

Abstract
Duchenne muscular dystrophy is a neuromuscular degenerative disorder caused by the absence of dystrophin protein. It is characterized by progressive muscle weakness and cycles of degeneration/regeneration accompanying chronic muscle damage and repair. Canine models of muscular dystrophy, including the dystrophin-deficient golden retriever muscular dystrophy (GRMD), are the most promising animal models for evaluation of potential therapies, however canine-specific molecular tools are limited. In particular, few immune reagents for extracellular epitopes marking canine satellite cells (muscle stem cells) are available. We generated an antibody to the satellite cell marker syndecan-4 that identifies canine satellite cells. We then characterized isolated satellite cells from GRMD muscle and wildtype muscle by several in vitro metrics, and surprisingly found no significant differences between the two populations. We discuss whether accumulated adverse changes in the muscle environment rather than cell-intrinsic defects may be implicated in the eventual failure of satellite cell efficacy in vivo.
Keywords: Syndecan-4, Satellite cells, GRMD, Muscular dystrophy
1. Introduction
Repair or replacement of skeletal muscle after injury or during pathological muscle degeneration/regeneration is mediated by satellite cells, a population of resident tissue-specific stem cells. Normally quiescent in uninjured tissue, in response to damage-associated factors such as hepatocyte growth factor (HGF) [1,2] or nitric oxide (NO) [3] satellite cells will activate and exit their resting position between the external lamina and sarcolemma of their host muscle fiber. They will re-enter the cell cycle, begin to express members of the MyoD family of muscle-specific transcription factors, and eventually differentiate and fuse either with each other or existing myofibers to repair and replace damaged muscle tissue (reviewed in [4,5]). The capacity of mammalian satellite cells for multiple cycles of repair is extensive; in experiments done on rats [6] and mdx dystrophic mice [7], over 50 successive rounds of injury-induced repair have been documented. This would be expected, as the role of these cells is to provide replacement cells throughout the entire lifespan.
Duchenne muscular dystrophy, a severe myopathy caused by mutations in the dystrophin gene, is characterized by progressive muscle weakness and chronic muscle degeneration and regeneration, eventually resulting in death. The dystrophin gene is located on the X chromosome, resulting in affected males and carrier females, and comprises the largest gene in the mammalian genome at over 2.2 million base pairs [8]. While DMD is an inherited disorder, it also occurs spontaneously in one-quarter to one-third of patients [9], due to the very large size of the dystrophin gene; over 1100 distinct mutations in the dystrophin gene have been identified to date, most of which result in deleterious symptoms [10]. Spontaneous mutations in the dystrophin gene occur in animals as well.
While multiple models of DMD exist in mice, including the mdx mouse [11] (which was a spontaneous mutation) and various versions of the dystrophin knockout mouse, the phenotype in mice is significantly less severe than that of human patients. This has been proposed to be due to increased regenerative capacity [12] and/or or compensation by utrophin, a related protein [13]. However, other animal models exist in which progression of the disease more closely follows what has been observed in human patients. Because the dystrophin gene is conserved in both size and function among mammals, dystrophinopathies arising from spontaneous mutations during gametogenesis in other animals such as dogs and cats have been reported; as with human cases, there are multiple different mutant alleles that lead to clinical signs of disease. The dog, in particular, has emerged as a valuable model for research on DMD pathology and therapy. Due to the common genetic basis of the disease in dog and human, GRMD (golden retriever muscular dystrophy) [14], GSHPMD (German shorthaired pointer muscular dystrophy) [15] and other inbred dystrophic dog lines descended from animals with spontaneous mutations have been extensively used in preclinical settings, particularly for cell and gene therapy studies.
As the progenitor cell population responsible for muscle repair after damage due to injury or disease, and the most likely cellular vector for therapeutic interventions, the status of satellite cells with respect to their overall number, proliferation capacity, gene expression, myogenic potential, etc. is of interest in both acute muscle regeneration and disease models. Because of their dispersion and rarity within the muscle tissue (only 1–6% of muscle-associated cells [5]) as well as the difficulty of longitudinal analysis of such a population in vivo, most studies of satellite cell activity are done in either single fiber culture or mass cell culture. By examining isolated satellite cells ex vivo, significant insight has been gained into both intracellular and extracellular molecular mechanisms regulating murine satellite cell activity (reviewed in [4,5]). More recently, protocols which identify populations of primary satellite cells for analysis or engraftment using fluorescence-activated cell sorting (FACS) based on extracellular epitope markers have provided significant advances in satellite cell biology [16–19]. However, a technical drawback to performing similar analyses in the dog has been the relative scarcity of compatible molecular tools for satellite cell identification and analysis compared to other model systems, although monoclonal antibodies specific to isoforms of neural cell adhesion molecule (NCAM) have previously been used to identify and/or isolate proliferating [20] and some differentiating [21] canine satellite cells. The most widely-recognized molecular marker of quiescent satellite cells in other systems such as mouse is the Pax7 transcription factor [22]; while the frequently used mouse monoclonal antibody directed against the chicken homolog of Pax7 [23] reacts well with canine muscle tissue in immunohistological sections (see Section 3), its usefulness for satellite cell isolation and analysis is limited due to the constraints noted above, as well as the requirement for fixation and permeablization of the sample. Pax7 expression is also lost as satellite cell-derived myoblasts transition to the committed and differentiated states [24] so its utility as a marker of satellite cells decreases in culture or under conditions of ongoing satellite cell-mediated myogenesis. Thus, immune reagents that recognize extracellular epitopes marking the entire canine satellite cell population at all stages of quiescence and activation would be desirable, particularly for studies in which viable satellite cells must be isolated for expansion in culture and/or molecular assays such as microarray or biochemical analysis.
