

Abstract
Congenital vertebral malformations (CVM) pose a significant health problem because they can be associated with spinal deformities, such as congenital scoliosis and kyphosis, in addition to various syndromes and other congenital malformations. Additional information remains to be learned regarding the natural history of congenital scoliosis and related health problems. Although significant progress has been made in understanding the process of somite formation, which gives rise to vertebral bodies, there is a wide gap in our understanding of how genetic factors contribute to CVM development. Maternal diabetes during pregnancy most commonly contributes to the occurrence of CVM, followed by other factors such as hypoxia and anticonvulsant medications. This review highlights several emerging clinical issues related to CVM, including pulmonary and orthopedic outcome in congenital scoliosis. Recent breakthroughs in genetics related to gene and environment interactions associated with CVM development are discussed. The Klippel-Feil syndrome which is associated with cervical segmentation abnormalities is illustrated as an example in which animal models, such as the zebrafish, can be utilized to provide functional evidence of pathogenicity of identified mutations.
Key Words: Congenital vertebral malformation, Hemifacial microsomia, Klippel-Feil syndrome, Maternal diabetes, Spondylocostal dysostosis, Spondylothoracic dysostosis, Thoracic insufficiency syndrome, VACTERL syndrome
Congenital Vertebral Malformations Definition, Pathogenesis and Epidemiology
Congenital segmentation defects resulting in congenital vertebral malformations (CVM) are etiologically heterogeneous with poorly understood environmental and genetic factors contributing to their occurrence. CVM in humans are associated with significant health problems including kyphosis, scoliosis, neck and back pain, disability, cosmetic disfigurement, pulmonary compromise, and functional distress. Although prior estimates indicate prevalence between 0.13–0.5 1/1000 live births, more recent information indicates that the incidence of CVM in the general population is unknown as many people who are asymptomatic do not present for medical care [Wynne-Davies, 1975; Brand, 2008].
Vertebral malformations shown in figure 1 represent defects of formation, such as a hemivertebrae (half of a vertebrae) butterfly or wedge shaped vertebrae, or defects of segmentation such as a vertebral bar (an abnormality of vertebral separation during development). Vertebral malformations may represent an isolated finding, occur in association with other renal, cardiac or spinal cord malformations, or occur as part of an underlying syndrome or chromosomal abnormality. Frequently encountered syndromes associated with CVM include Klippel-Feil syndrome (short neck, low posterior hairline and fusion of cervical vertebrae), Alagille syndrome (peripheral pulmonic stenosis, cholestasis and facial dysmorphism), spondylocostal dysostosis (short trunk dwarfism, multiple vertebral and rib defects), spondylothoracic dystrophy (short trunk dwarfism, multiple vertebral and rib defects with posterior rib fusion), Goldenhar syndrome (associated with craniofacial anomalies including microtia and epibulbar dermoids), and VACTERL association (Vertebral malformations, Anal atresia, Cardiac malformations, Tracheo-Esophageal fistula, Renal and radial anomalies and Limb defects). A list of representative syndromes is indicated in table 1. A classification scheme for vertebral malformations was recently proposed by members of the International Consortium for Vertebral Anomalies and Scoliosis [Offiah et al., 2010]. Additional studies are needed to determine whether molecular genetic mechanisms can be associated with various CVM phenotypes.
Fig. 1.

Schematic diagram of spine illustrating defects of formation (wedge and hemivertebrae) and segmentation (vertebral bar and block vertebrae). Reprinted with permission from Michael J. McMaster, MD [J R Coll Surg Edinb 2002;47:475–480].
Table 1.
Some syndromes that include segmentation defects of the vertebrae [Reproduced with permission of Informa UK Ltd. from Giampietro et al., 2008]
| Syndrome | OMIM | Gene(s) |
|---|---|---|
| Acrofacial dysostosis* | 263750 | |
| Aicardi* | 304050 | |
| Alagille | 118450 | JAGGED1, NOTCH2 |
| Anhalt* | 601344 | |
| Atelosteogenesis III | 108721 | FLNB |
| Campomelic dysplasia | 211970 | SOX9 |
| Casamassima-Morton-Nance* | 271520 | |
| Caudal regression* | 182940 | |
| Cerebro-facio-thoracic dysplasia* | 213980 | |
| CHARGE | 214800 | CHD7 |
| ‘Chromosomal’ | ||
| Currarino | 176450 | HLXB9 |
| DeLa Chapelle* | 256050 | |
| DeGeorge/Sedlackova | 188400 | Microdeletion, 22q11.2, 10p14–p13 |
| Dysspondylochondromatosis* | ||
| Femoral hypoplasia-unusual facies* | 134780 | |
| Fibrodysplasia ossificans progressive | 135100 | ACVR1 |
| Fryns-Moerman* | ||
| Goldenhar* | 164210 | |
| (Oculo-auriculo-vertebral spectrum) | ||
| Holmes-Schimke* | 308310 | |
| Incontinentia Pigmenti | 308300 | NEMO |
| Kabuki* | 147920 | MLL2 |
| Kaufman-McKusick | 236700 | MKKS |
| KBG Syndrome* | 148050 | |
| Klippel-Feil* | 148900 | ?PAXl, GDF6 |
| Larsen | 150250 | FLNB |
| Lower mesodermal agenesis* | ||
| Maternal diabetes* | ||
| MURCS Association* | 601076 | |
| Multiple Pterygium Syndrome | 265000 | CHRNG |
| OEIS Syndrome* | 258040 | |
| Phaver* | 261575 | |
| Rapadilino | 266280 | RECQL4 |
| Robinow | 268310 | ROR2 |
| Rolland-Desbuquois* | 224400 | |
| Rokitansky Sequence* | 277000 | ?WNT4 |
| Silverman | 224410 | HSPG2 |
| Simpson-Golabi-Behmel | 312870 | GPC3 |
| Sirenomelia* | 182940 | |
| Spondylocarpotarsal Synostosis | 272460 | FLNB |
| Spondylocostal Dysostosis | 277300 | DLL3, MESP2, LFNG |
| Spondylothoracic Dysotosis* | 277300 | MESP2 |
| Thakker-Donnai* | 227255 | |
| Toriello* | ||
| Urioste* | ||
| VATER/VACTERL* | 192350 | |
| Verloove-Vanhorick* | 215850 | |
| Wildevanck* | 314600 | |
| Zimmer* | 301090 |
Underlying cause not known.
Vertebral bodies are derived from somites through a repetitive process of budding off from the presomitic mesoderm mediated by a cyclical process involving the Wnt, FGF and Notch signaling pathways. A ‘clock and wavefront’ model for somitogenesis was originally proposed to account for somitogenesis [Cooke and Zeeman, 1976]. The ‘clock’ represents an oscillator connecting presomitic mesodermal cells, and the ‘wave’ represents a region of ‘rapid cellular change’ resulting in transition to somite development occurs, mediated by some type of gradient. The process of somitogenesis is illustrated in figure 2. Disruption of this process as further described below can result in CVM occurrence.
Fig. 2.

