL-17RA-deficient mice.
FIGURE 5.

Cytokine response to influenza challenge. Cytokines measured at 2 and 6 dpi. BALB/c (□) vs IL-17RA−/− (■). *, p < 0.05 (n = 4/group/time point; A).
We next sought to determine whether this decrease in inflammation resulted in lower levels of lung injury. Measurement of total protein in the BALF suggests decreased capillary leak in the IL-17RA knockout mice (Fig. 6A). Moreover, consistent with less lung injury, levels of LDH in the BALF was significantly lower in IL-17RA knockout mice (Fig. 6B). In the context of lower levels of inflammation, these data suggest that the protection seen in the IL-17RA knockout mice may be due to lower levels of immunopathology.
FIGURE 6.

Markers of lung injury. Total protein (A) and LDH activity (B) in the BALF. □, BALB/c mice; ■, IL-17RA−/− mice. *, p = 0.01; **, p = 0.01 (n = 4/group/time point).
Protection is independent of TNF-α and IL-6
Previously published studies have identified a role for TNF-α in pulmonary immunopathology using a transgenic model of influenza infection, as mice lacking TNFR p55 had decreased mortality and weight loss (42, 43). Because we observed a decrease in TNF-α levels in our knockout mice, we sought to determine whether reduced TNF production was responsible for the protective phenotype that we observed in IL-17RA−/− mice. To investigate this, TNFR p55/p75 double-knockout mice (29) were challenged with an LD90 of PR/8. These mice are commercially available, but have only been backcrossed four times; therefore, B6.129 mice were used as controls. Additionally, we infected IL-17RA−/− mice on a C57BL/6 background (24) along with wild-type controls to demonstrate that the phenotype described was not strain specific. In contrast to the IL-17RA−/− mice, TNFR p55/p75 double-knockout mice showed significant weight loss which was similar to observations in wild-type mice (Fig. 7A). Moreover, in contrast to IL-17RA−/− mice, TNFR p55/p75 double-knockout mice had similar levels of total protein in the BALF (Fig. 7B), compared with wild-type control mice. Additionally, levels of LDH were unchanged (supplemental Fig. 2A). This suggests that the phenotype observed in the IL-17RA knockout mice is independent of TNFR signaling. It is also important that G-CSF was unchanged and KC was elevated in the double knockouts (supplemental Fig. 2B), indicating that the reductions in these growth factors and chemokines among IL-17RA−/− mice was not TNF dependent.
FIGURE 7.

Protection is not dependent on TNF-α or IL-6. A, Weight loss among B6.129 and p55/p75 (n = 6/group) following influenza challenge. For comparison, IL-17RA−/− on a C57BL/6 background and wild-type controls (n = 4/group) are also provided. B, Lung injury assessed by total protein in the BALF on day 6. C, BALB/c mice were treated with either anti-IL-6 (n = 4) or control IgG (n = 4) before challenge and again on 3 dpi, weight loss monitored for 6 days and lavaged on day 6 (D) to measure total protein in the BALF. *, p < 0.05.
Similarly, we chose to test whether the decrease in IL-6 levels in the IL-17RA mice was responsible for this phenotype. Anti-IL-6 or control IgG was administered to BALB/c mice at the time of influenza challenge and again at 3 dpi, at a dosage of 3 μg/g body weight. This Ab has been characterized by the manufacturer as capable of neutralizing recombinant murine IL-6 activity by >95% at concentrations between 0.5 and 2.0 μg/ml, and has been successfully used by other groups to neutralize IL-6 in other models (44, 45). In our model, the anti-IL-6 treated mice showed no differences in weight loss compared with controls (Fig. 7C), nor did they show any differences in total protein in the BALF at 6 dpi (Fig. 7D). Likewise, levels of LDH were unchanged (supplemental Fig. 3A). There were no differences in G-CSF or KC after IL-6 neutralization (supplemental Fig. 3, B and C), illustrating that these proteins are also independent of IL-6. To rule out the possibility that the dosage of neutralizing Ab was insufficient, IL-6 was measured in lung homogenate at 6 dpi and found to be drastically reduced (supplemental Fig. 3D). Additionally, free anti-IL-6 was measured in the lung homogenates, and it was found that free concentrations of neutralizing Ab were present in excess of IL-6 (supplemental Fig. 3E). This, combined with previous reports using this Ab at comparable dosages, suggests that the dosage of Ab used in our studies was sufficient. Taken together, these data indicate that the decrease in IL-6 levels observed in the IL-17RA knockout mice is not responsible for the reduced cachexia.
