

Abstract
Steroid-resistant asthma comprises an important source of morbidity in patient populations. TH17 cells represent a distinct population of CD4+ Th cells that mediate neutrophilic inflammation and are characterized by the production of IL-17, IL-22, and IL-6. To investigate the function of TH17 cells in the context of Ag-induced airway inflammation, we polarized naive CD4+ T cells from DO11.10 OVA-specific TCR-transgenic mice to a TH2 or TH17 phenotype by culturing in conditioned medium. In addition, we also tested the steroid responsiveness of TH2 and TH17 cells. In vitro, TH17 cytokine responses were not sensitive to dexamethasone (DEX) treatment despite immunocytochemistry confirming glucocorticoid receptor translocation to the nucleus following treatment. Transfer of TH2 cells to mice challenged with OVA protein resulted in lymphocyte and eosinophil emigration into the lung that was markedly reduced by DEX treatment, whereas TH17 transfer resulted in increased CXC chemokine secretion and neutrophil influx that was not attenuated by DEX. Transfer of TH17 or TH2 cells was sufficient to induce airway hyperresponsiveness (AHR) to methacholine. Interestingly, AHR was not attenuated by DEX in the TH17 group. These data demonstrate that polarized Ag-specific T cells result in specific lung pathologies. Both TH2 and TH17 cells are able to induce AHR, whereas TH17 cell-mediated airway inflammation and AHR are steroid resistant, indicating a potential role for TH17 cells in steroid-resistant asthma.
The CD4+ Th cells are classified as TH1, TH2, and TH17 based on their cytokine expression profile. Classically, recruitment of TH2 lymphocytes that secrete IL-4, IL-5, IL-9, and IL-13 accompanied by eosinophil recruitment to the airways has been considered integral to the pathogenesis of asthma. Inflammatory cell recruitment to the lung results in tissue damage, mucus hypersecretion, bronchoconstriction, and airway hyperresponsiveness (AHR).3 Glucocorticoid use attenuates airway eosinophilia by inducing eosinophil apoptosis and inhibiting the response to IL-5 and GM-CSF survival signals (1, 2). The broad distribution of the glucocorticoid receptor in the airways suggests there are multiple cell targets including not only inflammatory cells, such as mast cells and the aforementioned eosinophil, but epithelial and airway smooth muscle cells (3). Studies have shown that 50% of asthma cases are noneosinophilic and in these cases CXCL8-mediated neutrophil inflammation in the airways is predominant (4).
TH17 cells are a distinct population of CD4+ T cells that secrete IL-17A, IL-17F, and IL-22 (5, 6). The importance of TH17 cells in neutrophilic inflammation lies in the ability of IL-17 to induce granulopoiesis, neutrophil chemotaxis, and the antiapoptotic properties of G-CSF (7, 8). IL-17 is increased in the sputum of asthmatic patients and correlates with CXCL8 levels and the number of neutrophils in the sputum. PBMCs from both atopic and nonatopic asthmatics secrete IL-17 in response to anti-CD3 and anti-CD28 Ab stimulation (9). Apoptosis of neutrophils is inhibited by glucocorticoids (10) and numerous studies have suggested noneosinophilic asthma is associated with poor response to corticosteroid treatment (11, 12). Based on this, we hypothesized that TH17 cells mediate airway inflammation and hyperresponsiveness associated with noneosinophilic asthma and are not responsive to glucocorticoid treatment.
Models using adjuvant-induced Ag sensitization with alum have shown that IL-17A and IL-17RA KO mice have reduced delayed-type hypersensitivity reactions in tissue (13) and reduced priming of TH2-immune responses in lung lymph nodes (Ref. 14 and our unpublished observations). This may be due to the recently described role of IL-17 in germinal center formation and setting up chemokine gradients in lymph nodes (15). Thus, upon Ag challenge, IL-17RA KO mice have shown reduced recruitment of both neutrophils and eosinophils into the airway (14). Whether this is due to a role for IL-17RA in T cell priming in the lymph nodes or in the effector phase after Ag challenge remains unclear (14). Moreover it remains unclear from the studies published to date whether TH17 cells in and of themselves mediate airway hyperresponsiveness and goblet cell hyperplasia independent of Th1 or TH2 cells. Based on these data, we used a model that had previously been used to study the effect of TH2 cells in inducing airway inflammation and airway hyperresponsiveness utilizing adoptive transfer of polarized T cell populations to SCID mice (16). SCID mice were chosen to isolate the contribution of the transferred cells as opposed to contaminating endogenous CD4+ T cell populations. Additionally, this model does not use adjuvant priming in vivo which has recently been questioned in terms of its relevance to airway sensitization, which is what occurs in human allergen sensitization.
Presently, we report that in vitro-polarized TH17 cells are non-responsive to glucocorticoids. Adoptive transfer of these cells in a model of Ag-induced airway inflammation resulted in increased CXC chemokine secretion and G-CSF in the lung that was associated with neutrophil influx to the airways. Treatment of TH17 cell reconstituted mice with dexamethasone (DEX) did not alter airway inflammation but did attenuate TH2-induced airway inflammation. Reconstitution of mice with either TH2 or TH17 cells resulted in increased AHR to methacholine challenge. Surprisingly, DEX treatment significantly reduced AHR in mice that received TH2 cells but not in TH17-reconstituted animals. These data demonstrate the significance of TH17 cellular responses in airway disease and implicate these cells as having a role in steroid-resistant asthma.
Materials and Methods
Mice
Six- to 8-wk-old, male BALB/c mice and SCID mice were purchased from The National Cancer Institute. C.DO11.10 TCR-transgenic mice were obtained from The Jackson Laboratory. IL-17R null mice have been previously described (17). All animals were housed in a pathogen-free environment and given food and water ad libitum. All experiments were approved by the Children’s Hospital of Pittsburgh Animal Research and Care Committee.