Illustration of the process of somite formation from the presomitic mesoderm (PSM) in the chick embryo. a Paired somites are formed in a periodic fashion every 90 minutes. b A molecular clocked linked to segmentation is identified by dynamic and periodic expression of the cyclic genes in the PSM. Top: The expression of Lunatic Fringe mRNA appears as a wave sweeping across the whole PSM once during each somite formation as illustrated by in situ hybridization in this 17-somite-old chick embryo. PSM cells as illustrated by the dot undergo a phase of upregulation of the cycling genes followed by a phase of downregulation of these genes each time a somite forms. Bottom: As shown in this schematic representation of the progression of somitogenesis in the embryo, the cycles of expression of the cyclic genes will last while the cells remain in the PSM, which corresponds approximately to the time to form 12 somites in the chick embryo. These PSM cells undergo 12 oscillations of the expression of the cycling genes. Reproduced with permission from Giampietro et al., 2008.
Natural History and Alteration by Surgical Management
The risk of developing scoliosis and its subsequent severity is a function of several factors including the type, number and location of CVM [McMaster and Ohtsuka, 1982]. Defects of formation including block, wedge and simple hemivertebrae (incarcerated, unsegmented) have a low risk for curve progression. Semi-/fully segmented multi hemivertebrae have a greater likelihood of progression. Mixed defects such as a unilateral unsegmented bar and contralateral hemivertebrae tend to be localized to the thoracic region and have a more aggressive course with respect to curve progression. Little prognostic information is available for complex mixed pattern types of anomalies, which is mainly due to the scarcity of published systematic analyses of follow-up data.
The presence of rib abnormalities in addition to vertebral abnormalities compromises development and growth of the lungs. Patients with congenital scoliosis were originally surgically treated by fusion procedures which resulted in a short trunk with minimal spinal curvature. Scoliosis related thoracic insufficiency syndrome or ‘the inability of the thorax to support normal respiration or lung growth’ has been a consequence of surgical fusion procedures to treat congenital scoliosis [Campbell et al., 2003]. In one retrospective study of 21 patients with congenital scoliosis who underwent spinal fusion prior to the age of 10 years, forced vital capacity (FVC), forced expiratory volume (FEV1), vital capacity, and total lung capacity were significantly lower as compared to healthy children [Vitale et al., 2008]. One study investigated pulmonary function in a cohort of 28 patients with scoliosis who had surgical fusion procedures prior to the age of 9 years [Karol et al., 2008]. This group had an average predicted FVC of 48.2% (range 27–86%) with 10 patients having severe restrictive lung disease as evidenced by a FVC of <50%. Eight patients between the ages of 12.4–22.8 years with thoracic heights between 18 and 22 cm had an average predicted FVC of 63% (86%), with 10 patients having severe restrictive lung disease as evidenced by a FVC of <50% (range 42–99%) including 2 with a FVC of <50%. Four patients ranging in age between 13.6 and 19.1 years, with thoracic heights measuring between 22 and 28 cm had an average FVC of 85.2% (range 80–91%), with none having a FVC of <50%. These results indicated that a shorter thoracic spine is associated with a lower FVC and a greater risk for pulmonary restrictive disease. While some surgeons use a thoracic spine measurement of 22 cm or greater as a cut-off for performing scoliosis surgery, there are no studies which demonstrate the clinical importance of this in the unfused congenital scoliotic spine.
Alternatives to fusion procedures include ‘growing rods’ or ‘vertical expandable prosthetic titanium rib’ which allow the spine and thorax to grow. Disadvantages to ‘growing rod’ treatment include the need for multiple surgeries to allow for periodic lengthening of the growing spine, imposing an economic and a psychological burden on families [Akbarnia and Emans, 2010]. Complications include rod fracture, skin breakage, wound complication, malalignment, and brachial plexus problems with an observed complication rate of 19% per procedure [Bess et al., 2010]. Optimizing preoperative nutrition, careful choice of the optimal surgical technique and careful soft tissue handling technique can help to minimize these complications. Thus far, it appears that the final results of treatment are a function of the underlying diagnosis, condition of the spine and chest wall, and the instrumentation used. Prospective studies are needed to obtain prognoses regarding the spine at the end of treatment.
Parental Risk Factors Associated with CVM
Occurrence of CVM in animal models and humans has been associated with various maternal exposures during pregnancy, including alcohol use [Treadwell et al., 1982], anticonvulsant medications including valproic acid [Vorhees, 1987; Menegola et al., 1996; Holmes et al., 2001], hyperthermia [Breen et al., 1999], maternal insulin-dependent diabetes mellitus, and gestational diabetes [Passarge and Lenz, 1966; Aberg et al., 2001; Martínez-Frías et al., 1998]. Exposure to phenytoin during pregnancy has been associated with CVM in mice [Loughnan et al., 1973]. Single nucleotide polymorphisms (SNPs) in GLUT1 and other glucose metabolizing genes such as HK1 and LEPR are hypothesized to be associated with patterns of malformations observed in diabetic embryopathy. Reactive oxygen species has been proposed as a mechanism for altered somitogenesis in infants of diabetic mothers [Alexander and Tuan, 2010]. CVM have been observed in laboratory animals exposed to I (Kr)-blockers (class III anti-arrhythmic agent), fumonisins (environmental toxins produced Fusarium moniliforme, F. verticilliodes, F. proliferatum, and other Fusarium species of molds), zinc deficient diet, as well as the organophosphate pesticide chlopyrifos, during pregnancy [Hickory et al., 1979; Sköld et al., 2001; Tian et al., 2005]. Exposure of juvenile fourhorn sculpin, Myoxocephalus quadricornis L. to tetrachloro-1,2-benzoquinone, a component in bleached kraft mill effluents, resulted in fish with vertebral deformities and abnormal mechanical vertebral properties [Bengtsson et al., 1988].
Alterations in HOX-mediated gene expression, which is important for vertebral positional specificity, are mediated by exposure to carbon monoxide [Farley et al., 2001] and boric acid [Wéry et al., 2003]. Retinoic acid, a vitamin A analogue, has been observed to cause homeotic transformations in mice and axial skeletal truncation [Owen et al., 2009]. Inhibition of nitric oxide production or addition of nitric oxide to developing chick embryos results in increased axial skeletal defects and areas correlated with apoptosis [Alexander et al., 2007]. Cigarette smoking during pregnancy has been reported to be associated with low birth weight, decreases in successive births and behavioral deficits that can be replicated by carbon monoxide alone in animal models [Fichtner et al., 1990; Bnait and Seller, 1995]. Anoxic damage to somites and the generation of reactive oxygen species by cigarette smoke could potentially contribute to the development of CVM.
Congenital scoliosis has been reported to occur in monozygotic twins [Kaspiris et al., 2008]. Monozygotic and dizygotic twinning is associated with an increased risk for congenital malformations [Corsello and Piro, 2010]. Assisted reproductive technology (ART) is linked with occurrence of congenital malformations and syndromes including Prader-Willi, Angelman and Beckwith-Wiedemann syndromes [Niemitz and Feinberg, 2004]. Epigenetic factors including methyl donor content of the growth media have been suggested as a possible mechanism of their occurrence in ART pregnancies, which is consistent with observations that nutritional factors have been implicated for their occurrence in non-ART pregnancies. The relatively sporadic nature of CVM, similar to other birth defects, makes epigenetic factors another plausible mechanism for investigation. Little information exists regarding the relative contribution of environmental factors to the development of CVM and congenital scoliosis.