IL-17RA mice have reduced neutrophil emigration and lipid oxidation
IL-17 is responsible for the induction of several granulopoietic factors and chemokines that can result in an influx of neutrophils (24, 26). Examination of the cells recovered in the BALF demonstrated that the IL-17RA knockout mice had a drastic reduction in the number of neutrophils recruited to the airway (Fig. 8A). In contrast, BAL macrophages and lymphocytes were not significantly reduced (Fig. 8A). Other groups have previously reported that CCR2+ monocyte recruitment to the lung is capable of inducing immunopathology (46 – 48), particularly through TRAIL-mediated apoptosis of epithelial cells (49). However, because we observed minimal differences in macrophage recruitment, coupled with no significant decrease in CCR2 ligand MCP-1 (supplemental Fig. 4), it is unlikely that this mechanism contributes significantly to the observed phenotype in the IL-17RA knockout mice.
FIGURE 8.

Differences in neutrophils and oxidized phospholipids. A, IL-17RA−/− mice (■) show decreases in the number of neutrophils in the BAL compared with BALB/c controls (□), but no differences in macrophages or lymphocytes (n = 4/group/time point). B, Pentamer staining at 10 dpi show that there is no difference in the numbers of HA-specific or NP-specific CD8+ T cells (n = 8/group). C, The numbers of IFN-γ positive CD4+ and CD8+ T cells are decreased in the IL-17RA−/− mice at 10 dpi. n = 4/group; *, p = 0.02; **, p = 0.005. D, MPO activity is decreased in the lung homogenates, and trends lower in the BALF, of the IL-17RA−/− mice compared with wild-type (WT) controls (n = 5/group/time point). *, p = 0.03; **, p = 0.01. E, Levels of oxidized phospholipids, measured by ELISA, are lower in IL-17RA−/− mice than in BALB/c controls. n = 5/group/time point.***, p = 0.02.
Further, examination of the HA- and NP-specific CD8+ T cell population revealed no differences at 10 dpi, indicating that the recruitment of these cells is unaffected in the IL-17RA knockout animals (Fig. 8B). At 6 dpi, there were also no differences in the NP-specific CD8+ T cells (BALB/c: 8.6 × 104 ± 1.0 × 104, IL-17RA−/−: 6.9 × 104 ± 1.5 × 104) and HA-specific cells were not detected. Further, because we detected a difference in the amount of IFN-γ in the lung, we chose to explore whether there might be a difference in production of IFN-γ by CD8+ or CD4+ T cells. By intracellular staining, we found statistically significant decreases in the number of both CD8+IFN+ and CD4+IFN+ cells (Fig. 8C). This suggests an inhibition of IFN-γ production and/or a biasing away from Th1/Tc1 polarization in the IL-17RA−/− mice.
Excessive neutrophil recruitment has been demonstrated in models of severe influenza infections, including H5N1 and a reconstructed 1918 virus, and is associated with more severe immunopathology (5, 11). Because of this, we chose to study the differences in neutrophils more closely. The activity of MPO, an enzyme found most abundantly in neutrophils and responsible for generating reactive oxygen species that can lead to oxidative damage (50), was decreased in lung homogenates of the IL-17RA-deficient mice when compared with BALB/c controls. Further, MPO activity trended to be lower in the BALF cell pellets of these animals as well (Fig. 8D). On the basis of these findings and the established role of oxidized phospholipids in the pathogenesis of acute lung injury (21), we hypothesized that the IL-17RA−/− mice might be accumulating fewer oxidized phospholipids, which would in turn explain the decrease in lung injury seen in these mice during influenza infection.
