

Abstract
Schistosomiasis caused by Schistosoma spp. is a serious public health concern, especially in sub-Saharan Africa. Praziquantel is the only drug currently administrated to treat this disease. However, praziquantel-resistant parasites have been identified in endemic areas and can be generated in the laboratory. Therefore, it is essential to find new therapeutics. Antioxidants are appealing drug targets. In order to survive in their hosts, schistosomes are challenged by reactive oxygen species from intrinsic and extrinsic sources. Schistosome antioxidant enzymes have been identified as essential proteins and novel drug targets and inhibition of the antioxidant response can lead to parasite death. Because the organization of the redox network in schistosomes is significantly different form that in humans, new drugs are being developed targeting schistosome antioxidants. In this paper the redox biology of schistosomes is discussed and their potential use as drug targets is reviewed. It is hoped that compounds targeting parasite antioxidant responses will become clinically relevant drugs in the near future.
Keywords: Schistosoma, drug development, antioxidants, glutathione, thioredoxin, thioredoxin glutathione reductase
Schistosomiasis (also known as bilharzia) is caused by blood-dwelling flatworms of the genus Schistosoma. Schistosomiasis is the second most important human parasitic disease after malaria, with an estimated 200 million people infected and greater than 200,000 deaths annually in tropical and subtropical areas [1–3]. Nearly 800 million people are at risk of infection in seventy-two counties [1]. In addition, schistosome infections lead to largely underreported chronic disabilities and morbidities, such as caloric malnutrition, growth stunting, anemia, and poor school performance, which lead to decreased quality of life and perpetuation of poverty [1–3]. The chronic morbidities associated with schistosomiasis can be exacerbated by co-infections with other helminths (parasitic worms, e.g., hookworms) [4] and schistosome infections can have significant impacts on the susceptibility and transmission of other infections, e.g., HIV [5, 6] and malaria [7, 8], and on immune responses to childhood vaccines [9]. Furthermore, S. haematobium is considered as a group 1 carcinogen leading to the development of urinary bladder cancer [10–12]. Intestinal schistosomiasis has been linked to hepatocellular carcinoma and colorectal cancer [13, 14], though this is not definitive.
Five species of Schistosoma parasitize humans including S. mansoni, S. japonicum, S. haematobium, S. intercalatum, and S. mekongi; the first three species have the widest geographic distribution whereas infections with the last two species only occur locally [15]. The life cycle of Schistosoma ssp. is complex [16, 17] and is divided into sexual and asexual cycles. In the asexual cycle, eggs are released into water with feces or urine of infected individuals. Miracidia hatch from the eggs and then locate and infect snails, the intermediate hosts. Within the snail the miracidium develops into a sporocyst, in which thousands of cercariae develop through asexual reproduction. The mature cercariae are released from the snails into water. Humans exposed to water contaminated with cercariae become infected when cercariae penetrate directly into their skin. In the process, the cercariae lose their bifurcated tails and become schistosomula. Schistosomes, unlike most parasitic flatworms, which are hermaphrodites, are dioecious and in humans they start their sexual cycle. After a few days in the skin, larval parasites enter the general circulation and are carried to the lungs (5–7 days post infection) and then they migrate to the liver where they undergo rapid development, mature, and pair. Paired S. mansoni, S. japonicum, S. intercalatum, and S. mekongi move to the mesenteric veins, whereas S. haematobium migrates to the perivesical and periurethral vessels. Pathology is caused by eggs produced by paired worms; a worm pair can produce 300–2000 eggs each day [18]. Eggs are deposited in the lumen of the vein and then transverses host tissues encapsulated in an immune-generated granuloma; half of eggs pass into bladder mucosa (S. haematobium) or intestinal mucosa (other species) and are then excreted into environments via urine or feces. The remainder of the eggs is trapped in different organs. In S. mansoni and S. japonicum infections eggs accumulate mainly in the liver while in S. haematobium infections they accumulate in the bladder wall and rectal and genital tissues. The trapped eggs are attacked by host immune cells, thereby forming tissue granulomas resulting in inflammation and scarring [19]. The granulomas are responsible for tissue-damaging pathology associated with schistosomiasis.
Control of transmission of schistosomiasis through reduction of snail densities is possible, but has been abandoned due to the expense and environmental problems associated with the widespread use of molluscicides [20]. In the mid-1980s, the World Health Organization (WHO) began using chemotherapy to control morbidity due to schistosomiasis [15]. Over the last few decades, several drugs have been used to treat the disease [16, 21]. Here, we provide brief overview of the previous and current antischistosomal drugs.
Praziquantel (PZQ, 2-(cyclohexylcarbonyl)-1,2,3,6,7,11b-hexahydro-4H-pyrazino(2,l-alpha)isoquinoline-4-one) (Figure 1) is essentially the only drug currently administrated to treat schistosomiasis. Commercial PZQ contains a racemic mixture of levo (−) and dextro (+) isomers; only the levo isomer has antischistosomal activity [22, 23]. PZQ is active against all schistosome species [16]; humans tolerate high doses of PZQ with little to no toxic side effects [24, 25]. It is thought that the antischistosomal activity of PZQ is due to the disruption of Ca2+ homeostasis in the parasites: PZQ treatment in vitro results in an increase the influx of Ca2+ [26, 27]. PZQ specifically targets the interface between α1 and β in the voltage-gate Ca2+ channels from schistosomes, and not in mammalian channels, leading to parasite death [26]. However, other studies suggest that calcium influx itself may not be the sole cause of the schistosomicidal activity of PZQ; pre-incubation with cytochalasin D, which completely suppresses the killing activity of PZQ, has no effect on calcium uptake in schistosomes exposed to PZQ [28]. Furthermore, even though larval parasites are largely insensitive to PZQ, a large calcium influx occurred in these worms after exposure to PZQ. Additional suggested mechanisms of PZQ action are that it causes damage to the worm’s surface (tegument) leading to changes in antigen presentation and the host’s immune responses against the parasite [16] and that it inhibits adenosine and uridine uptake in schistosomes leading to worm death [29].
Figure 1.

Structures of antischistosomal drugs previously and currently used in clinical practice.
An alternative to PZQ is oxamniquine (6-hydroxymethyl-2-isopropyl-aminomethyl-7-nitro-1,2,3,4-tetrahydroquinoline) (Figure 1). Because it is only active against S. mansoni [21] and is more expensive than PZQ, its use has been restricted to Brazil and other countries in South American [30]. It should be noted that in many endemic areas S. mansoni has developed oxamniquine resistance [31, 32]. It has been postulated that oxamniquine is a pro-drug and requires a sulfotransferase activity to convert it to its active form [33]; this activity appears to be present only in S. mansoni. The mechanism of action proposed is that the resultant oxamniquine ester binds covalently to and inactivates macromolecules in S. mansoni [21, 34].
The organophosphate metrifonate (2,2,2-trichloro-1-hydoxylethyl dimethyl phosphonate) (Figure 1) is effective only against S. haematobium [16, 21]. Its metabolite, dichlorvos (Figure 1), has been shown to exhibit its antischistosomal activity by inhibiting acetylcholinesterase, thereby paralyzing the parasites [35]. It has been proposed that metrifonate is effective against S. haematobium due to differences in anatomical location between schistosome species. Paralysis by metrifonate results in S. haematobium worms being forced by the blood flow to the lungs where it is trapped [36]. In contrast, paralysis of S. mansoni and S. japonicum results in a shift to portal vein and liver and after recovery from the affects of the drug, the worms are able to migrate back to the mesenteric veins [21, 36]. Additionally, it has been suggested that S. mansoni has higher glutathione S transferase (GST) activity, which detoxifies dichlorvos resulting in drug resistance [37].
Lucanthone (miracil D, 1-{[2-(diethylamino)ethyl]amino}-4-methyl-9H-thioxanthen-9-one; Figure 1) was initially found to display antischistosomal activity against S. mansoni and S. haematobium (but not S. japonicum) only when it was administrated orally, suggesting that biotransformation by the host results in activation of the pro-drug [21]. Later, it was found that hycanthone (Figure 1), a major metabolite of lucanthone, is more active against schistosomes [38] and hycanthone replaced lucanthone in clinical practice thereafter. Like oxamniquine, activation of lucanthone by enzymatic mechanisms leads to a reactive species capable of alkylating parasite macromolecules [39, 40]. The activating enzymes are present only in drug sensitive schistosomes. However, it was observed that hycanthone can cause acute hepatic toxicity and could be a carcinogen [21, 41]. In addition, the parasites developed drug resistance in both animal models and clinical practice [42–45] and, therefore, this drug is no longer used in clinical therapy.
Artemisinin (Figure 1) and its derivatives are widely used to treat malaria. Thirty years ago it was found also to be active against schistosome infections [46]. Since artemisinin has low solubility in water or oil, its water-soluble artesunate and oil-soluble artemether (Figure 1) derivatives were developed [47]. In animal experiments, artemether has been shown to be effective against S. mansoni, S. japonicum, and S. haematobium [20, 48, 49]. The juvenile stage of schistosomes, which is less sensitive to treatment with PZQ, (e.g., 5-21-day-old S. japonicum and 7-28-day-old S. mansoni) is most sensitive to artemether [20, 48]. In humans, oral administration of artemether can decrease incidence of infection of S. japonicum, S. mansoni and S. haematobium [20, 50, 51], although other studies have found lower efficacy suggesting that different dosing for the two parasite infections may be needed [52]. However, the drug should be used with caution, especially in monotherapy, in malaria-endemic areas to prevent the development of artemisinin-resistant malaria parasites [53]. There is evidence that malaria control programs using artesunate combination therapies may have the added bonus of reduction in schistosome infections [54]. Artemisinin derivatives induce morphological alternations and damage to the tegument of schistosomula and adult worms [20, 55], may affect the glycolysis of the parasites, and/or can interact with heme in the worm gut leading to its conversion to an unstable species capable of generating reactive oxygen species leading to worm death [20, 56].
Antimony potassium tartrate (tartar emetic) (Figure 1), introduced in 1918, and other trivalent antimonials, were used to treat schistosomiasis for over 50 years. However, these are toxic drugs and cause significant side effects including nausea and vomiting, diarrhea, fever, muscle and joint pains, skin rashes, hepatitis and headaches. The mechanism of antimonial compounds has been proposed: it inhibits phosphofructokinase which is involved in glycolysis [16, 21, 57]. Recently antimony potassium tartrate has been found to inhibit thioredoxin glutathione reductase with nanomolar efficiency [58], indicating that its mechanism of action requires further study.
Niridazole (Figure 1) is more effective against S. haematobium than S. mansoni or S. japonicum [16, 21, 57].The mechanism of action of niridazole has been suggested: the drug requires nitroreduction to generate reactive intermediates which covalently bind to the macromolecules of schistosomes [59, 60].
Praziquantel is the only drug currently administrated to treat schistosomiasis. Although PZQ resistance does not appear to be clinically relevant today, it is not good policy to rely on only one drug. Indeed, PZQ-resistant parasites have been selected for in laboratories and identified in some endemic areas [25, 30]. Thus, it is essential to develop new therapeutics for schistosomiasis treatment to replace PZQ should resistance become widespread or to be used in combination with PZQ to delay the evolution of resistance. There is compelling evidence that schistosome antioxidants are essential parasite proteins and suitable drug targets. Therefore, in this paper we will discuss redox biology of Schistosoma and its potential for the development of new antischistosomal drugs.
Reactive oxygen species
Reactive oxygen species (ROS) include the superoxide anion (•O2−), hydrogen peroxide (H2O2), singlet oxygen (1O2−), and hydroxyl radical (•OH). Superoxide anion cannot cross membranes because of its negative charge. Superoxide anion is considered to be a primary ROS because it can interact with other molecules to form secondary ROS [61]. Superoxide anion can disproportionate to form H2O2 and oxygen in aqueous solutions, especially at lower pH or can be dismutated catalytically by superoxide dismutase (SOD) [62]. Hydrogen peroxide can cross biological membranes and although it is less chemically active than the superoxide anion, hydroxyl radials can be generated from it in the presence of transition metals, e.g., Fe (II) (Fenton reaction; Reaction 1) and Cu (I). The product, ferric ion (Fe III), then interacts with superoxide anion to return to ferrous ion (Fe II) (Reaction 2). The net reaction of Reaction 1 and 2 is called Haber-Weiss reaction (Reaction 3). Hydroxyl radicals are highly reactive reacting with various macromolecules such as DNA, lipids and proteins in cells. The chemistry of ROS has been recently reviewed [61, 63, 64].
| (1) (Fenton reaction) |
| (2) |
| (3) (Haber-Weiss reaction) |
Surviving in human hosts, schistosomes are challenged by ROS generated by different mechanisms in the parasite as well as by activated host immune cells. ROS is generated by the mitochondrial respiratory system: 1–3% of electrons passing through the respiratory chain react with oxygen before the terminal oxidase resulting in the formation of superoxide anion [65]. In addition, hemoglobin is digested in parasite gut generating Fe (II) protoporphyrin IX, which subsequently can be oxidized to Fe (III) protoporphyrin IX by oxygen producing superoxide anions [66, 67]. Furthermore, ROS generated by immune cells also challenges schistosome worms. For example, it has been shown that parasites at schistosomula stage are killed by host immune cells through an antibody-dependent cell-mediated cytotoxicity in which host white cells (e.g., eosinophils) generate ROS (e.g., superoxide anions and hydroxyl radicals) through an oxidative burst to kill the parasites [68–70].
In order to neutralize ROS, most organisms have an antioxidant network composed of enzymatic and non-enzymatic components. Enzymatic antioxidants include SOD, catalase, and members of the glutathione and thioredoxin systems. Non-enzymatic antioxidants are redox active small molecules such as ascorbic acid, uric acid, and vitamin E. As indicated earlier, superoxide anion can be dismutated to H2O2 and O2 by SOD (Reaction 4). Hydrogen peroxide can be removed by catalase (Reaction 5), glutathione peroxidase (GPx) and peroxiredoxin (Prx) (Reactions 6 and 7). Reducing equivalents for GPx and Prx are provided by glutathione (GSH) or thioredoxin (Trx), respectively, via pathways with dedicated flavoenzyme oxidoreductases, glutathione reductase (GR) and thioredoxin reductase (TrxR). Reducing equivalents are ultimately derived from NADPH.
| (4) |
| (5) |
| (6) |
| (7) |
Antioxidants in schistosomes
Compared to their human hosts, schistosomes have limited antioxidant capacity. The parasites lack catalase [71] and schistosome GPx proteins have lower activity with hydrogen peroxide [72]. Therefore, schistosome worms appear to be more sensitive to hydrogen peroxide than humans and redox pathways are thought to be weak points in parasite biology and good drug targets [70]. Redox activity is developmentally regulated in schistosomes and antioxidants such as GST, cytosolic SOD (CT-SOD), peptide-containing SOD (SP-SOD) and GPx have their lowest expression in the schistosomula and their expression increases during development in the mammalian host resulting in increased resistance of the parasites to hydrogen peroxide [73, 74]. Generally, based on the abundance of respective mRNAs, the expression of antioxidant proteins is: GST > CT-SOD > SP-SOD > GPx [74]. Therefore, during development in humans larval parasites are susceptible to ROS generated in the skin and lung by activated immune cells, whereas adult worms are more resistant to ROS because they have higher levels of antioxidants [70].