Nonteratogentic doses of heat (<2°C) result in the recruitment of heat shock proteins which provide protection for proteins against damage by teratogenic doses of heat (>2°C). This is accomplished through the attachment to uncovered active sites, thus preventing their binding with other functionally impaired aggregate proteins [Bennett, 2010]. The exact mechanism responsible for altered somitogenesis associated with heat is uncertain. Hyperthermia inhibits the cell cycle and induces apoptosis. Hyperthermia may alter Notch/Delta signaling pathway proteins and result in abnormal vertebral patterning.
There are no reported studies which describe the relative contribution of maternal exposures during pregnancy to CVM development. In a series of 206, 244 live births, still births and elective terminations, a total of 5 cases of isolated hemivertebrae and 22 cases of hemivertebrae with other birth defects were identified [Holmes, 2012]. The most common maternal exposure associated with CVM was maternal diabetes (5 cases) followed by twinning (2 cases). An odds ratio of 12.8 (95% CI, 4.4–36.6; p < 0.001) for maternal risk factors including maternal insulin-dependent diabetes mellitus, valproic acid, alcohol, smoking, hyperthermia, twin gestation, ART, and in vitro fertilization during pregnancy was obtained on a pilot retrospective chart review by our multicenter group consisting of 228 cases with CVM between the ages of 0 and 50 years and 268 controls with normal spine morphology and minimal scoliosis (unpublished data). These results support a hypothesis for an association between the above maternal exposure factors during pregnancy and CVM and have implications for development of prevention strategies. Further prospective studies are needed to quantify association between CVM and specific maternal exposure factors.
Classification of CVM
A simplified and unified classification scheme for CVM is essential for clinicians and researchers to describe individual and collective CVM from both a phenotypic and genetic etiologic vantage point. A number of classification schemes for CVM have been proposed which have individually emphasized different components associated with CVM occurrence including mode of inheritance [Mortier et al., 1996], developmental mechanism associated with CVM [Aburakawa et al., 1996; Takikawa et al., 2006] and syndromic diagnosis of CVM (i.e. spondylocostal dysostosis, Klippel-Feil, etc.) [Klippel and Feil, 1912; Thomsen et al., 1997; Takikawa, et al., 2006]. A pilot classification system for CVM has recently been proposed by the International Consortium for Vertebral Anomalies and Scoliosis and is outlined algorithmically in figure 3 [Offiah et al., 2010]. A vertebral segmentation defect (VSD) may be defined as single (SVSD) or multiple (MVSD). A SVSD can be associated with a known syndrome such as hemifacial microsomia or VACTERL association. MVSD can be generalized in which 10 or greater contiguous vertebral bodies are involved, representing a defined phenotype such as spondylocostal dysostosis or spondylothoracic dysostosis, or an undefined phenotype. Alternatively, MSVD may have a regional distribution and be associated with a defined or undefined phenotype. The International Consortium for Vertebral Anomalies and Scoliosis has recommended that indiscriminate terminology such as ‘Jarcho Levin syndrome’ not be used, since prior usage of this term has been associated with a wide range of inconsistent skeletal features. The proposed classification system was found to have a high degree of inter-observer reliability and provides a means for future cohort genetic analysis of similar CVM phenotypes.
Fig. 3.

Schematic algorithm for International Consortium for Vertebral Anomalies and Scoliosis (ICVAS) congenital vertebral malformation classification. Reproduced with permission from Giampietro et al., 2008.
Monogenic CVM
Two monogenic forms of CVM associated with mutations in Notch signaling genes have been identified. Spondylocostal dysostosis is an autosomal recessive disorder, with rare autosomal dominant inheritance, associated with contiguous vertebral segmentation defects in addition to rib abnormalities (fig. 4). Affected individuals have short stature, with a shortened trunk and protuberant abdomen. Scoliosis and mild respiratory compromise are associated features. Using synteny conversion analysis, mutations in DLL3, a Notch pathway signaling gene, were identified in Arab-Israeli and Pakistani kindreds [Bulman et al., 2000]. Mutations in other Notch signaling pathway genes have subsequently been identified, including MESP2 [Whittock, et al., 2004], LFNG [Sparrow et al., 2006] and HES7 [Sparrow et al., 2010]. DLL3 mutations are usually associated with morphologically abnormal vertebral bodies characterized by a smooth, round contour or ‘pebble beach’ sign [Turnpenny et al., 2003]. One affected individual with a compound heterozygous mutation in HES7(158D/V186Y) was noted to have hypoplasia of the left vertebral artery.
Fig. 4.
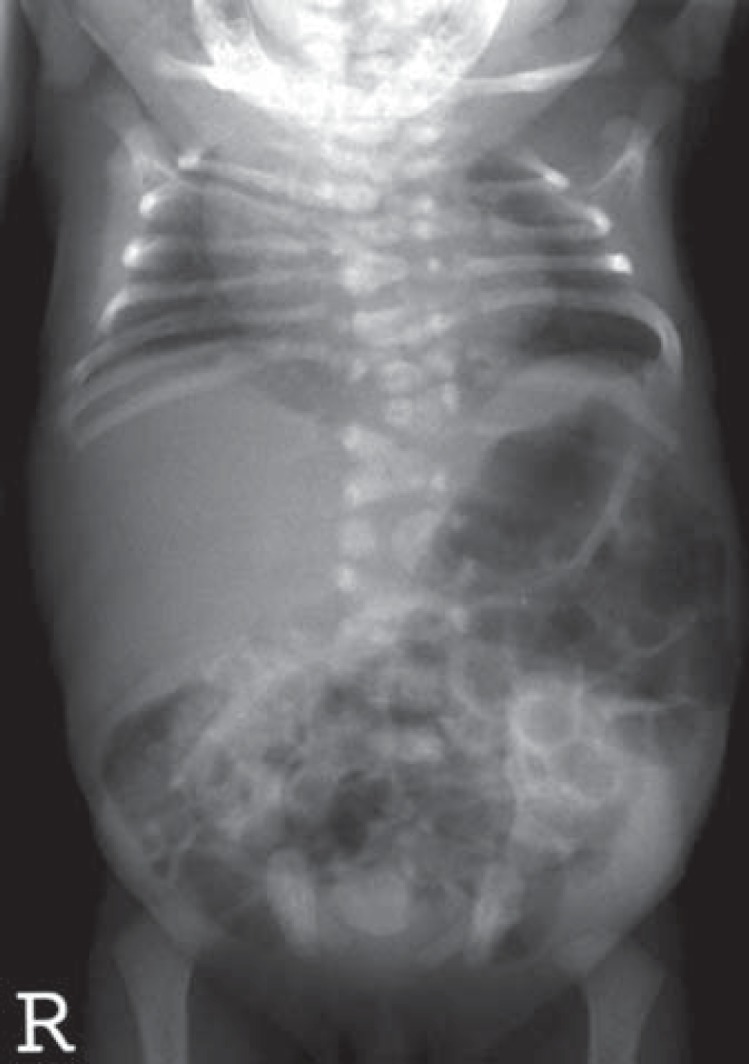
Radiograph illustrating contiguous vertebral malformations with asymmetric rib malformations, characteristic of spondylocostal dysostosis. Photograph courtesy of Peter D. Turnpenney, MD, Royal Devon and Exeter Hospital.