To measure oxidized phospholipids, we adsorbed BALF onto 96-well ELISA plates, normalized for the amount of protein in the sample. We then probed with EO6, a mAb that specifically recognizes the phosphocholine head of phospholipids that contain an oxidized sn2 side chain but does not recognize nonoxidized phospholipids (31). It has been previously demonstrated that oxidized phospholipids that are recognized by EO6 are proinflammatory (51). We found that the IL-17RA knockout mice had significantly decreased levels of oxidized phospholipids (Fig. 8E) in BALF compared with wild-type mice at 6 dpi. Because oxidized phospholipids have previously been shown to play a critical role in acute lung injury in both acute viral infections as well as in acid aspiration, we investigated whether IL-17RA signaling was also critical for the generation of oxidized phospholipids and lung injury in acid aspiration.
To test this hypothesis, we chose to examine a model of aspiration pneumonia wherein hydrochloric acid is instilled into the airway. After HCl challenge in the airway, IL-17RA−/− mice had a significant decrease in neutrophil influx at 24 h postchallenge (supplemental Fig. 5A) as well as a decrease in the amount of oxidized phospholipids (supplemental Fig. 5B) in BALF.
We next decided to specifically test the role of neutrophils in lipid oxidation and lung injury by depleting neutrophils in the HCl aspiration model using anti-Gr1. As stated above, anti-Gr1 has been shown to also deplete a subpopulation of dendritic cells as well as CD8+ T cells (18, 19), making it difficult to interpret data in the influenza model; however, due to the short time course of HCl model, depletion of other subsets would less likely play a role apart from depletion of neutrophils. Administration of anti-Gr1 24 h before HCl challenge resulted in a highly significant decrease in neutrophils in the BALF at 24 h postchallenge similar to the IL-17RA knockout mice (supplemental Fig. 4A). Importantly, anti-Gr1 also resulted in a significant decrease in the level of oxidized phospholipids in the BALF (supplemental Fig. 4B), indicating that neutrophils are important in generating oxidized phospholipids, and that the decrease in neutrophils in our model is sufficient to explain the decrease in the accumulation of oxidized phospholipids in the airways.
Discussion
The data presented in this report provide support for the concept that IL-17RA signaling plays a key role in the process of lung injury. We observed that IL-17RA knockout mice recruited fewer neutrophils to the airway in response to challenge with either influenza A virus or hydrochloric acid and that this decrease in neutrophils results in lower amounts of oxidized phospholipids. Others have previously shown that oxidized phospholipids play a key role in acute lung injury by serving as an endogenous ligand for TLR4 and signaling through the TRIF-TNFR-associated factor-6 pathway (21). Our data show that IL-17RA signaling regulates the amount of oxidized phospholipids in the airway, likely by regulating tissue burdens of neutrophils, which are critical sources of reactive oxygen species.
IL-17RA-deficient mice are more susceptible to certain extra-cellular bacterial infections of the lung (27, 52), in part due to defective neutrophil migration into the lung. However, the role of IL-17RA in primary pulmonary influenza host response has not been evaluated. Because IL-17 has been shown to regulate ligands for CXCR2 such as KC and LIX (critical for neutrophil recruitment) as well as the ligands for CXCR3, IFN-γ-induced monokine, and IFN-γ-inducible protein-10 (important for Th1 cell recruitment; Ref. 53), it was important to determine whether IL-17RA signaling was critical for viral clearance and virus-specific CD8+ T cell recruitment. Additionally, CD8+ T cells are a contributor to lung damage in response to influenza challenge (54, 55). Although IL-17RA−/− mice showed a trend toward higher viral burdens on day 6, these mice ultimately cleared the virus and recovered. Consistent with these findings, there was no significant differences in recruitment of virus specific CD8+ T cells either for NP on day 6 or 10 or HA on day 10. For these latter experiments, it was necessary to challenge mice with a sublethal dose (so as to avoid survival bias) and examine the Ag-specific population at 10 dpi because CD8+ T cells do not peak until 10 dpi. There is a difference in IFN-γ+CD8+ T cells, suggesting that there may be an inhibition of CD8+ T cells to produce IFN-γ, or perhaps a biasing toward other Tc subsets. It is tempting to say that this may contribute to the slight delay in viral clearance, but other studies have demonstrated little requirement for IFN-γ in efficient clearance of primary influenza challenge (56, 57).