Abs and reagents
Cell culture
Recombinant mouse (rm) IL-2, rmIL-4, rmIL-6, rmIL-23, porcine TGF-β, anti-mouse IL-4 (clone 30340.11), and anti-mouse IFN-γ (clone 37895) were purchased from R&D Systems.
Immunofluorescence
Rabbit anti-mouse glucocorticoid receptor (GR; Affinity BioReagents) and goat anti-rabbit IgG conjugated to Alexa Fluor 555 (Invitrogen) were used.
Western blot
Polyclonal rabbit anti-mouse GR (Affinity BioReagents), mouse monoclonal IgG1 anti-chicken β-actin (Santa Cruz Biotechnology), goat anti-rabbit IgG conjugated to alkaline phosphatase (AP; Bio-Rad), and goat anti-mouse IgG conjugated to AP (Santa Cruz Biotechnology) were used.
In vitro differentiation of Th cell subsets
CD4+ T cells from the spleens and lymph nodes of C.DO11.10 TCR-transgenic mice were enriched by negative selection using magnetic beads (Miltenyi Biotec). CD4+CD62L+CD25− cells were sorted on a FACSAria (BD Biosciences); purity was consistently >99%. Cells were cultured with irradiated APCs pulsed with OVA peptide 323–339 (OVA323–339) under polarizing conditions as previously reported (18). Briefly, medium was supplemented with 20 U/ml IL-2, 5 ng/ml rmIL-4, and 10 μg/ml anti-IFN-γ (for TH2 polarization), or 10 ng/ml rmIL-23, 1 ng/ml TGF-β, 20 ng/ml rmIL-6, 10 μg/ml anti-IL-4, and anti-IFN-γ (for TH17 polarization). Cultures were split 1:2 on day 3 and harvested on day 6. The total number of cells was determined by counting on a hemocytometer; viable cells were defined by trypan blue exclusion. APCs alone were CD4neg or CD4dim and proliferating CD4+ T cells were CD4bright and stained positively with KJ1-26 (eBioscience) and anti-Do11.01 TCR Ab. CD4bright cells were selected for immunohistochemistry studies.
In vitro assays
To confirm the phenotype of the Th cell subsets, cells were stimulated with in vitro with PMA/ionomycin for ELISPOT assays (R&D Systems) or anti-CD3/anti-CD28-coated microbeads (Dynal) for cytokine assays. Where indicated, cells were pretreated with increasing doses of DEX (Sicor Pharmaceuticals) for 2 h before the addition of microbeads. After 36 h in culture, cell supernatants were collected and stored at −80° until analysis. To measure NFAT activity of polarized T cells, primary culture mouse T cells (1 × 106/1.5 ml) were transfected with 4 μg of NFAT-luciferase plasmid (gift from S. Gaffen, State University of New York, Buffalo, NY) and 1 μg of SV40-Renilla. Transfection was conducted with the Mouse T Cell Nucleofector Kit (Amaxa Biosystems) using the X-005 program on the Nucleofector II instrument (Amaxa Biosystems). Following transfection, T cells were cultured for 18 h and were then treated with or without 1 μM DEX for 2 h before treatment with anti-CD3/anti-CD28-coated Dynabeads (Invitrogen Dynal) for 6 h at 37°C. After stimulation, luciferase activity was determined using the Dual-luciferase Reporter Assay System (Promega). NFAT-luciferase levels were normalized to SV40-Renilla as an internal transfection control.
Immunocytochemistry
Superfrost Plus Micro Slides (VWR International) of 100,000 cells CD4+ cells (selected by Miltenyi beads) were fixed in 100% ice-cold methanol for 10 min. Slides were stained with rabbit anti-mouse GR and visualized with goat anti-rabbit IgG conjugated to Alexa Fluor 555 (Invitrogen) as previously described (19). Accumulation of GR in the nucleus was observed by epifluorescence illumination with a Zeiss Axioplan 2 microscope equipped with a ×63, 1.4 aperture oil immersion objective. Quantitation of fluorescence was performed by analyzing regions of interests (drawn over the 4′,6-diamidino-2-phenylindole nuclear signal) of 100 cells captured with a charge-coupled device camera (Intelligent Imaging Innovations) and relative intensity values were determined and averaged with SlideBook software (Intelligent Imaging Innovations).
Western blot analysis
Nuclear and cytoplasmic protein fractions were extracted from differentiated T cells after DEX treatment using the Nuclear Extract Kit (Active Motif) according to the manufacturer’s instructions. Protein concentration was determined using a BCA Protein Assay Kit (Pierce Biotechnology). AP development was visualized by the AP Conjugate Substrate Kit (Bio-Rad) according to the manufacturer’s instructions.
Adoptive transfer model
BALB/c SCID mice were challenged with OVA protein (50 μg/mouse, intratracheal (i.t.); Sigma-Aldrich) on day −1. On day 0, 1 × 106 differentiated Th cells were adoptively transferred by retro-orbital injection. Mice were challenged with OVA for 3 consecutive days after cell transfer (50 μg/mouse/day, i.t.). Animals were sacrificed 24 h after the last airway challenge. Where indicated, mice were treated with 2.5 mg/kg DEX or PBS control 2 h before cell transfer on day 0 and before OVA challenge on day 2. In separate experiments, wild-type (WT) BALB/c mice and IL-17R null mice on a BALB/c background underwent the same adoptive transfer and OVA challenge protocol.
Bronchoalveolar lavage (BAL) fluid and lung tissue collection
Lungs were lavaged with five 1-ml volumes of calcium- and magnesium-free PBS. The supernatant from the first 1-ml aliquot was stored at −80°C for later analysis. Cell pellets from both aliquots were pooled in 1 ml and total white blood cells in BAL fluid were counted using a particle counter (Z1; Beckman Coulter). Cytospins of 100,000 cells were prepared using a Shandon Cytospin 4 (Thermo Electron), slides were stained with HEMA 3 (Fisher Scientific), and differentials were quantified by counting 100 cells.
