

Abstract
Mutations found in PTEN-induced putative kinase 1 (PINK1), a putative mitochondrial serine/threonine kinase of unknown function, have been linked to autosomal recessive Parkinson’s disease. It is suggested that mutations can cause a loss of PINK1 kinase activity and eventually lead to mitochondrial dysfunction. In this report, we examined the subcellular localization of PINK1 and the dynamic kinetics of PINK1 processing and degradation. We also identified cytosolic chaperone heat-shock protein 90 (Hsp90) as an interacting protein of PINK1 by PINK1 co-immunoprecipitation. Immunofluorescence of PINK1 protein and mitochondrial isolation show that the precursor form of PINK1 translocates to the mitochondria and is processed into two cleaved forms of PINK1, which in turn localize more to the cytosolic than mitochondrial fraction. The cleavage does not occur and the uncleaved precursor stays associated with the mitochondria when the mitochondrial membrane potential is disrupted. Metabolic labeling analyses show that the PINK1 processing is rapid and the levels of cleaved forms are tightly regulated. Furthermore, cleaved forms of PINK1 are stabilized by Hsp90 interaction as the loss of Hsp90 activity decreases PINK1 level after mitochondrial processing. Lastly, we also find that cleaved forms of PINK1 are degraded by the proteasome, which is uncommon for mitochondrial proteins. Our findings support a dual subcellular localization, implying that PINK1 can reside in the mitochondria and the cytosol. This raises intriguing functional roles that bridge these two cellular compartments.
Keywords: heat-shock protein 90, mitochondria, Parkinson’s disease, PTEN-induced putative kinase 1, valinomycin
Parkinson’s disease (PD) is a progressive neurodegenerative disorder involving a number of brainstem nuclei with a preponderance of dopaminergic neuronal loss in the substantia nigra pars compacta. PD affects about 0.3–1% of population, with an average age of onset of 55 and markedly increased prevalence and incidence with advancing age (de Lau and Breteler 2006) making it the second most common neurodegenerative disease after Alzheimer’s disease. The sporadic form accounts for most PD cases and the genetic component has been only recently appreciated (Gasser 2005). Although familial PD accounts for 3–10% of total cases, the convergence of implicated mechanisms from genetic mutations and environmental factors has vastly facilitated the studies on PD pathogenesis.
In 2004, mutations in PINK1 were identified in families with early onset PD with an autosomal recessive inheritance pattern (Valente et al. 2004). PINK1 is a 581 amino acid protein with a mitochondrial localization signal (MLS) and a functional serine/threonine kinase domain (Beilina et al. 2005; Silvestri et al. 2005; Sim et al. 2006). Recently, it was shown that in vivo phosphorylation of Omi/HtrA2 and TNF-receptor associated protein-1 are PINK1 dependent (Plun-Favreau et al. 2007; Pridgeon et al. 2007). So far, many homozygous mutations throughout PINK1 have been identified and represent the second most common recessive mutations in PD (Abou-Sleiman et al. 2006; Djarmati et al. 2006; Zadikoff et al. 2006; Tan and Skipper 2007). Furthermore, many heterozygous mutations of PINK1 have been noted in late-onset PD patients, suggesting a possible role of PINK1 mutation as a susceptibility factor (Klein et al. 2007). The current consensus is that PINK1 acts as a cytoprotective protein. In cell survival studies, PINK1 overexpression decreased staurosporine-induced apoptosis, and knockdown of PINK1 by small interfering RNA increased vulnerability to rotenone and MPP+ toxicity in SH-SY5Y cells (Deng et al. 2005; Petit et al. 2005). PC12 cells are more susceptible to oxidative stress when TNF-receptor associated protein-1 phosphorylation is reduced because of the loss of PINK1 (Pridgeon et al. 2007). An in vivo study showed that over-expression of wild-type (WT) PINK1 or PINK1 kinase domain in mouse neurons protected the neurons from MPP+ toxicity (Haque et al. 2008). In contrast, mouse PINK1 knockdown models using conditional RNAi (Zhou et al. 2007) or by conventional knockout (Kitada et al., 2007; Xiaoxi Zhuang, personal communication) methods did not induce dopaminergic neuron death or behavioral impairments, although PINK1 knockout in Drosophila showed cell death and mitochondrial defects in muscles and dopaminergic neurons (Clark et al. 2006; Park et al. 2006; Wang et al. 2006; Yang et al. 2006).
The functional role of PINK1 in the mitochondria is unclear and current localization data suggests that PINK1 localizes both to the mitochondria and the cytosol (Beilina et al. 2005; Silvestri et al. 2005; Pridgeon et al. 2007). Cytosolic localization of endogenous PINK1 is shown recently (Takatori et al. 2008; Weihofen et al. 2008), where the subcellular distribution of PINK1 can be influenced by Cdc37/heat-shock protein 90 (Hsp90) chaperones and parkin. Additional evidence supporting a cytosolic localization comes from PINK1 ubiquitination and accumulation when the proteasome activity is inhibited (Muqit et al. 2006; Tang et al. 2006) and its presence in Lewy bodies of PD brains (Gandhi et al. 2006). Functional rescue of PINK1-deficiency by parkin over-expression in Drosophila also suggests a potential cytosolic localization of PINK1 for the interaction (Park et al. 2005; Clark et al. 2006). Furthermore, cytosolic expression of PINK1 kinase domain also demonstrated a protective role against MPP+ (Haque et al. 2008). Many questions remain about how PINK1 is localized to the cytosol when it contains a functional mitochondrial targeting signal and how PINK1 is processed in cells. The detection of multiple PINK1 isoforms (Petit et al. 2005; Takatori et al. 2008) also begs the question of how each isoform is produced and regulated.
Therefore, in this study, we further characterize PINK1 processing, localization, and stability to gain insight into the potential mechanisms for the observed dual localization. We utilize a metabolic labeling technique to probe PINK1 processing and stability and perform biochemical fractionation to study PINK1 subcellular localization. We report that precursor PINK1 associates with mitochondria while an abundance of cleaved PINK1 forms are found in the cytosol. The processing of PINK1 precursor requires an intact mitochondrial membrane potential and the stability of PINK1 cleaved forms requires cytosolic chaperone Hsp90 activity. Loss of interaction with Hsp90 results in PINK1 degradation by the proteasome. Our combined data raises interesting questions regarding the role of mitochondria in PINK1 cell biology.