The slightly higher viral titers in the IL-17RA−/− mice were not unexpected, given that it has previously been shown that neutrophil depletion has a deleterious effect on clearance of influenza (11, 15, 16). However, this seemed to be in apparent conflict with the fact that they had decreased morbidity and mortality. This can be explained by studies that have demonstrated that during highly virulent influenza infections, excessive neutrophil infiltrates were associated with more immunopathology (11). This led us to investigate differences in inflammation and lung damage as a mechanism for the survival phenotype we observed.
We observed that the IL-17RA knockout mice did indeed have less lung injury, as indicated by less total protein and LDH activity in the BALF. Upon histological examination, it was apparent that the IL-17RA knockout mice had a substantial decrease in the amount of inflammation in the lung. This pathology was less severe than pathology described in fatal human influenza infections (58). One reason for this may be differences in the PR/8 model, in which perivascular and peribronchial inflammation is common, whereas human influenza is marked by alveolar infiltrates and bronchial desquamation. More importantly, we collected histopathology at 6 dpi to coincide with morbidity as measured by weight loss, but also to avoid survival bias, as mortality became an issue at 7 dpi and later. In contrast, the human pathology that has been described come mainly from autopsy material, and may often be complicated by secondary bacterial infections (58).
The differences in inflammation between wild-type and IL-17RA-deficient mice correlated well with differences in many inflammatory cytokines. Importantly, TNF-γ, IL-1γ, and IL-6, three proinflammatory cytokines that are generally elevated in severe influenza infection as part of a cytokine storm (5, 59), all either trended lower or were significantly reduced in the IL-17RA−/− animals. One potential explanation for the reduced levels of TNF-α and IL-1β is that neutrophils are a source of both TNF-α and IL-1β (8, 9, 60). Because the cytokine storm has been hypothesized to play a role in the immunopathology of the more virulent influenza infections and the fact that in certain models of arthritis, IL-17 has a TNF-α- and IL-1β- independent role in tissue inflammation (61, 62), we chose to examine whether TNF-α or IL-6 could account for the reduced lung injury and cachexia that we observed in IL-17RA−/− mice.
Neutralization of IL-6 or genetic ablation of TNF p55/p75 receptor signaling failed to reduce acute lung injury or cachexia associated with influenza infection, demonstrating that the reduced lung injury in IL-17RA−/− mice is independent of the differences observed in the levels of these cytokines. The lack of a role for TNFR signaling is in apparent contrast to similar experiments that have previously been reported using HA-transgenic models of influenza. In those studies, loss of p55 conferred a protective effect when HA-specific CD8+ T cells were transferred into HA-transgenic hosts (43). The differences between those experiments and our data may be due to the cellular source and timing of TNF production. In the transgenic experiments, adoptively transferred CD8+ T cells are the source of the TNF-α. In our infectious model, CD8+ T cells exist at low numbers throughout the first 6 days of infection and thus are less likely to contribute to our phenotype. Further, another report examining the role of TNF-α found no phenotype in p55/p75 double knockouts following challenge with H5N1 (59).
Having eliminated the possibility of a decreased cytokine storm and decreased CD8+ T cells as causes of the protection, we next investigated the differences in neutrophil recruitment. IL-17 plays a key role in neutrophil proliferation and migration, primarily by induction of granulopoietic factors and chemokines (22–26, 63). Further, it has been demonstrated that genetic ablation of CXCR2 results in decreased neutrophil migration to the lung in response to influenza (64), and IL-17 is responsible for the regulation of some CXCR2 ligands (24). We found that differences in KC were most apparent at 2 dpi whereas differences in G-CSF were detected at 6 dpi. This may be due to the fact that IL-17 regulates chemokine gene expression, including KC, through stabilization of mRNA (65), whereas its effects on G-CSF might be at the level of transcription (66). Therefore, in the IL-17RA−/− model, it is reasonable to expect that changes in chemokine levels due to deficiencies in IL-17 would be observed sooner than changes in G-CSF.