Superoxide oxide dismutase
As described above, SOD proteins (EC 1.15.1.1) catalyze the production of hydrogen peroxide from superoxide anion (Reaction 4). SODs are classified based on their location: cytosolic SOD (SOD-1/CT-SOD), mitochondrial SOD (SOD-2) and extracellular SOD (SOD-3/SP-SOD) [75]. In addition, based on the type of metals bound in their active sites, SODs are classified into two further groups, the Cu/Zn SOD class, and the Mn-SOD and Fe -SOD class. Cu/Zn-SODs are found in the cytosol and secretions of eukaryotic cells whereas Mn-SODs and Fe-SODs are usually found in prokaryotic cells and subcellular organelles of eukaryotic cells [75, 76]. Two Cu/Zn SOD isoforms have been identified in S. mansoni: CT-SOD and SP-SOD [77–79]. A hydrophobic leader sequence is present in SP-SOD as well as an N-linked glycosylation site, which are also found in human SOD-3 [77]. In terms of sequence identify, CT-SOD is more similar to human SOD-1 than its own extracellular form [80]. The 3D-structure of S. mansoni CT-SOD is similar to SODs from other species including humans, having a flattened, eight-stranded, Greek-keyβ-barrel in its structural core. The active site is located in the bottom of a shallow channel formed by two loops from β-barrel; the function of the shallow channel is to guide substrates to access the active site via the electrostatic interaction between the channel and substrates [80, 81]. There are differences between S. mansoni CT-SOD and other SODs in the residues present in the electrostatic loop of S. mansoni CT-SOD, suggesting that the loop is a potential drug target [80]. CT-SOD was found to be express in all life stages of S. mansoni and reach the highest expression in adult worms [77]. CT-SOD and SP-SOD were found to be expressed in tegument and gut epithelium in adult male S. mansoni; the presence of these enzymes in tegument and gut epithelium may protect the parasites from oxidative stress generated from the immune cells of hosts [74]. Recently it was found that SP-SOD is also expressed in reproductive system in female worms, specifically in immature regions of the vitelline glands, suggesting that this enzyme may be associated with reproduction functions [82].
Studies have been performed to investigate whether vaccine potential of SOD. DNA vaccination with CT-SOD and SP-SOD has been shown to exhibit protection of 44–60 % and 22–45%, respectively against cercariae challenges [83]. DNA vaccination with CT-SOD resulted in 36–43% decreases in worm burdens in mice challenged by adult worms that were surgically transferred into mesenteric veins; such protection was mediated by Th1-type host immune responses [84]. Mostly importantly, it has been shown that even though CT-SOD from S. mansoni shares high similarity with the human ortholog, antibodies from CT-SOD-immunized mouse and human exposed to naturally S. mansoni cannot recognize non-denatured human SOD (i.e., no cross-activity) suggesting that vaccines targeting CT-SOD would be expected to induce minimal host-protein reactivity [85].
The glutathione system
Glutathione (GSH), which exists ubiquitously in all domains of life, is the tripeptide γ-L-glutamyl-L-cysteinyl-glycine, with an uncommon gamma bond between glutamate and cysteine. GSH is the most abundant antioxidant in cells [86]. Because of the chemical properties of sulfur in cysteine, GSH can directly neutralize hydroxyl radicals [87]. Also, GSH can pass reducing equivalents to GPx and GST to detoxify reactive oxygen species. Cellular GSH levels are maintained through a balance between its synthesis, its degradation, its utilization as a conjugating agent, and its recycling from the oxidized form, GSH disulfide (GSSG). In many organisms, the last process (GSSG + 2H+ + 2e− → 2GSH) is carried out by GSH reductases (GR) with reducing equivalents from NADPH. However, schistosomes lack an authentic GR and GSSG reduction is carried out by a unique, hybrid enzyme, thioredoxin glutathione reductase (TGR) [88]. Disruption of the glutathione system is known to trigger S. mansoni death. Indeed, the anthelmintic oltipraz (OPZ; Figure 1) causes oxidative stress, decreased GSH concentration and glutathione reductase activity, and increased GPx activity [89, 90]. The drug is also known to have an effect on GST activity, although results of different studies are somewhat contradictory. Treatment of worms with OPZ can produce a decrease in GST activity resulting from the non-competitive inhibition of GST activity by OPZ [89, 91] or an increase in GST activity in a concentration and time-dependant manner [90], depending on the study. Furthermore, OPZ is an inhibitor of schistosome TGR [58]. Overall, these effects are thought to increase parasite susceptibility to oxidative stress and to be responsible for the antischistosomal action of OPZ.
γ-glutamylcysteine synthetase and glutathione synthase
De novo GSH biosynthesis proceeds via a two-step pathway involving the enzymes γ-glutamate cysteine ligase (GCL, also known as γ-glutamylcysteine synthetase, EC 4.4.1.1), the rate-limiting step, and GSH synthase (GS, EC 4.2.1.22). GCL catalyses the ATP-dependent condensation of L-cysteine and L-glutamate to form the dipeptide γ-glutamylcysteine. GS catalyses the condensation of γ-glutamylcysteine and glycine to yield GSH (Figure 2). Both enzymes have been identified in the genomes of S. mansoni [92] and S. japonicum [93] but to date they have not been characterized. Both GS and GCL of S. mansoni and S. japonicum display less than 60% identity with their human orthologs. Their potential as drug targets is worth investigating as has been done for the parasite Plasmodium falciparum, where it was found that de novo GSH appears to be essential to the parasite [94]. In addition, in P. falciparum GCL may play a role in drug resistance: higher levels of GSH, driven by high levels of GCL activity confer a more effective detoxification of heme and thus may be a major reason for resistance to chloroquine [95]. Depletion of GSH might be a chemotherapeutic strategy for malaria treatment, and GCL is proposed as a potential drug target. Chemotherapeutic effects of buthionine sulfoximine (BSO), an inhibitor of GSH synthesis, on trypanosomiasis has been reported [96]. In Schistosoma, it is not known whether GSH depletion results in parasite death. Cultured ex vivo worms can survive in the presence of the specific GSH synthesis inhibitor buthionine sulfoximine [97][Rigouin and Williams, unpublished]. However, during infection when the organism requires high levels of reduced GSH to fight oxidative stress, the depletion of GSH would likely affect parasite survival as suggested by studies with OPZ and with TGR (see below).
Figure 2.

Enzymes involved in the glutathione metabolism. Synthesis of glutathione. 1: condensation of L-cysteine (C) and L-glutamate (γE) by γ-glutamate cysteine ligase (CGL) to form the dipeptide γ-glutamylcysteine (γEC). 2: condensation of γ-glutamylcysteine (γEC) and glycine (G) by glutathione synthase (GS) to yield GSH (γECG). Detoxification processes. 3: conjugation of GSH to electrophilic compounds (X) catalyzed by glutathione S-transferase (GST). 4: cleavage of γ-glutamate from GSH or GSH conjugates (γECxG) by γ-glutamyl transferase (γGT) yielding respectively CG and CxG. 5: cleavage of glycine from GSH or GSH conjugates by phytochelatin synthase (PCS) to give respectively phytochelatins (γ(EC)nG) and γEC-X. 6: reduction of H2O2 and lipid hydroperoxides (LOOH) to water and the corresponding alcohols (LOH) and water with GSH by glutathione peroxidase and peroxiredoxins.
Glutathione peroxidase
Glutathione peroxidase (EC 1.11.1.9) catalyzes the reduction of hydrogen peroxide and/or organic peroxides to water or the corresponding alcohols using reduced GSH (Figure 2, reaction 6 and Figure 3). Many GPx are selenoproteins: they contain a selenocysteine (Sec) residue in the active site that is required for full catalytic activity [98]. Recently several non-selenium GPx, showing different cellular activities, have been identified [99]. The superfamily of GPx comprises ubiquitous enzymes widely distributed in Nature as isomeric forms; their classification is based on their amino-acid sequence, substrate specificity, and subcellular localization [98].
Figure 3.
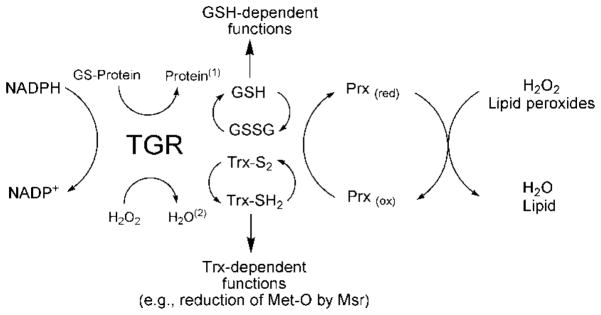
The antioxidant protein network in Schistosoma mansoni. TGR catalyzes deglutathionylation reactions of proteins and peptides(1). In addition, TGR not only reduces GSSG and oxidized Trx but also low molecular weight compounds (e.g., hydrogen peroxide)(2) utilizing NADPH. Prx proteins can receive electrons from either Trx or GSH to neutralize H2O2 and lipid peroxides.
In S. mansoni two selenium-containing class-4 GPx isoforms have been described: GPx-1, a monomeric protein of about 20 kDa [100] and GPx-2 [72], having respectively 53% and 55% identity with the human GPx4 protein. Kinetic analysis and substrate specificity of schistosome Gpx-1 showed that the enzyme is involved in the reduction of lipid hydroperoxides rather than inorganic peroxides, displaying similar activity as mammalian phospholipid-hydroperoxide glutathione peroxidase GPx4 [101]. Because S. mansoni GPx-2 has high sequence similarity with S. mansoni GPx-1 and mammalian phospholipid hydroperoxide GPx4, it is predicted to also have higher specificity activity toward phospholipid peroxides and poor reactivity toward H2O2 [72]. Other studies have shown GPx-1 to be developmentally regulated, with higher specific activities being found in the tegument-enriched Nonidet P-40 extract of adult worms and significantly higher activity with cumene hydroperoxide than with hydrogen peroxide [102]. Therefore, because of this peculiar activity and its localization to the tegument and gut epithelium [103], it is likely that schistosome GPx play an exclusive role in the protection of biological membranes from oxidative damage. GPx-1 has been studied as a vaccine candidate. Although immunization with GPx-1 resulted in a 43.4% reduction in worm burdens when mice were challenged with S. mansoni cercariae, no protection was observed against adult worms in existing infections [103]. Nevertheless, it has been proposed that because of its localization and involvement in parasite membrane protection, GPx should act as a first line of enzymatic cellular defense against tegument attack by host immune cells [103]. For this reason, it is likely that by inhibiting GPx the sensitivity of adult worms to oxidative stress would be enhanced.
The structure of S. mansoni GPx-1 has been determined providing insights into its mechanism of catalysis and confirming the schistosome GPx are most closely related to the mammalian GPx4 class [104]. Like other GPx4 enzymes, S. mansoni GPx-1 is a monomeric protein. The cysteine residue in the active site of the Sec43Cys mutant crystallized undergoes spontaneous oxidation to a sulfonic acid. This property of the schistosome enzyme, not seen in the structure of a similar human GPx4 mutant, combined with differences in the surface charge and hydrophobicity suggests special redox/catalytic properties of the worm enzyme that may support the rational design of specific inhibitors [104].
Glutathione S-transferase
The antioxidant molecule GSH also plays an important role in parasite defenses via the activity of GSTs (EC 2.5.1.18). These enzymes carry out a wide range of functions from the detoxification of exogenous electrophilic compounds (xenobiotics) (Figure 2, reaction 3), the inactivation of endogenous metabolites products of oxidative stress (lipid hydroperoxide and reactive carbonyl), the biosynthesis of signaling molecules (prostaglandins, leukotrienes), to the transport of ligands [105]. In general, GSTs share the same tertiary and quaternary dimeric structure. The monomer is made of two domains: the N-terminal region, which carries the GSH binding site and the C-terminal region responsible for the binding of the hydrophobic substrate [106]. The superfamily of GSTs comprises ubiquitous enzymes distributed in nature as isomeric forms. GST classification has first been established with mammalian enzymes and is based on several criteria including localization, amino acid nucleotide sequence, immunological, kinetic, and tertiary quaternary structural properties. The review of Sheehan [107] indicates the criteria on which GSTs have been classified thus far and highlights the fact that identification of new GSTs in groups like insects, plants, and helminths raised independent classification because of their unusual properties and overlapping activities.
GSTs have been detected in all helminths and are the most extensively characterized enzymes of detoxification pathways. Because parasitic helminths appear to lack or have very low levels of the phase I detoxification enzymes (cytochromes P450, cytochrome b5) [108], GSTs are likely to be the major enzymes involved in detoxification [109, 110]. Therefore, their study has raised real interest for the development of antischistosomal vaccines and drugs. In S. mansoni, four GST genes have been identified, GST1/2/3 of subunit size 28 kDa and two 26 kDa isoforms [37, 111–113], both belonging to the mu class, but also with similarities to the alpha and pi class GSTs, as well as one GST of the omega class [54]. In S. japonicum, at least two isoforms of GST have been identified that correspond to 28 kDa GST and 26 kDa GSTs of S. mansoni [112]. Only the 28 kDa isoform of S. haematobium has been identified [114]. These isoforms display different kinetic properties; their relative reactivity with a number of substrates and inhibitors varies, suggesting a distinct function of each isoform in the parasite. As demonstrated by Walker et al., [115], whereas the main isoform, S. mansoni 28 kDa GST, displays strong conjugation activity with xenobiotic substrates and reactive carbonyls products of lipid peroxidation, its shows only limited abilities to act as GSH peroxidase with lipid hydroperoxides. The 26 kDa GST isoforms seems to be more effective catalysts of reactive carbonyl-conjugation. Activities with hydroperoxides and reactive carbonyls show that, as with GPxs, GSTs are part of the biological response of the parasite to counteract oxidative stress. Girardini et al. [116] demonstrated that while the S. mansoni omega GST isoform has very low activity toward classical GSTs substrates, it has significant GSH-dependent dehydroascorbate reductase and thiol transferase activities. Moreover, GSTs are known to have binding functions: they are inhibited by a range of endogenous hydrophobic ligands such as bilirubin and haematin [115]. This binding function could be a passive detoxification mechanism of intracellular compounds or could involved GSTs in the transport of ligands. This provides evidence that GSTs interfere in vivo with a wide range of compounds that can modulate its activity.
The contribution of the different isoforms to a given metabolic pathway in schistosomes is reinforced by their different localization in the parasite. While both major isoforms (28 kDa GST and 26 kDa GST) are located in the tegument and in subtegumentary parenchymal cells, the 26 kDa GST is present in the cytoplasmic digitations localized in the apical chamber delineated by the flame cell body and the 28 kDa GST is present in immature germinal cells in both sexes and the ootype in the female genital system [117, 118].Therefore, it is likely that GST inhibition would disrupt metabolism in several key areas.
GSTs have been widely investigated because of their immunogenic character. The antigenic and protective properties of S. mansoni 28 kDa GST were discovered in 1987 by Balloul et al., [119] and has led since to its development as a vaccine candidate. The leading vaccine candidate, S. haematobium 28 kDa GST, has completed Phase I and II clinical trials but has yet to progress to Phase III trials [120]. Experiments in a variety of animal models of schistosome infection have demonstrated partial protection against infection by S. japonicum, S. mansoni, S. haematobium, and S. bovis [121–123]. Studies attempting to investigate the mechanism of immune response to S. mansoni 28 kDa GST revealed that at least a part of its protective effect could be due to immune-mediated inactivation of the enzyme. Functional analysis revealed that IgA antibodies to the protein displayed potent neutralizing activity on the enzymatic properties of the molecule leading to impaired schistosome fecundity, limited egg laying, and hatching capacity [124]. Furthermore, S. mansoni 28 kDa GST may play a crucial role in the production of prostaglandin D2, which may be important in the regulation of the immune response during schistosomiasis by altering the response of activated Langerhans cells in the epidermis during early phases of infection [125]. These data suggest that the inhibition of S. mansoni 28 kDa GST could lead to the stimulation of protective immunity against schistosomes. This raises the point that GSTs should be envisaged not only as an antigenic molecule, but also as a potential drug targets.
Because GSTs belongs to the family of xenobiotic metabolizing enzymes, GST activity may protect parasites from toxic effects of anthelmintics. Induction of GST occurs when worms are treated with drugs and might represent a defense strategy [126]. Because GSTs display a wide range of activities and substrate specificities, the different types of interaction between an anthelmintic compound and a GST can occur. An anthelmintic can be inhibitor of a GST or modulator of GST activity and compounds have been described to have such an effect on schistosome GSTs: the drug oltipraz interacts with thiol group of the GST and inhibits the transferase activity but not the peroxidase activity of the enzyme [91]. Artemether can decrease S. japonicum GST activity and at a lesser extent to SOD activity, suggesting that increasing schistosome oxidative stress might be responsible for the known antischistosomal action of the compound [127]. Artemether may be a non-substrate ligand processed by GST, through GST’s known role as an intracellular transporter could either potentiate the drug’s cytotoxicity or passively detoxify the compound. It has been shown that PZQ can bind schistosome GSTs [128], but in vitro studies have demonstrated that PZQ does not inhibit GST activity [129]. Alternatively, an anthelmintic may be a substrate for GST and its conjugation to GSH and further enzymatic processing would lead to its inactivation, degradation, and secretion. In this case GST affects the efficacy of the pharmacotherapy of the drug. Developing GST inhibitors would prevent any side effects attributed to drug metabolites leading to pharmacodynamic changes of the compound. Such inhibitor drugs would be use as a complement agent to an anthelmintic and would help to overcome drug resistance as well as increase drug efficacy.