Spondylothoracic dysostosis (STD) is an autosomal recessive disorder of vertebral segmentation characterized by short stature with increased thoracic anterior posterior diameter and a characteristic radiographic appearance consisting of posterior rib fusion, also referred to as a ‘crab like thorax’ (fig. 5) [Moseley and Bonforte, 1969]. Because of the short thoracic cage, there is some degree of respiratory compromise. STD has a prevalence in the Puerto Rican population of 1/12,000. STD is caused by mutations in the MESP2 gene, with a suggestion of a founder effect E103X (p.Glu103X) among Puerto Ricans [Cornier et al., 2008]. The degree of respiratory compromise is more severe in STD, with approximately 25% of affected children surviving into adolescence and adulthood. Diminished lung volume and chest wall stiffness are contributing factors which lead to the development of thoracic insufficiency syndrome in STD.
Fig. 5.

Radiograph illustrating contiguous vertebral malformations with symmetric posterior rib fusion, indicative of spondylothoracic dysostosis. Reproduced with permission [Expert Opin Med Diagn 2008;2:1107–1121].
Genetic Approaches to Study CVM
Because CVM and associated syndromes usually represent sporadic occurrences, even within a particular family identification of causal genetic factors is challenging. A panel of genes associated with vertebral patterning defects including PAX1, DLL3, SLC35A3, WNT3A, TBX6, and T (Brachyury) were sequenced by our group in 50 patients with heterogeneous types of CVMs [Ghebranious et al., 2006, 2007, 2008; Giampietro et al., 2005, 2006]. A mutation (c.1013C>T) resulting in an alanine to valine change was found at amino acid position 338 in the T (Brachyury) gene in 3 affected patients in this cohort that was not present among 886 chromosomes in the CEPH diversity panel [Ghebranious et al., 2008]. Collective maternal pregnancy exposure histories impacting 2 of the patients included diabetes, valproic acid and clomiphene. The third affected individual had no history of deleterious maternal exposures during pregnancy. The phenotypes of these patients were all distinct and included cervical and thoracic CVM and sacral agenesis. This mutation had previously been described in another individual with sacral agenesis with no history of maternal diabetes during pregnancy [Papapetrou et al., 1999]. Although no mutations in TBX6 were identified in the previously described patient series, polymorphisms of the somite patterning gene TBX6, specifically, rs2289292 (located at exon 8 and the only tagging SNP) and rs380962 (located at the 5′UTR and a functional SNP) may have an important role in the pathogenesis of congenital scoliosis in the Chinese Han population [Fei et al., 2010]. Both SNPs exhibited strong linkage disequilibrium.
CVM is a complex condition mediated by genetic, epigenetic and environmental influences. Gestational hypoxia in Hes7+/– and Mesp2+/– mice results in an increase in the number and severity of CVM in mice. This effect is mediated by abnormal FGF signaling resulting in altered somitogenesis, providing evidence that an environmental trigger such as hypoxia can potentiate CVM occurrence in a genetically susceptible background [Sparrow et al., 2012]. The observation that increased DNA methylation can alter the phenotypic expression of tail kinks in the axin fused mouse (AxinFu) supports an epigenetic contribution to CVM occurrence [Waterland et al., 2006].
Whole exome sequence (WES), and whole genome sequence (WGS) platforms represent suitable methodologies for the identification of candidate gene sequence variants and copy number variants (CNV). WES analyzes approximately 1% of the entire genome and highlights identification of sequence variation in the coding and splice site regions in annotated genes identifying approximately 20,000 sequence variants. WGS is capable of uncovering all genetic and genomic variations including single nucleotide variants (SNV) and CNV, identifying approximately 3.5 million sequence variants [Gonzaga-Jauregui et al., 2012]. A variety of filtering algorithms including elimination from primary consideration of sequence variants present in databases such as dbSNP and 1,000 Genomes Project database are implemented. Among coding variants, decreasing priority is given to nonsense, frameshift, splice-site, and missense mutations. Predictive inheritance modeling (dominant, recessive) and computer prediction in conjunction with disease specific information help to enable further refinement. For instance, evidence for localization of vertebral patterning genes identified in mice, Xenopus and chickens, and in synteny blocks supports a hypothesis for conservation of vertebral patterning genes among amniotes [Giampietro et al., 2012]. SNV identified in patterning genes previously identified in model organisms should be sought initially, although the advantage of WES and WGS is the ability to identify novel genes and pathways associated with disease. Following identification of a narrowed and focused list of candidate genes, functional confirmation is necessary. WES is applicable for the identification of SNV in highly penetrant mendelian disease phenotypes, whereas WGS has applications for both mendelian and complex phenotype identification in addition to sporadic phenotypes which are the result of de novo CNVs or SNVs.
Oculo-Auriculo-Vertebral Spectrum (Hemifacial Microsomia)
Salient features of oculo-auriculo-vertebral spectrum (OAVS) include unilateral microtia, craniofacial asymmetry, mandibular hypoplasia, ocular epibulbar dermoid, and CVM [Cohen et al., 1989]. Additional features include congenital heart defects, cleft lip with or without cleft palate and congenital renal malformations. OAVS overlaps with other syndromes including Treacher Collins syndrome (associated with microtia, lower eyelid colobomas and mandibular hypoplasia), Fanconi Anemia (radial ray abnormalities, short stature, elevated diepoxy butane induced chromosome breakage) and VACTERL association. Presently there is no unifying etiology for OAVS, although with evidence supporting vascular disruption [Cousley et al., 2002], maternal diabetes [Wang et al., 2002] in addition to other teratogenetic agents including retinoic acid [Lammer et al., 1985] and thalidomide [Smithells, 1963]. Twelve of 86 (14%) patients with hemifacial microsomia studied using high density olignonucleotide microarray CGH technology were identified as having a CNV including 4 patients with deletions and/or 8 patients with duplications ranging between 2.3–2.8 Mb in size [Rooryck et al., 2010]. Of the 3 patients with CVM who had CNV, one patient had a duplication involving 20p12.2. The ANKRD5 gene was present within this region and is not known to have any known function in somite formation; a second patient had a coincident isodicentric Y chromosome, and the third had a paternally inherited 9q34.11 duplication. None of the genes involved in the 9q34.11 have any known function with respect to vertebral body development. The absence of a unifying CNV abnormality supports a hypothesis for genetic heterogeneity of OAVS.