Consistent with the role that IL-17 signaling plays in promoting a neutrophil response, we observed reduced lung neutrophils following influenza infection in the IL-17RA knockout mice. Considering the mechanisms by which neutrophils are capable of causing tissue damage, one likely mechanism of lung injury is through the generation of reactive oxygen species and, specifically, the oxidation of phospholipids (21).
In support of that hypothesis, we found reduced MPO activity in both the BAL cell pellet and lung homogenate in IL-17RA knockout mice as well as a significantly lower level of oxidized phospholipids in the BALF. The combined decrease in neutrophil emigration and oxidized phospholipids is a likely explanation for the decrease in lung injury in the setting of influenza infection.
To confirm the role of IL-17RA signaling and the role of neutrophils in the generation of oxidized phospholipids we used a non-infectious model of acid aspiration, Again, in this model IL-17RA was required for neutrophil recruitment, generation of oxidized phospholipids, and ultimately, higher levels of lung injury. The specific role of neutrophils in the generation of oxidized phospholipids in this model was shown using anti-Gr1 depletion of neutrophils. Taken together, these data strongly implicate IL-17RA signaling in neutrophil recruitment and the generation of oxidized phospholipids in both influenza and acid aspiration. Moreover, in the setting of influenza infection, lung injury due to IL-17RA signaling is independent of TNF-α or IL-6. These data support the potential therapeutic manipulation of IL-17 or IL-17RA in acute lung injury.
Somewhat related, it has also previously been shown that TLR3-deficient mice have a similar phenotype with decreased inflammation, decreased weight loss, and decreased mortality despite slightly higher viral titers (67). Similar to oxidized phospholipids signaling through TLR4, TLR3 signals through TRIF and not MyD88 (68). This provides further evidence that TRIF signaling may play a key role in ALI.
IL-17 signaling has not been extensively studied in viral models, despite the fact that the cytokine has been detected in the setting of viral infection (69, 70). A recent report has described the generation of IL-17+CD8+ T cells (termed Tc17 cells) in response to influenza. These cells, when generated in vivo and transferred to mice before influenza challenge, were protective, although these cells lacked granzyme B and perforin. Additionally, in apparent contrast to our study, Ab neutralization of IL-17 reduced protection from heterosubtypical challenge (40). This may be due to a role for IL-17 in the memory response, something that we do not examine in the present study.
Although our mouse model of influenza A/PR/8 infection is well characterized, it is important that it may differ from infection with other strains or from human influenza. However, our observations are similar to those made by others in a corneal HSV-1 model (71), in which it was shown that IL-17RA knockout mice have decreased neutrophil infiltration that correlated with a decrease in corneal opacity, suggesting less tissue damage, at 24 h postchallenge. In contrast to our model, this effect was transient, with the IL-17RA knockouts having similar levels of neutrophils and corneal pathology by 4 dpi. These data seem to suggest that therapeutic modulation of IL-17 signaling may be beneficial not only in ALI but also in treating immunopathology associated with viral infections of other organs.
Supplementary Material
Acknowledgments
We acknowledge Dr. Joseph Witztum (University of California, San Diego, CA) for kindly providing the EO6 mAb. We also acknowledge the advice and technical assistance provided by Amy Magill, Lynne Bauer, and Alison Logar.