Because of the various activities carried out by GSTs, their targeted inhibition may be advantageous as it could deprive parasites of their major defense against oxidative stress, prevent the synthesis or the transport of metabolites essential for the parasite survival, prevent xenobiotic elimination that would cause harm or potentiate antihelmintic drugs, as well as modulate the immunological response of the host to the parasite. However, no studies have reported the identification and analysis of GST inhibitors against schistosome infections. This may be because, although GST inhibitors have been identified [130], GST inhibitors that are relatively nontoxic, isoenzyme specific, and active in vivo have not yet been developed. Primary amino acid sequence alignment of helminth GSTs with mammalian GSTs indicates that non-mammalian GST superfamilies are represented in these parasites. Since S. mansoni 28kDa GST and 26kDa GST exhibit a combination of alpha-, mu- and pi-type amino acid sequence similarities and biochemical characteristics it may prove possible to inhibit both enzymes selectively with respect to mammalian pi class, the major isoenzymes in human extrahepatic tissues [115]. The increasing information available on crystal structures and activity properties helps the rational design of specific inhibitors. The three dimensional structure for the main isoforms of S. haematobium (28 kDa GST) and S. japonicum (26 kDa GST) have been elucidated. In all cases, structural comparisons of the parasitic enzymes to their mammalian homologues gives evidence on the conservation of the GSH-bonding site but highlights many structural differences in the xenobiotic binding sites, which provide opportunities to develop specific inhibitors[128, 131–133]. Jao et al. [134] have combined both structure-based drug design and the concept of polyvalency to discover a series of potent S. japonicum GST inhibitors, but the compounds have yet to be tested against parasites. The same group has further evaluated the participation of a set of compounds in GSH conjugate formation through NMR based-technique and uncovered specific potent inhibitors of S. japonicum GST [135]. Interestingly, high throughput assays are well established to measure GST activity, for example, Yasgar and colleagues [136] have described the utility of a luminescent assay by profiling a select group of inhibitors against mouse GST A4-4, human GSTs A1-1, M1-1, P1-1, and the S. japonicum 26 kDa GST.
Phytochelatin synthase
Another enzyme involved in GSH metabolism, likely to be important but still under-investigated in helminths, is phytochelatin synthase (PCS). Thus far, PCS have been widely studied in plants, yeast, algae, and fungi [137–141], and more recently they have been identified in bacteria [142, 143] and animals, especially in worms [144–147]. Surprisingly, many animals, but not vertebrates, have PCS genes. PCS is a γ-glutamylcysteine dipetidyltranspeptidase (EC 2.3.2.15) displaying a bi-functional activity. Analysis of the 3D structure of Nostoc PCS showed that the enzyme belongs to the papain superfamily of cysteine proteases with conservation of the catalytic triad of cysteine, histidine and aspartate and potential mechanism of catalysis involving an alkylated enzyme intermediate [148]. The enzyme displays two activities depending on the substrate available: it acts as a peptidase using GSH S-conjugates as substrates, catalyzing the removal of the glycine residue and yielding the corresponding γGlu-Cys-S-conjugates and it acts as a transpeptidase using GSH as a substrate in the synthesis of GSH-derived peptides called phytochelatins [149] (Figure 2, reaction 5). Due to this bi-functionality, the role of PCS in GSH metabolism is of great interest. Because PCS is absent from the human genome, PCS may be a suitable drug target for the treatment of diseases caused by pathogens expressing PCS. Its potential as a target for schistosomiasis treatment has been recently discussed [147].
Peptidases involved in xenobiotic metabolism?
In mammals, after conjugation of GSH to electrophilic compounds by GSTs, GSH-S-conjugates may be excreted or further metabolized to the mercapturic acid derivatives by enzymes of the xenobiotic degradation pathway. In helminths, and more particularly in schistosomes, because there is a lack of evidence regarding the involvement of phase I detoxification enzymes in xenobiotic metabolism [110, 150], it is thought that schistosomes use mainly the hydrolytic pathway involving GST conjugation activity to eliminate xenobiotic rather than oxidation/reduction reactions catalyzed by cytochrome P450s. In this regard, it is likely that enzymes acting downstream of GST, in tandem or independently, namely γ-glutamyltranspeptidase (γ-GT, EC 2.3.2.2) and PCS [151], lead to xenobiotic metabolism and could play a major role in xenobiotic elimination. γ-GT catalyzes the transfer of the γ-glutamyl moiety of GSH or GSH S-conjugates to an acceptor, which may be an amino acid, a peptide, or water (Figure 2, reaction 4). Such activity has been described in the worms Ascaris suum [152], Moniezia expansa, and Necator americanus [153]. The gene encoding the γGT has been identified in S. mansoni [92] and S. japonicum [93], but to date, no study has described its activity or its role in GSH S-conjugate hydrolysis in schistosomes species or other helminthes. The gene encoding PCS has been identified in S. mansoni [147] and S. japonicum. Although the role of PCS as a GSH S-conjugate hydrolytic enzyme in Schistosoma species and in animals generally is unknown, its involvement in the detoxification pathway in plants and yeasts has been reported [154, 155]. Current studies on recombinant S. mansoni PCS provide evidence for hydrolytic activity of the enzyme on GSH S-conjugates and an increase in PCS mRNA levels is seen when worms are cultured with drugs (Rigouin and Williams, unpublished results). Inhibition of either γGT or PCS may lead to the accumulation of toxin-derived compounds that could be harmful to the parasite, establishing these peptidases as novel potential targets for drug development, especially for PCS which has no equivalent in human. In addition, these enzymes are interesting targets to prevent drug resistance in parasites, as described above for GSTs.
Are transpeptidases involved in metal scavenging?
Essential heavy metals such as copper and zinc are required cofactors in redox reactions, ligand interactions and a number of other reactions. However, non essential heavy metals, such as arsenic, cadmium, lead, and mercury, are highly reactive and can be toxic through the displacement of endogenous metal cofactors from their binding sites in proteins and in the formation of reactive oxygen species through the Fenton reaction or by the inhibition of enzymes involved in reducing oxidative stress (see preceding text). Organisms must be able to control the cellular uptake and respond to the accumulation of heavy metals. This is accomplished by producing metal-binding ligands, most notably the metallothioneins and the phytochelatins. Schistosomes do not appear to synthesize metallothioneins or to have genes encoding them. This suggests that other proteins or factors play important functions in metal availability in schistosomes; PCS may be the major enzyme responsible for parasite protection against metal toxicity [147]. Phytochelatins (PCs) synthesized by PCS are a condensation of GSH moieties with the structure (γ-Glu-Cys)n-Gly, where n ≥ 2. A number of studies implicate the activity of PCs in heavy metal detoxification: PCs are immediately produced after exposure to a range of heavy metal ions [141] and confer to the metal-sensitive organism a tolerance or a resistance to the element. Organisms deficient in PCS display hypersensitivity to cadmium or become unable to tolerate cadmium toxicity, for instance, in C. elegans silencing PCS expression by RNA interference leads to a cadmium-hypersensitive phenotype[145].
Recombinant expression of S. mansoni PCS in a yeast system leads to an increase in cadmium tolerance (< 50 μM to > 1,500 μM) due to the synthesis of phytochelatins [147]. The role of metals in the induction of PCS expression differs among organisms, but the metal that seems unequivocally involved is cadmium. The function of PCS in schistosome biology is not clear as there is no evidence that schistosomes are exposed to significant amounts of cadmium in their definitive host. However, schistosomes degrade host hemoglobin and may use phytochelatins to neutralize heme toxicity. As indicated above, reduced iron in heme liberated from hemoglobin can react with oxygen to generate oxygen radicals and other toxic reactive species. Although schistosomes produce hemozoin, a crystalline, particulate form of heme, to avoid heme toxicity [156] it is speculated that iron flux must be tightly regulated. The mechanism of iron uptake and metabolism is still not well understood but has been proposed to be a potential target for novel therapeutics against schistosomes [157, 158]. In addition, although the mechanism of action of artemether against schistosomes is still under investigation, it is believed to have iron-dependant antiparasitic activity. The cleavage of artemether into an active metabolite responsible for parasite killing is induced by hemin or another iron-containing molecule [127]. Furthermore, evidence that phytochelatins could play a role in iron homeostasis in the parasite come from its increased expression in worms cultured in the presence of iron [147]. This hypothesis is supported by the recent discovery that GSH plays a vital role in iron metabolism in that an iron starvation-like response constitutes the near-unique genome-wide consequence of GSH depletion in yeast [159]. These investigators propose that GSH is split between an essential function in iron metabolism and a thiol-redox maintenance function that serves as a backup of the thioredoxin pathway but requires much higher levels of GSH [159]. If this is true of schistosomes, it is likely that, as with GSH, phytochelatins may be involved in metal detoxification and/or homeostasis. Therefore, depriving the parasite of such a defense or control system would be deleterious for the parasite.
The thioredoxin system
Thioredoxin
Thioredoxin proteins are small, ~100 amino acids, and act as electron vectors in a number of redox reaction via the reversible oxidation and reduction of the reactive-site cysteine pair (Figure 3). Thioredoxin from S. mansoni has been found to be expressed in all stages of worm development tested and to be present in the egg secretory products [160]. Two isoforms of Trx-1 exist in different life stages: an adult form and a juvenile form with an N-terminal extension of four amino acid residues (Gln-Leu-Val-Ile) [161]. Based on the genome of S. mansoni, there are two additional Trx genes, a second cytoplasmic Trx and a mitochondria-targeted Trx (Williams, unpublished results). Recently, the structure of S. mansoni Trx-1 has been explored and found to be similar to Trx proteins from other organisms with a typical Trx fold [161]. Like other Trxs, Trx-1 is able to reduce insulin in the presence of dithiothreitol (ΔA650nm=0.35 min−1, cf. 0.41 min−1 for human Trx), whereas lower activity was found when the reducing agent was GSH [160, 161]. It has been shown that Trx-1 is able to reduce GSSG with kcat values of 0.085 s−1 [161] and 0.014 s−1 [162] when GSSG concentrations are infinite. However, in dipteran insects (e.g., Drosophila melanogaster and Anopheles gambiae) where GR is absent, a high ratio of GSH to GSSG is maintained by the nonenzymatic reduction of GSSG by Trx (a rate constant of 170 M−1 s−1 (or 0.01 μM−1 min−1) for D. melanogaster Trx reacting with approximately 20 μM GSSG) [163, 164]. These results indicate that in schistosomes Trx would only reduce GSSG under extreme oxidative stress; therefore, it is not clear whether reduction of GSSG catalyzed by Trx-1 is physiologically relevant in S mansoni. In contrast, in Echinococcus granulosus, a tapeworm parasite with a redox network similar to that found in schistosomes [165], it has been shown that that Trx is able to reduce GSSG and to catalyze deglutathionylation reactions at high concentrations of GSSG in which the activity of its TGR would be inhibited [166]. These results suggest that redox pathways may vary in different Platyhelminthes parasites.
Schistosome Trx-1 has only two cysteine residues, both occurring in its active site (Trp-Cys-Gly-Pro-Gly), whereas human cytosolic Trx-1 has three additional cysteine residues (Cys-62, Cys-69 and Cys-73) outside of its active site (Cys-32/Cys-35). These three additional cysteine residues have distinct biological functions. For example, Cys-62 and Cys-69 can form an intramolecular disulfide bond under oxidative stress; this mechanism can transiently impair Trx activity [167]. Because the redox potential of Cys-62/Cys-69 is more negative than Cys-32/Cys-35 in the active site, the disulfide bond between Cys-62 and Cys-69 is formed after the cysteine pair in the active site is oxidized. The formation of the intramolecular disulfide (Cys-62 and Cys-69) results in the disruption of Trx folding, which further prevents the interaction of Trx with TrxR (or other substrates). This mechanism could prevent overoxidation of Trx, which may lead to the loss of activity of Trx-1 under oxidative stress [167]. In addition, mammalian Trx-1 forms a homodimer via an intermolecular disulfide via Cys-73. The dimerization has been proposed to be a regulatory mechanism [168]. Furthermore, Trx can regulate the intracellular concentration of nitric oxide via S-nitrosylation of Cys-69 [169]. Because S. mansoni Trx-1 has no noncatalytic cysteine residues it may be more susceptible to oxidative and nitrosative stress than human Trx-1. S. mansoni Trx provide electrons to oxidized Prx during the neutralization of H2O2 and other hydroperoxides [170]. It has been suggested that Trx-1 and Prx-1 protect eggs trapped in granulomas from oxidative stress resulting from the host’s immune reactions [160]. After passing reducing equivalents to its various substrates, oxidized schistosome Trx proteins can be reduced by TGR [88].
Thioredoxin glutathione reductase
In many organisms oxidized Trx and GSSG are reduced by TrxR and GR, respectively. In S. mansoni and other flatworm parasites (e.g., E. granulosus, Taenia crassiceps, and Fasciola hepatica), authentic TrxR and GR proteins are absent [88, 165]. Instead, TGR (EC 1.8.1.9), a multifunction enzyme, is able to reduce both GSSG and Trx (Figure 3). Mammalian cells also have TGR; however, it is associated with reproduction and less important in general redox protection in most cells. Mammalian TGR is predominately expressed in testis and its proposed function is to facilitate sperm maturation with GPx-4 [171–173]. Cloning of S. mansoni TGR lead to the prediction that the enzyme was a selenoprotein [88]. The absence of authentic TrxR and GR proteins and the central role of TGR in the redox biology of schistosomes was established using biochemical approaches. Immunoprecipitation of TGR with antibodies produced against the recombinant protein removed GR, TrxR, and glutaredoxin (Grx) activities, while Western blotting of the precipitated proteins identified a single protein of the size expected for TGR (larger than known GR, TrxR, and Grx proteins), suggesting that TGR was responsible for all activities in the worm [88]. Using the specific inhibitor auranofin, reduction of both GSSG and oxidized Trx in worm homogenates was equally inhibited, again suggesting that a single protein was responsible for both activities [88]. The absence of authentic GR and TrxR genes was confirmed by the S. mansoni genome sequence [92].
Like selenoprotein TrxRs [174], S. mansoni TGR has broad substrate reactivity reducing H2O2, sodium selenite, lipoamide, and tert-butyl hydroperoxide [58]. Recently it has been shown that the Sec597Cys mutant of S. mansoni TGR is able to reduce these substrates, albeit with lower efficiencies, showing that the selenocysteine is important but not essential for the activity of TGR [175]. Unlike mammalian TrxR however, S. mansoni TGR is unable to reduce ubiquinone or dehydroascorbic acid [58, 174, 176]. S. mansoni TGR also catalyzes deglutathionylation reactions [58, 175]. Under oxidative stress and resulting high GSSG concentrations, cysteine residues in proteins can become S-glutathionylated. This is thought to protect redox-sensitive cysteines from becoming irreversibly over-oxidized to sulfinic or sulfonic acids with consequent loss of protein activity [177]. Deglutathionylation (Grx) reactions are important regulatory mechanisms in cells to restore glutathionylated proteins to full activity after return to normal cellular redox status.
Because TGR in schistosomes plays a central role in redox homeostasis, redox regulation, and maintaining reduced pools of glutathione and thioredoxin for numerous essential biochemical processes, it is undoubtedly important to the survival of the parasites. Indeed, TGR has been validated by both chemical and reverse genetic approaches as an essential protein in S. mansoni. Exposing cultured, ex vivo adult worms to 10 μM auranofin, a potent TGR inhibitor [88], inhibits TGR activities, reduces the ratio of reduced to oxidized glutathione, and leads to worm unpairing and death [58]. Skin-stage parasites and juvenile liver-stage parasites are killed by 5 μM auranofin. In addition, decreased worm burdens were found in infected mice treated with auranofin (59–63% decrease). RNAi silencing of TGR in cultural, larval parasites led to decreases in TGR activity and parasite survival [58]. This affect was seen in parasites cultured both aerobically and anaerobically, indicating that TGR function is broader than simply protecting worms from redox stress. The specificity of auranofin and RNA silencing was shown by a synergistic effect of their combined activity against cultured parasites [58].
