

Abstract
The DEAD box protein family member DDX3 was previously identified as an inhibitor of death receptor-mediated extrinsic apoptotic signaling. However, there had been no studies of the role of DDX3 in regulating the other major type of apoptosis, intrinsic apoptotic signaling, which was examined here. Intrinsic apoptosis was induced in MCF-7 cells by treatment with staurosporine, a general kinase inhibitor, thapsigargin, which induces endoplasmic reticulum (ER) stress, and camptothecin, which causes DNA damage. Each of these treatments caused time-dependent activation of caspase-7, the predominant executioner caspase in these cells. Depletion of DDX3 using shRNA did not alter apoptotic responses to staurosporine or thapsigargin. However, caspase-7 activation induced by camptothecin was regulated by DDX3 in a manner dependent on the functional status of p53. Depletion of DDX3 abrogated camptothecin-induced caspase-7 activation in MCF-7 cells expressing functional wild-type p53, but oppositely potentiated camptothecin-mediated caspase activation in cells expressing mutant or non-functional p53, which was accompanied by increased activation of the extrinsic apoptotic signaling initiator caspase-8. In MCF-7 cells, depletion of DDX3 reduced by more than 50% camptothecin-induced p53 accumulation, and this effect was blocked by inhibition of the proteasome with MG132, indicating that DDX3 regulates p53 not at expression level but rather its stabilization after DNA damage. Co-immunoprecipitation experiments demonstrated that DDX3 associates with p53, and overexpression of DDX3 was sufficient to double the accumulation of p53 in the nucleus after DNA damage. Thus, DDX3 associates with p53, increases p53 accumulation, and positively regulates camptothecin-induced apoptotic signaling in cells expressing functional wild-type p53, whereas in cells expressing mutant or non-functional p53 DDX3 inhibits activation of the extrinsic apoptotic pathway to reduce caspase activation. These results demonstrate that DDX3 not only regulates extrinsic apoptotic signaling, as previously reported, but also selectively regulates intrinsic apoptotic signaling following DNA damage.
Keywords: DDX3, p53, apoptosis, DNA damage, camptothecin
Background
DDX3 is a member of the DEAD box family of proteins, named for a conserved D-E-A-D sequence, that contain putative ATPase and RNA helicase domains [1, 2, 3]. There is limited knowledge about the functions of DDX3, which were originally linked to actions of the hepatitis C and human immunodeficiency viruses [4-6]. DDX3 is a nucleo-cytoplasmic shuttling protein [5, 7], associates with the Sp1 transcription factor in the nucleus, and contributes to regulating cyclin D1, cyclin E1, and p21 [8, 9, 10, 11]. Recently, several findings linked DDX3 to cancer. DDX3 levels were decreased in hepatocellular carcinomas [8, 9], although another study reported the opposite relationship [12], DDX3 levels increased in carcinogen-treated MCF-7 cells [13], DDX3 impedes death receptor-induced apoptosis [14, 15], and DDX3 regulates Snail and affects cancer cell motility and proliferation (16). These findings raised the possibility that DDX3 may have regulatory influences on the development, progression, or treatment of certain cancers.
Many cancers contain mutated p53, a tumor suppressor transcription factor that is activated by a wide variety of cellular stresses, notably DNA damage [17, 18, 19]. p53 is a rapidly turning-over protein that is usually maintained at low levels by rapid degradation through ubiquitin-dependent proteolysis [20]. Post-translation modifications, such as phosphorylation, and interactions with other proteins are important regulators of the stabilization and activation of the normally short-lived p53 [21, 22]. Activation leads to accumulation of p53 in the nucleus where it regulates the expression of genes and ultimately leads to two well-defined cellular responses, cell cycle arrest and apoptosis.
Herein we report that DDX3 contributes to regulating apoptotic signaling and the accumulation of p53 after DNA damage. DDX3 promotes the retention and accumulation of p53 in the nucleus, which co-immunoprecipitation experiments suggest may be mediated by the association of DDX3 with p53. In cells expressing functional wild-type p53, DDX3 positively regulates DNA damage-induced caspase activation, but in cells expressing nonfunctional p53, DDX3 inhibits DNA damage-induced caspase activation.