Lungs were harvested following the collection of lavage fluid. The right lungs were homogenized in 1 ml of PBS containing 0.05% Triton X-100 and Complete Protease Inhibitor (Roche). Homogenate was centrifuged at 12,000 × g for 15 min and supernatant was stored at −80°C for later cytokine analysis.
Cytokine analysis
Cell supernatants, BAL fluid, and lung homogenate (LH) samples were analyzed for protein levels of G-CSF, IL-4, IL-5, IL-6, IL-13, IL-17, IFN-γ, and KC using a Luminex multiplex suspension cytokine array (Linco) according to the manufacturer’s instructions. The data were analyzed using Bio-Plex Manager software (Bio-Rad). IL-22 was measured using a commercial ELISA (R&D Systems) following instructions provided by the manufacturer.
Overexpression of IL-4 and IL-17 in the lungs
Adenovirus expressing IL-4, IL-17, or luciferase as a control was given at 108 PFU i.t. on day 0 and AHR was determined 72 h later. Overexpression of IL-4, IL-13, and IL-17 was determined by Luminex in both the BAL fluid and LH of recipient mice.
Determination of AHR
Airway responsiveness to methacholine challenge was determined on day 4 as previously described using anesthetized, intubated mice connected to a computer-controlled small-animal mechanical ventilator (flexiVent; SCI-REQ) (20). Airway resistance values (Rn) were calculated in response to progressive concentrations of methacholine administered by i.v. injection.
Statistical analysis
Data are reported as mean ± SEM. GraphPad Prism version 4.0 was used to calculate p values using one-way ANOVA with a Tukey multiple comparison posttest. A value of p < 0.05 was considered statistically significant.
Results
TH17 cells are resistant to DEX treatment in vitro
Studies have shown that glucocorticoids inhibit the production of IL-4, IL-5, and IL-13 from TH2 cells (21). To test the sensitivity of TH17 cells to glucocorticoids, naive CD4+ T cells from DO11.10 TCR-transgenic mice were differentiated in the presence of IL-6, IL-23, TGF-β, anti-IL-4, and anti-IFN-γ (18). As a positive control, naive CD4+ T cells were polarized toward a TH2 phenotype by conditioning with IL-2, IL-4, and anti-IFN-γ. After 6 days in culture, polarization was confirmed by PMA/ionomycin stimulation and ELISPOT analysis (Fig. 1, A and B) and secreted cytokine analysis (Fig. 1, C–F). Only TH17 cells produced IL-17 (Fig. 1A), whereas in TH2 conditions the precursor frequency of IL-4-producing T cells was 5.6-fold greater in TH2 cells compared with T cells grown in TH17 conditions (Fig. 1B). Cells were also harvested on day 6 and treated with increasing doses of DEX for 2 h before stimulation with anti-CD3/anti-CD28-coated microbeads. As expected, TH2 cells secreted IL-4 (data not shown), IL-5, and IL-13 (Fig. 1, C and D) following CD3/CD28 stimulation and TH17 cells secreted high levels of IL-17 and IL-22 (Fig. 1, E and F). Although both IL-5 and IL-13 generation from TH2 cells were inhibited at all doses of DEX studied, TH17 cell cytokine production was not sensitive to DEX treatment at any dose tested. Apoptosis was analyzed by annexin V and 7-ami-noactinomycin D staining and >85% of the TH2 and TH17 cells were annexin V and 7-aminoactinomycin negative (viable) after 1 μM DEX treatment (data not shown).
FIGURE 1.

TH17 cells are not sensitive to DEX treatment in vitro. CD4+CD62L+CD25− naive T cells isolated from DO11.10 OVA TCR-transgenic mice were cultured with WT BALB/c splenocytes that had been pulsed with OVA323–339 under polarizing conditions. On day 6, cells were collected and stimulated with PMA/ionomycin for precursor frequency by ELISPOT or pretreated for 2 h with the indicated doses of DEX before stimulation with CD3/CD28 beads + IL-2. A, Spot frequencies of IL-17-producing T cells. B, Spot frequencies of IL-4-producing T cells. Cells were plated under the following conditions: T cells indicate un-stimulated T cells cultured without bead stimulation; 1 μM DEX indicates T cells pretreated with 1 μM DEX but not stimulated with CD3/CD28 microbeads; beads indicates untreated T cells stimulated with CD3/CD28 microbeads; μM DEX + bead indicates T cells pretreated with the indicated dose of DEX and stimulated with CD3/CD28 microbeads. Cell culture supernatant was collected at 36 h after stimulation and the levels of TH2 and TH17 cytokines were determined by multiplex suspension array assay. C, IL-5 levels from TH2 cells. D, IL-13 levels from TH2 cells. E, IL-17 levels from TH17 cells. F, IL-22 levels from TH17 cells. All data are graphed as mean ± SEM for n = 5–8;*, p < 0.01.
DEX treatment inhibits Ag-specific TH2-mediated airway inflammation but not TH17-mediated airway inflammation in an adoptive transfer model
To determine the Ag-specific immune response elicited by effector TH17 cells in vivo, an adoptive transfer model of airway Ag-induced inflammation was used (Fig. 2A). In vitro-polarized TH cell subsets were transferred i.v. to SCID mice on a BALB/c background that had been challenged with OVA protein i.t. 1 day before cell transfer to promote cell migration to the airways (16). Following cell transfer, mice were challenged with OVA i.t. for 3 consecutive days and sacrificed on day 4, 24 h after the last OVA challenge. Mice were treated with DEX (2.5 mg/kg) or PBS control by i.p. injection 2h before cell transfer on day 0 and again 2 h before OVA challenge on day 2. Control mice reconstituted with TH2 cells that were challenged with OVA had increased levels of IL-5 and IL-13 in LH (Fig. 2, B and C). Mice transferred TH2 cells without OVA challenge or scid mice challenged with OVA without T cell transfer had LH levels of IL-5 and IL-13 that were <50 pg/ml (data not shown). Treatment with DEX significantly inhibited the lung levels of IL-5 and IL-13 (Fig. 2, B and C). Groups reconstituted with TH17 cells showed an increase in IL-17, G-CSF, and the mouse homolog of CXCL8 KC in LH that was not observed in mice receiving TH2 cells (Fig. 2, D–F). Mice transferred TH17 cells without OVA challenge or scid mice challenged with OVA without T cell transfer had LH levels of IL-17 and G-CSF that were <100 pg/ml; KC levels were <500 pg/ml in these control mice (data not shown). Concurrent with the in vitro glucocorticoid resistance, DEX treatment did not attenuate the TH17-induced inflammatory cytokine response; in fact, there was a trend toward increased levels of KC in the LH of DEX-treated TH17-reconstituted mice compared with the TH17 PBS control.