Experimental procedures
cDNAs and antibodies
Wild-type human PINK1 cDNA from Origene Technologies (Rockville, MD, USA) was amplified by PCR with primers sense 5′-AAGATTCAATGGCGGTGCGACAGGCG-3′ and antisense 5′-ATAGGATTACAGGGCTGCCCTCCATGA-3′. The PCR product was cloned into p3XFLAG-CMV14 (Sigma, St Louis, MO, USA), flanked by EcoRI and BamHI restriction enzyme sites. The final expression plasmid was sequence verified and named PINK1-flag. The following antibodies were purchased commercially. Anti-PINK1 (BC100-494) 1 : 2500 (Novus Biological, Littleton, CO, USA), anti-FLAG 1 : 100 (Sigma), anti-FLAG M2 1 : 2500 (Sigma), anti-Hsp90β 1 : 250 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-Hsp90 1 : 5000 (BD Biosciences, San Jose, CA, USA), anti-β actin 1 : 5000 (Sigma), anti-cytochrome c (cyt c) 1 : 1000 (Cell Signaling Technology, Beverly, MA, USA), anti-p38 mitogen-activated protein kinase 1 : 1000 (Cell Signal Technologies), anti-cytochrome oxidase (COX IV) 1 : 200 (Invitrogen), anti-mouse IgG-horseradish peroxidase (HRP) 1 : 5000 (Jackson Immuno-Research, West Grove, PA, USA), anti-goat IgG-HRP 1 : 1000 (Promega, Madison, WI, USA), and protein A-HRP 1 : 2500 (Sigma).
Transfection
Transfection method was performed according to manufacture’s protocol (Invitrogen). Briefly, HeLa cells were plated onto 60-mm2 tissue culture dishes at 90% confluency at the time of transfection; 1–2 μg of cDNA was diluted in 250 μL OPTI-MEM1 reduced serum medium (OPTI-MEM) (Invitrogen). Five microliter of Lipofectamine 2000 was diluted in 250 μL of OPTI-MEM. The mixture of cDNA and Lipofectamine 2000 was added to cells in OPTI-MEM. The transfection media was replaced by Dulbecco’s modified Eagle’s medium (DMEM) growth media 6 h after transfection. Cells were subjected to experiments 48 h following transfection. For stable over-expression, HeLa cells were selected in G418 for 2 weeks and the clones were pooled to avoid clonal variations.
Metabolic labeling
Following transfection, cells were starved for 30 min in 1% foetal bovine serum containing DMEM lacking methionine or cysteine (Invitrogen). Appropriate drug treatments were carried out during starvation, pulse, and chase period. For pulse labeling, 2 mL of DMEM containing 250 μCi/mL of 35S-met/cys trans-label (MP Biomedical, Irvine, CA, USA) was added to the cells for 3 min. For chase, the dishes were placed on ice and washed with cold DMEM containing 1 mM methionine, 1 mM cysteine, and appropriate drug, and chased in warm media for the indicated time. Cells were lysed in immunoprecipitation (IP) buffer [150 mM NaCl, 50 mM Tris–HCl, 0.5% Nonidet P-40 (Sigma), 0.5% sodium deoxycholate, 5 mM EDTA, 0.2 5 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail]. Cell lysates were centrifuged at 13 000 g for 2 min, and 0.25% sodium dodecyl sulfate (SDS) was added to the supernatant and pre-cleared with 40 μL of protein A beads (Pierce, Rockford, IL, USA) for 30 min. Five microliter of the final supernatant was used for TCA counts. Equal radioactive counts per sample were used to IP. All experiments were repeated three times and quantification was performed with Image J software (National Institute of Health, USA). Half-life of PINK1 isoforms were estimated by fitting a one-phase exponential decay curve to the data by non-linear regression using Prism 5 software (GraphPad Software Inc., San Diego, CA, USA).
Immunoprecipitation
Radiolabeled lysates were incubated with 5 μL of rabbit anti-FLAG antibody (Sigma) overnight, and 50 μL of protein A beads were added to each sample for 30 min. The beads were washed three times with IP buffer containing 0.25% SDS for 15 min each. The beads were then loaded on a 1 M sucrose cushion to remove protein aggregates. The beads were resuspended in 40 μL of 2x SDS sample buffer and boiled for 5 min prior to loading on SDS–HEPES polyacrylamide gel electrophoresis (PAGE).
Co-immunoprecipitation
Co-IP experiments followed the methods described previously (Shen et al. 2002). Briefly, cells were lysed in 1% Triton X-100 buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, protease inhibitor cocktail, and PMSF). Lysates were rotated for 1 h at 4°C and then cleared by centrifugation at 13 000 g for 10 min at 4°C. PINK1-flag and Hsp90β were immunoprecipitated by rabbit anti-FLAG antibodies (1 : 100) and goat anti-Hsp90β antibodies (1 : 50), respectively, for 2 h at 4°C. IP is described above, except the co-IP wash buffer is the 1% Triton X-100 buffer. Proteins were resolved on SDS–HEPES PAGE.
Mitochondrial isolation
Mitochondria were isolated from HeLa cells according to manufacturer’s protocol (Pierce) with minor modifications. Briefly, the cells were trypsinized and harvested. A Dounce homogenizer was used to lyse the cells. After removing the nuclear fraction, the crude supernatant was spun at 3000 g for 20 min to pellet the intact mitochondria. The mitochondrial pellet was resuspended in IP buffer (with 0.25% SDS, protease inhibitors, and PMSF) to collect mitochondrial proteins. For each fractionation, equal amounts of soluble cytosolic protein and mitochondrial protein were concentrated using PAGEprep (Pierce). Proteins were resolved on SDS–HEPES PAGE.