The syndecans, which in mammals comprise a four-member family of type 1 transmembrane proteoglycans, share homology only with each other within their cytoplasmic domains, and each has an extracellular domain that is unique in the genome (reviewed in [25]. Syndecan-3 and syndecan-4 have both been shown to mark satellite cells in the mouse; syndecan-4 in particular has proven to be a useful tool for marking myogenic stem/progenitor cells at all stages of activation (while syndecan-4 is comparatively broadly expressed in the body, in muscle tissue its expression is limited to satellite cells) [18,26]. We previously raised an antibody that specifically detects syndecan-4 of murine origin [27]; however, testing in other species has shown that this antibody does not recognize human, canine, chick or zebrafish syndecan-4 (Cornelison and others, unpublished data).
We generated an antibody against the canine homolog of syndecan-4 with the goal of developing a reagent that would facilitate isolation and analysis of canine satellite cells. We then confirmed its ability to identify primary canine satellite cells, using cells derived from both a wild-type adult dog and a GRMD dog euthanized at 10 months of age due to poor cardiac function, anorexia and severe loss of muscle mass. Surprisingly, in the course of this analysis we observed that using conventional qualitative and quantitative in vitro measures of muscle stem cell identity, proliferative capacity, and myogenic differentiation potential, cells from these two animals are phenotypically indistinguishable from each other under both growth and differentiation conditions. We hypothesize that either instead of or in addition to an accumulated deficit in satellite cell function, the later stages of human and canine muscular dystrophy may involve accumulation of abnormalities in the muscle that render it refractory to satellite cell-mediated repair. The generation of an antibody to the extracellular domain of canine syndecan-4 should also prove useful in future identification, purification, and analysis of satellite cells in disease and therapy studies done in the dog model.
2. Materials and methods
2.1. Anti-canine syndecan-4
The cDNA encoding the extracellular domain of canine syndecan-4 (nt 76–458 of XM_543017.2) was isolated by RT-PCR (forward primer 5′GGG GAT CCG AGT CGA TCC GAG AGA CCG AAG TCA TCG 3′, reverse primer 5′CGA ATT CAC CTC TGT CCT CTC AAA GAT GTT GCC GCC 3′) from total RNA extracted from canine muscle. The product was cut at BamHI and EcoRI sites engineered into the primers and cloned into pRSET-A (Invitrogen) and validated by sequencing. The expression vector was transformed into BL21-STAR cells and a single clone was picked and grown in liquid culture. The cells were induced with imidazole to produce 6× His-tagged epitope, which was purified over a Ni++ slurry (ProBond, Invitrogen). Purified recombinant protein was used to produce chicken polyclonal antibodies (Aves Labs, Tigard OR). The antibody specifically recognizes the immunogenic peptide as well as a 22 kDa band in programmed but not unprogrammed rabbit reticulocyte lysate.
2.2. Canine satellite cells
Primary satellite cells were isolated from canine muscle using the protocol we have established for mouse satellite cells [27]. Wild type cells are from the cranial tibial muscle of a 1-year-old female euthanized after surgery and GRMD cells are from the cranial tibial muscle of a 9-month-old GRMD female euthanized on veterinarian’s recommendation due to the severity of the dystrophic phenotype. Briefly, we removed 1 cm3 muscle samples from each dog, post-euthanasia, and physically dissociated the tissue by mincing, followed by enzymatic digestion with collagenase type I (Worthington). Adjacent 1 cm3 samples from each animal were harvested at the same time and frozen on dry ice for sectioning. Liberated cells are preplated for 24 h, then nonadherent cells are passaged to gelatin-coated plates for expansion. Cells are maintained at 37 °C in a humidified incubator at 5% CO2; growth medium is Ham’s F-12 supplemented with 15% horse serum, 0.5 nM recombinant human FGF2, and penicillin/streptomycin. Differentiation medium is Kaighn’s F-12 supplemented with 2% horse serum and penicillin/streptomycin.