Results
Apoptosis induced by DNA damage is selectively affected by DDX3
Since DDX3 was recently found to be anti-apoptotic towards extrinsic death receptor-induced apoptosis [15], we tested if DDX3 also modulates intrinsic apoptosis. Three stimuli that activate intrinsic apoptotic signaling by different mechanisms were examined: staurosporine, a kinase inhibitor well-known to induce intrinsic apoptosis in essentially all types of cells [23], thapsigargin, which inhibits the Ca++-ATPase in the endoplasmic reticulum (ER) to cause ER stress-induced apoptosis [24], and camptothecin, a topoisomerase 1 inhibitor that causes DNA damage resulting in p53-mediated apoptosis [25]. Apoptotic signaling was detected by measuring activation of effector caspases 3 or 7 and cleavage of their substrate PARP, and regulation by DDX3 was examined by using cells in which the level of DDX3 was knocked down by shRNA. Treatment with 1 μM staurosporine caused time-dependent activation of caspase-7 and cleavage of PARP in MCF-7 cells, and these responses were not altered in cells with the level of DDX3 knocked down (Figure 1A). MCF-7 cells treated with 1 μM thapsigargin also displayed time-dependent activation of caspase-7 and concomitant PARP cleavage, and these responses were not changed by knockdown of DDX3 (Figure 1B). In contrast to the other two insults, knocking down DDX3 resulted in reduced caspase-7 activation and PARP cleavage in MCF-7 cells after treatment with 10 μM camptothecin (Figure 1C). These results indicate that DDX3 does not universally affect intrinsic apoptotic signaling, as it does extrinsic apoptotic signaling, but DDX3 selectively regulates intrinsic apoptotic signaling induced by DNA damage.
Figure 1. DDX3 protects MCF-7 cells from DNA damage-induced apoptosis but potentiates it in HeLa cells.


Wild-type (WT) and DDX3 knockdown (KD) stable MCF-7 cells were treated for the indicated times with (A) 1 μM staurosporine, (B) 1 μM thapsigargin (after overnight preincubation in serum free media), or (C) 10 μM camptothecin (after overnight preincubation in serum free media). (D) HeLa cells and (E) MDA-MB-231 cells were treated with 10 μM camptothecin for the indicated time points (after overnight preincubation in serum free media). Cell extracts were collected and immunoblotted for active caspase-3/7, cleaved PARP, DDX3, and β-actin as a loading control. (F) MCF-7, (G) HeLa and (H) MDA-MB231 wild-type (WT) and DDX3 knockdown (KD) cells were treated with 10 μM Adriamycin for the indicated time points, followed by immunoblotting for active caspase-3/7, cleaved-PARP, DDX3 and β-actin. Experiments were repeated at least two times with similar results.
DDX3 promotes camptothecin-induced apoptosis in cells expressing functional wild-type p53 but impedes apoptosis in cells with nonfunctional p53
To test if the modulatory effect of DDX3 involves p53, a critical mediator of DNA damage-induced apoptotic signaling, apoptosis was measured in HeLa cells, which contain wild-type p53 that is functionally disabled due to the presence of the human papilloma virus E6 protein (23, 24) compared with MCF-7 cells that express active wild-type p53. Caspase-3 activation and PARP cleavage increased time-dependently after treatment of HeLa cells with 10 μM camptothecin (Figure 1D). Surprisingly, these apoptotic signals were amplified in cells with DDX3 knocked down, similar to previous reports that DDX3 inhibits extrinsic apoptotic signaling (14, 15). In addition, we tested another p53 mutant breast cancer cell line MDA-MB-231 that expresses mutant p53. MDA-MB-231 displayed a response similar to HeLa cells, as the knockdown of DDX3 facilitated apoptosis signaling indicated by activation of caspase-3 and PARP cleavage induced by camptothecin treatment (Figure 1E), further demonstrating that DDX3 inhibits DNA damage-induced apoptosis in cells with non-functional p53. Adriamycin, a topoisomerase II inhibitor, was used as another DNA damaging agent to further examine the effect of DDX3. MCF-7, HeLa and MDA-MB-231 cells, with DDX3 or with DDX3 knocked down, were treated with Adriamycin (10 μM) for 24 or 48 hr. Apoptosis was reduced by knocking down DDX3 in MCF-7 cells, but was increased in HeLa and MDA-MB-231 cells (Figure 1F-G). These results indicate that selective regulation by DDX3 of DNA damage-induced caspase activation is dependent on the functional status of p53.