Immunocytochemistry
HeLa cells transfected with PINK1-flag were fixed in p-formaldehyde (4% for 5 min) and then washed three times in 0.1% Triton X- 100. Antigen retrieval was performed by incubating coverslips in 50 mM Tris-buffered saline, pH 7.5, at 95°C for 20 min, followed by three washes in phosphate-buffered saline. Non-specific immunoreactivity was blocked with 10% goat serum. Cultures were incubated overnight at 4°C in phosphate-buffered saline containing a polyclonal FLAG (1 : 250 dilution) antibody and a monoclonal COX IV or Hsp90 (1 : 250 dilution) antibody. Immunoreactivity to FLAG was amplified and detected using an Alexa 488 conjugate of a goat anti-rabbit IgG antibody and COX IV and Hsp90 were amplified with Alexa 563 conjugate of a goat anti-mouse IgG antibody. The cells were imaged using a 150×, 1.4 NA objective, and optical slices through the cultures were obtained using the 488 and 543 nm lines, respectively, of an Olympus Optical (Tokyo, Japan) Fluoview 200 laser scanning confocal microscope.
Western blot analysis
Protein quantification was carried out using the bicinchoninic acid method (Pierce). Immobilon polyvinylidene difluoride membrane was used in western blotting (Millipore Corporation, Bedford, MA, USA). After wet transfer, membrane was rinsed briefly with water. For radioactive samples, the membrane was air-dried and exposed to storage phosphor imager (GE Healthcare, Piscataway, NJ, USA) and scanned with Molecular Imager (Bio-Rad Laboratories, Hercules, CA, USA). For non-radioactive samples, the membrane was blocked for 2 h in Tris-buffered saline containing 5% milk, and 0.1% Tween 20. Primary antibodies were incubated for overnight in blocking buffer, and secondary antibodies were incubated in 25°C for 1 h in blocking buffer. The membrane was then developed with enhanced chemiluminescence reagents (GE Healthcare) and imaged with ChemiGenius Bio-Imaging system (Syngene, Frederick, MD, USA).
Chemicals
All chemicals are from Sigma, unless noted.
Results
PINK1 is rapidly processed by the mitochondria
The N-terminal mitochondrial signal of PINK1 is predicted to have multiple sites for processing by computer programs such as MitoProt, TargetP, and SignalP. Cleavage at these potential sites can generate multiple PINK1 isoforms from the full length precursor (FL). Currently, it is ambiguous exactly where, and how many times, PINK1 precursor is cleaved to generate PINK1 isoforms. Silvestri et al. (2005) demonstrated that one possible cleavage site is at position 77, while others observed additional processing, potentially downstream of amino acid 77 (Petit et al. 2005; Takatori et al. 2008). In our experiments, we observed two cleaved forms of PINK1, named Δ1 and Δ2 on western blot analysis of HeLa cells over-expressing untagged PINK1 and PINK1-flag constructs detected with Novus anti-PINK1 antibodies (Fig. 1a). Only the FL form of the endogenous PINK1 was detectible at a very low level in the vector control lane, and endogenous Δ1 and Δ2 isoforms were below detection limit. However, Novus anti-PINK1 antibodies detected endogenous PINK1 isoforms when HeLa cells were treated with valinomycin or epoxomicin (Fig. 1a), demonstrating the physiological validity of these PINK1 forms.
Fig. 1.

PINK1 processing is dependent on mitochondrial membrane potential. (a) Anti-PINK1 antibody was used to detect endogenous PINK1 as well as exogenous PINK1 in HeLa cells. Anti-PINK1 antibody detects over-expressed untagged or FLAG-tagged PINK1 and also detects the endogenous PINK1 when cells are treated with 1 μM epoxomicin or 1 μM valinomycin for 24 h. The accumulation of endogenous PINK FL and Δ1 confirms the expression pattern of the exogenous PINK1. The right panel shows an overexposure image to demonstrate the faint Δ2 band. (b) Stably transfected HeLa cells were treated with 1 μM valinomycin for 24 h and PINK1-flag steady state level was examined by western blotting with anti-Flag antibody. PINK1 FL accumulation is observed with valinomycin treatment, similar to the endogenous PINK1 FL in panel (a). (c and d) Metabolic labeling of HeLa cells transiently transfected with PINK1-flag. Proteins were labeled with 35S-met/cys trans-label for 3 min, chased with cold methionine and cysteine at various times noted in the figure, and analyzed by immunoprecipitation and exposure to phosphorscreen. Mitochondrial membrane potential was dissipated with 1 μM valinomycin during the starvation, pulse, and chase period. (e and f) Levels of each isoform of PINK1-flag were normalized as percentage of the total amount of labeled PINK1 (all three isoforms) present at the end of the labeling period and graphed over time. The data from three independent labeling experiments are graphed with mean ± SEM and curve fitted with a one-phase exponential decay. A representative figure from each experiment is shown in (c and d).
PTEN-induced putative kinase 1 processing by the mitochondria was previously suggested by several authors (Valente et al. 2004; Beilina et al. 2005; Silvestri et al. 2005; Abou-Sleiman et al. 2006) and demonstrated in vitro by Silvestri et al. (2005) when they utilized purified mitochondria to show the generation of a cleaved form of PINK1. First we tested the idea that PINK1 processing is dependent on the mitochondrial potential gradient in HeLa cells stably overexpressing PINK1-flag. To address this, we dissipated the mitochondrial membrane potential needed for protein import with valinomycin. As shown in Fig. 1b, steady state level of PINK1 FL accumulated after valinomycin treatment, suggesting that proteolysis of FL was impeded. We then investigated the dynamics of mitochondrial processing and the kinetics of PINK1 FL, Δ1, and Δ2. To address this, we performed a 3-min 35S-methionine/cysteine pulse chase experiment in HeLa cells over-expressing PINK1-flag cDNA. Under control conditions, the estimated half-life of PINK1 FL is about 27 min (Table 1, Fig. 1c and e). Conversion of FL to Δ1 and Δ2 was rapid as Δ1 and Δ2 were detected after the initial 3 min pulse. PINK1 Δ1 had an estimated half-life of 30 min (Table 1). PINK1 Δ2 was quite stable and did not show significant degradation for the duration of up to 120 min, despite the fact that the amount of Δ2 generated was a small fraction (about 20%) of the total synthesized FL. This stability of Δ2 form accounts for the fact that this is one of the predominant forms in the steady state. This also suggests that the level of PINK1 Δ2 is tightly regulated. We then wanted to confirm by metabolic labeling that FL is the precursor to Δ1 and Δ2. Treatment with valinomycin abrogated the formation of Δ1 and Δ2 (Fig. 1d and f) and led to the increased half-life of PINK1 FL, consistent with the accumulation of PINK1 FL in the steady state (Fig. 1b). This data demonstrates that Δ1 and Δ2 are the cleaved products of FL and that mitochondrial integrity is critical in PINK1 processing.