2.3. Western blotting
Immunogen, in vitro translated canine syndecan-4 (TnT, Promega) or lysate of proliferating canine satellite cells in modified RIPA buffer (50 mM Tris pH 7.4, 1% NP-40, 0.25% Na deoxycholate, 150 mM NaCl, 1 mMEDTA) supplemented with protease inhibitors (Roche) were electrophoresed on 4–12% gradient polyacrylamide gels (Invitrogen) and transferred overnight to PVDF membrane (Millipore). Membranes were blocked in Starting Block (Pierce) then incubated overnight at 4° with anti-canine-syndecan-4 diluted 1:1500 in Starting Block. Membranes were washed in TBS containing 0.1% Tween-20 then incubated with goat anti-chicken HRP (Santa Cruz) at 1:10,000 for 1 h at room temperature. Secondary antibody binding was visualized using SuperSignal West (Pierce).
2.4. Antibodies and staining
Cells were plated on glass coverslips coated with 0.66% gelatin and allowed to proliferate for 48 h. Half of the coverslips were fixed in 4% paraformaldehyde, half were washed in PBS and switched to differentiation media for an additional 72 h before fixing. Coverslips with proliferating or differentiated cells were blocked in 10% BlokHen II (Aves) in water for 1 h, rinsed with PBS and blocked in 10% goat serum in PBS for 1 h. They were then incubated with primary antibodies or 10% goat serum as a control overnight, rinsed with PBS, and incubated with secondary antibodies for 1 h at RT. Coverslips were washed and mounted, nuclei were visualized with DAPI. Frozen sections (cut from the block of tissue adjacent to the sample from which cells were isolated) were postfixed in 4% paraformaldehyde prior to staining. Primary antibodies used were: anti-canine-syndecan-4, 1:1500; anti-Pax7 (Developmental Studies Hybridoma Bank) at 1:10; anti-phospho histone 3 (Santa Cruz) at 1:100; anti-MyoD (Novocastra) at 1:10; anti-myogenin (F5D, Developmental Studies Hybridoma Bank) at 1:5; anti-MHC (Developmental Studies Hybridoma Bank) neat; anti-dystrophin (Santa Cruz) at 1:50; anti-NCAM (Chemicon) at 1:100. Images were collected and analyzed using Slidebook (Intelligent Imaging Innovations).
2.5. Flow cytometry
Proliferating cells were removed from the plate with warmed PBS and fixed for 10 min in ice-cold 4% paraformaldehyde. Fixed cells were washed in PBS, blocked in 10% normal goat serum in PBS, then incubated overnight at 4 °C with primary antibody or with 10% serum alone as a control. Cells were washed twice in PBS then incubated with secondary antibody for 45 min at RT, then analyzed on an Accuri C6 flow cytometer. Gates were set such that less than 3% of cells stained with secondary antibodies alone were counted. Syndecan-4 (chicken) was analyzed in the green channel, NCAM (rat) was analyzed in the far-red channel, and Pax7, pH3, or MyoD (all mouse) were analyzed in the red channel.
2.6. Motility assay
Cell motility in two dimensions was assayed by time-lapse videomiscoscopy. Cells were plated on gelatin-coated wells and allowed to adhere, then monitored for 24 h with one image acquired every 10 min. Images were stacked in IPLab and analyzed in MetaMorph; individual cell tracings of 50 cells/genotype were collected.
3. Results
3.1. Cloning of canine syndecan-4 coding sequence
To provide a template with which to generate a recombinant protein fragment, we isolated the mRNA coding for syndecan-4 from canine muscle tissue. Using primers written to the published genomic sequence, we isolated and sequenced a 926-bp cDNA corresponding to the entire coding sequence of syndecan-4, as well as 51 bp upstream of the translation start site and 23 bp downstream of the translation stop site; when compared to mouse and human syndecan-4 coding sequences, the canine sequence shares approximately 80% identity at both the DNA and the protein level (Fig. 1). This sequence was then used to design primers that would amplify the coding sequence for the extracellular domain downstream from the signal sequence and upstream of the transmembrane domain, flanked by convenient restriction sites.
Fig. 1.