To further examine how DDX3 regulates camptothecin-induced apoptosis differently in cells with functional wild-type and mutant p53, p38 activation was examined because the p38 and p53 signaling pathways can mutually regulate each other in response to DNA damage (26) and the p38 pathway has been reported to be involved in the extrinsic apoptotic pathway where DDX3 is known to be regulatory (27). Following camptothecin treatment, DDX3 knockdown attenuated the activation of p38 in MCF-7 cells with active p53 (Figure 2A), but enhanced p38 activation in HeLa cells with nonfunctional p53 (Figure 2B), as determined by immunoblotting the active phosphorylated level of p38.
Figure 2. DDX3 regulates DNA damage-induced apoptosis in a manner dependent on the functional status of p53.


p-p38, p38 and p-JNK levels were measured in wild-type (WT) and DDX3 knockdown (KD) (A) MCF-7 cells and (B) HeLa cells following treatment with 10 μM camptothecin for the indicated time points. Intact procaspase-8 (55/53 kDa) and cleaved 43/41 kDa caspase-8 and β-actin levels were measured after HeLa (C) and BJAB (D) cells were treated with 10 μM camptothecin for the indicated time points. Experiments were repeated at least two times with similar results. Cell survival was examined after treatment with 0, 0.1, 1, and 10 μM camptothecin for 24 hr in (E) MCF-7 cells, (F) HeLa cells, and (G) MDA-MB-231 cells with DDX3 (WT) and with DDX3 knocked down (KD). Means ± SEM; n=3; *p<0.05.
We previously reported that DDX3 depletion promotes caspase-8 activation induced by stimulation of death receptors [15]. Since the caspase-8-mediated extrinsic apoptotic pathway has been reported to be activated following DNA damage [28-35], we tested if this was modulated by DDX3. Caspase-8 was activated, and this was increased by DDX3 depletion, following DNA damage in HeLa cells (Figure 2C), and type I BJAB cells that express mutant, nonfunctional p53 (Figure 2D). Although this is unlikely to be the sole action of DDX3 since increased caspase-3 activation was detected earlier than caspase-8 activation, these results are consistent with previous reports that death receptors can be activated after DNA damage and that knocking down DDX3 promotes death receptor-induced apoptotic signaling. Furthermore, cell survival was measured in MCF-7 (Figure 2E), HeLa (Figure 2F) and MDA-MB-231 cells (Figure 2G). Knocking down DDX3 increased cell survival in MCF-7 cells with functional p53, but reduced cell survival in HeLa cells with nonfunctional p53 and in MDA-MB-231 cells with mutant p53.
DDX3 knockdown reduces p53 accumulation in MCF-7 cells expressing functional wild-type p53
Since p53 is a central mediator of cellular responses to DNA damage, we tested if DDX3 modulates the level of p53 in response to DNA damage caused by treatment with camptothecin in MCF-7 cells expressing functional wild-type p53. The cellular level of p53 increased time-dependently following camptothecin treatment, and this accumulation of p53 was diminished in cells with depleted DDX3 (Figure 3A) to a similar extent as caspase-7 activation was reduced by DDX3 depletion (Figure 1C). In contrast, DDX3 knockdown did not change p53 levels in the presence or absence of camptothecin in HeLa cells expressing nonfunctional p53 (Figure 3B) and in MDA-MB-231 cells expressing mutant p53 (Figure 3C). These results indicate that DDX3 may promote camptothecin-induced caspase activation by promoting the accumulation of functional p53. Furthermore, comparison of basal and camptothecin-induced p53 levels in two lines of MCF-7 cells with different extents of DDX3 knockdown induced with two different shRNA sequences showed that there was a direct relationship between less DDX3 and lower levels of p53 (Figure 3D), suggesting that DDX3 supports accumulation of functional wild-type p53. Quantitation of p53 levels from three individual experiments demonstrated that knocking down DDX3 resulted in a 52 ± 8% decrease in basal p53 and a 66 ± 5% decrease in p53 after 4 hr of camptothecin treatment (Figure 3E). This relationship was further confirmed by treating MCF-7 cells with etoposide, a topoisomerase II inhibitor, which resulted in much less p53 accumulation in MCF-7 cells with DDX3 knocked down than in wild-type cells (Figure 3F).