Klippel-Feil Syndrome
The majority of cases of Klippel-Feil syndrome (short neck, low posterior hairline and fusion of cervical vertebrae) represent sporadic occurrences within a family. However, Klippel-Feil syndrome may represent a familial occurrence in which multiple family members are affected. Autosomal dominant, autosomal recessive and X-linked forms of Klippel-Feil syndrome have been reported. A summary of the different classification schemes is indicated in table 2. The Wildervanck syndrome refers to a constellation of features including Klippel-Feil syndrome, congenital hearing loss and Duane retraction syndrome (limitation of abduction with narrowing of the palpebral fissure and retraction of the globe) [Wildervank, 1978].
Table 2.
Summary of features associated with Klippel-Feil syndrome
| Reference | Cervical fusion abnormalities | Thoracic fusion abnormalities | Lumbar fusion abnormalities | Other malformations | Inheritance |
|---|---|---|---|---|---|
| Klippel et al., 1912 | short neck, low posterior hairline, absence of cervical vertebrae | N/A | N/A | N/A | sporadic |
| Feil, 1919 |
|
groups I and III | group III | Sprengel deformity | N/A |
| Heisenger et al., 1982; MacEwen et al., 1972 |
|
N/A | N/A | N/A | N/A |
| Clarke et al., 1995 |
|
N/A | N/A |
|
|
| Manaligod et al., 1999 | cervical fusion | thoracic fusion | N/A | N/A | N/A |
| Samartzis et al., 2006 |
|
N/A | N/A | N/A | N/A |
KF = Klippel-Feil syndrome; N/A = not applicable.
Mirror movements, or the involuntary movement of the one extremity mimicking the opposite extremity, with a central mirror serving as a reference point, reflecting the image of the voluntary extremity to the opposite side, is a phenomenon which has been found to be associated with Klippel-Feil syndrome [Gunderson and Solitare, 1968; Gardner, 1979; Rasmussen, 1993; Royal et al., 2002]. One neuroanatomic basis for mirror movements is hypothesized to be related to variations in the normal pathways of descending corticospinal tracts including the crossed lateral corticospinal tract, uncrossed anterior corticospinal tract and anterolateral corticospinal tract. An association has been demonstrated between cervicomedullary neuroschisis and mirror movements in cases of Klippel-Feil syndrome [Royal et al., 2002]. Other hypotheses include delayed resolution following a CNS insult or loss of normal control pathways. Neuroschisis could alter the path of descending corticospinal fibers, the majority of which cross at the cervicomedullary junction. This would require recruitment of unbranched pathways with induction of dual branching at more inferior sites. To test the hypothesis that mutations in genes associated with axon migration may contribute to the occurrence of mirror movements, a series of genes associated with aberrant ocular motor and corticospinal axon path development were sequenced in a patient with Wildervanck syndrome, mirror movements and neuroschisis, including ROBO3, CHN1, HOXA1, DCC and GDF6 [Hogen et al., 2012]. No coding mutations were identified in any of these genes suggesting that mutations in regulatory factors or other genes may contribute to this patient's clinical features.
A mutation at a highly conserved region in the BMP ligand GDF6 gene c.866T>C was identified in both sporadic and familial forms of Klippel-Feil syndrome [Tassabehji et al., 2008]. The variable expressivity observed in affected family members and incomplete penetrance observed in GDF6 knockout mice suggest GDF6 thresholds for spine development are subject to modification by environmental factors and may vary between individuals and within different spinal regions. An autosomal dominant mutation (R266C) in GDF3 has been identified in one family with ocular defects including retinal and iris coloboma and CVM [Ye et al., 2010]. Zebrafish morpholinos demonstrated retinal colobomas and trunk shortening with vertebral malformations.
Adults with Klippel-Feil syndrome are prone to degenerative changes in intervertebral discs, which can be visualized as a low-intensity signal on T2 weighted MRI images [Guille et al., 1995]. Additionally, findings include disc protrusion, osteophytes, syringomyelia, and narrowing at the level of the craniovertebral junction. Symptoms may include pain, weakness, numbness, and increased reflexes. MRI findings have revealed degenerative changes in the disc in 100% of patients studied (n = 22); however, there are no studies which delineate the progression of degenerative changes, which presumably could begin during childhood [Guille et al., 1995]. While cervical degenerative disk disease is not present in every child, in some children it may be evident at an early age [Ulmer et al., 1993; Allsopp et al., 2001]. The cause(s) for this are not known and a primary defect of formation has been postulated [Ulmer et al., 1993].
Relation of Congenital Scoliosis to Idiopathic Scoliosis
Multiple lines of evidence support an etiologic relationship between congenital and idiopathic scoliosis. As defined by the Scoliosis Research Society (http://www.srs.org), idiopathic scoliosis (IS) is defined by a lateral curvature of the spine of 10° or greater as measured on plain radiograph. Detailed pedigree analysis performed on 237 families in which an affected child had congenital scoliosis demonstrated a family history of IS in 17.3% of these families [Purkiss et al., 2002]. Linkage analysis in 52 families with IS revealed a significant linkage peak on chromosome 8q12 (multipoint LOD 2.77; p = 0028). Over transmission of the rs4738824 polymorphism, in the CHD7 gene in patients with IS was observed. Replacement of the A allele of this polymorphism with the G allele of this polymorphism predicts disruption of a possible binding site for caudal-type (cdx) homeodomain-containing transcription factors. Mutations in CHD7, a chromeodomain helicase DNA binding protein, are associated with CHARGE syndrome (coloboma of the eye, heart defects, atresia of the choanae, retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness) [Vissers et al., 2004]. CHD7 may act postnatally to alter spinal growth during the adolescent growth spurt. In zebrafish, Chd7 is expressed in somites, brain, eye, and otic vesicle. The Chd7 gene is necessary for the proper symmetric expression of critically important somitogenesis associated genes which are located downstream from Wnt including cdx1a, dlc, her7,mespa, and ripply. Morpholino knockdown of CHD7 resulted in zebrafish embryos which had tail kinks and a progressively shortened axis [Jacobs-McDaniels and Albertson, 2011]. Chd7 plays an important role in somitogenesis as highlighted by a lack of distinct somite boundary formation and abnormal expression of ephrin B2a, an important segment polarity gene when it is knocked down in zebrafish [Patten et al., 2012].
Conclusions and Direction for Future Studies
Advances in genetic technology are providing more robust tools for deciphering genetic etiologies associated with CVM development. The current challenges faced today by researchers include phenotypic and genetic heterogeneity, and determining the potential pathogenicity of identified mutations. The use of bioinformatics filtering strategies and animal models such as zebrafish which enable screening of larger numbers of mutations should facilitate refinement of the large number of potential candidate genes identified through WES analysis. Since CVM are associated with multiple syndromes and birth defects, research continues to shed light on genetic causes associated with CVM related conditions. To provide optimal care and advance research, a multidisciplinary approach consisting of orthopedic surgery, clinical genetics, developmental biology, human genetics, otolaryngology, pulmonology, and cardiology are necessary. Epidemiologic and animal studies have identified risk factors associated with CVM occurrence during pregnancy. A future challenge will be to determine the interplay between genetic, epigenetic and environmental factors contributing to CVM occurrence. Improved understanding of the multiple etiologic factors contributing to the development of CVM should enable targeted screening for families at risk and improved prevention strategies.
Acknowledgement
We would like to thank the Scoliosis Research Society and acknowledge the technical assistance of Gail Pearsall.