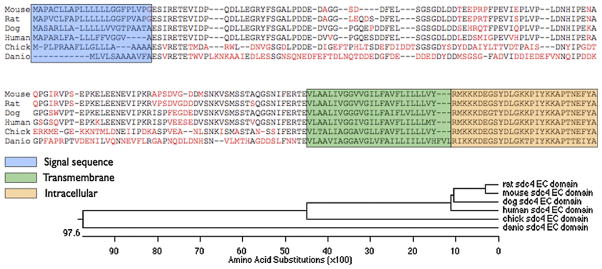
Alignment of syndecan-4 protein sequences for commonly-used model organisms (including human). Amino acids shown in red in the extracellular domain are nonconserved compared to the concensus. The phylogenetic tree depicts amino acid divergence among sequences.
3.2. Generation of canine-specific anti-syndecan-4
To prospectively identify canine satellite cells in either muscle sections or primary cell culture, we raised a polyclonal antibody to the extracellular domain of canine syndecan- 4. A similar antibody raised against the murine epitope has been useful in identification of satellite cells in mouse via indirect immunohistochemistry or fluorescence- activated flow cytometry [18,19]. However, that antibody does not detect canine syndecan-4 (Fig. 2) necessitating production of a canine-specific reagent.
Fig. 2.

Anti-canine-syndecan-4 reacts with syndecan-4 epitope and protein. (a) Western blot of serial 10-fold dilutions ranging from 10 μg to 100 pg of bacterially-expressed canine syndecan-4 extracellular domain. (b) Western blot of 1/10 reaction of in vitro translated (TnT, Promega) full-length canine syndecan-4 protein (lane 1) and 1/10 reaction of unprogrammed lystate (lane 2). (c) Western blot of 60 μg of canine satellite cell lysate.
A bacterially-expressed fusion protein corresponding to the fragment of canine syndecan-4 protein between the signal sequence and the transmembrane domain was used as immunogen to produce a chicken polyclonal antibody (Aves Labs). We first tested the antibody in Western blot to determine its specificity for (1) the bacterially-expressed extracellular domain epitope it was raised against; (2) in vitro translated full-length canine syndecan-4, which would contain the core protein but would not be decorated by heparan sulfate; and (3) lysate of primary canine satellite cells, treated with heparitinase to remove the large, heterogeneous, and highly-charged heparan sulfate chains. We found that the antibody will reliably detect as little as 100 pg of epitope (Fig. 2a), and specifically detects full-length in vitro translated protein (Fig. 2b). When we queried wild type canine satellite cell lysate, we were able to detect two bands of ~22 and ~45 kDa, which correspond to the predicted molecular weight of the canine syndecan-4 core protein monomer and a possible homodimer (Fig. 2c). From these results, we conclude that the antibody we raised specifically detects syndecan-4 in canine samples, and should therefore be a useful tool for identification of satellite cells in vivo or in vitro.
3.3. Identification of canine satellite cells
To confirm that the syndecan-4 antibody could identify quiescent satellite cells in section by indirect immunohistochemistry, we obtained cryosections from the muscle of a healthy dog as well from a 10-month-old GRMD dog euthanized due to clinical signs associated with the disease. Following brief paraformaldehyde fixation, both sets of sections were stained with an antibody to laminin (to indicate the myofiber external lamina) and costained with either anti-syndecan-4 or anti-Pax-7. Nuclei were visualized with DAPI. We identified multiple views that contained cells that meet our criteria for satellite cells in sections (beneath the external lamina, containing a single nucleus, and expressing either syndecan-4 or Pax-7) (Fig. 3). We then asked if there were differences in quiescent satellite cell number in GRMD vs. wildtype samples. By Pax7 staining, we identified similar numbers of satellite cells in both sample types (55.5 ± 3.5 and 53.7 ± 4.6 cells/mm2 in 10 μm sections of wild type and GRMD, respectively).
Fig. 3.

Anti-syndecan-4, as well as anti-Pax7, identifies quiescent canine satellite cells in fixed frozen sections. Frozen sections of wild type or GRMD cranial tibial muscle stained for laminin (red) to identify myofiber boundaries. Panels are costained (green) for either syndecan-4 (top row) or Pax-7 (bottom row). Scale bar = 50 μm. Insets: 2× magnified fields containing stained satellite cells indicated by arrowheads.