In order to identify compounds specifically targeting S. mansoni TGR, it is necessary to better understand its catalytic mechanisms. Because of its high sequence similarity (~50%) and active sites that are virtually identical to other dimeric NADPH-oxidoreductases such as GR and TrxR [58, 175, 178–180], S. mansoni TGR is thought to have similar catalytic mechanisms. TGR is a homodimeric flavoprotein with a head-to-tail monomer arrangement with each monomer containing both TrxR and Grx domains [181]. As has been proposed for mammalian TGR [172, 179], the active sites of S. mansoni TGR are thought to be contributed by residues from both monomer subunits: an FAD, an adjacent redox-active cysteine pair (Cys-154 and Cys-159) in the TrxR domain and a cysteine pair (Cys-28 and Cys-31) in the Grx domain from one subunit act in concert with the C-terminal cysteine-selenocysteine pair (Cys-596 and Sec-597) of the other subunit. It is thought that during the catalytic cycle reducing equivalents from NADPH are passed to FAD, then to the Cys-154/Cys-159 couple, then to Cys-596′/Sec-597′ in the other subunit. This redox-active couple is required for the reduction of Trx as a truncated form in which Sec-597 and Gly-598 are missing lacks any TrxR activity [181]. The reduced C-terminal active site is then proposed to pass electrons to a substrate (e.g., oxidized Trx) or to the Cys-28/Cys-31 couple in the other subunit [175, 182]. Through structural and modeling studies it has been suggested that the C-terminal tail containing Cys-596′/Sec-597′ undergoes conformational changes to allow electron transfer to Cys-28/Cys-31 [181–183]. Spectroscopic analyses of anaerobic NADPH titrations support this hypothesis [175]. DTNB (5,5′-dithiobis-(2-nitrobenzoic acid); Ellman’s reagent) is used as a model substrate to determine TrxR activity of authentic TrxR enzymes [174]. It was found that S. mansoni TGR can reduce DTNB via its Cys-596/Sec-597 active site (as in authentic TrxR proteins) as well as at its Cys-28/Cys-31 active site [175].
Authentic TrxR proteins cannot reduce GSSG [174, 184]. This suggests that in TGR proteins GSSG reduction occurs at the Grx domain cysteine couple (Cys-28/Cys-31) and/or at the cysteine couple proximal to the NADPH binding site (Cys-154/Cys-159), which occurs within an amino acid sequence that is identical to that found in many authentic GR proteins [175, 181, 185]. In order to determine the site of GSSG reduction, S. mansoni TGR proteins with cysteine to alanine or cysteine to serine mutations were generated and characterized. The GR activity of the Cys28Ala variant was only 3–4 % of wild-type activity, while the Cys31Ala mutant had ~20% and the Cys31Ser mutant had >100% of wild-type GR activity, respectively, indicating that Cys-28 is the residue responsible for the nucleophilic attack on GSSG. The role of Cys-31 appears to be to facilitate the formation of the nucleophilic thiolate on Cys-28. The Cys-154/Cys-159 couple may provide the residual GR activity in the Cys28Ala variant as the electrostatic environment around this redox couple is similar to that in GR proteins and may facilitate the binding of GSSG [175, 181, 185]. In several characterized TGR proteins high concentrations of GSSG result in a transient inhibition of GSSG reduction and a delay in attaining full GR activity [178, 180, 186]. The lag time in reaching full activity could be eliminated by the addition of GSH to reactions leading to the hypothesis that the lag was due to glutathionylation of cysteines in TGR [178, 180]. Proteomic analysis of TGR from E. granulosus isolated during this lag phase found that Cys-88 and Cys-354 were glutathionylated [180]. However, these two cysteine residues are relevant to the structural integrity of TGR, are unlikely to be involved in the catalytic cycle of TGR, and are not found in all TGR proteins that show this lag effect [175, 186]. Kinetic analyses of the S. mansoni Cys-31 mutants found that the lag time in GR activity disappeared entirely, suggesting that the lag may be due to glutathionylation on Cys-31 [175]. Under oxidative stress (high GSSG/GSH) glutathionylation of key cysteine residues in TGR proteins may be a mechanism to prevent their overoxidation thus preventing potential permanent loss of activity. Mammalian TGR proteins have a Cys-Xaa-Xaa-Ser motif is the Grx domain. To the best of our knowledge, no lag in GR activities have been found in mammalian TGR proteins. Therefore, the active site of Grx domain of mammalian TGRs may be more susceptible to oxidative inactivation. However, because TrxR and GR are present in mammalian cells, loss of TGR activity would be compensated by these enzymes.
As mentioned previously, unlike TrxR and GR, TGR also catalyzes deglutathionylation reactions. The deglutathionylation activities of wild-type TGR and its Grx-domain variants have been characterized. Two catalytic mechanisms are involved in deglutathionylation reactions of TGR: the active site in the Grx domain can be reduced by TrxR domain of the dimer or by free GSH [175]. At high ratios of GSH/GSSG (i.e., reducing conditions), TGR undergoes a monothiol mechanism. The thiol anion on Cys-28 undergoes nucleophilic attack on the mixed-disulfide between GSH and proteins (or peptides), forming a glutathionylated Cys-28 TGR intermediate and free proteins/peptides. The glutathionylated Cys-28 is then resolved by GSH, generating GSSG and free Cys-28. At low ratios of GSH/GSSG (i.e., oxidizing conditions), the TGR Cys-28–peptide intermediate is resolved by Cys-31, thereby forming an intramolecular disulfide (Cys-28–Cys-31). The mixed disulfide is then reduced by electrons from NADPH via the TrxR domain of the protein. These mechanisms suggest that TGR is able to catalyze deglutathionylation reactions when the parasites are challenged by ROS and under oxidative stress. Recently, Bonilla et al., found that the Grx activity of TGR from E. granulosus requires both Sec and the Grx domain and catalyzes deglutathionylation only via a monothiol mechanism. The difference between the deglutathionylation reactions of E. granulosus TGR and S. mansoni TGR may be explained by different distances between cysteines in the Grx active sites of the two proteins. In E. granulosus TGR, the distance between Cys-31 and Cys-34 is longer than that between Cys-28 and Cys-31 in S. mansoni TGR and, therefore, the accessibility of Cys-34 to Cys-31 may be somewhat limited in E. granulosus TGR [166, 175].
Because S. mansoni TGR has been validated as an essential protein and drug target, studies have been undertaken to identify TGR inhibitors. Because active sites of TGR are similar to that of mammalian TrxR and GR, known inhibitors of these proteins may be useful starting points to identify TGR inhibitors. Gold-containing compounds (e.g., auranofin, aurothioglucose, aurothiomalate), which are good mammalian TrxR inhibitors [187, 188], inhibit schistosome TGR [58]. However, while platinum drugs RMA 19 and RMA 35 inhibit mammalian TrxR, only RMA35 but not RMA19 inhibits S. mansoni TGR [58]. This indicates that differences in structural and catalytic proprieties between TrxR and TGR exist and could be used for the design of TGR-specific inhibitors. Structural analyses of auranofin-S. mansoni TGR complexes found that gold was removed from auranofin and bound at three different sites in the TGR protein: (1) Cys154/Cys-159, the redox center adjacent to FAD, (2) the putative NADPH binding site, and (3) complexed with Cys-520/Cys-574 [189]. The last cysteine couple had not previously been implicated in the catalytic activity of TGR. Recent studies show that this cysteine pair is involved in the structure integrity of schistosome TGR, rather than catalysis [175]. In addition, several studies suggest that inhibition of selenoproteins by gold and platinum containing compounds is selenocysteine-dependent, i.e., they are prodrugs that require selenocysteine for conversion into active species [188, 190–194] and that specific reactivity with TrxR and not with GR was due to the absence of selenocysteine in GR [187]. However, selenocysteine was found not to be essential for inhibition of S. mansoni TGR by auranofin. Furthermore, a selenocysteine-deficient variant of S. mansoni TGR and yeast GR were inhibited by auranofin, but at much lower rates than selenocysteine-containing TGR [175, 189]. The kinetics of inhibition of selenocysteine-deficient TGR and GR could be increased to wild-type levels in the presence of exogenous selenium [189]. Further studies have found that in addition to auranofin, oxadiazole 2-oxides and antimony potassium tartrate can inhibit wild-type TGR and the Sec597Cys variant with the same efficiency, confirming that selenocysteine is not essential for inhibitory effects of many drugs [175].
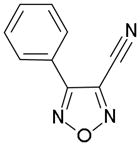
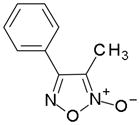
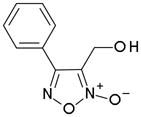
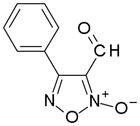
In order to identify compounds inhibiting the redox pathway of schistosomes a quantitative high-throughput screen has been performed against the reconstructed pathway shown [195–197]. In the assay, S. mansoni Prx-2 uses GSH as an electron donor to reduce H2O2. The resulting GSSG is reduced by TGR utilizing NADPH and the reaction progress was monitored by following the change in NADPH fluorescence (Figure 4). The coupled enzymatic assay was miniaturized and used to screen 70,000 compounds in the NIH Molecular Libraries Small Molecules Repository [197]. Once compounds were found to inhibit the coupled reaction, Prx-2 and TGR target-deconvolution assays were used to identify the target of the compounds. Derivatives of phosphinic amides, oxadiazole-2-oxides and isoxazolones were found to inhibit TGR with low μM to nM inhibitory constants [197]. The efficacies and activities of phosphinic amides and oxadiazole-2-oxides were further tested [198]. 4-phenyl-1,2,5-oxadiazole-3-carbonitrile-2-oxide (4-phenyl-3-furoxancarbonitrile or furoxan) was identified to be a irreversible inhibitor of S. mansoni TGR. Most importantly, furoxan resulted in 100% parasite death at 10 μM within 24 hours and within 120 hours at 2 μM. In addition, it is active against multiple developmental stages of S. mansoni and adult worms of S. japonicum and S. haematobium. Furthermore, treatment of skin-stage parasites, juvenile, liver-stage parasites and adult worms with furoxan resulted in highly significantly decreased worm burdens in infected mice [198]. Nitric oxide released from furoxan through the action of TGR has been demonstrated to S-nitrosylate cysteine residues of TGR, resulting in its inhibition and parasite death [198, 199]. A SAR (structure-activity relationship) study found that the N-oxide is essential for furoxan activity and worm killing as the furazan analogue is inactive and that the analogue with the N-oxide regiochemically transposed is also inactive (Table 1) [199]. Replacing the 3-cyano group of furoxan with a groups that are less electronegative (3-methyl or 3-hydroxymethyl) also results in loss of compound activity (Figure 5 and Table 1) [199]. Although more electronegative substitutions (3-formyl, 3-carboxyl) in this position are more active than furoxan as TGR inhibitors, these analogs have no worm-killing activity, probably due to limitations in bioavailability (Table 1). The 3-carboxamide derivative of furoxan retains significant activity and may have better drug properties than the 3-cyano analog, but this remains to be determined. Electron-withdrawing groups in R2 position also slightly enhance potency (Figure 5) [199]. Currently, a number of furoxan analogs are being characterized with respect to their inhibition of TGR, worm killing capacity, cytotoxicity, stability to microsomal breakdown, and drug (ADME) properties.
Figure 4.
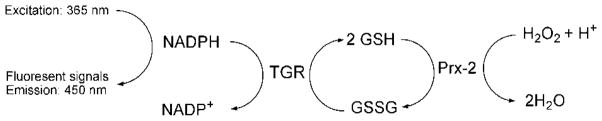
Assays used in high throughput drug screen. A coupled enzymatic assay was used in order to screen compounds which are able to inhibit TGR and/or Prx-2. Prx-2 catalyzes conversion of hydrogen peroxide to water utilizing GSH. The resultant GSSG from the previous reaction is recycled to GSH by TGR using NADPH. The consumption rates of NADPH were monitored with fluorescence of NADPH (excitation: 365 nm and emission: 450 nm). The figure is adapted from [198].
Table 1.
The activities of oxidazole-2-oxides against TGR enzyme and cultured ex vivo adult S. mansoni worms. The table and data were adapted from [199].
| Compound | TGR | Worms |
|---|---|---|
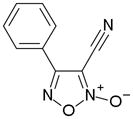
|
Active | Active |
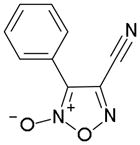
|
Inactive | Inactive |
|
Inactive | Inactive |
|
Inactive | Inactive |
|
Inactive | Inactive |
|
Active | Inactive |
|
Active | Inactive |
Figure 5.
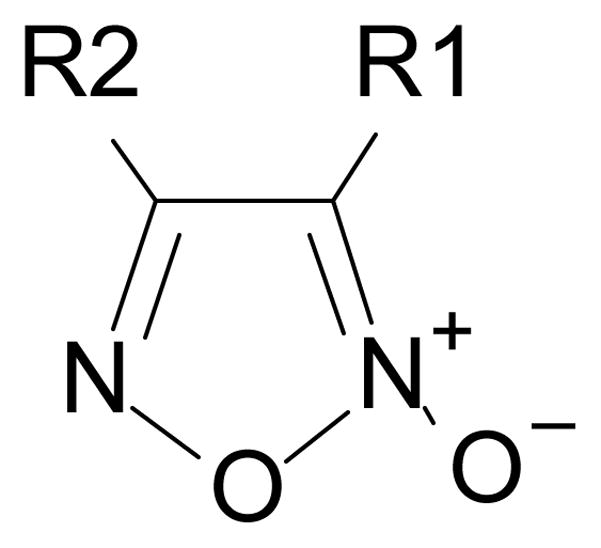
The general structure of oxidazole-2-oxides. The presence of electron-withdrawing groups at positions R1 and R2 are essential to the activity of oxidazole-2-oxides.
Peroxiredoxins
As mentioned previously, schistosomes have limited capacity to neutralize H2O2 because they lack catalase and their GPx proteins have low activity with H2O2 [72, 170]. It has been proposed that schistosomes use Prx (EC 1.11.1.15) to reduce H2O2 and, along with GPx and GST, to neutralize lipid hydroperoxides (Figure 2, reaction 6 and Figure 3) [72, 200]. Three Prx classes with distinct catalytic mechanisms have been identified in a multitude of organisms: the classic 2-Cys Prxs, atypical 2-Cys Prxs and 1-Cys Prxs [201, 202]. The three Prx genes found in S. mansoni encode typical 2-Cys Prx proteins and their sequences are highly similar (ca. 65% similarity at the amino acid level) [72, 170]. S. mansoni Prx-3 is predicated to be targeted to the mitochondria and is found specifically in mitochondrial fractions of adult worms [170]. Based on transcript abundance, S. mansoni Prx-1 is the generally the most highly expressed species, with highest expression in eggs and adult female worms. Prx-2 expression is very low in cercariae, but its expression increases significantly within 24 hours of transformation and gradually increases during worm development. Prx-3 expression is low in all stages with the highest expression found in adult female worms [72].
The catalytic mechanism of typical 2-Cys Prx has been investigated. Typical 2-Cys Prx proteins form homodimers and function in multimeric forms of five or six dimers (decamers and dodecamers, respectively) [203–205]. These decamers/dodecamers form a higher-order, toroid structures [204, 205]. Oligomerization of the multimeric forms results in stacks of toroids and changes Prx activity from a peroxidase to a stress-regulated chaperone [206, 207]. In the peroxidase catalytic cycle of typical 2-Cys Prx proteins, the thiol group of the N-terminal peroxidative cysteine is oxidized to a sulfenic acid after reacting with H2O2. Subsequently, the C-terminal resolving cysteine from the other subunit reacts with the sulfenic acid of the peroxidative cysteine forming an intermolecular disulfide. The resulting mixed disulfide is resolved by Trx in most cases [201, 202, 208]. Interestingly, S. mansoni Prx-2 and Prx-3 can use both Trx and GSH as electron donors whereas Prx-1 has better activity with Trx [170]. In addition, S. mansoni Prx-1 demonstrates a typical Prx kinetic behavior (ping-pong, unsaturable) whereas Prx-2 and Prx-3 display unusual kinetics [170]. A C-terminal extension of ~22 amino acids exists in both S. mansoni Prx-2 and Prx-3, which is absent from Prx-1, and has been shown to confer the unusual kinetic behavior and increased activity with GSH [170].