Footnotes
Abbreviations used in this paper: ALI, acute lung injury; ARDS, acute respiratory distress syndrome; TRIF, Toll/IL-1R domain-containing adapter-inducing IFN-β; ROI, reactive oxygen intermediate; BAL, bronchoalveolar lavage; BALF, BAL fluid; LDH, lactate dehydrogenase; NP, nucleocapsid protein; HA, hemagglutinin; dpi, days postinfection; KC, CXCL1; MPO, myeloperoxidase.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31:S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 2.Lew TW, Kwek TK, Tai D, Earnest A, Loo S, Singh K, Kwan KM, Chan Y, Yim CF, Bek SL, et al. Acute respiratory distress syndrome in critically ill patients with severe acute respiratory syndrome. JAMA. 2003;290:374–380. doi: 10.1001/jama.290.3.374. [DOI] [PubMed] [Google Scholar]
- 3.Beigel JH, Farrar J, Han AM, Hayden FG, Hyer R, de Jong MD, Lochindarat S, Nguyen TK, Nguyen TH, Tran TH, et al. Avian influenza A (H5N1) infection in humans. N Engl J Med. 2005;353:1374–1385. doi: 10.1056/NEJMra052211. [DOI] [PubMed] [Google Scholar]
- 4.Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, Garcia-Sastre A. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77– 80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 5.Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 6.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abraham E, Kaneko DJ, Shenkar R. Effects of endogenous and exogenous catecholamines on LPS-induced neutrophil trafficking and activation. Am J Physiol. 1999;276:L1–L8. doi: 10.1152/ajplung.1999.276.1.L1. [DOI] [PubMed] [Google Scholar]
- 8.Faggioni R, Gatti S, Demitri MT, Delgado R, Echtenacher B, Gnocchi P, Heremans H, Ghezzi P. Role of xanthine oxidase and reactive oxygen intermediates in LPS- and TNF-induced pulmonary edema. J Lab Clin Med. 1994;123:394–399. [PubMed] [Google Scholar]
- 9.Parsey MV, Tuder RM, Abraham E. Neutrophils are major contributors to intraparenchymal lung IL-1β expression after hemorrhage and endotoxemia. J Immunol. 1998;160:1007–1013. [PubMed] [Google Scholar]
- 10.Parsey MV, Kaneko D, Shenkar R, Abraham E. Neutrophil apoptosis in the lung after hemorrhage or endotoxemia: apoptosis and migration are independent of interleukin-1beta. Chest. 1999;116:67S– 68S. doi: 10.1378/chest.116.suppl_1.67s. [DOI] [PubMed] [Google Scholar]
- 11.Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol. 2005;79:14933–14944. doi: 10.1128/JVI.79.23.14933-14944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White MR, Tecle T, Crouch EC, Hartshorn KL. Impact of neutrophils on antiviral activity of human bronchoalveolar lavage fluid. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1293–1299. doi: 10.1152/ajplung.00266.2007. [DOI] [PubMed] [Google Scholar]
- 13.Tecle T, White MR, Gantz D, Crouch EC, Hartshorn KL. Human neutrophil defensins increase neutrophil uptake of influenza A virus and bacteria and modify virus-induced respiratory burst responses. J Immunol. 2007;178:8046– 8052. doi: 10.4049/jimmunol.178.12.8046. [DOI] [PubMed] [Google Scholar]
- 14.Salvatore M, Garcia-Sastre A, Ruchala P, Lehrer RI, Chang T, Klotman ME. α-Defensin inhibits influenza virus replication by cell-mediated mechanism(s) J Infect Dis. 2007;196:835– 843. doi: 10.1086/521027. [DOI] [PubMed] [Google Scholar]
- 15.Fujisawa H. Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J Virol. 2008;82:2772–2783. doi: 10.1128/JVI.01210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tate MD, Brooks AG, Reading PC. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir Res. 2008;9:57. doi: 10.1186/1465-9921-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tumpey TM, Chen SH, Oakes JE, Lausch RN. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J Virol. 1996;70:898–904. doi: 10.1128/jvi.70.2.898-904.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuzaki J, Tsuji T, Chamoto K, Takeshima T, Sendo F, Nishimura T. Successful elimination of memory-type CD8+ T cell subsets by the administration of anti-Gr-1 monoclonal antibody in vivo. Cell Immunol. 2003;224:98–105. doi: 10.1016/j.cellimm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 19.de Oca RM, Buendia AJ, Del Rio L, Sanchez J, Salinas J, Navarro JA. Polymorphonuclear neutrophils are necessary for the recruitment of CD8+ T cells in the liver in a pregnant mouse model of Chlamy-dophila abortus (Chlamydia psittaci serotype 1) infection. Infect Immun. 2000;68:1746–1751. doi: 10.1128/iai.68.3.1746-1751.