3.4. In vitro comparison of primary satellite cells from wild type and dystrophic muscle: identity, proliferation, and differentiation
Primary satellite cells were mechanically and enzymatically isolated from samples of muscle adjacent to the areas shown in frozen section above according to our conventional protocol developed for mouse [28], then expanded in adherent culture on gelatin-coated plates in the presence of 15% horse serum and 0.5 nM FGF-2 (to maintain proliferation). These satellite cell-derived myoblasts were then either replated on gelatin-coated coverslips for immunostaining, or gently detached for flow cytometric analysis. Sets of coverslips were either maintained in proliferation conditions, or switched to 2% horse serum in the absence of FGF2 to promote differentiation; after three days under these conditions coverslips were fixed and stained with antibody to syndecan-4 and costained with antibodies directed against either: Pax7, to identify syndecan-4+ve cells coexpressing the satellite cell lineage marker; phosphorylated histone 3 (pH3), to identify syndecan-4+ve cells in M phase of the cell cycle; the early myogenic regulatory transcription factor MyoD and the late myogenic regulatory transcription factor myogenin, to identify syndecan-4+ve cells committed to myogenesis; or myosin heavy chain, to assess terminal differentiation. In parallel, we used flow cytometric analysis to quantify the fraction of satellite cells derived from wild type or GRMD samples positive for Pax7, pH3, or MyoD. While live unfixed canine satellite cell progeny can be successfully identified with anti-syndecan-4 by flow cytometry (data not shown), all samples described here were briefly fixed in paraformaldehyde to permit costaining for these nuclear antigens. We have previously shown that a monoclonal antibody to NCAM (Chemicon) marks mouse satellite cell progeny that have committed to myogenic differentiation [22]; we find that this antibody can also be used to mark differentiating canine myocytes (data not shown). Because of significant changes in morphology associated with myogenic differentiation, FACS and flow cytometry are problematic on differentiating myoblasts; we therefore concentrated on markers associated with proliferation-competent myoblasts and excluded satellite cell progeny already committed to differentiation. We noted that the fraction of satellite cell progeny (syndecan-4 positive; blue bars in Fig. 4f) that had not committed to terminal differentiation (syndecan-4 positive, NCAM negative; red bars in Fig. 4f) was similar in all samples, ranging from 74.9% to 79.2% of all syndecan-4 positive cells. We then asked what percentage of that compartment (proliferation-competent myoblasts) also scored positive for either Pax7, phosphohistone 3, or MyoD in each genotype.
Fig. 4.

Markers of satellite cell identity, proliferation, and differentiation are equivalently distributed in wildtype and GRMD satellite cells in culture. (a) Under proliferation conditions, syndecan-4 and Pax7 are coexpressed by both wildtype and GRMD cells. (b) Under proliferation conditions, mitotic satellite cells identified by anti-syndecan-4 from wildtype and GRMD cultures react with anti-phosphohistone 3 (arrowheads). (c) Under both proliferation and differentiation conditions, satellite cells identified by anti-syndecan-4 from both wildtype and GRMD cultures express MyoD. (d and e) Under proliferation conditions, few syndecan-4 positive satellite cells from either wildtype or GRMD cultures express myogenin or myosin heavy chain, while after three days under differentiation conditions both genotypes display nuclear myogenin and cytoplasmic myosin heavy chain in differentiated myotubes. Myotubes from both genotypes are also qualitatively similar (size, myonuclear number, morphology, etc.). (f) Flow cytometric analysis of mononuclear cells using the immune reagents shown in a–c; distribution of syndecan-4+ve NCAM−ve cells (proliferation-competent myoblasts) is quantitatively similar in all samples. Percentage of proliferation-competent myoblasts that costain for either Pax7, pH3, or MyoD are equivalent in both genotypes. Blue bars, % S4+ (all satellite cell progeny); red bars, % S4+ve NCAM−ve (proliferation-competent myoblasts); green bars, % S4+ve NCAM−veother+ve (proliferation-competent myoblasts positive for Pax7 (first two samples), pH3 (center two samples) or MyoD (last two samples).
3.4.1. Identity
We found that in adherent culture, all cells identified as satellite cells by Pax7 immunoreactivity also stained positively with our syndecan-4 antibody (Fig. 4a). By flow cytometry, greater than 99% of the proliferation-competent myoblast pool (S4+ve NCAM−ve) expressed Pax7 in both genotypes (green bars, first two samples in Fig. 4f), recapitulating the immunohistochemical staining on adherent cells. We therefore conclude that syndecan-4 staining identifies canine satellite cell-derived myoblasts after isolation and culture, and that there is no defect in lineage specification in the GRMD satellite cells.
3.4.2. Proliferation
In adherent culture, syndecan-4 positive cells with characteristic anaphase staining for pH3 were observed in both wild type and GRMD samples (arrowheads, Fig. 4b). By flow cytometry, we find that the fraction of proliferation-competent myoblasts staining positive for pH3 is equivalent between wild type and GRMD cultures, with 5.0% of S4+ve NCAM−ve wild type cells and 5.1% of S4+ve NCAM−ve GRMD cells staining positive for pH3 (green bars, center two samples in Fig. 4f). Clone size in adherent culture is also comparable (data not shown). We therefore conclude that, in ex vivo culture, satellite cells derived from a wildtype dog and an affected GRMD dog do not display a marked difference in proliferation capacity.