In addition to its toxic effects, H2O2 is an important signaling molecule [209]. In addition to their role as antioxidants Prx proteins have been shown to play important functions to regulate H2O2 signaling. Active Prx proteins maintain low resting levels of H2O2. When H2O2 is produced during signaling events, Prx are inactivated through the overoxidation of their peroxidatic cysteine to a sulfinic acid, leading to focally increased H2O2 levels and signal transduction can proceed; Prx effectively act as a molecular floodgate [210–212]. Wood and coworkers [211] postulated that an internal loop containing a Gly-Gly-Leu-Gly motif and the C-terminal helix containing a Tyr-Phe motif pack next to each other and bury the active site helix containing the peroxidatic cysteine stabilizing the oxidized enzyme and preventing easy reduction of the peroxidatic cysteine sulfenic acid (-SOH), which can then be oxidized by H2O2 to the inactive sulfinic acid (-SO2H) form. Analysis of the schistosome Prx sequences indicated that the Gly-Gly-Leu-Gly motif is present in all three schistosome Prx sequences [170]. Although the C-terminal helix and its Tyr-Phe motif are present in Prx-2 and Prx-3, it is absent from Prx-1. Analysis of the proteins propensity to oxidative inactivation found that Prx-1 was resistant and Prx-2 was sensitive. Transfer of the C-terminal 22 amino acids with the Tyr-Phe motif of Prx-2 to Prx-1 converted Prx-1 into an overoxidation-sensitive enzyme. Likewise, C-truncated Prx-2, which lacks the C-terminal helix and Tyr-Phe motif, is a robust enzyme, with resistance to oxidative inactivation similar to Prx1. Since Prx-1 may protect schistosome eggs from oxidative stress [213], it should be less sensitive to oxidative inactivation in the oxidizing extracellular environment of the granuloma.
Three Prx genes have also been identified in S. japonicum: Prx-1 and Prx-2 are cytosolic forms and Prx-3 is a putative mitochondrial form [214]. Prx from S. japonicum have high sequence identity with their orthologs from S. mansoni (85–90%). All three forms are expressed in the egg, cercariae, and adult stages. S. japonicum Prx-1 is more highly expressed in eggs than in cercariae and adult worms, while its Prx-2 in predominately expressed in adult worms [214]. As in S. mansoni, S. japonicum Prx-1 is secreted by eggs and may protect eggs from oxidative stress [160, 214]. In addition, it was found that S. japonicum Prx-1 is in worm tegument, whereas Prx-2 is in sub-tegumental layers, parenchyma, vitelline, and gut epithelial tissues in adult worms [214]. It was shown that Prx-1, but not Prx-2, in schistosomula can neutralize exogenous hydroperoxides and organic peroxides; however, neither S. japonicum Prx is able to neutralize nitric oxide [215].
Methionine sulfoxide reductase
Under oxidative stress conditions some methionine (Met) residues in proteins may become oxidized to methionine sulfoxide (Met-O), the formation of which may impair the structural integrity and activity of proteins. Methionine sulfoxide reductase proteins (Msr) reduce Met-O back to Met with electrons from Trx (Figure 6). Msr proteins have been found to be important in a variety of different organisms to manage oxidative stress and repair damaged proteins [216–218]. Also, it has been proposed that Met residues on proteins may act as ROS scavengers. After reacting with ROS, Met can be converted to Met-O; the resultant Met-O can be re-reduced to Met by Msr [218, 219]. Met-O residues exist in two stereoisomeric forms, the S epimer (Met-S-O) and the R epimer (Met-R-O) and Msr proteins are classified into two types based on their substrate stereospecificity. Typically, MsrA forms reduce peptide-Met-S-O and free Met-S-O, while MsrB proteins reduce peptide-Met-R-O and have week activity with free Met-R-SO [218, 220]. Two MsrB-like genes have been found in S. mansoni [220]. While in humans two types of MsrB proteins have been identified, selenocysteine- and Cys-containing MsrB forms [221, 222], both MsrB proteins in S. mansoni (MsrB1 and MsrB2) are Cys-containing. MsrB1 has two cysteine residues in the active site whereas the active site of MsrB2 has one cysteine residue [220]. The catalytic mechanisms of S. mansoni MsrB1 and MsrB2 have been proposed. In MsrB1, the catalytic cysteine residue interacts with Met-O, generating a sulfenic acid intermediate on the MsrB1 catalytic cysteine residue releasing free or protein bound Met. The second (recycling) cysteine of MsrB1 then attacks the sulfenic acid intermediate to form an intramolecular disulfide. The disulfide in MsrB1 can then be resolved by reduced Trx. In MsrB2 the resolving cysteine is replaced by a threonine, and therefore, Trx must directly reduce the sulfenic acid intermediate on the catalytic cysteine residue [220, 223]. Both MsrB1 and MsrB2 have a predicted, conserved zinc-binding motif essential to maintain their proper 3D structure [220, 224]. MsrB1 and MsrB2 appear to have different substrate specificities with MsrB1 preferring free Met-S-SO and MsrB2 preferring peptide-Met-S-SO [220]. MsrB1 and MsrB2 are expressed in all parasite development stages investigated with the highest expression seen in eggs and the lowest expression in schistosomula. Elevated expression of Msr proteins in eggs may help to neutralize ROS in the granuloma produced by the immune responses of hosts [220]. Similarity searches of the S. mansoni genome fail to identify any MsrA orthologs. It remains to be determined if one of the MsrB proteins or if a phylogenetically unrelated protein(s) in the S. mansoni genome have MsrA-like activity. It is also not known if S. mansoni Msr proteins are suitable drug targets.
Figure 6.

The function of methionine sulfoxide reductase (Msr) in the redox network of Schistosoma mansoni. Reactive oxygen species (ROS) can convert methionine (the free amino acid as shown here or in a protein) to methionine sulfoxide. This may lead to inactivation of protein function. Reduction of methionine sulfoxide and reactivation of the protein and prevention of higher oxidation states of methionine can be carried out by Msr using reduced thioredoxin (Trx(red)) as the electron donor producing oxidized thioredoxin (Trx(ox)). Reduction of Trx(ox) by thioredoxin glutathione reductase (TGR) is accompanied by the consumption of NADPH.
Concluding remarks
Schistosomiasis causes serious public health issues in much of the developing world. Even though PZQ is an effective treatment for this disease, there is a constant threat that the parasites may evolve resistance to PZQ eliminating the main control measure. There is little interest by pharmaceutical companies in developing drugs for schistosomiasis and other neglected tropical diseases because of the low financial return. Because schistosomes reside in an aerobic environment and are under oxidative stress they require adequate antioxidant responses to neutralize ROS. Because the schistosome antioxidant network differs significantly and is highly restricted compared to its human host, it may be the parasite’s Achilles heel. Indeed, several recent studies indicate that antioxidants in schistosomes are good drug targets and drug development based on these antioxidants is in process. In this paper, we comprehensively discuss biochemical aspects of schistosome antioxidants and their potential as drug targets. Future studies should be directed to provide a better understanding of the schistosome redox network and toward the identification of effective molecules targeting schistosome redox enzymes.
Acknowledgments
This work is supported by US NIH grants AI065622 and AI081107 (DLW).
List of abbreviations
- GPx
Glutathione peroxidase
- GR
glutathione reductase
- GSH
glutathione
- GST
glutathione S-transferase
- Msr
methionine sulfoxide reductase
- OPZ
oltipraz
- PCS
phytochelatin synthase
- Prx
peroxiredoxin
- PZQ
Praziquantel
- SOD
superoxide dismutase
- TGR
thioredoxin glutathione reductase
- Trx
thioredoxin
- TrxR
thioredoxin reductase
References
- 1.Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6:411–25. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]
- 2.King CH, Dangerfield-Cha M. The unacknowledged impact of chronic schistosomiasis. Chronic Illn. 2008;4:65–79. doi: 10.1177/1742395307084407. [DOI] [PubMed] [Google Scholar]
- 3.King CH. Parasites and poverty: the case of schistosomiasis. Acta Trop. 2010;113:95–104. doi: 10.1016/j.actatropica.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ezeamama AE, McGarvey ST, Acosta LP, et al. The synergistic effect of concomitant schistosomiasis, hookworm, and trichuris infections on children’s anemia burden. PLoS Negl Trop Dis. 2008;2:e245. doi: 10.1371/journal.pntd.0000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Secor WE, Sundstrom JB. Below the belt: new insights into potential complications of HIV-1/schistosome coinfections. Curr Opin Infect Dis. 2007;20:519–23. doi: 10.1097/QCO.0b013e3282e9ac03. [DOI] [PubMed] [Google Scholar]
- 6.Hotez PJ, Fenwick A, Kjetland EF. Africa’s 32 cents solution for HIV/AIDS. PLoS Negl Trop Dis. 2009;3:e430. doi: 10.1371/journal.pntd.0000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diallo TO, Remoue F, Schacht AM, et al. Schistosomiasis co-infection in humans influences inflammatory markers in uncomplicated Plasmodium falciparum malaria. Parasite Immunol. 2004;26:365–9. doi: 10.1111/j.0141-9838.2004.00719.x. [DOI] [PubMed] [Google Scholar]
- 8.Sangweme DT, Midzi N, Zinyowera-Mutapuri S, Mduluza T, Diener-West M, Kumar N. Impact of schistosome infection on Plasmodium falciparum Malariometric indices and immune correlates in school age children in Burma Valley, Zimbabwe. PLoS Negl Trop Dis. 2010;4:e882. doi: 10.1371/journal.pntd.0000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Labeaud AD, Malhotra I, King MJ, King CL, King CH. Do antenatal parasite infections devalue childhood vaccination? PLoS Negl Trop Dis. 2009;3:e442. doi: 10.1371/journal.pntd.0000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schistosomes liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; Lyon. 7–14 June 1994; 1994. pp. 1–241. [PMC free article] [PubMed] [Google Scholar]
- 11.Rollinson D. A wake up call for urinary schistosomiasis: reconciling research effort with public health importance. Parasitology. 2009;136:1593–610. doi: 10.1017/S0031182009990552. [DOI] [PubMed] [Google Scholar]
- 12.Fried B, Reddy A, Mayer D. Helminths in human carcinogenesis. Cancer Lett. 2011;305:239–49. doi: 10.1016/j.canlet.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Ishii A, Matsuoka H, Aji T, et al. Parasite infection and cancer: with special emphasis on Schistosoma japonicum infections (Trematoda) A review Mutat Res. 1994;305:273–81. doi: 10.1016/0027-5107(94)90247-x. [DOI] [PubMed] [Google Scholar]
- 14.OEHS, Hamid HK, Mekki SO, Suleiman SH, Ibrahim SZ. Colorectal carcinoma associated with schistosomiasis: a possible causal relationship. World J Surg Oncol. 2010;8:68. doi: 10.1186/1477-7819-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keiser J, Utzinger J. Artemisinins and synthetic trioxolanes in the treatment of helminth infections. Curr Opin Infect Dis. 2007;20:605–12. doi: 10.1097/QCO.0b013e3282f19ec4. [DOI] [PubMed] [Google Scholar]
- 16.Ribeiro-dos-Santos G, Verjovski-Almeida S, Leite LC. Schistosomiasis--a century searching for chemotherapeutic drugs. Parasitol Res. 2006;99:505–21. doi: 10.1007/s00436-006-0175-2. [DOI] [PubMed] [Google Scholar]
- 17.Domling A, Khoury K. Praziquantel and schistosomiasis. ChemMedChem. 2010;5:1420–34. doi: 10.1002/cmdc.201000202. [DOI] [PubMed] [Google Scholar]
- 18.Cheever AW, Macedonia JG, Mosimann JE, Cheever EA. Kinetics of egg production and egg excretion by Schistosoma mansoni and S. japonicum in mice infected with a single pair of worms. Am J Trop Med Hyg. 1994;50:281–95. doi: 10.4269/ajtmh.1994.50.281. [DOI] [PubMed] [Google Scholar]
- 19.Schramm G, Haas H. Th2 immune response against Schistosoma mansoni infection. Microbes Infect. 2010;12:881–8. doi: 10.1016/j.micinf.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Utzinger J, Xiao S, N’Goran EK, Bergquist R, Tanner M. The potential of artemether for the control of schistosomiasis. Int J Parasitol. 2001;31:1549–62. doi: 10.1016/s0020-7519(01)00297-1. [DOI] [PubMed] [Google Scholar]
- 21.Cioli D, Pica-Mattoccia L, Archer S. Antischistosomal drugs: past, present ... and future? Pharmacol Ther. 1995;68:35–85. doi: 10.1016/0163-7258(95)00026-7. [DOI] [PubMed] [Google Scholar]
- 22.Xiao SH, Catto BA. Comparative in vitro and in vivo activity of racemic praziquantel and its levorotated isomer on Schistosoma mansoni. J Infect Dis. 1989;159:589–92. doi: 10.1093/infdis/159.3.589. [DOI] [PubMed] [Google Scholar]
- 23.Wu MH, Wei CC, Xu ZY, et al. Comparison of the therapeutic efficacy and side effects of a single dose of levo-praziquantel with mixed isomer praziquantel in 278 cases of schistosomiasis japonica. Am J Trop Med Hyg. 1991;45:345–9. doi: 10.4269/ajtmh.1991.45.345. [DOI] [PubMed] [Google Scholar]
- 24.Leopold G, Ungethum W, Groll E, Diekmann HW, Nowak H, Wegner DH. Clinical pharmacology in normal volunteers of praziquantel, a new drug against schistosomes and cestodes. An example of a complex study covering both tolerance and pharmacokinetics. Eur J Clin Pharmacol. 1978;14:281–91. doi: 10.1007/BF00560463. [DOI] [PubMed] [Google Scholar]
- 25.Caffrey CR. Chemotherapy of schistosomiasis: present and future. Curr Opin Chem Biol. 2007;11:433–9. doi: 10.1016/j.cbpa.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 26.Kohn AB, Anderson PA, Roberts-Misterly JM, Greenberg RM. Schistosome calcium channel beta subunits. Unusual modulatory effects and potential role in the action of the antischistosomal drug praziquantel. J Biol Chem. 2001;276:36873–6. doi: 10.1074/jbc.C100273200. [DOI] [PubMed] [Google Scholar]
- 27.Kohn AB, Roberts-Misterly JM, Anderson PA, Khan N, Greenberg RM. Specific sites in the Beta Interaction Domain of a schistosome Ca2+ channel beta subunit are key to its role in sensitivity to the anti-schistosomal drug praziquantel. Parasitology. 2003;127:349–56. doi: 10.1017/s003118200300386x. [DOI] [PubMed] [Google Scholar]
- 28.Pica-Mattoccia L, Orsini T, Basso A, et al. Schistosoma mansoni: lack of correlation between praziquantel-induced intra-worm calcium influx and parasite death. Exp Parasitol. 2008;119:332–5. doi: 10.1016/j.exppara.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Angelucci F, Basso A, Bellelli A, Brunori M, Pica Mattoccia L, Valle C. The anti-schistosomal drug praziquantel is an adenosine antagonist. Parasitology. 2007;134:1215–21. doi: 10.1017/S0031182007002600. [DOI] [PubMed] [Google Scholar]
- 30.Doenhoff MJ, Cioli D, Utzinger J. Praziquantel: mechanisms of action, resistance and new derivatives for schistosomiasis. Curr Opin Infect Dis. 2008;21:659–67. doi: 10.1097/QCO.0b013e328318978f. [DOI] [PubMed] [Google Scholar]