FIGURE 2.

In vivo cytokine and chemokine profiles induced by TH17 cell transfer and allergen provocation are not attenuated by DEX. A, SCID mice on a BALB/c background were challenged with OVA protein (50 μg/mouse) i.t. on day −1. On day 0, mice were pretreated with DEX (2.5 mg/kg, i.p.) or PBS control 2 h before cell transfer (1 × 106 in vitro-polarized TH cells). Mice were subsequently challenged with 50 μg/mouse OVA for 3 consecutive days. Animals were pretreated with DEX on day 2, 2 h before challenge. Mice were sacrificed on day 4, 24 h after the last airway challenge. Cytokine and chemokine levels in the BAL fluid (data not shown, unless indicated) and LH (B–E) were determined by multiplex suspension cytokine array. Data are graphed as mean ± SEM for n = 4 – 8;*, p < 0.05 TH2_PBS vs TH2_DEX.
Transfer of either TH2 or TH17 cells resulted in specific cellular influx into the airways. We initially assessed lymphocyte recruitment into the lung 24 h after adoptive transfer. Both transfer of TH2 cells and TH17 cells resulted in 0.15 + 0.06 × 106 and 0.18 + 0.07 × 106 CD3/CD4+ cells in BAL (p = NS). However, after three challenges with OVA, differential counting of BAL fluid cytospins showed that TH2 reconstitution resulted in airway inflammation consisting of both eosinophils and lymphocytes and to a lesser extent polymorphonuclear neutrophils (Fig. 3, A–C). Mice transferred TH2 cells without OVA challenge or scid mice challenged with OVA without T cell transfer had BAL eosinophil counts that were <0.1 × 106/ml (data not shown). The influx of lymphocytes and eosinophils was highly sensitive to DEX (Fig. 3, A and B), whereas the small numbers of polymorphonuclear neutrophils in the BAL were not inhibited by DEX treatment (Fig. 3C). As expected, TH17 transfer resulted in a primarily neutrophilic airway response (Fig. 3C). Mice transferred TH17 cells without OVA challenge or scid mice challenged with OVA without T cell transfer had BAL neutrophil counts that were <0.2 × 106/ml (data not shown). In contrast to mice receiving TH2 cells, DEX treatment exacerbated airway neutrophilia in TH17-reconstituted mice.
FIGURE 3.

TH2- and TH17-mediated airway inflammation and AHR. BAL fluid differential was determined by counting at least 100 cells from cytospins prepared of 100,000 cells. Data for A, lymphocytes; B, eosinophils; C, neutrophils; and D, macrophages are graphed as mean ± SEM percentage of total cells for n = 4 – 6; *, p < 0.05 PBS vs DEX in same transfer group; #, p < 0.05 TH2_PBS vs TH17_PBS.
To determine whether the airway responses of reconstituted mice were mediated by IL-17 per se or another product of TH17 cells, we adoptively transferred TH17 cells into IL-17R KO mice on a BALB/c background and challenged them with OVA 1 day before and for 3 consecutive days after cell transfer as before (Fig. 2A). IL-17R KO mice had significantly attenuated KC, G-CSF, and IL-6 responses compared with WT BALB/c controls (Fig. 4, A–C). Consistent with the decrease in CXC chemokine production in IL-17R KO mice, there was a significant decrease in the number of neutrophils in the airways (Fig. 4D). Again, there was not a significant increase in eosinophil recruitment to the lung in either WT or IL-17R KO mice transferred TH17 cells and challenged with OVA (Fig. 4E). These data indicate that the inflammatory cytokine response and cellular influx associated with TH17 cell transfer in this model system is mediated primarily by IL-17 signaling through the IL-17R.
FIGURE 4.
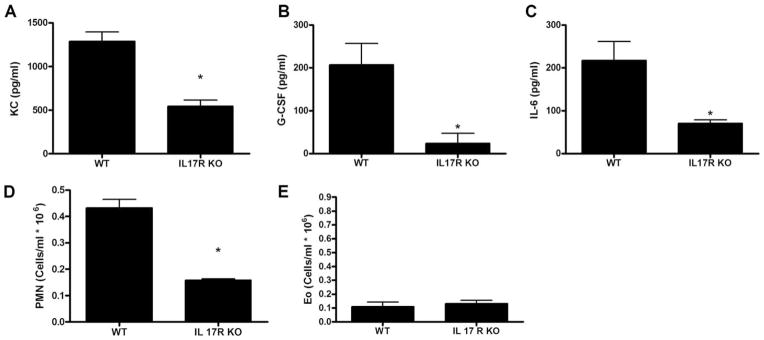
The inflammatory response associated with TH17 cell transfer is mediated by IL-17. TH17 cells were transferred to IL-17R KO mice and BALB/c controls that had been challenged with OVA 1 day before transfer. Mice were challenged with 50 μg of OVA per mouse for 3 consecutive days after transfer. BAL fluid differential and LH levels of cytokines and chemokines 24 h after the last airway challenge were determined. Levels of KC (A), G-CSF (B), and IL-6 (C) in the LH were determined by multiplex suspension cytokine array. BAL fluid neutrophils (D), and eosinophils (E) are expressed as cells/ml × 106. Data are graphed as mean ± SEM for n = 4 – 6; *, p < 0.05 compared with WT control.