3.4.3. Differentiation
We looked at markers of myogenic commitment and differentiation, as well as terminal differentiation markers and cell fusion indices. When we asked what fraction of proliferation-competent myoblasts express the myogenic regulatory transcription factor (MRF) MyoD, indicative of early stages of myogenesis, we detected expression in the nuclei of both mononuclear cells in proliferation medium and multinucleated cells switched to low-serum medium for three days to promote differentiation (Fig. 4c). By flow cytometry, 96.6% of S4+ve NCAM−ve wild type cells and 96.7% of S4+ve NCAM−ve GRMD cells are also positive for MyoD (green bars, last two samples in Fig. 4f). In adherent culture, expression of myogenin, an MRF associated with commitment to terminal differentiation, was largely absent in proliferating cells but strongly associated with fused nuclei in differentiated myotubes (Fig. 4d). Terminal differentiation in myocytes results in upregulation of muscle structural proteins such as myosin heavy chain and fusion of single myocytes into multinucleate myotubes. When we stained for myosin heavy chain we saw equivalent levels of well-organized myosin in both genotypes (Fig. 4e); we also noted a decrease in syndecan-4 expression after terminal differentiation and fusion, as is seen in murine myotubes in culture [26]. We therefore conclude that no primary defect in myogenic differentiation exists ex vivo in satellite cells derived from a GRMD animal.
3.4.4. Migration and motility
While it is not as frequently assessed as proliferation or differentiation, motility of activated satellite cells within injured muscle may also be required for accumulation of necessary numbers of potential myocytes, at least in the case of localized injuries. Failure of engrafted myogenic cells to emigrate from the injection site is also a current difficulty in cell-based therapies [29]. While we do not as yet have the capacity to measure satellite cell migration in vivo, particularly in a large animal model, we can measure simple motility in two dimensions in tissue culture: we have recently used such techniques to identify factors regulating murine satellite cell motility [30]. We therefore compared motility of wild type and GRMD satellite cells over a 24-hour period by timelapse microscopy and found that motility was virtually identical between the two genotypes. In monoculture on a laminin substrate, the average velocity for wild type cells was 54 ± 3 μm/h, while GRMD cells’ average velocity under the same conditions was 53 ± 2 μm/h. Qualitative differences in motility were also undetectable (data not shown).
4. Discussion
The ability of skeletal muscle to regenerate is rivaled only by that of the liver, however there are several physiological states that decrease the efficiency of satellite cell-mediated muscle regeneration. In particular, patients with Duchenne muscular dystrophy are thought to undergo a significant decline in satellite cell capacity after onset of disease symptoms [31], which manifests in increasing severity of muscle wasting and loss of muscle function. However, no explanation for how the loss of dystrophin protein would lead directly to defects in satellite cells (which do not themselves express dystrophin) has yet been validated.
In other mammalian model systems such as the mouse, significant insight has been gained into satellite cell gene expression, signal transduction, engraftment capacity and lineage potential by isolation and analysis of primary satellite cells via immune reagents specific to satellite cell extracellular epitopes. We originally set out to generate such an antibody to the canine syndecan-4 protein, to facilitate identification and purification of canine satellite cells, in particular for use in studies of canine muscular dystrophy models. We cloned the syndecan-4 cDNA from canine tissue and produced a fusion protein consisting of the extracellular domain (which is unique in the genome) and a 6× His tag for purification, then used the fusion protein as immunogen to produce a chicken polyclonal antibody. This reagent identifies both the fusion protein and the native protein by Western blot, and specifically stains the surface of canine satellite cells in culture; it also identifies canine satellite cells by flow cytometry or FACS. We therefore used it to identify canine satellite cells derived from either wildtype or GRMD muscle and compared the satellite cells on several common metrics.
Surprisingly, given the disease state of the GRMD cell donor, we found no overt differences in satellite cell physiology between genotypes as measured by: proliferation rate, clonogenicity, expression of Pax7 or myogenic regulatory transcription factors, incorporation into multinucleate myotubes, expression of terminal differentiation proteins, or motility in two dimensions. These in vitro results suggested that there is no major deficit in satellite cell function from dystrophic animals when they are removed from the dystrophic muscle environment, even if the animal presents with clinical signs of severe disease warranting euthanasia. This parallels earlier work in which few differences in cellular morphology were found in culture between cells taken from much younger (2–17 weeks of age) wild type or X-linked muscular dystrophy animals [32]. These unexpected findings are both encouraging, as they suggest that satellite cell capacity may not be exhausted or significantly decreased even after prolonged degeneration/regeneration cycles in a progressive disease setting, and problematic, because if the affected muscle environment is a significant contributor to the decrease in satellite cell function in vivo then therapies aimed at enhancing satellite cell activity in tissue where these changes have already occurred may not be effective. A significant question to be answered, therefore, is which aspects of the local and/or systemic environment are deleteriously affecting satellite cell function?