References
- Aberg A, Westbom L, Källén B. Congenital malformations among infants whose mothers had gestational diabetes or preexisting diabetes. Early Hum Dev. 2001;61:85–95. doi: 10.1016/s0378-3782(00)00125-0. [DOI] [PubMed] [Google Scholar]
- Aburakawa K, Harada M, Otake S. Clinical evaluations of the treatment of scoliosis. Orthop Surg Trauma. 1996;39:55–62. [Google Scholar]
- Akbarnia BA, Emans JB. Complications of growth-sparing surgery in early onset scoliosis. Spine (Phila Pa 1976) 2010;35:2193–2204. doi: 10.1097/BRS.0b013e3181f070b5. [DOI] [PubMed] [Google Scholar]
- Alexander PG, Tuan RS. Role of environmental factors in axial skeletal dysmorphogenesis. Birth Defects Res C Embryo Today. 2010;90:118–132. doi: 10.1002/bdrc.20179. [DOI] [PubMed] [Google Scholar]
- Alexander PG, Chau L, Tuan RS. Role of nitric oxide in chick embryonic organogenesis and dysmorphogenesis. Birth Defects Res A Clin Mol Teratol. 2007;79:581–594. doi: 10.1002/bdra.20386. [DOI] [PubMed] [Google Scholar]
- Allsopp GM, Griffiths S, Sgouros S. Cervical disc prolapse in childhood associated with Klippel-Feil syndrome. Childs Nerv Syst. 2001;17:69–70. doi: 10.1007/s003810000377. [DOI] [PubMed] [Google Scholar]
- Bengtsson BE, Larsson A, Bengtsson A, Renberg L. Sublethal effects of tetrachloro-1,2-benzoquinone–a component in bleachery effluents from pulp mills–on vertebral quality and physiological parameters in fourhorn sculpin. Ecotoxicol Environ Saf. 1988;15:62–71. doi: 10.1016/0147-6513(88)90043-7. [DOI] [PubMed] [Google Scholar]
- Bennett GD. (2010). Hyperthermia: Malformations to chaperones. Birth Defect Res B Dev Reprod Toxicol. 2010;89:279–288. doi: 10.1002/bdrb.20254. [DOI] [PubMed] [Google Scholar]
- Bess S, Akbarnia BA, Thompson GH, Sponseller PD, Shah SA, et al. Complications of growing-rod treatment for early-onset scoliosis: analysis of one hundred and forty patients. J Bone Joint Surg Am. 2010;92:2533–2543. doi: 10.2106/JBJS.I.01471. [DOI] [PubMed] [Google Scholar]
- Bnait KS, Seller MJ. Ultrastructural changes in 9-day-old mouse embryos following maternal tobacco smoke inhalation. Exp Toxicol Pathol. 1995;47:453–461. doi: 10.1016/S0940-2993(11)80327-1. [DOI] [PubMed] [Google Scholar]
- Brand MC. Examination of the newborn with congenital scoliosis: focus on the physical. Adv Neonatal Care. 2008;8:265–273. doi: 10.1097/01.ANC.0000338016.03040.6b. [DOI] [PubMed] [Google Scholar]
- Breen JG, Claggett TW, Kimmel GL, Kimmel CA. Heat shock during rat embryo development in vitro results in decreased mitosis and abundant cell death. Reprod Toxicol. 1999;13:31–39. doi: 10.1016/s0890-6238(98)00056-2. [DOI] [PubMed] [Google Scholar]
- Bulman M, Kusumi K, Frayling TM, McKeown C, Garrett C. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet. 2000;4:438–441. doi: 10.1038/74307. [DOI] [PubMed] [Google Scholar]
- Campbell RM, Jr, Smith MD, Mayes TC, Mangos JA, Willey-Courand DB, et al. The characteristics of thoracic insufficiency syndrome associated with fused ribs and congenital scoliosis. J Bone Joint Surg Am. 2003;85-A:399–408. doi: 10.2106/00004623-200303000-00001. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Jr, Rollnick BR, Kaye CI. Oculoauriculovertebral spectrum: an updated critique. Cleft Palate J. 1989;26:276–286. [PubMed] [Google Scholar]
- Cooke J, Zeeman EC. A clock and wavefront model for control of the number of repeated structures during animal morphogenesis. J Theor Biol. 1976;58:455–476. doi: 10.1016/s0022-5193(76)80131-2. [DOI] [PubMed] [Google Scholar]
- Cornier AS, Staehling-Hampton K, Delventhal KM, Saga Y, Caubet JF, et al. Mutations in the MESP2 gene cause spondylothoracic dysostosis/Jarcho-Levin syndrome. Am J Hum Genet. 2008;82:1334–1341. doi: 10.1016/j.ajhg.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello G, Piro E. The world of twins: an update. J Matern Fetal Neonatal Med 23 Suppl. 2010;3:59–62. doi: 10.3109/14767058.2010.508218. [DOI] [PubMed] [Google Scholar]
- Cousley R, Naora H, Yokoyama M, Kimura M, Otani H. Validity of the Hfm transgenic mouse as a model for hemifacial microsomia. Cleft Palate Craniofac J. 2002;39:81–92. doi: 10.1597/1545-1569_2002_039_0081_vothtm_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- Farley FA, Loder RT, Nolan BT, Dillon MT, Frankenburg EP, et al. Mouse model for thoracic congenital scoliosis. J Pediatr Orthop. 2001;21:537–540. [PubMed] [Google Scholar]
- Fei Q, Wu Z, Wang H, Zhou X, Wang N, et al. The association analysis of TBX6 polymorphism with susceptibility to congenital scoliosis in a Chinese Han population. Spine (Phila Pa 1976) 2010;35:983–988. doi: 10.1097/BRS.0b013e3181bc963c. [DOI] [PubMed] [Google Scholar]
- Fichtner RR, Sullivan KM, Zyrkowski CL, Trowbridge FL. Racial/ethnic differences in smoking, other risk factors, and low birth weight among low-income pregnant women, 1978–1988. MMWR CDC Surveill Summ. 1990;39:13–21. [PubMed] [Google Scholar]
- Gardner WJ. Klippel-Feil syndrome, iniencephalus, anencephalus, hindbrain hernia and mirror movements: overdistention of the neural tube. Childs Brain. 1979;5:361–379. doi: 10.1159/000119832. [DOI] [PubMed] [Google Scholar]
- Ghebranious N, Burmester JK, Glurich I, McPherson E, Ivacic L, et al. Evaluation of SLC35A3 as a candidate gene for human vertebral malformations. Am J Med Genet A. 2006;140:1346–1348. doi: 10.1002/ajmg.a.31307. [DOI] [PubMed] [Google Scholar]
- Ghebranious N, Raggio CL, Blank RD, McPherson E, Burmester JK, et al. Lack of evidence of WNT3A as a candidate gene for congenital vertebral malformations. Scoliosis. 2007;2:13. doi: 10.1186/1748-7161-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghebranious N, Blank RD, Raggio CL, Staubli J, McPherson E, et al. A missense T (Brachyury) mutation contributes to vertebral malformations. J Bone Miner Res. 2008;23:1576–1583. doi: 10.1359/jbmr.080503. [DOI] [PubMed] [Google Scholar]
- Giampietro PF, Raggio CL, Reynolds CE, Shukla SK, McPherson E, et al. An analysis of PAX1 in the development of vertebral malformations. Clin Genet. 2005;68:448–453. doi: 10.1111/j.1399-0004.2005.00520.x. [DOI] [PubMed] [Google Scholar]