Ag-induced AHR following adoptive transfer of TH17 cells is not attenuated by DEX treatment
Having established the airway inflammation induced by TH17 cell transfer, we wanted to determine whether TH17 cells are sufficient to induce mucus hyperplasia and AHR at a level comparable to that observed following transfer of TH2 cells. A significant hallmark of allergic airway disease is mucus hyperplasia; to address this, LH expression of gob5 was determined by real-time PCR. Data are normalized to control mice that had been challenged with OVA on days −1, 1, 2, and 3 but received a mock PBS transfer on day 0. TH2 transfer resulted in a marked significant increase in gob5 gene expression in the lung (Fig. 5A), which was decreased in the DEX treatment group (Fig. 5B). Expression of gob5 in TH17 cell transfer groups was also significantly elevated over mock-transferred animals (200-fold increase compared with OVA-challenged mice without cell transfer) but was ~10-fold lower than that of the TH2 transfer groups (Fig. 5A). However, compared with the TH2 transfer groups, DEX treatment had no effect on gob5 gene expression in the TH17 cell transfer group. Expression of gob5 was confirmed by periodic acid-Schiff (PAS) staining of lung sections; Fig. 5B is representative sections from TH2 and TH17 cell recipient animals treated with PBS or DEX, respectively. Both TH2 and TH17 transfer groups exhibit positive PAS staining in airway epithelial cells.
FIGURE 5.

IL-17 is sufficient to induce mucus secretion and AHR. A, gob5 expression was determined in whole lung by quantitative real-time PCR and normalized to the housekeeping gene 18S. Data are graphed as mean ± SEM for n = 4; *, p < 0.05 vs TH2_PBS. B, Expression data were confirmed by PAS staining of lung sections from TH2, and TH17, transferred animals. C, An adenovirus overexpressing IL-4, IL-17, or luciferase control was given i.t. to WT BALB/c (108 PFU/mouse) and airway responsiveness to methacholine was determine on day 3. *, p < 0.05 AdIL-4 vs Adluc; τ, p < 0.5 AdIL-17 vs Adluc. D, Methacholine dose-response curves in control, TH2 vehicle-treated, or TH2 DEX-treated mice. Control mice received all OVA challenges but PBS i.v. at time of cell transfer to experimental groups. Data are graphed as mean ± SEM for n = 6 – 8; *, p < 0.05 for TH2 vs TH2 + DEX. E, Methacholine dose-response curves in control, TH17 vehicle- treated, or TH17 + DEX-treated mice. Control mice received all OVA challenges but PBS i.v. at time of cell transfer to experimental groups. Data are graphed as mean ± SEM for n = 6 – 8.
To date, there are no data to demonstrate whether TH17 cells are sufficient to induce AHR. IL-17 levels in the sputum correlate with AHR to methacholine in asthmatic and chronic bronchitis patients (11). To determine whether IL-17 was sufficient to induce AHR, an adenovirus (Ad) overexpressing IL-17 (AdIL-17) was administered i.t. to WT BALB/c mice and AHR to methacholine was measured 72 h later. As controls, separate groups of mice were given an adenovirus overexpressing luciferase (Adluc) or IL-4 (AdIL-4) which induces local IL-13 and increase in AHR in response to methacholine (mean IL-13 concentrations in AdIL-4-treated mice was 358 ± 68 pg/ml vs 5.6 ± 2.2 in Adluc-treated mice). Overexpression of IL-4 or IL-17 in the lung was sufficient to cause significant increases in airway resistance in response to methacholine challenge (Fig. 5C). AdIL-4 caused a greater shift in the methacholine dose-response curve with significantly higher airway resistance Rn) values in mice administered the mid-range dose of 12.5 mg/ml. At the 6.25-mg/ml dose and the 25-mg/ml dose, there was no difference between AdIL-4 and AdIL-17, but both were significantly higher than Adluc control mice. These data that show IL-17 overexpression is sufficient to induce AHR to methacholine in WT mice.
We next examined the AHR mediated by TH2 and TH17 cells in the adoptive transfer model of Ag-specific airway inflammation (Fig. 2A). AHR to methacholine was determined 24 h after the last airway challenge. Fig. 5D depicts the dose response of methacholine in control mice, mice that received TH2 cells treated with vehicle, or mice transferred TH2 cells treated with DEX. Ag-challenged mice that received TH2 cells and vehicle treatment showed a significant leftward shift of the methacholine dose-response curve compared with Ag-challenged control mice that did not receive T cells (Fig. 5D). This shift was significantly attenuated with DEX treatment (Fig. 5D). Ag-challenged mice that received TH17 cells and vehicle treatment also showed a significant leftward shift of the methacholine dose-response curve compared with Ag-challenged control mice that did not receive T cells (Fig. 5E). However, in contrast to TH2 cell-transferred mice, this shift was significantly attenuated with DEX treatment (Fig. 5E).
Mechanism of enhanced TH17 cellular responses to DEX treatment
Glucocorticoids exert their effects through binding the GR, resulting in nuclear translocation and transactivation of genes containing glucocorticoid-response elements (12). To determine whether TH17 cells showed a defect in GRα binding to DEX resulting in diminished translocation to the nucleus, GRα localization was visualized by immunocytochemistry in in vitro-polarized TH cell subsets (Fig. 6A). Fluorescent staining showed that the GRα is primarily cytoplasmic before treatment with DEX, and DEX treatment resulted in a significant increase in nuclear GRα in both cell types. This observation was confirmed by Western blot, which showed a decrease in cytoplasmic GRα content (Fig. 6B). Fluorescent microscopy analysis showed a significant increase in nuclear GRα in both TH2 and TH17 cells 30 min after DEX treatment (Fig. 6C). These results indicate that TH17 cells do express GRα and are not deficient in their ability to translocate GRα to the nucleus upon glucocorticoid treatment.
FIGURE 6.