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berliner JA, Leitinger N, Tsimikas S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res. 2009;50(Suppl):S207–S212. doi: 10.1194/jlr.R800074-JLR200. ePub December 22, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarzenberger P, Huang W, Ye P, Oliver P, Manuel M, Zhang Z, Bagby G, Nelson S, Kolls JK. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol. 2000;164:4783– 4789. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- 23.Schwarzenberger P, La Russa V, Miller A, Ye P, Huang W, Zieske A, Nelson S, Bagby GJ, Stoltz D, Mynatt RL, Spriggs M, Kolls JK. IL-17 stimulates granulopoiesis in mice: use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J Immunol. 1998;161:6383– 6389. [PubMed] [Google Scholar]
- 24.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, Polubinska A, Friess H, Gahl GM, Frei U, Jorres A. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GROα chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- 26.Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC, Skoogh BE, Linden A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- 27.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, Kolls JK. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25:335–340. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 28.Yu JJ, Ruddy MJ, Conti HR, Boonanantanasarn K, Gaffen SL. The interleukin-17 receptor plays a gender-dependent role in host protection against Porphyromonas gingivalis-induced periodontal bone loss. Infect Immun. 2008;76:4206– 4213. doi: 10.1128/IAI.01209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- 30.Itohara S, Mombaerts P, Lafaille J, Iacomini J, Nelson A, Clarke AR, Hooper ML, Farr A, Tonegawa S. T cell receptor δ gene mutant mice: independent generation of αβ T cells and programmed rearrangements of γδ TCR genes. Cell. 1993;72:337–348. doi: 10.1016/0092-8674(93)90112-4. [DOI] [PubMed] [Google Scholar]
- 31.Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, Witztum JL. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. 2000;105:1731–1740. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 33.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ley K, Smith E, Stark MA. IL-17A-producing neutrophil-regulatory Tn lymphocytes. Immunol Res. 2006;34:229–242. doi: 10.1385/IR:34:3:229. [DOI] [PubMed] [Google Scholar]
- 36.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 37.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 38.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doherty PC, Christensen JP. Accessing complexity: the dynamics of virus-specific T cell responses. Annu Rev Immunol. 2000;18:561–592. doi: 10.1146/annurev.immunol.18.1.561. [DOI] [PubMed] [Google Scholar]
- 40.Hamada H, Garcia-Hernandez L, de M, Reome JB, Misra SK, Strutt TM, McKinstry KK, Cooper AM, Swain SL, Dutton RW. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol. 2009;182:3469–3481. doi: 10.4049/jimmunol.0801814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beck MA, Nelson HK, Shi Q, Van Dael P, Schiffrin EJ, Blum S, Barclay D, Levander OA. Selenium deficiency increases the pathology of an influenza virus infection. FASEB J. 2001;15:1481–1483. [PubMed] [Google Scholar]
- 42.Zhao MQ, Stoler MH, Liu AN, Wei B, Soguero C, Hahn YS, Enelow RI. Alveolar epithelial cell chemokine expression triggered by antigen-specific cytolytic CD8+ T cell recognition. J Clin Invest. 2000;106:R49–R58. doi: 10.1172/JCI9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L, Yoon H, Zhao MQ, Liu J, Ramana CV, Enelow RI. Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-α by antiviral CD8+ T cells. J Immunol. 2004;173:721–725. doi: 10.4049/jimmunol.173.2.721. [DOI] [PubMed] [Google Scholar]
- 44.Riedemann NC, Neff TA, Guo RF, Bernacki KD, Laudes IJ, Sarma JV, Lambris JD, Ward PA. Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J Immunol. 2003;170:503–507. doi: 10.4049/jimmunol.170.1.503. [DOI] [PubMed] [Google Scholar]
- 45.Lang JE, Williams ES, Mizgerd JP, Shore SA. Effect of obesity on pulmonary inflammation induced by acute ozone exposure: role of interleukin-6. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1013–L1020. doi: 10.1152/ajplung.00122.2007. [DOI] [PubMed] [Google Scholar]