Table 1.
Half-life of PINK1 isoforms
| PINK1 isoform | Treatment | T½ (min) | T½ (min) 95% CI | Curve fit R2 value |
|---|---|---|---|---|
| FL | Control | 27.31 | 20.50–40.87 | 0.9171 |
| Δ1 | Control | 30.18 | 22.62–45.33 | 0.9043 |
| Δ2 | Control | > 120 | ND | 0.2946 |
| FL | MG-132 | 29.74 | 19.33–64.38 | 0.7527 |
| Δ1 | MG-132 | 57.31 | 37.60–120.5 | 0.7672 |
| Δ2 | MG-132 | > 120 | ND | 0.4486 |
| FL | 17-AAG | 31.61 | 20.55–68.43 | 0.7171 |
| Δ1 | 17-AAG | 24.7 | 16.48–49.28 | 0.7863 |
| FL | Valino | > 120 | ND | 0.1682 |
| FL | Valino + 17-AAG | 114.1 | 79.14–204.2 | 0.7266 |
PINK1, PTEN-induced putative kinase 1; FL, full length; 17-AAG, 17- (allylamino)-17-demethoxygeldanamycin; ND, not determined.
PINK1 localizes to mitochondria and cytosol
Previous data on PINK1 subcellular localization show both mitochondrial and cytosolic co-localization (Valente et al. 2004; Beilina et al. 2005; Silvestri et al. 2005; Takatori et al. 2008). As we observed two post-translationally cleaved forms of PINK1, we attempted to clarify the localization of our PINK1 construct by subcellular fractionation and confocal microscopy. We investigated the effect of valinomycin on PINK1 localization and asked whether valinomycin treatment compromises mitochondrial localization of PINK1. Using mitochondrial isolation, we observed under normal conditions that all three isoforms of PINK1-flag were detected in the mitochondrial fraction (cyt c positive) and in the cytosol (p38 mitogen-activated protein kinase positive), with the cleaved form PINK1 Δ2 most abundant in the cytosol (Fig. 2a). Valinomycin treatment increased PINK1 FL in the mitochondrial fraction and reduced the PINK1 levels in the cytosol, especially the Δ2 form (Fig. 2a). Similarly when we examined the endogenous PINK1 localization by mitochondrial isolation, we first noticed that endogenous PINK1 isoforms were found in both cytosolic and mitochondrial fractions under normal conditions. Then we observed that valinomycin treatment increased PINK1 FL in the mitochondrial fraction and epoxomicin treatment enhanced PINK1 Δ1 in the cytosolic fraction (Fig. 2b). These results from stable PINK1-flag over-expression and endogenous PINK1 demonstrate that PINK1 FL translocates to the mitochondria independent of mitochondrial membrane potential and disruption of mitochondrial membrane potential causes PINK1 FL to accumulate in the mitochondria. On the other hand, cleaved forms of PINK1 are primarily localized to the cytosol. The biochemical fractionation data are further supported by immunofluorescence microscopy. Under basal conditions, PINK1-flag exhibited partial colocalization with COX IV of the mitochondria and also Hsp90 in the cytosol (Fig. 3b–d and h–j). Cells treated with valinomycin showed a change in PINK1 immunofluorescence pattern to reflect the enhanced PINK1 co-localization with COX IV and a detectable loss in Hsp90 co-localization (Fig. 3e–g and k–m).
Fig. 2.

PINK1 localizes to the mitochondria and cytosol. (a) Mitochondrial isolation was performed with stably transfected HeLa cells under control or valinomycin treatment conditions and lysates were analyzed by western blotting. The cytosolic fraction is marked by the abundance of p38 mitogen-activated protein kinase (MAPK) and the mitochondrial fraction is marked by the abundance of cytochrome c. (b) Endogenous PINK1 localization in HeLa cells was examined by mitochondrial isolation under normal conditions or treated with epoxomicin or valinomycin. Similar to exogenous PINK1-flag, endogenous PINK1 FL increased with valinomycin and the cytosolic fraction of PINK1 Δ1 increased with epoxomicin.
Fig. 3.

Valinomycin leads to increased PINK1 mitochondrial co-localization. (a) Immunofluorescence control showing the specificity of FLAG staining comparing vector transfected to PINK1-flag transfected in HeLa cells. (b–m) Immunofluorescence staining of HeLa cells transiently transfected with PINK1-flag stained with FLAG (b, e, h, and k), COX IV (i and l), or Hsp90 (c and f). Merged green and red images show partial co-localization of PINK1 with both cytosol (d) and mitochondria (j) markers under control conditions and increased mitochondrial co-localization (m), with a loss of Hsp90 co-localization (g) when cells were treated with 1 μM valinomycin for 24 h prior to staining. The inset represents 8× zoomed image.
PINK1 binds to Hsp90 and Hsp90 stabilizes PINK1
We performed co-IP with anti-FLAG antibodies in HeLa cells over-expressing PINK1-flag to determine potential binding partners of PINK1. Samples were resolved on SDS–HEPES PAGE and the protein bands were visualized with silver stain. By comparing the staining pattern, we excised the band that was abundant and only observed in the PINK1-flag lane and not the vector lane. By mass spectrometry, we identified Hsp90β in the band, as a potential partner (data not shown). To confirm this result, we tested the specificity of the protein–protein interaction with 17-(allylamino)- 17-demethoxygeldanamycin (17-AAG), a chemical inhibitor of Hsp90 analogous to geldanamycin. 17-AAG competes for the ATP binding site in Hsp90 and leads to dissociation and eventual degradation of Hsp90 client proteins. We first examined if 17-AAG treatment decreased PINK1-flag level in HeLa cells stably over-expressing PINK1-flag. In a time course experiment ranging from 0 to 4 h, 17-AAG significantly decreased all PINK1 isoforms after 30 min (Fig. 4a and b). However, both PINK1 FL and Δ1 were not continually affected by 17-AAG between 30 min and 4 h. In contrast, 17-AAG continued to affect PINK1 Δ2 level throughout the 4 h period (Fig. 4a and b). As Δ2 has a very low rate of synthesis and maintains its relatively high steady state level because of it stability, this decline probably reflects instability of this isoform with 17-AAG treatment. Next, we examined if this loss of PINK1 level is due to the dissociation of PINK1 from Hsp90β and attempted to validate the specificity of interaction by co-IP. Proteasome inhibitor MG132 was added in the co-IP experiment to prevent 17-AAG-induced degradation of PINK1-flag. Although the expression pattern of PINK1-flag was changed, the combined optical densities of all forms of PINK1 were similar to the control group (further discussed below in Fig. 6). Immunoblot with Hsp90β antibody showed dissociation of Hsp90β from PINK1-flag (Fig. 4c left panel) without a change in total Hsp90β level (Fig. 4c right panel). These data suggest that Hsp90β plays an important role in PINK1 stability by protein-protein interaction.