Mechanism of TH17 cell resistance to DEX treatment. TH17 cells are not deficient in their ability to translocate GRα. A, GR staining by immunofluorescent microscopy in DEX-treated (1 μM) and nontreated TH2 and TH17 cells. B, Cytoplasmic protein fractions of DEX-treated and nontreated TH2 and TH17 cells were immunoblotted for GRα and β-actin. Samples were loaded in the following order: TH2_PBS, TH2_DEX, TH17_PBS, and TH17_DEX. C, Florescent intensity of nuclear GRα staining in TH2 and TH17 cells with and without DEX treatment. Data are graphed as mean ± SEM; *, p < 0.05 for DEX compared with no DEX conditions. D and E, TH2 (C) or TH17 (D) cells were transfected with NFAT-luciferase before stimulation with anti-CD3 and anti-CD28 microbeads (6 h) with or without pretreatment (2 h before beads) with DEX; *, p < 0.05 control (CNTRL) vs CD3/CD28; #, p < 0.05 CD3/CD28 vs CD3/CD28 + DEX.
Although the ability of GRα to bind glucocorticoids and translocate to the nucleus is intact, the defect may lie at the level of DNA binding. Suppression of IL-5 by glucocorticoids involves repression of GATA-3 signaling mediated by GRα binding to the IL-5 NFAT/AP-1-response element and subsequent recruitment of histone deacetylase (22). Studies have shown that IL-17 expression in mouse and human CD4+ T cells is dependent on the NFAT and MAPK pathways (23, 24). Activated GRα may be unable to repress NFAT binding to the IL-17 locus, which would explain the fact that IL-17 production is not attenuated by DEX. Although there are several mechanisms by which TH17 cells might prevent activated GRα from repressing NFAT activity, up-regulation of NFAT expression in response to DEX is one way by which the cell could mask the drug’s effects. To test this hypothesis, polarized TH2 and TH17 cells were transfected with a NFAT luciferase reporter plasmid and were stimulated with anti-CD3 and anti-CD28 microbeads in the presence or absence of DEX (Fig. 6, C and D). DEX significantly inhibited NFAT luciferase activity in TH17 cells, discounting the concept that NFAT activity is steroid independent in TH17 cells.
Discussion
We have shown that reconstitution of SCID mice with TH2 or TH17 cells in a model of Ag-induced airway inflammation lead to differential chemokine profiles and cellular influx to the airways. Adoptive transfer of TH17 cells resulted in increased levels of CXC chemokines and G-CSF in the BAL fluid and LH following Ag provocation. We do not believe this is due to homeostatic proliferation due to the short time frame of the transfer model (25) as well as similar findings were observed in WT mice (Fig. 4). Moreover, the experiment in WT mice vs IL-17RA KO mice demonstrated that IL-17RA signaling is required for the induction of KC and G-CSF, as well as neutrophil recruitment after TH17 T cell transfer (Fig. 4). This was associated with neutrophil influx to the airways and is exacerbated by DEX treatment. Animal-transferred TH2 cells in this model exhibit a typical TH2 phenotype characterized by the influx of lymphocytes and eosinophils to the airways and are sensitive to DEX treatment. The chemokine and inflammatory responses elicited by TH17 cell transfer primarily involves signaling through the IL-17R. This is consistent with the recent study by Liang et al. (26) that showed that IL-22, another product of TH17 cells, played a limited role in neutrophil recruitment in an adoptive transfer model compared with IL-17A or IL-17A/F heterodimer. Despite differential chemokine and cellular responses, both TH2 and TH17 recipient animals exhibited increased AHR and mucus secretion. TH17 cells secrete IL-17A and F, IL-6, IL-22, and TNF-α (5). Transfer of TH17 cells to IL-17R KO mice showed a substantial decrease in CXC chemokine expression, G-CSF, IL-6 and lung neutrophilia confirming that the TH17 cells are migrating to the airways to initiate a response and IL-17R signaling is necessary for the phenotype associated with their transfer. However, the individual contributions of IL-17A or IL-17F homodimers and IL-17A/F heterodimers in AHR remains to be determined.
It has been suggested that IL-17 modulates allergic airway inflammation by dampening Th2 cytokine responses (14, 27). One study showed that Ab neutralization of IL-17 before OVA challenge in sensitized mice resulted in decreased neutrophil influx to the airways but increased airway eosinophilia that those authors attributed to enhanced serum and BAL fluid IL-5 levels in these animals (27). We found that the transfer of TH17 cells to WT BALB/c mice resulted in very low and variable Th2 cytokine responses and the IL-17R KO mice did not exhibit an exacerbated Th2 response. One explanation of these divergent findings is that IL-17 signaling is required for the initiation of Ag-mediated airway inflammation but functions to dampen Th2 responses thereafter. This was suggested by studies showing that IL-17R KO mice exhibited decreased airway eosinophilia and IL-5 in an OVA-induced model of pulmonary inflammation, while treating WT animals with exogenous IL-17 reduced Th2 cytokine levels in BAL and LH and eosinophil influx (14). The former IL-17R KO data are in agreement with what we have found in our model of TH17-induced airway inflammation. To date, the ability of TH17 cells to directly regulate Th2 cells has not been shown. However, regulation between Th2 and TH17 may not be as important as the possibility that these Th cell populations are mediating distinct endophenotypes of asthma. Asthma can be classified as atopic (allergic) and nonallergic. Patients with nonatopic asthma do not have elevated IgEs, tend to have more airway neutrophilia, and are clinically steroid resistant (12). The data presented in our model system support a role for TH17 cells in nonatopic disease.