It is well-established that satellite cell activity and efficiency in young versus aged animals can be altered by factors deriving from sources outside the muscle itself; for example, classical studies of heterochronic transplants [33] and more recent heterochronic parabiosis experiments [34] have demonstrated that satellite cells from a young donor exposed to an aged cellular environment show a decrease in proliferation and differentiation capacity, while cells from an old animal can be ‘rejuvenated’ by exposure to a young environment or circulation. This effect has been shown to rely on levels of signaling components such as Delta in the circulation [35], which alters satellite cell proliferation. No such experiments have been done to assess the effect of circulating factors on satellite cell efficacy per se during disease, however such an effect would be consistent with current research (reviewed in [36]). Since muscle breakdown in muscular dystrophy is accompanied by chronic inflammation, it is possible the presence of inflammatory cytokines is negatively affecting satellite cell activity. Alternatively, in both human patients and canine models, structural changes in the muscle including fibrosis, interstitial and intracellular lipid deposits, and discrepancies in myofiber caliber accumulate with disease progression. These changes may cumulatively render the muscle refractory to repair by satellite cells, independent of the state of the satellite cells themselves. In either case, an awareness of this effect will ideally result in improved satellite cell-based therapies in the future.
Acknowledgments
The authors with to thank Dr. FA Mann and Dr. Dongsheng Duan for wildtype and dystrophic canine muscle samples.
Footnotes
The ability to isolate primary satellite cells has proven valuable in studies of satellite cell-specific gene expression, cell signaling, and cell activity in other model organisms such as mouse. It is our hope that additional studies into the satellite cell contribution to neuromuscular disease progression and therapies will be aided by the development of the anti-canine-syndecan-4, and we would welcome requests for its distribution to other researchers.
References
- 1.Tatsumi R, Anderson JE, Nevoret CJ, Halevy O, Allen RE. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev Biol. 1998;194:114–28. doi: 10.1006/dbio.1997.8803. [DOI] [PubMed] [Google Scholar]
- 2.Allen RE, Sheehan SM, Taylor RG, Kendall TL, Rice GM. Hepatocyte growth factor activates quiescent skeletal muscle satellite cells in vitro. J Cell Physiol. 1995;165:307–12. doi: 10.1002/jcp.1041650211. [DOI] [PubMed] [Google Scholar]
- 3.Anderson JE. A role for nitric oxide in muscle repair: nitric oxidemediated activation of muscle satellite cells. Mol Biol Cell. 2000;11:1859–74. doi: 10.1091/mbc.11.5.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi X, Garry DJ. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006;20:1692–708. doi: 10.1101/gad.1419406. [DOI] [PubMed] [Google Scholar]
- 5.Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol. 2001;91:534–51. doi: 10.1152/jappl.2001.91.2.534. [DOI] [PubMed] [Google Scholar]
- 6.Sadeh M, Czyewski K, Stern LZ. Chronic myopathy induced by repeated bupivacaine injections. J Neurol Sci. 1985;67:229–38. doi: 10.1016/0022-510x(85)90119-4. [DOI] [PubMed] [Google Scholar]
- 7.Luz MA, Marques MJ, Santo Neto H. Impaired regeneration of dystrophin-deficient muscle fibers is caused by exhaustion of myogenic cells. Braz J Med Biol Res. 2002;35:691–5. doi: 10.1590/s0100-879x2002000600009. [DOI] [PubMed] [Google Scholar]
- 8.Tennyson CN, Klamut HJ, Worton RG. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet. 1995;9:184–90. doi: 10.1038/ng0295-184. [DOI] [PubMed] [Google Scholar]
- 9.Tuffery-Giraud S, Beroud C, Leturcq F, et al. Genotype–phenotype analysis in 2405 patients with a dystrophinopathy using the UMD–DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009;30:934–45. doi: 10.1002/humu.20976. [DOI] [PubMed] [Google Scholar]
- 10.Flanigan KM, Dunn DM, von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–66. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–80. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 12.Itagaki Y, Saida K, Iwamura K. Regenerative capacity of mdx mouse muscles after repeated applications of myo-necrotic bupivacaine. Acta Neuropathol. 1995;89:380–4. doi: 10.1007/BF00309633. [DOI] [PubMed] [Google Scholar]
- 13.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–38. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 14.Kornegay JN, Tuler SM, Miller DM, Levesque DC. Muscular dystrophy in a litter of golden retriever dogs. Muscle Nerve. 1988;11:1056–64. doi: 10.1002/mus.880111008. [DOI] [PubMed] [Google Scholar]