Fig. 4.
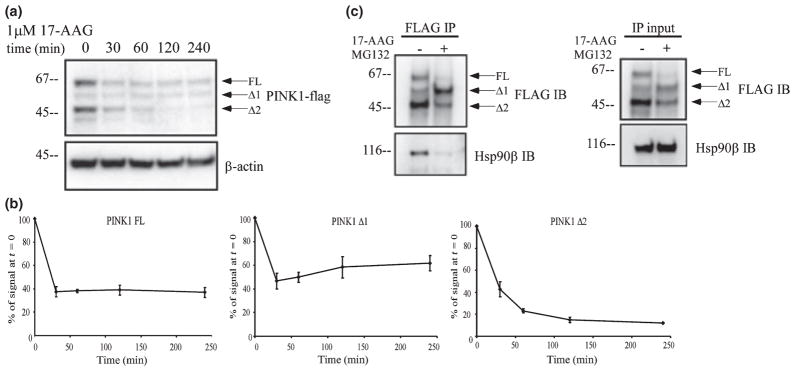
PINK1 interacts with Hsp90. (a) Stably transfected HeLa cells were treated for up to 4 h with 1 μM 17-AAG, an inhibitor of Hsp90 activity. 17-AAG treatment significantly decreased PINK1 Δ2 levels. (b) Quantification of each PINK1 isoform level from three independent 17-AAG experiments as represented in (a). Results are expressed as mean ± SEM. While expression of PINK1 Δ2 continued to decrease over time, level of PINK1 FL and Δ1 did not decrease after 30 min of 17-AAG treatment. (c) FLAG co-immunoprecipitation in HeLa cells transiently transfected with PINK1-flag. Cells were treated with 1 μM 17-AAG for 30 min; 1 μM MG132 was added to prevent 17-AAG-induced degradation. Lysates were immunoprecipitated with anti-flag antibodies and immunoblotted for flag or Hsp90β. Total lysates were used to detect Hsp90β and PINK1-flag level by western blot. 17-AAG led to a dissociation of PINK1-flag from Hsp90β.
Fig. 6.
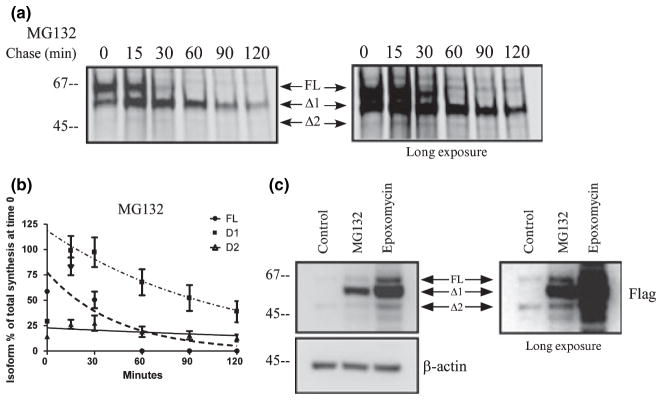
PINK1 is degraded by the proteasome. (a) Metabolic labeling of HeLa cells transiently transfected with PINK1-flag and treated with 1 μM MG132, a proteasome inhibitor. Proteins were labeled with 35S-met/cys trans-label for 3 min, chased with cold methionine and cysteine at various times noted in the figure, and analyzed by immunoprecipitation and exposure to phosphorscreen. An overexposed image is on the right to demonstrate the faint Δ2 band. (b) Levels of each isoform of PINK1-flag were normalized to the total amount of labeled PINK1 (all three isoforms) present at the end of the labeling period and graphed over time. The data from three independent labeling experiments are graphed with mean ± SEM and curve fitted with a one phase exponential decay. The representative figure from the experiment is shown in (a). Inhibiting proteasomal activity with MG132 increased PINK1 Δ1 half-life without affecting half-life of PINK1 FL or PINK1 Δ2 compared with Fig. 1e. (c) Stably transfected HeLa cells were treated for 24 h with 1 μM MG132 or 0.5 μM epoxomicin. PINK1-flag level was examined by western blot. Accumulation of endogenous PINK1 Δ1 after proteasome inhibition was also noted in Fig. 1a.
Hsp90 affects the stability of PINK1 cleaved forms
Steady state level can decrease either because of decreased protein synthesis or enhanced degradation. Therefore, we performed pulse chase experiments to study the stability of PINK1 and to confirm that 17-AAG decreases PINK1 cleaved forms primarily. Using metabolic labeling, 17- AAG minimally affected the kinetics of PINK1 FL or Δ1 (Fig. 5a and b), compared with the control conditions (Fig. 1c and e). On the other hand, 17-AAG treatment abrogated the detection of PINK1 Δ2 after the short 3-min synthesis. The data suggest that the absence of Hsp90 primarily destabilizes the newly formed Δ2 isoform and does not affect the stability of either PINK1 FL or Δ1. This is in agreement with the data shown in Fig 4c, where 30 min of 17-AAG treatment was able to reduce existing Δ2 expression to 50% compared with control. From the aforementioned data in Fig. 1, we know that valinomcyin leads to a persistent level of PINK1 FL and we asked whether this stability is mediated by Hsp90β. By metabolic labeling, the combined treatment of 17-AAG with valinomycin did not produce an effect on PINK1 FL stability (Fig. 5c and d), similar to that of valinomycin alone (Fig. 1d and f). Combined with data shown in Fig. 4, PINK1 Δ2 exhibits a greater dependence on Hsp90 for stability compared with FL or Δ1. This is consistent with the idea that Hsp90 predominantly stabilizes cleaved forms more than the precursor and supports the notion that PINK1 Δ2 is very susceptible to changes in its stability.