- 46.Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol. 2000;156:1951–1959. doi: 10.1016/S0002-9440(10)65068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol. 2008;180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 48.Wareing MD, Lyon A, Inglis C, Giannoni F, Charo I, Sarawar SR. Chemokine regulation of the inflammatory response to a low-dose influenza infection in CCR2−/− mice. J Leukocyte Biol. 2007;81:793– 801. doi: 10.1189/jlb.0506299. [DOI] [PubMed] [Google Scholar]
- 49.Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, Mack M, Kuziel WA, Corazza N, Brunner T, Seeger W, Lohmeyer J. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med. 2008;205:3065–3077. doi: 10.1084/jem.20080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinecke JW, Li W, Francis GA, Goldstein JA. Tyrosyl radical generated by myeloperoxidase catalyzes the oxidative cross-linking of proteins. J Clin Invest. 1993;91:2866–2872. doi: 10.1172/JCI116531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartvigsen K, Chou MY, Hansen LF, Shaw PX, Tsimikas S, Binder CJ, Witztum JL. The role of innate immunity in atherogenesis. J Lipid Res. 2009;50(Suppl):S388–S393. doi: 10.1194/jlr.R800100-JLR200. ePub December 22, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007;9:78– 86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 54.Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of interferon-γ by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol. 2001;158:119–130. doi: 10.1016/s0002-9440(10)63950-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moskophidis D, Kioussis D. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J Exp Med. 1998;188:223–232. doi: 10.1084/jem.188.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen HH, van Ginkel FW, Vu HL, Novak MJ, McGhee JR, Mestecky J. γ interferon is not required for mucosal cytotoxic T-lympho-cyte responses or heterosubtypic immunity to influenza A virus infection in mice. J Virol. 2000;74:5495–5501. doi: 10.1128/jvi.74.12.5495-5501.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bot A, Bot S, Bona CA. Protective role of γ interferon during the recall response to influenza virus. J Virol. 1998;72:6637– 6645. doi: 10.1128/jvi.72.8.6637-6645.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci USA. 2007;104:12479–12481. doi: 10.1073/pnas.0705289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gatti S, Faggioni R, Echtenacher B, Ghezzi P. Role of tumour necrosis factor and reactive oxygen intermediates in lipopolysaccharide-induced pulmonary oedema and lethality. Clin Exp Immunol. 1993;91:456– 461. doi: 10.1111/j.1365-2249.1993.tb05924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koenders MI, Lubberts E, van de Loo FA, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Kolls JK, Di Padova FE, Joosten LA, van den Berg WB. Interleukin-17 acts independently of TNF-α under arthritic conditions. J Immunol. 2006;176:6262– 6269. doi: 10.4049/jimmunol.176.10.6262. [DOI] [PubMed] [Google Scholar]
- 62.Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci USA. 2003;100:5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aujla SJ, Dubin PJ, Kolls JK. Th17 cells and mucosal host defense. Semin Immunol. 2007;19:377–382. doi: 10.1016/j.smim.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wareing MD, Shea AL, Inglis CA, Dias PB, Sarawar SR. CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral Immunol. 2007;20:369–378. doi: 10.1089/vim.2006.0101. [DOI] [PubMed] [Google Scholar]
- 65.Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135– 4141. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- 66.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 67.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 69.Kohyama S, Ohno S, Isoda A, Moriya O, Belladonna ML, Hayashi H, Iwakura Y, Yoshimoto T, Akatsuka T, Matsui M. IL-23 enhances host defense against vaccinia virus infection via a mechanism partly involving IL-17. J Immunol. 2007;179:3917–3925. doi: 10.4049/jimmunol.179.6.3917. [DOI] [PubMed] [Google Scholar]
- 70.Yue FY, Merchant A, Kovacs CM, Loutfy M, Persad D, Ostrowski MA. Virus-specific interleukin-17-producing CD4+ T cells are detectable in early human immunodeficiency virus type 1 infection. J Virol. 2008;82:6767– 6771. doi: 10.1128/JVI.02550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Molesworth-Kenyon SJ, Yin R, Oakes JE, Lausch RN. IL-17 receptor signaling influences virus-induced corneal inflammation. J Leukocyte Biol. 2008;83:401– 408. doi: 10.1189/jlb.0807571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.