- 31.Bruce JI, Dias LC, Liang YS, Coles GC. Drug resistance in schistosomiasis: a review. Mem Inst Oswaldo Cruz. 1987;82 (Suppl 4):143–50. doi: 10.1590/s0074-02761987000800025. [DOI] [PubMed] [Google Scholar]
- 32.Coles GC, Mutahi WT, Kinoti GK, Bruce JI, Katz N. Tolerance of Kenyan Schistosoma mansoni to oxamniquine. Trans R Soc Trop Med Hyg. 1987;81:782–5. doi: 10.1016/0035-9203(87)90032-0. [DOI] [PubMed] [Google Scholar]
- 33.Pica-Mattoccia L, Carlini D, Guidi A, Cimica V, Vigorosi F, Cioli D. The schistosome enzyme that activates oxamniquine has the characteristics of a sulfotransferase. Mem Inst Oswaldo Cruz. 2006;101 (Suppl 1):307–12. doi: 10.1590/s0074-02762006000900048. [DOI] [PubMed] [Google Scholar]
- 34.Mullner A, Helfer A, Kotlyar D, Oswald J, Efferth T. Chemistry and pharmacology of neglected helminthic diseases. Curr Med Chem. 2011;18:767–89. doi: 10.2174/092986711794480096. [DOI] [PubMed] [Google Scholar]
- 35.Bueding E, Liu CL, Rogers SH. Inhibition by metrifonate and dichlorvos of cholinesterases in schistosomes. Br J Pharmacol. 1972;46:480–7. doi: 10.1111/j.1476-5381.1972.tb08145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denham DA, Holdsworth RJ. The effect of metrifonate in vitro on Schistosoma haematobium and s. mansoni adults. Trans R Soc Trop Med Hyg. 1971;65:696. doi: 10.1016/0035-9203(71)90070-8. [DOI] [PubMed] [Google Scholar]
- 37.O’Leary KA, Tracy JW. Schistosoma mansoni: glutathione S-transferase-catalyzed detoxication of dichlorvos. Exp Parasitol. 1991;72:355–61. doi: 10.1016/0014-4894(91)90081-7. [DOI] [PubMed] [Google Scholar]
- 38.Rosi D, Peruzzotti G, Dennis EW, et al. Hycanthone, a new active metabolite of lucanthone. J Med Chem. 1967;10:867–76. doi: 10.1021/jm00317a025. [DOI] [PubMed] [Google Scholar]
- 39.Archer S, el-Hamouly W, Seyed-Mozaffari A, Butler RH, Pica-Mattoccia L, Cioli D. Mode of action of the schistosomicide hycanthone: site of DNA alkylation. Mol Biochem Parasitol. 1990;43:89–95. doi: 10.1016/0166-6851(90)90133-7. [DOI] [PubMed] [Google Scholar]
- 40.Pica-Mattoccia L, Archer S, Cioli D. Hycanthone resistance in schistosomes correlates with the lack of an enzymatic activity which produces the covalent binding of hycanthone to parasite macromolecules. Mol Biochem Parasitol. 1992;55:167–75. doi: 10.1016/0166-6851(92)90137-9. [DOI] [PubMed] [Google Scholar]
- 41.Hartman PE, Levine K, Hartman Z, Berger H. Hycanthone: a frameshift mutagen. Science. 1971;172:1058–60. doi: 10.1126/science.172.3987.1058. [DOI] [PubMed] [Google Scholar]
- 42.Rogers SH, Bueding E. Hycanthone resistance: development in Schistosoma mansoni. Science. 1971;172:1057–8. doi: 10.1126/science.172.3987.1057. [DOI] [PubMed] [Google Scholar]
- 43.de Souza Dias LC, de Jesus Pedro R, Deberaldini ER. Use of praziquantel in patients with schistosomiasis mansoni previously treated with oxamniquine and/or hycanthone: resistance of Schistosoma mansoni to schistosomicidal agents. Trans R Soc Trop Med Hyg. 1982;76:652–9. doi: 10.1016/0035-9203(82)90235-8. [DOI] [PubMed] [Google Scholar]
- 44.Cioli D, Pica-Mattoccia L, Moroni R. Schistosoma mansoni: hycanthone/oxamniquine resistance is controlled by a single autosomal recessive gene. Exp Parasitol. 1992;75:425–32. doi: 10.1016/0014-4894(92)90255-9. [DOI] [PubMed] [Google Scholar]
- 45.Pica-Mattoccia L, Dias LC, Moroni R, Cioli D. Schistosoma mansoni: genetic complementation analysis shows that two independent hycanthone/oxamniquine-resistant strains are mutated in the same gene. Exp Parasitol. 1993;77:445–9. doi: 10.1006/expr.1993.1104. [DOI] [PubMed] [Google Scholar]
- 46.Xiao SH, Keiser J, Chen MG, Tanner M, Utzinger J. Research and development of antischistosomal drugs in the People’s Republic of China a 60-year review. Adv Parasitol. 2010;73:231–95. doi: 10.1016/S0065-308X(10)73009-8. [DOI] [PubMed] [Google Scholar]
- 47.Golenser J, Waknine JH, Krugliak M, Hunt NH, Grau GE. Current perspectives on the mechanism of action of artemisinins. Int J Parasitol. 2006;36:1427–41. doi: 10.1016/j.ijpara.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 48.Xiao S, Utzinger J, Chollet J, Endriss Y, N’Goran EK, Tanner M. Effect of artemether against Schistosoma haematobium in experimentally infected hamsters. Int J Parasitol. 2000;30:1001–6. doi: 10.1016/s0020-7519(00)00091-6. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y, Xiao S, Tanner M, et al. Histopathological changes in juvenile Schistosoma haematobium harboured in hamsters treated with artemether. Acta Trop. 2001;79:135–41. doi: 10.1016/s0001-706x(01)00069-9. [DOI] [PubMed] [Google Scholar]
- 50.Utzinger J, N’Goran EK, N’Dri A, Lengeler C, Xiao S, Tanner M. Oral artemether for prevention of Schistosoma mansoni infection: randomised controlled trial. Lancet. 2000;355:1320–5. doi: 10.1016/s0140-6736(00)02114-0. [DOI] [PubMed] [Google Scholar]
- 51.N’Goran EK, Utzinger J, Gnaka HN, et al. Randomized, double-blind, placebo-controlled trial of oral artemether for the prevention of patent Schistosoma haematobium infections. Am J Trop Med Hyg. 2003;68:24–32. [PubMed] [Google Scholar]
- 52.Obonyo CO, Muok EM, Mwinzi PN. Efficacy of artesunate with sulfalene plus pyrimethamine versus praziquantel for treatment of Schistosoma mansoni in Kenyan children: an open-label randomised controlled trial. Lancet Infect Dis. 2010;10:603–11. doi: 10.1016/S1473-3099(10)70161-4. [DOI] [PubMed] [Google Scholar]
- 53.Utzinger J, Xiao SH, Tanner M, Keiser J. Artemisinins for schistosomiasis and beyond. Curr Opin Investig Drugs. 2007;8:105–16. [PubMed] [Google Scholar]
- 54.Boulanger D, Dieng Y, Cisse B, et al. Antischistosomal efficacy of artesunate combination therapies administered as curative treatments for malaria attacks. Trans R Soc Trop Med Hyg. 2007;101:113–6. doi: 10.1016/j.trstmh.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 55.Xiao S, Shen B, Chollet J, Utzinger J, Tanner M. Tegumental changes in adult Schistosoma mansoni harbored in mice treated with artemether. J Parasitol. 2000;86:1125–32. doi: 10.1645/0022-3395(2000)086[1125:TCIASM]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 56.Xiao SH, Wu YL, Tanner M, et al. Schistosoma japonicumIn vitro effects of artemether combined with haemin depend on cultivation media and appraisal of artemether products appearing in the media. Parasitol Res. 2003;89:459–66. doi: 10.1007/s00436-002-0786-1. [DOI] [PubMed] [Google Scholar]
- 57.Bueding E, Fisher J. Factors affecting the inhibition of phosphofructokinase activity of Schistosoma mansoni by trivalent organic antimonials. Biochem Pharmacol. 1966;15:1197–211. doi: 10.1016/0006-2952(66)90285-1. [DOI] [PubMed] [Google Scholar]
- 58.Kuntz AN, Davioud-Charvet E, Sayed AA, et al. Thioredoxin glutathione reductase from Schistosoma mansoni: an essential parasite enzyme and a key drug target. PLoS Med. 2007;4:e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tracy JW, Catto BA, Webster LT., Jr Reductive metabolism of niridazole by adult Schistosoma mansoni Correlation with covalent drug binding to parasite macromolecules. Mol Pharmacol. 1983;24:291–9. [PubMed] [Google Scholar]
- 60.Harder A. Chemotherapeutic approaches to schistosomes: current knowledge and outlook. Parasitol Res. 2002;88:395–7. doi: 10.1007/s00436-001-0588-x. [DOI] [PubMed] [Google Scholar]
- 61.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 62.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–28. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 63.Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 64.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 65.Arrigo AP. Gene expression and the thiol redox state. Free Radic Biol Med. 1999;27:936–44. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 66.Loria P, Miller S, Foley M, Tilley L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem J. 1999;339 (Pt 2):363–70. [PMC free article] [PubMed] [Google Scholar]
- 67.de Villiers KA, Egan TJ. Recent advances in the discovery of haem-targeting drugs for malaria and schistosomiasis. Molecules. 2009;14:2868–87. doi: 10.3390/molecules14082868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Capron A, Dessaint JP, Capron M, Ouma JH, Butterworth AE. Immunity to schistosomes: progress toward vaccine. Science. 1987;238:1065–72. doi: 10.1126/science.3317823. [DOI] [PubMed] [Google Scholar]
- 69.Callahan HL, Crouch RK, James ER. Helminth anti-oxidant enzymes: a protective mechanism against host oxidants? Parasitol Today. 1988;4:218–25. doi: 10.1016/0169-4758(88)90162-7. [DOI] [PubMed] [Google Scholar]
- 70.Loverde PT. Do antioxidants play a role in schistosome host-parasite interactions? Parasitol Today. 1998;14:284–9. doi: 10.1016/s0169-4758(98)01261-7. [DOI] [PubMed] [Google Scholar]
- 71.Mkoji GM, Smith JM, Prichard RK. Antioxidant systems in Schistosoma mansoni: correlation between susceptibility to oxidant killing and the levels of scavengers of hydrogen peroxide and oxygen free radicals. Int J Parasitol. 1988;18:661–6. doi: 10.1016/0020-7519(88)90101-4. [DOI] [PubMed] [Google Scholar]
- 72.Sayed AA, Cook SK, Williams DL. Redox balance mechanisms in Schistosoma mansoni rely on peroxiredoxins and albumin and implicate peroxiredoxins as novel drug targets. J Biol Chem. 2006;281:17001–10. doi: 10.1074/jbc.M512601200. [DOI] [PubMed] [Google Scholar]
- 73.Nare B, Smith JM, Prichard RK. Schistosoma mansoni: levels of antioxidants and resistance to oxidants increase during development. Exp Parasitol. 1990;70:389–97. doi: 10.1016/0014-4894(90)90122-s. [DOI] [PubMed] [Google Scholar]
- 74.Mei H, LoVerde PT. Schistosoma mansoni: the developmental regulation and immunolocalization of antioxidant enzymes. Exp Parasitol. 1997;86:69–78. doi: 10.1006/expr.1997.4150. [DOI] [PubMed] [Google Scholar]
- 75.Johnson F, Giulivi C. Superoxide dismutases and their impact upon human health. Mol Aspects Med. 2005;26:340–52. doi: 10.1016/j.mam.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 76.Hong Z, LoVerde PT, Thakur A, Hammarskjold ML, Rekosh D. Schistosoma mansoni: a Cu/Zn superoxide dismutase is glycosylated when expressed in mammalian cells and localizes to a subtegumental region in adult schistosomes. Exp Parasitol. 1993;76:101–14. doi: 10.1006/expr.1993.1012. [DOI] [PubMed] [Google Scholar]
- 77.Simurda MC, van Keulen H, Rekosh DM, LoVerde PT. Schistosoma mansoni: identification and analysis of an mRNA and a gene encoding superoxide dismutase (Cu/Zn) Exp Parasitol. 1988;67:73–84. doi: 10.1016/0014-4894(88)90010-0. [DOI] [PubMed] [Google Scholar]
- 78.da Silva AC, LePresle T, Capron A, Pierce RJ. Molecular cloning of a 16-kilodalton Cu/Zn superoxide dismutase from Schistosoma mansoni. Mol Biochem Parasitol. 1992;52:275–8. doi: 10.1016/0166-6851(92)90060-w. [DOI] [PubMed] [Google Scholar]
- 79.Hong Z, Kosman DJ, Thakur A, Rekosh D, LoVerde PT. Identification and purification of a second form of Cu/Zn superoxide dismutase from Schistosoma mansoni. Infect Immun. 1992;60:3641–51. doi: 10.1128/iai.60.9.3641-3651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cardoso RM, Silva CH, Ulian de Araujo AP, Tanaka T, Tanaka M, Garratt RC. Structure of the cytosolic Cu,Zn superoxide dismutase from Schistosoma mansoni. Acta Crystallogr D Biol Crystallogr. 2004;60:1569–78. doi: 10.1107/S0907444904016798. [DOI] [PubMed] [Google Scholar]
- 81.Mei H, Hirai H, Tanaka M, Hong Z, Rekosh D, LoVerde PT. Schistosoma mansoni: cloning and characterization of a gene encoding cytosolic Cu/Zn superoxide dismutase. Exp Parasitol. 1995;80:250–9. doi: 10.1006/expr.1995.1031. [DOI] [PubMed] [Google Scholar]
- 82.Cogswell AA, Collins JJ, 3rd, Newmark PA, Williams DL. Whole mount in situ hybridization methodology for Schistosoma mansoni. Mol Biochem Parasitol. 2011;178:46–50. doi: 10.1016/j.molbiopara.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shalaby KA, Yin L, Thakur A, Christen L, Niles EG, LoVerde PT. Protection against Schistosoma mansoni utilizing DNA vaccination with genes encoding Cu/Zn cytosolic superoxide dismutase, signal peptide-containing superoxide dismutase and glutathione peroxidase enzymes. Vaccine. 2003;22:130–6. doi: 10.1016/s0264-410x(03)00535-8. [DOI] [PubMed] [Google Scholar]
- 84.Cook RM, Carvalho-Queiroz C, Wilding G, LoVerde PT. Nucleic acid vaccination with Schistosoma mansoni antioxidant enzyme cytosolic superoxide dismutase and the structural protein filamin confers protection against the adult worm stage. Infect Immun. 2004;72:6112–24. doi: 10.1128/IAI.72.10.6112-6124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carvalho-Queiroz C, Cook R, Wang CC, et al. Cross-reactivity of Schistosoma mansoni cytosolic superoxide dismutase, a protective vaccine candidate, with host superoxide dismutase and identification of parasite-specific B epitopes. Infect Immun. 2004;72:2635–47. doi: 10.1128/IAI.72.5.2635-2647.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jocelyn PC, Kamminga A. The non-protein thiol of rat liver mitochondria. Biochim Biophys Acta. 1974;343:356–62. doi: 10.1016/0304-4165(74)90099-3. [DOI] [PubMed] [Google Scholar]
- 87.Sjoberg L, Eriksen TE, Revesz L. The reaction of the hydroxyl radical with glutathione in neutral and alkaline aqueous solution. Radiat Res. 1982;89:255–63. [PubMed] [Google Scholar]
- 88.Alger HM, Williams DL. The disulfide redox system of Schistosoma mansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Mol Biochem Parasitol. 2002;121:129–39. doi: 10.1016/s0166-6851(02)00031-2. [DOI] [PubMed] [Google Scholar]
- 89.Mkoji GM, Smith JM, Prichard RK. Glutathione redox state, lipid peroxide levels, and activities of glutathione enzymes in oltipraz-treated adult Schistosoma mansoni. Biochem Pharmacol. 1989;38:4307–13. doi: 10.1016/0006-2952(89)90530-3. [DOI] [PubMed] [Google Scholar]
- 90.El-Bassiouni EA, Helmy MH, Abdel-Hamid MA, Shohayeb MA, Ismail SS. In vitro effect of low concentrations of oltipraz on the antioxidant defence of mouse hepatocytes and Schistosoma mansoni worms. Br J Biomed Sci. 2004;61:15–21. doi: 10.1080/09674845.2004.11732640. [DOI] [PubMed] [Google Scholar]
- 91.Nare B, Smith JM, Prichard RK. Mechanisms of inactivation of Schistosoma mansoni and mammalian glutathione S-transferase activity by the antischistosomal drug oltipraz. Biochem Pharmacol. 1992;43:1345–51. doi: 10.1016/0006-2952(92)90512-h. [DOI] [PubMed] [Google Scholar]
- 92.Berriman M, Haas BJ, LoVerde PT, et al. The genome of the blood fluke Schistosoma mansoni. Nature. 2009;460:352–8. doi: 10.1038/nature08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu F, Lu J, Hu W, et al. New perspectives on host-parasite interplay by comparative transcriptomic and proteomic analyses of Schistosoma japonicum. PLoS Pathog. 2006;2:e29. doi: 10.1371/journal.ppat.0020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luersen K, Walter RD, Muller S. The putative gamma-glutamylcysteine synthetase from Plasmodium falciparum contains large insertions and a variable tandem repeat. Mol Biochem Parasitol. 1999;98:131–42. doi: 10.1016/s0166-6851(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 95.Platel DF, Mangou F, Tribouley-Duret J. Role of glutathione in the detoxification of ferriprotoporphyrin IX in chloroquine resistant Plasmodium berghei. Mol Biochem Parasitol. 1999;98:215–23. doi: 10.1016/s0166-6851(98)00170-4. [DOI] [PubMed] [Google Scholar]