Fig. 5.

Hsp90 affects the stability of PINK1 cleaved forms. (a and c) Metabolic labeling of HeLa cells transiently transfected with PINK1-flag and treated with 1 μM 17-AAG or 1 μM 17-AAG and 1 μM valinomycin. Proteins were labeled with 35S-met/cys trans-label for 3 min, chased with cold methionine and cysteine at various times noted in the figure, and analyzed by immunoprecipitation and exposure to phosphorscreen. The inhibitors were added to the cells during starvation, pulse, and chase period. (b and d) Levels of each isoform of PINK1-flag were normalized to the total amount of labeled PINK1 (all three isoforms) present at the end of the labeling period and graphed over time. The data from three independent labeling experiments are graphed with mean ± SEM and curve fitted with one-phase exponential decay. A representative figure from each experiment is shown in (a and c). PINK1 Δ2 is not detectable following 17-AAG treatment and persistent PINK1 FL level following valinomycin is not affected by 17-AAG compared with Fig. 1f.
PINK1 is degraded by the proteasome
Published findings link PINK1 turn-over to proteasome degradation by showing the accumulation of PINK1 after inhibiting the proteasome with MG132. During the pulse chase experiment, we noticed that PINK1 Δ1 was transient and asked whether it was degraded by the proteasome. Treatment with MG132 led to the accumulation of Δ1 without affecting either FL or Δ2 level in a 2-h chase period (Fig. 6a and b). When we examined the steady state level of PINK1 by western blot in HeLa cells stably over-expressing PINK1 and treated with MG132, we also observed Δ1 accumulation, whereas FL and Δ2 were less affected (Fig. 6c). We reasoned that MG132 might exhibit some non-specific effect on proteases because mitochondrial proteins are not commonly degraded by the ubiquitin-proteasome system. Therefore, we used epoxomicin, a selective irreversible inhibitor of the proteasome. Similar to MG132, epoxomicin caused an accumulation of PINK1 Δ1 and increased FL and Δ2 on a steady state level, suggesting that Δ1 is mainly degraded by the proteasome upon processing of FL (Fig. 6c). We also observed that endogenous PINK1 Δ1 accumulated with epoxomicin treatment (Figs 1a and 2b). These data underscore the notion that PINK1 processing can lead to rapid PINK1 degradation by the proteasome.
Discussion
PINK1 is processed in the mitochondria
Here, we reexamine the subcellular localization of PINK1 because PINK1 has been found in both the mitochondria and the cytosol in published literature, with emphasis given to the mitochondrial localization. The MLS of PINK1 has been shown to be sufficient to direct enhanced cyan fluorescent protein to the mitochondria when enhanced cyan fluorescent protein is tagged with the PINK1 MLS (Silvestri et al. 2005; Muqit et al. 2006; Takatori et al. 2008). This suggests that the MLS can direct PINK1 to the mitochondria for processing, but it does not necessarily imply that PINK1 will reside in the mitochondria after processing. Published electron microscopy and mitochondrial subfractionation data show that PINK1 is likely localized to the intermembrane space or tethered to the inner membrane (Silvestri et al. 2005; Muqit et al. 2006; Pridgeon et al. 2007). Consistent with published in vitro data, first we confirm that PINK1 processing is mitochondria dependent in an in vivo whole cell preparation. When the mitochondrial import is blocked by valinomycin, we observe increased accumulation of PINK1 precursor in the mitochondria, consistent with the notion that precursor PINK1 contains a signal for mitochondrial translocation and mitochondrial integrity is required for processing. The metabolic labeling data demonstrate that under normal conditions only a small fraction of synthesized PINK1 is processed into the Δ2 form, which is more stable and therefore accumulates at a higher steady state level than predicted from the small fraction of conversion into Δ2. This suggests that Δ2 PINK1 level is tightly regulated and also raises a possibility that the steady state Δ2 level can be easily modulated by various factors. For example, we observe that endogenous PINK1 Δ2 is at a very barely detectible level at a steady state (Figs 1 and 6) compared with FL or Δ1. This could reflect the low sensitivity of the available antibody against endogenous protein, a low endogenous PINK1 expression in cells, and/or potential differences in stability conferred by epitope-tagging of PINK1. As a result of the limitation of endogenous PINK1 antibodies for IP, we are not able to address the kinetics of endogenous PINK1 or untagged over-expressed PINK1 by metabolic labeling. We also observe that the formation of the PINK1 cleaved forms is rapid as PINK1 Δ1 and Δ2 are present at the end of the 3 min metabolic labeling and that Δ1 is rapidly degraded upon formation. Based on this, we hypothesize an independent formation of PINK1 Δ1 and Δ2 from the precursor rather than a sequential one, where Δ2 is the cleaved product of Δ1. However, the rapid mitochondrial processing of PINK1, which can be attributed to the optimal length of the MLS (Matouschek et al. 1997), impedes us from concluding that PINK1 processing is not sequential. More experiments on identifying the cleavage enzyme(s) and cleavage sites as well as a better kinetic resolution of PINK1 import and processing will help elucidate this observation.