Figure 3. DDX3 regulates p53 levels.

p53 and β-actin levels were measured in (A) MCF-7 , (B) HeLa and (C) MDA-MB-231 cells after wild-type (WT) and DDX3 knockdown (KD) cells were treated with 10 μM camptothecin for indicated the time points. D) Cell lysates of two lines of MCF-7 cells with different extents of DDX3 knockdown using shRNA sequence #1 or #4 were immunoblotted for p53 and DDX3 before and after treatment with 10 μM camptothecin for 4 hr. (E) p53 quantitative values are from basal and 4 hr camptothecin-treated samples and are means ± SEM; n=3; *p<0.05 comparing wild-type and DDX3 knockdown cells. (F) p53, DDX3, and β-actin levels were measured in MCF-7 wild-type and DDX3 knockdown cells after 24 hr treatment with 200 μM topoisomerase II inhibitor etoposide.
We examined the subcellular localization of p53 regulated by DDX3 in MCF-7 cells after DNA damage. Treatment with camptothecin caused accumulation of p53 in both the cytosol and nucleus, but with DDX3 knocked down p53 accumulated much less in both compartments (Figure 4). These results indicate DDX3 is an important factor in the accumulation of p53 after DNA damage, as little p53 can accumulate after DNA damage in the absence of DDX3, raising the possibility that DDX3 may associate with p53.
Figure 4. DDX3 regulates p53 accumulation after DNA damage.
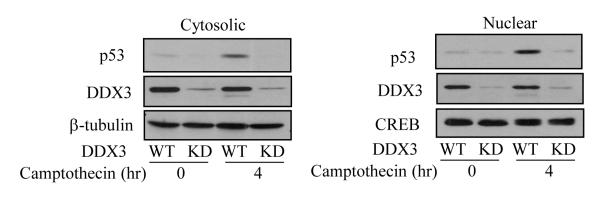
Cytosolic (left panel) and nuclear (right panel) fractions were prepared from MCF-7 wild-type or DDX3 knockdown cells after treatment with 10 μM camptothecin for 4 hr, followed by measurements of p53 and DDX3 levels. β-tubulin was used as a loading control for cytosolic fractions, and CREB for nuclear fractions. Experiments were repeated at least two times with similar results.
DDX3 associates with p53
To further test if DDX3 promotes the stabilization of p53, and if this occurs under non-apoptotic conditions, cells were treated with the proteasome inhibitor MG132 to cause accumulation of p53. MG132 treatment (10 μM; 5 hr) increased p53 levels equivalently in MCF-7 cells with or without knockdown of DDX3 (Figure 5A), indicating that depletion of DDX3 did not impair the accumulation of p53 stabilized by proteasome inhibition. Since DDX3 can bind glycogen synthase kinase-3 (GSK3) [15] and GSK3 can bind and regulate p53 [36], we also tested if treatment with the GSK3 inhibitor lithium altered the basal or MG132-stabilized levels of p53 with or without DDX3 knockdown. However, lithium treatment did not alter the effects of MG132 or DDX3 knockdown on the level of p53 (Figure 5A), suggesting that the effect of DDX3 is independent of GSK3.
Figure 5. DDX3 stabilizes and associates with p53.

(A) MCF-7 wild-type (WT) or DDX3 knockdown (KD) cells were treated with 10 μM MG132, 20 mM lithium, or both, for 5 hr, followed by measuring the levels of p53 and DDX3. (B) MCF-7 wild-type and DDX3 knockdown cells were treated with 10 μM MG132 for 5 hr, p53 was immunoprecipitated, and immunoprecipitants (bottom) and cell lysates (top) were immunoblotted. p53 was immunoprecipitated from 500 μg protein of cell lysates after camptothecin treatment for 0, 2 and 4 hr in MCF-7 (C) and MDA-MB-231 cells (D) , followed by immunoblotting DDX3 and p53, Experiments were repeated at least two times with similar results.