- 96.Arrick BA, Griffith OW, Cerami A. Inhibition of glutathione synthesis as a chemotherapeutic strategy for trypanosomiasis. J Exp Med. 1981;153:720–5. doi: 10.1084/jem.153.3.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ribeiro F, Coelho PM, Vieira LQ, Watson DG, Kusel JR. The effect of praziquantel treatment on glutathione concentration in Schistosoma mansoni. Parasitology. 1998;116 (Pt 3):229–36. doi: 10.1017/s0031182097002291. [DOI] [PubMed] [Google Scholar]
- 98.Toppo S, Vanin S, Bosello V, Tosatto SC. Evolutionary and structural insights into the multifaceted glutathione peroxidase (Gpx) superfamily. Antioxid Redox Signal. 2008;10:1501–14. doi: 10.1089/ars.2008.2057. [DOI] [PubMed] [Google Scholar]
- 99.Herbette S, Roeckel-Drevet P, Drevet JR. Seleno-independent glutathione peroxidases. More than simple antioxidant scavengers. FEBS J. 2007;274:2163–80. doi: 10.1111/j.1742-4658.2007.05774.x. [DOI] [PubMed] [Google Scholar]
- 100.Williams DL, Pierce RJ, Cookson E, Capron A. Molecular cloning and sequencing of glutathione peroxidase from Schistosoma mansoni. Mol Biochem Parasitol. 1992;52:127–30. doi: 10.1016/0166-6851(92)90042-i. [DOI] [PubMed] [Google Scholar]
- 101.Maiorino M, Roche C, Kiess M, et al. A selenium-containing phospholipid-hydroperoxide glutathione peroxidase in Schistosoma mansoni. Eur J Biochem. 1996;238:838–44. doi: 10.1111/j.1432-1033.1996.0838w.x. [DOI] [PubMed] [Google Scholar]
- 102.Mei H, Thakur A, Schwartz J, Lo Verde PT. Expression and characterization of glutathione peroxidase activity in the human blood fluke Schistosoma mansoni. Infect Immun. 1996;64:4299–306. doi: 10.1128/iai.64.10.4299-4306.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.LoVerde PT, Carvalho-Queiroz C, Cook R. Vaccination with antioxidant enzymes confers protective immunity against challenge infection with Schistosoma mansoni. Mem Inst Oswaldo Cruz. 2004;99:37–43. doi: 10.1590/s0074-02762004000900007. [DOI] [PubMed] [Google Scholar]
- 104.Dimastrogiovanni D, Anselmi M, Miele AE, et al. Combining crystallography and molecular dynamics: the case of Schistosoma mansoni phospholipid glutathione peroxidase. Proteins. 2010;78:259–70. doi: 10.1002/prot.22536. [DOI] [PubMed] [Google Scholar]
- 105.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 106.Torres-Rivera A, Landa A. Glutathione transferases from parasites: a biochemical view. Acta Trop. 2008;105:99–112. doi: 10.1016/j.actatropica.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 107.Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Precious WY, Barrett J. The possible absence of cytochrome P-450 linked xenobiotic metabolism in helminths. Biochim Biophys Acta. 1989;992:215–22. doi: 10.1016/0304-4165(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 109.Brophy PM, Barrett J. Glutathione transferase in helminths. Parasitology. 1990;100(Pt 2):345–9. doi: 10.1017/s0031182000061369. [DOI] [PubMed] [Google Scholar]
- 110.Cvilink V, Lamka J, Skalova L. Xenobiotic metabolizing enzymes and metabolism of anthelminthics in helminths. Drug Metab Rev. 2009;41:8–26. doi: 10.1080/03602530802602880. [DOI] [PubMed] [Google Scholar]
- 111.O’Leary KA, Tracy JW. Purification of three cytosolic glutathione S-transferases from adult Schistosoma mansoni. Arch Biochem Biophys. 1988;264:1–12. doi: 10.1016/0003-9861(88)90563-2. [DOI] [PubMed] [Google Scholar]
- 112.Wright MD, Harrison RA, Melder AM, Newport GR, Mitchell GF. Another 26-kilodalton glutathione S-transferase of Schistosoma mansoni. Mol Biochem Parasitol. 1991;49:177–9. doi: 10.1016/0166-6851(91)90140-2. [DOI] [PubMed] [Google Scholar]
- 113.O’Leary KA, Hathaway KM, Tracy JW. Schistosoma mansoni: single-step purification and characterization of glutathione S-transferase isoenzyme 4. Exp Parasitol. 1992;75:47–55. doi: 10.1016/0014-4894(92)90121-p. [DOI] [PubMed] [Google Scholar]
- 114.Trottein F, Godin C, Pierce RJ, et al. Inter-species variation of schistosome 28-kDa glutathione S-transferases. Mol Biochem Parasitol. 1992;54:63–72. doi: 10.1016/0166-6851(92)90095-2. [DOI] [PubMed] [Google Scholar]
- 115.Walker J, Crowley P, Moreman AD, Barrett J. Biochemical properties of cloned glutathione S-transferases from Schistosoma mansoni and Schistosoma japonicum. Mol Biochem Parasitol. 1993;61:255–64. doi: 10.1016/0166-6851(93)90071-5. [DOI] [PubMed] [Google Scholar]
- 116.Girardini J, Amirante A, Zemzoumi K, Serra E. Characterization of an omega-class glutathione S-transferase from Schistosoma mansoni with glutaredoxin-like dehydroascorbate reductase and thiol transferase activities. Eur J Biochem. 2002;269:5512–21. doi: 10.1046/j.1432-1033.2002.03254.x. [DOI] [PubMed] [Google Scholar]
- 117.Trottein F, Kieny MP, Verwaerde C, et al. Molecular cloning and tissue distribution of a 26-kilodalton Schistosoma mansoni glutathione S-transferase. Mol Biochem Parasitol. 1990;41:35–44. doi: 10.1016/0166-6851(90)90094-3. [DOI] [PubMed] [Google Scholar]
- 118.Porchet E, McNair A, Caron A, Kusnierz JP, Zemzoumi K, Capron A. Tissue expression of the Schistosoma mansoni 28 kDa glutathione S-transferase. Parasitology. 1994;109:565–72. doi: 10.1017/s0031182000076447. [DOI] [PubMed] [Google Scholar]
- 119.Balloul JM, Sondermeyer P, Dreyer D, et al. Molecular cloning of a protective antigen of schistosomes. Nature. 1987;326:149–153. doi: 10.1038/326149a0. [DOI] [PubMed] [Google Scholar]
- 120.Capron A, Riveau G, Capron M, Trottein F. Schistosomes: the road from host-parasite interactions to vaccines in clinical trials. Trends Parasitol. 2005;21:143–9. doi: 10.1016/j.pt.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 121.Auriault C, Gras-Masse H, Pierce RJ, et al. Antibody response of Schistosoma mansoni-infected human subjects to the recombinant P28 glutathione-S-transferase and to synthetic peptides. J Clin Microbiol. 1990;28:1918–24. doi: 10.1128/jcm.28.9.1918-1924.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boulanger D, Reid GD, Sturrock RF, et al. Immunization of mice and baboons with the recombinant Sm28GST affects both worm viability and fecundity after experimental infection with Schistosoma mansoni. Parasite Immunol. 1991;13:473–90. doi: 10.1111/j.1365-3024.1991.tb00545.x. [DOI] [PubMed] [Google Scholar]
- 123.Grzych JM, De Bont J, Liu J, et al. Relationship of impairment of schistosome 28-kilodalton glutathione S-transferase (GST) activity to expression of immunity to Schistosoma mattheei in calves vaccinated with recombinant Schistosoma bovis 28-kilodalton GST. Infect Immun. 1998;66:1142–8. doi: 10.1128/iai.66.3.1142-1148.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Grzych JM, Grezel D, Xu CB, et al. IgA antibodies to a protective antigen in human Schistosomiasis mansoni. J Immunol. 1993;150:527–35. [PubMed] [Google Scholar]
- 125.Herve M, Angeli V, Pinzar E, et al. Pivotal roles of the parasite PGD2 synthase and of the host D prostanoid receptor 1 in schistosome immune evasion. Eur J Immunol. 2003;33:2764–72. doi: 10.1002/eji.200324143. [DOI] [PubMed] [Google Scholar]
- 126.Vande Waa EA, Campbell CK, O’Leary KA, Tracy JW. Induction of Schistosoma mansoni glutathione S-transferase by xenobiotics. Arch Biochem Biophys. 1993;303:15–21. doi: 10.1006/abbi.1993.1249. [DOI] [PubMed] [Google Scholar]
- 127.Xiao SH, You JQ, Gao HF, et al. Schistosoma japonicum: effect of artemether on glutathione S-transferase and superoxide dismutase. Exp Parasitol. 2002;102:38–45. doi: 10.1016/s0014-4894(02)00145-5. [DOI] [PubMed] [Google Scholar]
- 128.McTigue MA, Williams DR, Tainer JA. Crystal structures of a schistosomal drug and vaccine target: glutathione S-transferase from Schistosoma japonica and its complex with the leading antischistosomal drug praziquantel. J Mol Biol. 1995;246:21–7. doi: 10.1006/jmbi.1994.0061. [DOI] [PubMed] [Google Scholar]
- 129.Milhon JL, Thiboldeaux RL, Glowac K, Tracy JW. Schistosoma japonicum GSH S-transferase Sj26 is not the molecular target of praziquantel action. Exp Parasitol. 1997;87:268–74. doi: 10.1006/expr.1997.4231. [DOI] [PubMed] [Google Scholar]
- 130.Mahajan S, Atkins WM. The chemistry and biology of inhibitors and pro-drugs targeted to glutathione S-transferases. Cell Mol Life Sci. 2005;62:1221–33. doi: 10.1007/s00018-005-4524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Trottein F, Vaney MC, Bachet B, et al. Crystallization and preliminary X-ray diffraction studies of a protective cloned 28 kDa glutathione S-transferase from Schistosoma mansoni. J Mol Biol. 1992;224:515–8. doi: 10.1016/0022-2836(92)91013-f. [DOI] [PubMed] [Google Scholar]
- 132.Rufer AC, Thiebach L, Baer K, Klein HW, Hennig M. X-ray structure of glutathione S-transferase from Schistosoma japonicum in a new crystal form reveals flexibility of the substrate-binding site. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:263–5. doi: 10.1107/S1744309105004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Baiocco P, Gourlay LJ, Angelucci F, et al. Probing the mechanism of GSH activation in Schistosoma haematobium glutathione-S-transferase by site-directed mutagenesis and X-ray crystallography. J Mol Biol. 2006;360:678–89. doi: 10.1016/j.jmb.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 134.Jao SC, Chen J, Yang K, Li WS. Design of potent inhibitors for Schistosoma japonica glutathione S-transferase. Bioorg Med Chem. 2006;14:304–18. doi: 10.1016/j.bmc.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 135.Lo WJ, Chiou YC, Hsu YT, et al. Enzymatic and nonenzymatic synthesis of glutathione conjugates: application to the understanding of a parasite’s defense system and alternative to the discovery of potent glutathione S-transferase inhibitors. Bioconjug Chem. 2007;18:109–20. doi: 10.1021/bc0601727. [DOI] [PubMed] [Google Scholar]
- 136.Yasgar A, Shultz J, Zhou W, et al. A high-throughput 1,536-well luminescence assay for glutathione S-transferase activity. Assay Drug Dev Technol. 2010;8:200–11. doi: 10.1089/adt.2009.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zenk MH. Heavy metal detoxification in higher plants--a review. Gene. 1996;179:21–30. doi: 10.1016/s0378-1119(96)00422-2. [DOI] [PubMed] [Google Scholar]
- 138.Ha SB, Smith AP, Howden R, et al. Phytochelatin synthase genes from Arabidopsis and the yeast Schizosaccharomyces pombe. Plant Cell. 1999;11:1153–64. doi: 10.1105/tpc.11.6.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cobbett C, Goldsbrough P. Phytochelatins and metallothioneins: roles in heavy metal detoxification and homeostasis. Annu Rev Plant Biol. 2002;53:159–82. doi: 10.1146/annurev.arplant.53.100301.135154. [DOI] [PubMed] [Google Scholar]
- 140.Tsuji N, Hirayanagi N, Okada M, et al. Enhancement of tolerance to heavy metals and oxidative stress in Dunaliella tertiolecta by Zn-induced phytochelatin synthesis. Biochem Biophys Res Commun. 2002;293:653–9. doi: 10.1016/S0006-291X(02)00265-6. [DOI] [PubMed] [Google Scholar]
- 141.Pal R, Rai JP. Phytochelatins: peptides involved in heavy metal detoxification. Appl Biochem Biotechnol. 2010;160:945–63. doi: 10.1007/s12010-009-8565-4. [DOI] [PubMed] [Google Scholar]
- 142.Rea PA, Vatamaniuk OK, Rigden DJ. Weeds, worms, and more. Papain’s long-lost cousin, phytochelatin synthase. Plant Physiol. 2004;136:2463–74. doi: 10.1104/pp.104.048579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tsuji N, Nishikori S, Iwabe O, et al. Characterization of phytochelatin synthase-like protein encoded by alr0975 from a prokaryote, Nostoc sp. PCC 7120. Biochem Biophys Res Commun. 2004;315:751–5. doi: 10.1016/j.bbrc.2004.01.122. [DOI] [PubMed] [Google Scholar]
- 144.Clemens S, Schroeder JI, Degenkolb T. Caenorhabditis elegans expresses a functional phytochelatin synthase. Eur J Biochem. 2001;268:3640–3. doi: 10.1046/j.1432-1327.2001.02293.x. [DOI] [PubMed] [Google Scholar]
- 145.Vatamaniuk OK, Bucher EA, Ward JT, Rea PA. A new pathway for heavy metal detoxification in animals. Phytochelatin synthase is required for cadmium tolerance in Caenorhabditis elegans. J Biol Chem. 2001;276:20817–20. doi: 10.1074/jbc.C100152200. [DOI] [PubMed] [Google Scholar]
- 146.Brulle F, Cocquerelle C, Wamalah AN, et al. cDNA cloning and expression analysis of Eisenia fetida (Annelida: Oligochaeta) phytochelatin synthase under cadmium exposure. Ecotoxicol Environ Saf. 2008;71:47–55. doi: 10.1016/j.ecoenv.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 147.Ray D, Williams DL. Characterization of the Phytochelatin Synthase of Schistosoma mansoni. PLoS Negl Trop Dis. 2011;5:e1168. doi: 10.1371/journal.pntd.0001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vivares D, Arnoux P, Pignol D. A papain-like enzyme at work: native and acyl-enzyme intermediate structures in phytochelatin synthesis. Proc Natl Acad Sci U S A. 2005;102:18848–53. doi: 10.1073/pnas.0505833102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Clemens S, Persoh D. Muti-tasking phytochelatin synthases. Plant Sci. 2009;117:266–71. [Google Scholar]
- 150.Precious WY, Barrett J. Xenobiotic metabolism in helminths. Parasitology today (Personal ed) 1989;5:156–60. doi: 10.1016/0169-4758(89)90080-x. [DOI] [PubMed] [Google Scholar]
- 151.Walker J, Barrett J. Parasite sulphur amino acid metabolism. Int J Parasitol. 1997;27:883–97. doi: 10.1016/s0020-7519(97)00039-8. [DOI] [PubMed] [Google Scholar]
- 152.Dass PD, Donahue MJ. gamma-Glutamyl transpeptidase activity in Ascaris suum. Mol Biochem Parasitol. 1986;20:233–6. doi: 10.1016/0166-6851(86)90103-9. [DOI] [PubMed] [Google Scholar]
- 153.Adcock HJ, Brophy PM, Teesdale-Spittle PH, Buckberry LD. Cysteine conjugate beta-lyase activity in three species of parasitic helminth. Int J Parasitol. 1999;29:543–8. doi: 10.1016/s0020-7519(99)00022-3. [DOI] [PubMed] [Google Scholar]
- 154.Blum R, Beck A, Korte A, et al. Function of phytochelatin synthase in catabolism of glutathione-conjugates. Plant J. 2007;49:740–9. doi: 10.1111/j.1365-313X.2006.02993.x. [DOI] [PubMed] [Google Scholar]
- 155.Wunschmann J, Krajewski M, Letzel T, et al. Dissection of glutathione conjugate turnover in yeast. Phytochemistry. 2010;71:54–61. doi: 10.1016/j.phytochem.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 156.Egan TJ. Haemozoin formation. Mol Biochem Parasitol. 2008;157:127–36. doi: 10.1016/j.molbiopara.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 157.Glanfield A, McManus DP, Anderson GJ, Jones MK. Pumping iron: a potential target for novel therapeutics against schistosomes. Trends Parasitol. 2007;23:583–8. doi: 10.1016/j.pt.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jones MK, McManus DP, Sivadorai P, et al. Tracking the fate of iron in early development of human blood flukes. Int J Biochem Cell Biol. 2007;39:1646–58. doi: 10.1016/j.biocel.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kumar C, Igbaria A, D’Autreaux B, et al. Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control. The EMBO journal. 2011;30:2044–56. doi: 10.1038/emboj.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Alger HM, Sayed AA, Stadecker MJ, Williams DL. Molecular and enzymatic characterisation of Schistosoma mansoni thioredoxin. Int J Parasitol. 2002;32:1285–92. doi: 10.1016/s0020-7519(02)00108-x. [DOI] [PubMed] [Google Scholar]