PINK1 stability requires Hsp90 interaction
Here, we describe evidence supporting PINK1 interaction with Hsp90 as a client protein, as published by Weihofen et al. (2008) and Moriwaki et al. (2008). Hsp90 is an essential cytosolic chaperone protein that consists of 1–2% of total cellular protein. The structure of Hsp90 comprises of a N-terminal ATPase domain for chaperone activity, a middle domain responsible for interacting with client proteins and co-chaperones, and a C-terminal domain that is important for dimerization (Prodromou and Pearl 2003). Inhibitors of Hsp90 ATPase activity, such as geldanamycin or its analog 17-AAG, lead to the rapid degradation of client proteins by the proteasome (Citri et al. 2006). There are α- and β-Hsp90 isoforms although little is known about the functional difference between the two. Functioning as a homodimer, Hsp90 binds to a diverse group of client proteins that are crucial in cell proliferation, survival, and development (Whitesell and Lindquist 2005). Interestingly, many Hsp90 clients are kinases as they require Hsp90 binding for stabilization of structure in the mature or active state (Caplan et al. 2007). The main consequences of Hsp90 interaction is the stabilization of PINK1, as the loss of Hsp90 activity leads to decreased PINK1 level. In contrast to the finding that Hsp90 affects the steady state levels of PINK1 FL more than that of PINK1 cleaved forms (Weihofen et al. 2008), our data suggests that Hsp90 participates in stabilizing the cleaved forms of PINK1 because inhibition of Hsp90 does not change the stability of PINK1 FL when further processing is blocked by valinomycin, when examined by pulse chase experiment. The steady state level of different PINK1 isoforms shown in the western blot also demonstrates more dramatic reduction of cleaved forms than FL when Hsp90 loses its activity. It is possible that FL is stabilized by other proteins or chaperones beside Hsp90, such as Cdc37 (Weihofen et al. 2008). This effect of the cytosolic chaperone Hsp90 on the cleaved form of PINK1, but not on FL PINK1 is also consistent with the cytosolic localization of the cleaved forms. Aside from the chaperone activity, Hsp90 recruits nuclear-encoded mitochondrial pre-proteins for mitochondrial import (Truscott et al. 2003). It is believed that Hsp90 facilitates import of pre-proteins containing an internal mitochondrial targeting signal (Truscott et al. 2003) by targeting the pre-proteins–chaperone complex to the translocase of the mitochondrial outer membrane complex (Young et al. 2003). Subsequently, the ATPase activity of Hsp90 is required for the release of client proteins for proper import (Fan et al. 2006). We do not rule out the possibility that Hsp90 also participates in PINK1 import into the mitochondria. How Hsp90 facilitates PINK1 import and processing requires further studies.
Proteasomal degradation of PINK1 supports a cytosolic localization
Combined with data showing more abundant presence of cleaved PINK1 in the cytosolic fraction than in the mitochondria and the binding of PINK1 to cytosolic chaperone Hsp90, PINK1 degradation by the proteasome strongly suggests that a cytosolic localization has an important functional role. The pulse chase experiments show that PINK1 becomes unstable once it is processed by the mitochondria. The unstable form is degraded by the proteasome as it accumulates in the presence of proteasomal inhibitors. Our data on cytosolic localization of PINK1 explains two previous reports of cleaved PINK1 being ubiquitinated and degraded by the proteasome (Muqit et al. 2006; Tang et al. 2006). It is known that Hsp90 client proteins are degraded by the ubiquitin-proteasome system when they lose Hsp90 interaction. However, it is unusual that a mitochondrial protein is a proteasome substrate. Classically, the MLS of nuclear-encoded mitochondrial pre-proteins is cleaved by mitochondrial matrix peptidases upon import. Once imported into the mitochondria, intramitochondrial proteolysis is responsible for protein turn-over (Bota and Davies 2001). This degradation machinery involves proteases that act specifically in their mitochondrial compartment. The question remains how PINK1 can be degraded by the proteasome if it resides in the cristae as reported (Silvestri et al. 2005). Here, we offer two ideas to explain proteasomal degradation of PINK1. The first possibility raises the hypothesis that PINK1 is imported into the mitochondria and can retrotranslocate out to the cytosol for degradation. It has been demonstrated that proteasomal degradation of endoplasmic reticulum (ER) proteins requires retrotranslocation from the endoplasmic reticulum to the cytosol. Although this degradation mechanism is not yet observed for mitochondrial proteins, a handful of mitochondrial proteins, for example, cyt c, smac/ Diablo, and Omi/HtrA2, are released to the cytosol with the proper stimulus These soluble proteins can exert a secondary function while in the cytosol. A similar prediction for a retrotranslocation mechanism can be made for PINK1 although we observe cytosolic PINK1 in the absence of a stimulus. The second idea suggests that PINK1 is not fully imported into the mitochondria whereby upon MLS processing, PINK1 is released into the cytosol. The length of the PINK1 MLS could span both mitochondrial membranes to allow cleavage by the matrix peptidases while the folded C-terminal domain still remains in the cytosol, as observed for some MLS-containing mitochondrial proteins (Wickner and Schekman 2005). The well-studied yeast protein fumerase is a good example of a protein displaying dual localization (Knox et al. 1998; Sass et al. 2001, 2003). In short, a single gene product of FUM1 gives rise to the fumerase precursor protein that contains an amino-terminal mitochondrial pre-sequence that is removed by the mitochondrial matrix peptidase upon mitochondrial import. Processing of the precursor generates two identical isoenzymes where about 20% are fully imported into the mitochondrial matrix and about 70–80% fumerase remains in the cytosol. No proteins in the mammalian system have been found to exhibit similar properties for dual targeting and PINK1 would represent the first mammalian example. If cleaved PINK1 is the functional form, the cytosolic fraction of PINK1 Δ2 implies that PINK1 has dual functional roles, one in the mitochondria and the other in the cytosol. The interaction of PINK1 with Hsp90 also corroborates a cytosolic function for PINK1. Recent data on the interaction of parkin and PINK1 (Clark et al. 2006; Park et al. 2006; Yang et al. 2006; Exner et al. 2007; Weihofen et al. 2008) could also be explained by published findings (Takatori et al. 2008; Weihofen et al. 2008) and our data that endogenous PINK1 resides in the cytosol. Interestingly, Haque et al. (2008) showed that expression of cytosolic PINK1 isoform exhibits a similar protective role against MPP+ as WT PINK1, adding more credence to a cytosolic function of PINK1 in addition to its mitochondrial role. More experiments are necessary to solve the mechanism of how PINK1 localizes to the cytosol after processing in the mitochondria and if PINK1 translocation from mitochondria to cytosol has functional significance.
Acknowledgments
We thank Drs Xiaoxi Zhuang, Linan Chen, LisaWon, Jim Mastrianni, and Ben Glick for their insightful discussions. This project is supported by Grants from US PHS NS043286, American Parkinson Disease Association, and Brain Frontier Project, MoST, ROK.