DEX treatment of TH17 cell-reconstituted animals resulted in increased numbers of neutrophils in the airways following allergen challenge. The mechanism of increased neutrophil numbers in DEX-treated animals reconstituted with TH17 cells is not clear. The expression of IL-17 and CXCL8 is increased in the sputum of asthmatic patients and positively correlates with each other and with neutrophil levels in the sputum (28). Our data suggest that increased levels of neutrophil chemoattractants and G-CSF, which has been shown to be an important survival factor for neutrophils, may be a mechanism of enhanced recruitment and survival of neutrophils following DEX treatment in TH17-reconstituted animals. This is fitting with published reports that corticosteroids down-regulate the CC chemokines CCL2 and CCL11 while exacerbating the CXCL8 response in the airways of asthmatic patients (29). This same study found that corticosteroids decreased airway eosinophilia but increased the number of neutrophils in the airways. Neutrophils are relatively steroid resistant compared with T cells and eosinophils and glucocorticoids enhance survival of neutrophils in vitro (10, 30). In vitro neutralization of another neutrophil survival factor GM-CSF did not alter neutrophil survival following glucocorticoid treatment (31). Glucocorticoids exert their effects through binding the GR, resulting in nuclear translocation and transactivation of genes containing glucocorticoid-response elements (32). It has been suggested that human neutrophils express higher levels of the dominant-negative isoform of the GR, GRβ (19), rendering them nonresponsive to glucocorticoids. However, murine cells do not express GRβ and we observed equivalent nuclear translocation of GR to the nucleus of Th2 and TH17 cells and a similar ability to inhibit NFAT transcriptional activation.
Multiple mechanisms of steroid-resistant asthma have been described. One study found that steroid-resistant asthmatics fell into two categories based on GR-binding and expression patterns; one group of patients exhibited increased GR number but reduced GR binding affinity for glucocorticoids, whereas another subset of steroid-resistant patients had a GR-binding affinity similar to that of steroid-sensitive patients, but reduced GR expression per cell (33). Future studies will need to examine GRα binding to regions within the Il17 and Il22 loci.
In contrast to the TH17 transfer model, in the Th2 transfer model both lymphocyte and eosinophil recruitment to the airways of Th2 recipient animals treated with DEX and AHR was reduced by DEX treatment. The reduction in lymphocytes may be due to differential DEX sensitivity of chemokines necessary for T cell recruitment or proliferation in vivo. We did not observe differences in T cell apoptosis of Th2 or TH17 cells in vitro. Both cells showed >85% viability after DEX treatment for 24 h (data not shown). However, this does not exclude a potential role for differential proliferation or apoptosis in vivo. AHR was significantly improved with DEX treatment, but not to the same levels as the effect of DEX on airway inflammation. AHR is a hallmark feature of asthma and can generally be separated into two categories, variable AHR that occurs during an allergen-induced late asthmatic response and persistent AHR. Although some studies link airway inflammation with AHR, it has been suggested that AHR, especially persistent AHR, can occur in the absence of airway inflammation (34). Tournoy et al. (35) showed AHR occurred independent of eosinophil influx to the airways in a model of house dust mite-mediated airway inflammation. Clinically, administration of a mAb to IL-5 to asthmatic patients also significantly decreased the number or eosinophils in the sputum without altering AHR. In our model system, AHR is mediated by the CD4+ cell populations transferred to the mice in a mast cell/IgE-independent manner. Mast cells are resident in vascularized tissue and express high levels of the IgE receptor FcεRI. IgE binding and Ag cross-linking activates mast cells to release inflammatory mediators including histamine, leukotrienes, cytokines. and chemokines, resulting in bronchoconstriction (37). Mast cells can potentiate Th2-mediated allergic inflammation through the release of IL-5 that is important in eosinophil survival and the release of histamine that directs dendritic cells to secrete IL-4-polarizing naive cells toward a Th2 phenotype (38, 39). Mast cells are sensitive to glucocorticoids; therefore, it is possible that a mast cell-dependent model system would show that Th2-induced AHR is more responsive to DEX treatment.
We have shown that reconstitution of SCID mice with TH17 cells in the setting of Ag provocation induces CXC chemokine and G-CSF secretion and neutrophil influx to the airways and is sufficient to induce mucus hyperplasia and AHR. In the setting of TH17 cell transfer, chemokine secretion, cellular influx to the airways, and AHR were not sensitive to DEX treatment in these studies. Conversely, in the setting of Th2 cell transfer, cytokine secretion, eosinophil influx to the airways, and AHR were sensitive to DEX. These data highlight the complex relationships between airway inflammation and AHR and support the concept that TH17 cells may be critical mediators of steroid-resistant airways inflammation and AHR.
Footnotes
This work was supported by National Heart, Ling, and Blood Institute Grant R01HL079142 and National Institute of Allergy and Infectious Disease Grant 5T32AI060525.