- 161.Boumis G, Angelucci F, Bellelli A, Brunori M, Dimastrogiovanni D, Erica Miele A. Structural and functional characterization of Schistosoma mansoni thioredoxin. Protein Sci. 2011;2011:1069–76. doi: 10.1002/pro.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Huang HH, Ballou DP, Williams CH, Williams DL. The catalytic mechanism of the glutaredoxin domain of thioredoxin glutathione reductase from Schistosoma mansoni. Proceedings of 17th International Symposium on Flavins and Flavoproteins; 2011 July 24–29; Berkeley, CA, USA. (In press) [Google Scholar]
- 163.Kanzok SM, Fechner A, Bauer H, et al. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science. 2001;291:643–6. doi: 10.1126/science.291.5504.643. [DOI] [PubMed] [Google Scholar]
- 164.Bauer H, Gromer S, Urbani A, Schnolzer M, Schirmer RH, Muller HM. Thioredoxin reductase from the malaria mosquito Anopheles gambiae. Eur J Biochem. 2003;270:4272–81. doi: 10.1046/j.1432-1033.2003.03812.x. [DOI] [PubMed] [Google Scholar]
- 165.Salinas G, Selkirk ME, Chalar C, Maizels RM, Fernandez C. Linked thioredoxin-glutathione systems in platyhelminths. Trends Parasitol. 2004;20:340–6. doi: 10.1016/j.pt.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 166.Bonilla M, Denicola A, Marino SM, Gladyshev VN, Salinas G. Linked thioredoxin-glutathione systems in Platyhelminth parasites: Alternative pathways for glutathione reduction and deglutathionylation. J Biol Chem. 2011;286:4959–67. doi: 10.1074/jbc.M110.170761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Watson WH, Pohl J, Montfort WR, et al. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278:33408–15. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- 168.Weichsel A, Gasdaska JR, Powis G, Montfort WR. Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure. 1996;4:735–51. doi: 10.1016/s0969-2126(96)00079-2. [DOI] [PubMed] [Google Scholar]
- 169.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol. 2002;4:743–9. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 170.Sayed AA, Williams DL. Biochemical characterization of 2-Cys peroxiredoxins from Schistosoma mansoni. J Biol Chem. 2004;279:26159–66. doi: 10.1074/jbc.M401748200. [DOI] [PubMed] [Google Scholar]
- 171.Sun QA, Wu Y, Zappacosta F, et al. Redox regulation of cell signaling by selenocysteine in mammalian thioredoxin reductases. J Biol Chem. 1999;274:24522–30. doi: 10.1074/jbc.274.35.24522. [DOI] [PubMed] [Google Scholar]
- 172.Sun QA, Kirnarsky L, Sherman S, Gladyshev VN. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc Natl Acad Sci U S A. 2001;98:3673–8. doi: 10.1073/pnas.051454398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Su D, Novoselov SV, Sun QA, et al. Mammalian selenoprotein thioredoxin-glutathione reductase. Roles in disulfide bond formation and sperm maturation. J Biol Chem. 2005;280:26491–8. doi: 10.1074/jbc.M503638200. [DOI] [PubMed] [Google Scholar]
- 174.Arner ES, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 1999;300:226–39. doi: 10.1016/s0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- 175.Huang HH, Day L, Cass CL, Ballou DP, Williams CH, Williams DL. Investigations on the Catalytic Mechanism of Thioredoxin Glutathione Reductase (TGR) from Schistosoma mansoni. Biochemistry. 2011;50:5870–82. doi: 10.1021/bi200107n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xia L, Nordman T, Olsson JM, et al. The mammalian cytosolic selenoenzyme thioredoxin reductase reduces ubiquinone. A novel mechanism for defense against oxidative stress. J Biol Chem. 2003;278:2141–6. doi: 10.1074/jbc.M210456200. [DOI] [PubMed] [Google Scholar]
- 177.Kalinina EV, Chernov NN, Saprin AN. Involvement of thio-, peroxi-, and glutaredoxins in cellular redox-dependent processes. Biochemistry (Mosc) 2008;73:1493–510. doi: 10.1134/s0006297908130099. [DOI] [PubMed] [Google Scholar]
- 178.Rendon JL, del Arenal IP, Guevara-Flores A, Uribe A, Plancarte A, Mendoza-Hernandez G. Purification, characterization and kinetic properties of the multifunctional thioredoxin-glutathione reductase from Taenia crassiceps metacestode (cysticerci) Mol Biochem Parasitol. 2004;133:61–9. doi: 10.1016/j.molbiopara.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 179.Sun QA, Su D, Novoselov SV, Carlson BA, Hatfield DL, Gladyshev VN. Reaction mechanism and regulation of mammalian thioredoxin/glutathione reductase. Biochemistry. 2005;44:14528–37. doi: 10.1021/bi051321w. [DOI] [PubMed] [Google Scholar]
- 180.Bonilla M, Denicola A, Novoselov SV, et al. Platyhelminth mitochondrial and cytosolic redox homeostasis is controlled by a single thioredoxin glutathione reductase and dependent on selenium and glutathione. J Biol Chem. 2008;283:17898–907. doi: 10.1074/jbc.M710609200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Angelucci F, Miele AE, Boumis G, Dimastrogiovanni D, Brunori M, Bellelli A. Glutathione reductase and thioredoxin reductase at the crossroad: the structure of Schistosoma mansoni thioredoxin glutathione reductase. Proteins. 2008;72:936–45. doi: 10.1002/prot.21986. [DOI] [PubMed] [Google Scholar]
- 182.Angelucci F, Dimastrogiovanni D, Boumis G, et al. Mapping the catalytic cycle of Schistosoma mansoni thioredoxin glutathione reductase by x-ray crystallography. J Biol Chem. 2010;285:32557–67. doi: 10.1074/jbc.M110.141960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Prast-Nielsen S, Huang HH, Williams DL. Thioredoxin-glutathione reductase: Its role in redox biology and potential as a target for drugs against neglected diseases. Biochim Biophys Acta. 2011;1810:1262–71. doi: 10.1016/j.bbagen.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Luthman M, Holmgren A. Rat liver thioredoxin and thioredoxin reductase: purification and characterization. Biochemistry. 1982;21:6628–33. doi: 10.1021/bi00269a003. [DOI] [PubMed] [Google Scholar]
- 185.Sharma M, Khanna S, Bulusu G, Mitra A. Comparative modeling of thioredoxin glutathione reductase from Schistosoma mansoni: a multifunctional target for antischistosomal therapy. J Mol Graph Model. 2009;27:665–75. doi: 10.1016/j.jmgm.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 186.Guevara-Flores A, Pardo JP, Rendon JL. Hysteresis in thioredoxin-glutathione reductase (TGR) from the adult stage of the liver fluke Fasciola hepatica. Parasitol Int. 2011;60:156–60. doi: 10.1016/j.parint.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 187.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem. 1998;273:20096–101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 188.Rigobello MP, Scutari G, Folda A, Bindoli A. Mitochondrial thioredoxin reductase inhibition by gold(I) compounds and concurrent stimulation of permeability transition and release of cytochrome c. Biochem Pharmacol. 2004;67:689–96. doi: 10.1016/j.bcp.2003.09.038. [DOI] [PubMed] [Google Scholar]
- 189.Angelucci F, Sayed AA, Williams DL, et al. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: structural and kinetic aspects. J Biol Chem. 2009;284:28977–85. doi: 10.1074/jbc.M109.020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Smith AD, Guidry CA, Morris VC, Levander OA. Aurothioglucose inhibits murine thioredoxin reductase activity in vivo. J Nutr. 1999;129:194–8. doi: 10.1093/jn/129.1.194. [DOI] [PubMed] [Google Scholar]
- 191.Cenas N, Nivinskas H, Anusevicius Z, Sarlauskas J, Lederer F, Arner ES. Interactions of quinones with thioredoxin reductase: a challenge to the antioxidant role of the mammalian selenoprotein. J Biol Chem. 2004;279:2583–92. doi: 10.1074/jbc.M310292200. [DOI] [PubMed] [Google Scholar]
- 192.Gromer S, Urig S, Becker K. The thioredoxin system--from science to clinic. Med Res Rev. 2004;24:40–89. doi: 10.1002/med.10051. [DOI] [PubMed] [Google Scholar]
- 193.Witte AB, Anestal K, Jerremalm E, Ehrsson H, Arner ES. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic Biol Med. 2005;39:696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 194.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–93. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Inglese J, Auld DS, Jadhav A, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103:11473–8. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lea WA, Jadhav A, Rai G, et al. A 1,536-well-based kinetic HTS assay for inhibitors of Schistosoma mansoni thioredoxin glutathione reductase. Assay Drug Dev Technol. 2008;6:551–5. doi: 10.1089/adt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Simeonov A, Jadhav A, Sayed AA, et al. Quantitative high-throughput screen identifies inhibitors of the Schistosoma mansoni redox cascade. PLoS Negl Trop Dis. 2008;2:e127. doi: 10.1371/journal.pntd.0000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sayed AA, Simeonov A, Thomas CJ, Inglese J, Austin CP, Williams DL. Identification of oxadiazoles as new drug leads for the control of schistosomiasis. Nat Med. 2008;14:407–12. doi: 10.1038/nm1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rai G, Sayed AA, Lea WA, et al. Structure mechanism insights and the role of nitric oxide donation guide the development of oxadiazole-2-oxides as therapeutic agents against schistosomiasis. J Med Chem. 2009;52:6474–83. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kwatia MA, Botkin DJ, Williams DL. Molecular and enzymatic characterization of Schistosoma mansoni thioredoxin peroxidase. J Parasitol. 2000;86:908–15. doi: 10.1645/0022-3395(2000)086[0908:MAECOS]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 201.Rhee SG, Kang SW, Chang TS, Jeong W, Kim K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life. 2001;52:35–41. doi: 10.1080/15216540252774748. [DOI] [PubMed] [Google Scholar]
- 202.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–52. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 203.Harris JR. Some negative contrast staining features of a protein from erythrocyte ghosts. J Mol Biol. 1969;46:329–35. doi: 10.1016/0022-2836(69)90425-2. [DOI] [PubMed] [Google Scholar]
- 204.Harris JR, Schroder E, Isupov MN, et al. Comparison of the decameric structure of peroxiredoxin-II by transmission electron microscopy and X-ray crystallography. Biochim Biophys Acta. 2001;1547:221–34. doi: 10.1016/s0167-4838(01)00184-4. [DOI] [PubMed] [Google Scholar]
- 205.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 206.Jang HH, Lee KO, Chi YH, et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–35. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 207.Moon JC, Hah YS, Kim WY, et al. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J Biol Chem. 2005;280:28775–84. doi: 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- 208.Chae HZ, Chung SJ, Rhee SG. Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem. 1994;269:27670–8. [PubMed] [Google Scholar]
- 209.Veal E, Day A. Hydrogen peroxide as a signaling molecule. Antioxid Redox Signal. 2011;15:147–51. doi: 10.1089/ars.2011.3968. [DOI] [PubMed] [Google Scholar]
- 210.Woo HA, Chae HZ, Hwang SC, et al. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–6. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 211.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–3. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 212.Kang SW, Chang TS, Lee TH, Kim ES, Yu DY, Rhee SG. Cytosolic peroxiredoxin attenuates the activation of Jnk and p38 but potentiates that of Erk in Hela cells stimulated with tumor necrosis factor-alpha. J Biol Chem. 2004;279:2535–43. doi: 10.1074/jbc.M307698200. [DOI] [PubMed] [Google Scholar]
- 213.Williams DL, Asahi H, Botkin DJ, Stadecker MJ. Schistosome infection stimulates host CD4(+) T helper cell and B-cell responses against a novel egg antigen, thioredoxin peroxidase. Infect Immun. 2001;69:1134–41. doi: 10.1128/IAI.69.2.1134-1141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kumagai T, Osada Y, Kanazawa T. 2-Cys peroxiredoxins from Schistosoma japonicum: the expression profile and localization in the life cycle. Mol Biochem Parasitol. 2006;149:135–43. doi: 10.1016/j.molbiopara.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 215.Kumagai T, Osada Y, Ohta N, Kanazawa T. Peroxiredoxin-1 from Schistosoma japonicum functions as a scavenger against hydrogen peroxide but not nitric oxide. Mol Biochem Parasitol. 2009;164:26–31. doi: 10.1016/j.molbiopara.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 216.Ruan H, Tang XD, Chen ML, et al. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc Natl Acad Sci U S A. 2002;99:2748–53. doi: 10.1073/pnas.032671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kantorow M, Hawse JR, Cowell TL, et al. Methionine sulfoxide reductase A is important for lens cell viability and resistance to oxidative stress. Proc Natl Acad Sci U S A. 2004;101:9654–9. doi: 10.1073/pnas.0403532101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lee BC, Dikiy A, Kim HY, Gladyshev VN. Functions and evolution of selenoprotein methionine sulfoxide reductases. Biochim Biophys Acta. 2009;1790:1471–7. doi: 10.1016/j.bbagen.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Moskovitz J. Roles of methionine suldfoxide reductases in antioxidant defense, protein regulation and survival. Curr Pharm Des. 2005;11:1451–7. doi: 10.2174/1381612053507846. [DOI] [PubMed] [Google Scholar]
- 220.Oke TT, Moskovitz J, Williams DL. Characterization of the methionine sulfoxide reductases of Schistosoma mansoni. J Parasitol. 2009;95:1421–8. doi: 10.1645/GE-2062.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kim HY, Gladyshev VN. Methionine sulfoxide reduction in mammals: characterization of methionine-R-sulfoxide reductases. Mol Biol Cell. 2004;15:1055–64. doi: 10.1091/mbc.E03-08-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kim HY, Gladyshev VN. Characterization of mouse endoplasmic reticulum methionine-R-sulfoxide reductase. Biochem Biophys Res Commun. 2004;320:1277–83. doi: 10.1016/j.bbrc.2004.06.078. [DOI] [PubMed] [Google Scholar]
- 223.Boschi-Muller S, Gand A, Branlant G. The methionine sulfoxide reductases: Catalysis and substrate specificities. Arch Biochem Biophys. 2008;474:266–73. doi: 10.1016/j.abb.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 224.Kumar RA, Koc A, Cerny RL, Gladyshev VN. Reaction mechanism, evolutionary analysis, and role of zinc in Drosophila methionine-R-sulfoxide reductase. J Biol Chem. 2002;277:37527–35. doi: 10.1074/jbc.M203496200. [DOI] [PubMed] [Google Scholar]