- Giampietro PF, Raggio CL, Reynolds C, Ghebranious N, Burmester JK, et al. DLL3 as a candidate for vertebral malformations. Am J Med Genet A. 2006;140:2447–2453. doi: 10.1002/ajmg.a.31509. [DOI] [PubMed] [Google Scholar]
- Giampietro PF, et al. Molecular diagnosis of vertebral segmentation disorders in humans. Expert Opin Med Diagn. 2008;2:1107–1121. doi: 10.1517/17530059.2.10.1107. [DOI] [PubMed] [Google Scholar]
- Giampietro PF, Raggio CL, Blank RD. Use of synteny conversion in identification of candidate genes for somitogenesis in humans. OJO. 2012;2:62–68. [Google Scholar]
- Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med. 2012;63:35–61. doi: 10.1146/annurev-med-051010-162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guille JT, Miller A, Bowen JR, Forlin E, Caro PA. The natural history of Klippel-Feil syndrome: clinical, roentgenographic, and magnetic resonance imaging findings at adulthood. J Pediatr Orthop. 1995;15:617–626. doi: 10.1097/01241398-199509000-00014. [DOI] [PubMed] [Google Scholar]
- Gunderson CH, Solitare GB. Mirror movements in patients with the Klippel-Feil syndrome. Neuropathologic observations. Arch Neurol. 1968;18:675–679. doi: 10.1001/archneur.1968.00470360097009. [DOI] [PubMed] [Google Scholar]
- Hickory W, Nanda R, Catalanotto FA. Fetal skeletal malformations associated with moderate zinc deficiency during pregnancy. J Nutr. 1979;109:883–891. doi: 10.1093/jn/109.5.883. [DOI] [PubMed] [Google Scholar]
- Hogen T, Chan WM, Riedel E, Brüning R, Chang HH, et al. Wildervanck's syndrome and mirror movements: a congenital disorder of axon migration? J Neurol. 2012;259:761–763. doi: 10.1007/s00415-011-6239-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes LB. Common Malformations. New York: Oxford University Press; 2012. Vertebral Anomalies: Hemivertebrae; pp. 283–289. [Google Scholar]
- Holmes LB, Harvey EA, Coull BA, Huntington KB, Khoshbin S, et al. The teratogenicity of anticonvulsant drugs. N Engl J Med. 2001;344:1132–1138. doi: 10.1056/NEJM200104123441504. [DOI] [PubMed] [Google Scholar]
- Jacobs-McDaniels NL, Albertson RC. Chd7 plays a critical role in controlling left-right symmetry during zebrafish somitogenesis. Dev Dyn. 2011;240:2272–2280. doi: 10.1002/dvdy.22722. [DOI] [PubMed] [Google Scholar]
- Karol LA, Johnston C, Mladenov K, Schochet P, Walters P, Browne RH. Pulmonary function following early thoracic fusion in non-neuromuscular scoliosis. J Bone Joint Surg Am. 2008;90:1272–1281. doi: 10.2106/JBJS.G.00184. [DOI] [PubMed] [Google Scholar]
- Kaspiris A, Grivas TB, Weiss HR. Congenital scoliosis in monozygotic twins: case report and review of possible factors contributing to its development. Scoliosis. 2008;3:17. doi: 10.1186/1748-7161-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel M, Feil A. Un cas d'absence des vertebres cervicales. Nouv Icon Salpet. 1912;25:223. [Google Scholar]
- Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313:837–841. doi: 10.1056/NEJM198510033131401. [DOI] [PubMed] [Google Scholar]
- Loughnan PM, Gold H, Vance JC. Phenytoin teratogenicity in man. Lancet. 1973;1:70–72. doi: 10.1016/s0140-6736(73)90467-4. [DOI] [PubMed] [Google Scholar]
- Martínez-Frías ML, Bermejo E, Rodríguez-Pinilla E, Prieto L, Frías JL. Epidemiological analysis of outcomes of pregnancy in gestational diabetic mothers. Am J Med Genet. 1998;78:140–145. doi: 10.1002/(sici)1096-8628(19980630)78:2<140::aid-ajmg8>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- McMaster M, Ohtsuka K. The natural history of congenital scoliosis. A study of two hundred and fifty-one patients. J Bone Joint Surg Am. 1982;64:1128–1147. [PubMed] [Google Scholar]
- Menegola E, Broccia ML, Nau H, Prati M, Ricolfi R, Giavini E. Teratogenic effects of sodium valproate in mice and rats at midgestation and at term. Teratog Carcinog Mutagen. 1996;16:97–108. doi: 10.1002/(SICI)1520-6866(1996)16:2<97::AID-TCM4>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Mortier GR, Lachman RS, Bocian M, Rimoin DL. Multiple vertebral segmentation defects: analysis of 26 new patients and review of the literature. Am J Med Genet. 1996;61:310–319. doi: 10.1002/(SICI)1096-8628(19960202)61:4<310::AID-AJMG3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Moseley JE, Bonforte RJ. Spondylothoracic dysplasia–a syndrome of congenital anomalies. Am J Roentgenol Radium Ther Nucl Med. 1969;106:166–169. doi: 10.2214/ajr.106.1.166. [DOI] [PubMed] [Google Scholar]
- Niemitz EL, Feinberg AP. Epigenetics and assisted reproductive technology: a call for investigation. Am J Hum Genet. 2004;74:599–609. doi: 10.1086/382897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offiah A, Alman B, Cornier AS, Giampietro PF, Tassy O, et al. Pilot assessment of a radiologic classification system for segmentation defects of the vertebrae. Am J Med Genet A. 2010;152A:1357–1371. doi: 10.1002/ajmg.a.33361. [DOI] [PubMed] [Google Scholar]
- Owen MH, Ryan LM, Holmes LB. Effects of retinoic acid on dominant hemimelia expression in mice. Birth Defects Res A Clin Mol Teratol. 2009;85:36–41. doi: 10.1002/bdra.20528. [DOI] [PubMed] [Google Scholar]
- Papapetrou C, Drummond F, Reardon W, Winter R, Spitz L, Edwards YH. A genetic study of the human T gene and its exclusion as a major candidate gene for sacral agenesis with anorectal atresia. J Med Genet. 1999;36:208–213. [PMC free article] [PubMed] [Google Scholar]
- Passarge E, Lenz W. Syndrome of caudal regression in infants of diabetic mothers: observations of further cases. Pediatrics. 1966;37:672–675. [PubMed] [Google Scholar]
- Patten SA, Jacobs-McDaniels NL, Zaouter C, Drapeau P, Albertson RC, Moldovan F. Role of Chd7 in zebrafish: a model for CHARGE syndrome. PloS One. 2012;7:e31650. doi: 10.1371/journal.pone.0031650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkiss SB, Driscoll B, Cole WG, Alman B. Idiopathic scoliosis in families of children with congenital scoliosis. Clin Orthop Relat Res. 2002:27–31. doi: 10.1097/00003086-200208000-00005. [DOI] [PubMed] [Google Scholar]