Abbreviations used in this paper: AHR, airway hyperresponsiveness; Ad, adenovirus; BAL, bronchoalveolar lavage; DEX, dexamethasone; GR, glucocorticoid receptor; i.t., intratracheal; LH, lung homogenate; KO, knockout; rm, recombinant mouse; WT, wild type; PMN, polymorphonuclear neutrophil; PAS, periodic acid-Schiff.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Wallen N, Kita H, Weiler D, Gleich GJ. Glucocorticoids inhibit cytokine-mediated eosinophil survival. J Immunol. 1991;147:3490–3495. [PubMed] [Google Scholar]
- 2.Lamas AM, Leon OG, Schleimer RP. Glucocorticoids inhibit eosinophil responses to granulocyte-macrophage colony-stimulating factor. J Immunol. 1991;147:254–259. [PubMed] [Google Scholar]
- 3.Barnes PJ. Distribution of receptor targets in the lung. Proc Am Thorac Soc. 2004;1:345–351. doi: 10.1513/pats.200409-045MS. [DOI] [PubMed] [Google Scholar]
- 4.Douwes J, Gibson P, Pekkanen J, Pearce N. Non-eosinophilic asthma: importance and possible mechanisms. Thorax. 2002;57:643–648. doi: 10.1136/thorax.57.7.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 6.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 8.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashimoto T, Akiyama K, Kobayashi N, Mori A. Comparison of IL-17 production by helper T cells among atopic and nonatopic asthmatics and control subjects. Int Arch Allergy Immunol. 2005;137(Suppl 1):51–54. doi: 10.1159/000085432. [DOI] [PubMed] [Google Scholar]
- 10.Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils: separation of survival and activation outcomes. J Immunol. 1995;154:4719–4725. [PubMed] [Google Scholar]
- 11.Pavord ID. Non-eosinophilic asthma and the innate immune response. Thorax. 2007;62:193–194. doi: 10.1136/thx.2006.065805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pavord ID, Brightling CE, Woltmann G, Wardlaw AJ. Non-eosinophilic corticosteroid unresponsive asthma. Lancet. 1999;353:2213–2214. doi: 10.1016/S0140-6736(99)01813-9. [DOI] [PubMed] [Google Scholar]
- 13.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 14.Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, Fossiez F, Ryffel B, Schnyder B. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 16.Hansen G, Berry G, DeKruyff RH, Umetsu DT. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest. 1999;103:175–183. doi: 10.1172/JCI5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–528. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 19.Strickland I, Kisich K, Hauk PJ, Vottero A, Chrousos GP, Klemm DJ, Leung DY. High constitutive glucocorticoid receptor β in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J Exp Med. 2001;193:585–593. doi: 10.1084/jem.193.5.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bates JH, Irvin CG. Measuring lung function in mice: the phenotyping uncertainty principle. J Appl Physiol. 2003;94:1297–1306. doi: 10.1152/japplphysiol.00706.2002. [DOI] [PubMed] [Google Scholar]
- 21.Braun CM, Huang SK, Bashian GG, Kagey-Sobotka A, Lichtenstein LM, Essayan DM. Corticosteroid modulation of human, antigen-specific Th1 and Th2 responses. J Allergy Clin Immunol. 1997;100:400–407. doi: 10.1016/s0091-6749(97)70255-0. [DOI] [PubMed] [Google Scholar]
- 22.Jee YK, Gilmour J, Kelly A, Bowen H, Richards D, Soh C, Smith P, Hawrylowicz C, Cousins D, Lee T, Lavender P. Repression of interleukin-5 transcription by the glucocorticoid receptor targets GATA3 signaling and involves histone deacetylase recruitment. J Biol Chem. 2005;280:23243–23250. doi: 10.1074/jbc.M503659200. [DOI] [PubMed] [Google Scholar]
- 23.Liu XK, Lin X, Gaffen SL. Crucial role for nuclear factor of activated T cells in T cell receptor-mediated regulation of human interleukin-17. J Biol Chem. 2004;279:52762–52771. doi: 10.1074/jbc.M405764200. [DOI] [PubMed] [Google Scholar]
- 24.Liu XK, Clements JL, Gaffen SL. Signaling through the murine T cell receptor induces IL-17 production in the absence of costimulation, IL-23 or dendritic cells. Mol Cells. 2005;20:339–347. [PubMed] [Google Scholar]
- 25.Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, Iwakura Y, Sakaguchi N, Sakaguchi S. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med. 2007;204:41–47. doi: 10.1084/jem.20062259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, Goldman SJ, Dunussi-Joannopoulos K, Williams CM, Wright JF, Fouser LA. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179:7791–7799. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- 27.Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, Mathieu C, Ceuppens JL. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 28.Bullens DM, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, Ceuppens JL. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukakusa M, Bergeron C, Tulic MK, Fiset PO, Al Dewachi O, Laviolette M, Hamid Q, Chakir J. Oral corticosteroids decrease eosinophil and CC chemokine expression but increase neutrophil, IL-8, and IFN-γ-inducible protein 10 expression in asthmatic airway mucosa. J Allergy Clin Immunol. 2005;115:280–286. doi: 10.1016/j.jaci.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 30.Schleimer RP. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc Am Thorac Soc. 2004;1:222–230. doi: 10.1513/pats.200402-018MS. [DOI] [PubMed] [Google Scholar]
- 31.Cox G, Austin RC. Dexamethasone-induced suppression of apoptosis in human neutrophils requires continuous stimulation of new protein synthesis. J Leukocyte Biol. 1997;61:224–230. doi: 10.1002/jlb.61.2.224. [DOI] [PubMed] [Google Scholar]
- 32.Winoto A, Littman DR. Nuclear hormone receptors in T lymphocytes. Cell. 2002;109(Suppl):S57–S66. doi: 10.1016/s0092-8674(02)00710-9. [DOI] [PubMed] [Google Scholar]
- 33.Sher ER, Leung DY, Surs W, Kam JC, Zieg G, Kamada AK, Szefler SJ. Steroid-resistant asthma: cellular mechanisms contributing to inadequate response to glucocorticoid therapy. J Clin Invest. 1994;93:33–39. doi: 10.1172/JCI116963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cockcroft DW, Davis BE. Mechanisms of airway hyperresponsiveness. J Allergy Clin Immunol. 2006;118:551–559. doi: 10.1016/j.jaci.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 35.Tournoy KG, Kips JC, Schou C, Pauwels RA. Airway eosinophilia is not a requirement for allergen-induced airway hyperresponsiveness. Clin Exp Allergy. 2000;30:79–85. doi: 10.1046/j.1365-2222.2000.00772.x. [DOI] [PubMed] [Google Scholar]
- 36.Leckie MJ, ten Brinke A, Khan J, Diamant Z, O’Connor BJ, Walls CM, Mathur AK, Cowley HC, Chung KF, Djukanovic R, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyperresponsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 37.Galli SJ. Complexity and redundancy in the pathogenesis of asthma: reassessing the roles of mast cells and T cells. J Exp Med. 1997;186:343–347. doi: 10.1084/jem.186.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mazzoni A, Young HA, Spitzer JH, Visintin A, Segal DM. Histamine regulates cytokine production in maturing dendritic cells, resulting in altered T cell polarization. J Clin Invest. 2001;108:1865–1873. doi: 10.1172/JCI13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bressler RB, Lesko J, Jones ML, Wasserman M, Dickason RR, Huston MM, Cook SW, Huston DP. Production of IL-5 and granulocyte-macrophage colony-stimulating factor by naive human mast cells activated by high-affinity IgE receptor ligation. J Allergy Clin Immunol. 1997;99:508–514. doi: 10.1016/s0091-6749(97)70078-2. [DOI] [PubMed] [Google Scholar]