Abbreviations used
- 17-AAG
17-(allylamino)-17-demethoxygel-danamycin
- COX
cytochrome oxidase
- cyt c
cytochrome c
- DMEM
Dulbecco’s modified Eagle’s medium
- FL
full length
- HRP
horseradish peroxidase
- Hsp90
heat-shock protein 90
- IP
immunoprecipitation
- MLS
mitochondrial localization signal
- OPTI-MEM
OPTI-MEM1 reduced serum medium
- PAGE
polyacrylamide gel electrophoresis
- PD
Parkinson’s disease
- PINK1
PTEN-induced putative kinase 1
- PMSF
phenylmethylsulfonyl fluoride
- SDS
sodium dodecyl sulfate
- WT
wild-type
References
- Abou-Sleiman PM, Muqit MM, McDonald NQ, et al. A heterozygous effect for PINK1 mutations in Parkinson’s disease? Ann Neurol. 2006;60:414–419. doi: 10.1002/ana.20960. [DOI] [PubMed] [Google Scholar]
- Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bota DA, Davies KJ. Protein degradation in mitochondria: implications for oxidative stress, aging and disease: a novel etiological classification of mitochondrial proteolytic disorders. Mitochondrion. 2001;1:33–49. doi: 10.1016/s1567-7249(01)00005-8. [DOI] [PubMed] [Google Scholar]
- Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Citri A, Harari D, Shohat G, et al. Hsp90 recognizes a common surface on client kinases. J Biol Chem. 2006;281:14361–14369. doi: 10.1074/jbc.M512613200. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Deng H, Jankovic J, Guo Y, Xie W, Le W. Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem Biophys Res Commun. 2005;337:1133–1138. doi: 10.1016/j.bbrc.2005.09.178. [DOI] [PubMed] [Google Scholar]
- Djarmati A, Hedrich K, Svetel M, Lohnau T, Schwinger E, Romac S, Pramstaller PP, Kostic V, Klein C. Heterozygous PINK1 mutations: a susceptibility factor for Parkinson disease? Mov Disord. 2006;21:1526–1530. doi: 10.1002/mds.20977. [DOI] [PubMed] [Google Scholar]
- Exner N, Treske B, Paquet D, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan AC, Bhangoo MK, Young JC. Hsp90 functions in the targeting and outer membrane translocation steps of Tom70-mediated mitochondrial import. J Biol Chem. 2006;281:33313–33324. doi: 10.1074/jbc.M605250200. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Muqit MM, Stanyer L, et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain. 2006;129:1720–1731. doi: 10.1093/brain/awl114. [DOI] [PubMed] [Google Scholar]
- Gasser T. Genetics of Parkinson’s disease. Curr Opin Neurol. 2005;18:363–369. doi: 10.1097/01.wco.0000170951.08924.3d. [DOI] [PubMed] [Google Scholar]
- Haque ME, Thomas KJ, D’Souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci USA. 2008;105:1716–1721. doi: 10.1073/pnas.0705363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6:652–662. doi: 10.1016/S1474-4422(07)70174-6. [DOI] [PubMed] [Google Scholar]
- Knox C, Sass E, Neupert W, Pines O. Import into mitochondria, folding and retrograde movement of fumarase in yeast. J Biol Chem. 1998;273:25587–25593. doi: 10.1074/jbc.273.40.25587. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- Matouschek A, Azem A, Ratliff K, Glick BS, Schmid K, Schatz G. Active unfolding of precursor proteins during mitochondrial protein import. EMBO J. 1997;16:6727–6736. doi: 10.1093/emboj/16.22.6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki Y, Kim YJ, Ido Y, Misawa H, Kawashima K, Endo S, Takahasi R. L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci Res. 2008 doi: 10.1016/j.neures.2008. 01.006. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Abou-Sleiman PM, Saurin AT, et al. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98:156–169. doi: 10.1111/j.1471-4159.2006.03845.x. [DOI] [PubMed] [Google Scholar]
- Park J, Kim SY, Cha GH, Lee SB, Kim S, Chung J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 2005;361C:133–139. doi: 10.1016/j.gene.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Petit A, Kawarai T, Paitel E, et al. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J Biol Chem. 2005;280:34025–34032. doi: 10.1074/jbc.M505143200. [DOI] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Pearl LH. Structure and functional relationships of Hsp90. Curr Cancer Drug Targets. 2003;3:301–323. doi: 10.2174/1568009033481877. [DOI] [PubMed] [Google Scholar]
- Sass E, Blachinsky E, Karniely S, Pines O. Mitochondrial and cytosolic isoforms of yeast fumarase are derivatives of a single translation product and have identical amino termini. J Biol Chem. 2001;276:46111–46117. doi: 10.1074/jbc.M106061200. [DOI] [PubMed] [Google Scholar]
- Sass E, Karniely S, Pines O. Folding of fumarase during mitochondrial import determines its dual targeting in yeast. J Biol Chem. 2003;278:45109–45116. doi: 10.1074/jbc.M302344200. [DOI] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- Sim CH, Lio DS, Mok SS, Masters CL, Hill AF, Culvenor JG, Cheng HC. C-terminal truncation and Parkinson’s disease-associated mutations down-regulate the protein serine/ threonine kinase activity of PTEN-induced kinase-1. Hum Mol Genet. 2006;15:3251–3262. doi: 10.1093/hmg/ddl398. [DOI] [PubMed] [Google Scholar]
- Takatori S, Ito G, Iwatsubo T. Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett. 2008;430:13–17. doi: 10.1016/j.neulet.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Hum Mutat. 2007;28:641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- Tang B, Xiong H, Sun P, et al. Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson’s disease. Hum Mol Genet. 2006;15:1816–1825. doi: 10.1093/hmg/ddl104. [DOI] [PubMed] [Google Scholar]
- Truscott KN, Brandner K, Pfanner N. Mechanisms of protein import into mitochondria. Curr Biol. 2003;13:R326–R337. doi: 10.1016/s0960-9822(03)00239-2. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, Oldham S, Xia K, Wang J, Bodmer R, Zhang Z. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci USA. 2006;103:13520–13525. doi: 10.1073/pnas.0604661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet. 2008;17:602–616. doi: 10.1093/hmg/ddm334. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Wickner W, Schekman R. Protein translocation across biological membranes. Science. 2005;310:1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- Zadikoff C, Rogaeva E, Djarmati A, Sato C, Salehi-Rad S, St George-Hyslop P, Klein C, Lang AE. Homozygous and heterozygous PINK1 mutations: considerations for diagnosis and care of Parkinson’s disease patients. Mov Disord. 2006;21:875– 879. doi: 10.1002/mds.20854. [DOI] [PubMed] [Google Scholar]
- Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007;3:242–250. doi: 10.7150/ijbs.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]