- 15.Schatzberg SJ, Olby NJ, Breen M, et al. Molecular analysis of a spontaneous dystrophin ‘knockout’ dog. Neuromuscul Disord. 1999;9:289–95. doi: 10.1016/s0960-8966(99)00011-5. [DOI] [PubMed] [Google Scholar]
- 16.Montarras D, Morgan J, Collins C, et al. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–7. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- 17.Fukada S, Uezumi A, Ikemoto M, et al. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells. 2007;25:2448–59. doi: 10.1634/stemcells.2007-0019. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka KK, Hall JK, Troy AA, Cornelison DD, Majka SM, Olwin BB. Syndecan-4-expressing muscle progenitor cells in the SP engraft as satellite cells during muscle regeneration. Cell Stem Cell. 2009;4:217–25. doi: 10.1016/j.stem.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conboy MJ, Cerletti M, Wagers AJ, Conboy IM. Immuno-analysis and FACS sorting of adult muscle fiber-associated stem/precursor cells. Methods Mol Biol. 2010;621:165–73. doi: 10.1007/978-1-60761-063-2_11. [DOI] [PubMed] [Google Scholar]
- 20.Prattis SM, Gebhart DH, Dickson G, Watt DJ, Kornegay JN. Magnetic affinity cell sorting (MACS) separation and flow cytometric characterization of neural cell adhesion molecule-positive, cultured myogenic cells from normal and dystrophic dogs. Exp Cell Res. 1993;208:453–64. doi: 10.1006/excr.1993.1267. [DOI] [PubMed] [Google Scholar]
- 21.Michal J, Xiang Z, Davenport G, Hayek M, Dodson MV, Byrne KM. Isolation and characterization of canine satellite cells. In Vitro Cell Dev Biol Anim. 2002;38:467–80. doi: 10.1290/1071-2690(2002)038<0467:IACOCS>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 22.Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–86. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 23.Ericson J, Morton S, Kawakami A, Roelink H, Jessell TM. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell. 1996;87:661–73. doi: 10.1016/s0092-8674(00)81386-0. [DOI] [PubMed] [Google Scholar]
- 24.Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol. 2004;166:347–57. doi: 10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmermann P, David G. The syndecans, tuners of transmembrane signaling. FASEB J. 1999;13:S91–S100. doi: 10.1096/fasebj.13.9001.s91. [DOI] [PubMed] [Google Scholar]
- 26.Cornelison DDW, Filla MS, Stanley HM, Rapraeger AC, Olwin BB. Syndecan-3 and syndecan-4 specifically mark skeletal muscle satellite cells and are implicated in satellite cell maintenance and muscle regeneration. Dev Biol. 2001;239:79–94. doi: 10.1006/dbio.2001.0416. [DOI] [PubMed] [Google Scholar]
- 27.Cornelison DDW, Wilcox-Adelman SA, Goetinck PF, Rauvala H, Rapraeger AC, Olwin BB. Essential and separable roles for Syndecan-3 and Syndecan-4 in skeletal muscle development and regeneration. Genes Dev. 2004;18:2231–6. doi: 10.1101/gad.1214204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capkovic KL, Stevenson S, Johnson MC, Thelen JJ, Cornelison DDW. Neural cell adhesion molecule (NCAM) marks adult myogenic cells committed to differentiation. Exp Cell Res. 2008;314:1553–65. doi: 10.1016/j.yexcr.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peault B, Rudnicki M, Torrente Y, et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007;15:867–77. doi: 10.1038/mt.sj.6300145. [DOI] [PubMed] [Google Scholar]
- 30.Siegel AL, Atchison K, Fisher KE, Davis GE, Cornelison DD. 3D timelapse analysis of muscle satellite cell motility. Stem Cells. 2009;27:2527–38. doi: 10.1002/stem.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blau HM, Webster C, Pavlath GK, Chiu CP. Evidence for defective myoblasts in Duchenne muscular dystrophy. Adv Exp Med Biol. 1985;182:85–110. doi: 10.1007/978-1-4684-4907-5_7. [DOI] [PubMed] [Google Scholar]
- 32.Valentine BA, Chandler SK, Cummings JF, Cooper BJ. In vitro characteristics of normal and dystrophic skeletal muscle from dogs. Am J Vet Res. 1991;52:104–7. [PubMed] [Google Scholar]
- 33.Carlson BM, Faulkner JA. Muscle transplantation between young and old rats: age of host determines recovery. Am J Physiol. 1989;256:C1262–6. doi: 10.1152/ajpcell.1989.256.6.C1262. [DOI] [PubMed] [Google Scholar]
- 34.Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–4. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 35.Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–7. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 36.Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:R345–53. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]