- Rasmussen P. Persistent mirror movements: a clinical study of 17 children, adolescents and young adults. Dev Med Child Neurol. 1993;35:699–707. doi: 10.1111/j.1469-8749.1993.tb11715.x. [DOI] [PubMed] [Google Scholar]
- Rooryck C, Souakri N, Cailley D, Bouron J, Goizet C, et al. Array-CGH analysis of a cohort of 86 patients with oculoauriculovertebral spectrum. Am J Med Genet A. 2010;152A:1984–1989. doi: 10.1002/ajmg.a.33491. [DOI] [PubMed] [Google Scholar]
- Royal SA, Tubbs RS, D'Antonio MG, Rauzzino MJ, Oakes WJ, et al. Investigations into the association between cervicomedullary neuroschisis and mirror movements in patients with Klippel-Feil syndrome. AJNR Am J Neuroradiol. 2002;23:724–729. [PMC free article] [PubMed] [Google Scholar]
- Sköld AC, Wellfelt K, Danielsson BR. Stage-specific skeletal and visceral defects of the I(Kr)-blocker almokalant: further evidence for teratogenicity via a hypoxia-related mechanism. Teratology. 2001;2001:292–300. doi: 10.1002/tera.1084. [DOI] [PubMed] [Google Scholar]
- Smithells RW. The incidence of limb and ear defects since the withdrawal of thalidomide. Lancet. 1963;1:1095–1097. doi: 10.1016/s0140-6736(63)92129-9. [DOI] [PubMed] [Google Scholar]
- Sparrow DB, Chapman G, Wouters MA, Whittock NV, Ellard S, et al. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am J Hum Genet. 2006;78:28–37. doi: 10.1086/498879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow DB, Sillence D, Wouters MA, Turnpenny PD, Dunwoodie SL. Two novel missense mutations in HAIRY-AND-ENHANCER-OF-SPLIT-7 in a family with spondylocostal dysostosis. Eur J Hum Genet. 2010;18:674–679. doi: 10.1038/ejhg.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow DB, Chapman G, Smith AJ, Mattar MZ, Major JA, et al. A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell. 2012;149:295–306. doi: 10.1016/j.cell.2012.02.054. [DOI] [PubMed] [Google Scholar]
- Takikawa K, Haga N, Maruyama T, Nakatomi A, Kondoh T, et al. Spine and rib abnormalities and stature in spondylocostal dysostosis. Spine (Phila Pa 1976) 2006;31:E192–197. doi: 10.1097/01.brs.0000208166.61618.8f. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Fang ZM, Hilton EN, McGaughran J, Zhao Z, et al. Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum Mutat. 2008;29:1017–1027. doi: 10.1002/humu.20741. [DOI] [PubMed] [Google Scholar]
- Thomsen M, Schneider U, Weber M, Johannisson R, Niethard FU. Scoliosis and congenital anomalies associated with Klippel-Feil syndrome types I-III. Spine (Phila Pa 1976) 1997;22:396–401. doi: 10.1097/00007632-199702150-00008. [DOI] [PubMed] [Google Scholar]
- Tian Y, Ishikawa H, Yamaguchi T, Yamauchi T, Yokoyama K. Teratogenicity and developmental toxicity of chlorpyrifos. Maternal exposure during organogenesis in mice. Reprod Toxicol. 2005;20:267–270. doi: 10.1016/j.reprotox.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Treadwell SJ, Smith DF, Macleod PJ, Wood BJ. Cervical spine anomalies in fetal alcohol syndrome. Spine (Phila Pa 1976) 1982;7:331–334. doi: 10.1097/00007632-198207000-00002. [DOI] [PubMed] [Google Scholar]
- Turnpenny P, Whittock N, Duncan J, Dunwoodie S, Kusumi K, Ellard S. Novel mutations in DLL3, a somitogenesis gene encoding a ligand for the Notch signalling pathway, cause a consistent pattern of abnormal vertebral segmentation in spondylocostal dysostosis. J Med Genet. 2003;40:333–339. doi: 10.1136/jmg.40.5.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer JL, Elster AD, Ginsberg LE, Williams DW., 3rd Klippel-Feil syndrome: CT and MR of acquired and congenital abnormalities of cervical spine and cord. J Comput Assist Tomogr. 1993;17:215–224. [PubMed] [Google Scholar]
- Vitale MG, Matsumoto H, Bye MR, Gomez JA, Booker WA, et al. A retrospective cohort study of pulmonary function, radiographic measures, and quality of life in children with congenital scoliosis: an evaluation of patient outcomes after early spinal fusion. Spine (Phila Pa 1976) 2008;33:1242–1249. doi: 10.1097/BRS.0b013e3181714536. [DOI] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, et al. Mutations in a new member of the chromeodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Vorhees CV. Teratogenicity and developmental toxicity of valproic acid in rats. Teratology. 1987;35:195–202. doi: 10.1002/tera.1420350205. [DOI] [PubMed] [Google Scholar]
- Wang R, Martínez-Frías ML, Graham JM., Jr Infants of diabetic mothers are at increased risk for the oculo-auriculo-vertebral sequence: a case-based and case-control approach. J Pediatr. 2002;141:611–617. doi: 10.1067/mpd.2002.128891. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Dolinoy DC, Lin JR, Smith CA, Shi X, Tahiliani KG. Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis. 2006;44:401–406. doi: 10.1002/dvg.20230. [DOI] [PubMed] [Google Scholar]
- Wéry N, Narotsky MG, Pacico N, Kavlock RJ, Picard JJ, Gofflot F. Defects in cervical vertebrae in boric acid-exposed rat embryos are associated with anterior shifts of hox gene expression domains. Birth Defects Res A Clin Mol Teratol. 2003;67:59–67. doi: 10.1002/bdra.10031. [DOI] [PubMed] [Google Scholar]
- Whittock NV, Sparrow DB, Wouters MA, Sillence D, Ellard S, et al. Mutated MESP2 causes spondylocostal dysostosis in humans. Am J Hum Genet. 2004;74:1249–1254. doi: 10.1086/421053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildervank L. The cerrvico-oculo-acusticus syndrome. In: Vinken P, Bruyn G, Myrianthopoulous N, editors. Handbook of Clinical Neurology: Congenital Malformations of the Spine and Spinal Cord. vol 32. New York: North Holland Publishing Co; 1978. [Google Scholar]
- Wynne-Davies R. Congenital vertebral anomalies: aetiology and relationship to spina bifida cystica. J Med Genet. 1975;12:280–288. doi: 10.1136/jmg.12.3.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Berry-Wynne KM, Asai-Coakwell M, Sundaresan P, Footz T, et al. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum Mol Genet. 2010;19:287–298. doi: 10.1093/hmg/ddp496. [DOI] [PubMed] [Google Scholar]