Co-immunoprecipitation experiments were used to test if there is an association between DDX3 and p53. p53 was immunoprecipitated from wild-type and DDX3 knockdown MCF-7 cells, with and without stabilization of p53 with MG132 treatment (10 μM; 5 hr). These measurements showed that DDX3 co-immunoprecipitated with p53, and more DDX3 co-immunoprecipitated with p53 in cells with increased p53 levels caused by MG132 treatment (Figure 5B). We also tested if DDX3 associates with p53 following camptothecin-induced DNA damage. After treatment of MCF-7 cells with camptothecin for 2, and 4 hr, p53 accumulated and DDX3 co-immunoprecipitated with p53 (Figure 5C). These results suggest that DDX3 binding to p53 contributes to the accumulation of p53. Although, DDX3 interacted with mutant p53 in untreated MDA-MB-231 cells, the interaction was reduced by camptothecin treatment (Figure 5D). We hypothesize that DDX3 interacts with mutant p53 in the nucleus in the absence of DNA damage, but dissociation upon DNA damage allows DDX3 to translocate to death receptors where it attenuates caspase-8 activation and apoptotic signaling.
DDX3 promotes nuclear functional wild-type p53 accumulation and activates its downstream target p21 expression in response to DNA damage
Conversely to knocking down DDX3, we tested if overexpression of DDX3 using an adenovirus construct was capable of increasing p53 levels. For these experiments human neuroblastoma SH-SY5Y cells were used because nearly 100% of cells are infected with adenoviruses. In SH-SY5Y cells, which express functional wild-type p53, camptothecin treatment (10 μM; 4 hr) increased cytosolic and nuclear levels of p53 (Figure 6A). Overexpression of DDX3 caused a 2-fold increase in nuclear p53 after camptothecin treatment, but did not alter the cytosolic level of p53 (Figure 6B). Thus, DDX3 appears to be a limiting factor in regulating the extent of nuclear p53 accumulation after DNA damage. Furthermore, p21 as a well-characterized downstream target of p53 transactivation was examined in MCF-7 cells with depletion of DDX3 and in SH-SY5Y with overexpression of DDX3. The level of p21 was increased in response to DNA damage as expected, and this was abrogated by DDX3 depletion (Figure 6C) and further increased by DDX3 overexpression (Figure 6D).
Figure 6. DDX3 stabilizes nuclear p53and increases p21 in response to DNA damage.

(A) Cytosolic (left panels) and nuclear (right panels) fractions were prepared from SH-SY5Y wild-type (WT) cells and SH-SY5Y cells after DDX3 overexpression (OE) by transient adenovirus infection with or without camptothecin (10 μM; 4 hr) treatment, and immunoblotted for p53 and DDX3. β-tubulin and CREB were used as cytosol and nuclear loading controls, respectively. (B) p53 quantitative values are from basal and 4 hr camptothecin-treated cytosolic and nuclear samples in SH-SY5Y wild-type (WT) and DDX3 over-expressing (OE) cells. means ± SEM; n=3; *p<0.05 (C) Wild-type and DDX3 knockdown MCF-7 cells were treated with 10 μM camptothecin for 0, 2 and 3 hr, followed by p21 and DDX3 level measurements. (D) Wild-type SH-SY5Y cells and DDX3-overexpressing SH-SY5Y cells were treated with camptothecin for 0, 2 and 3 hr, and protein levels of p21 and DDX3 were measured. Experiments were repeated three times with similar results.
Discussion
Recent reports have linked DDX3 to signaling events important in carcinogenesis and chemotherapy, as discussed in the Introduction. Those links were extended in this study with the identification of two novel regulatory effects of DDX3 following camptothecin-induced DNA damage. First, DDX3 was found to oppositely regulate DNA damage-induced caspase activation in cells expressing functional wild-type p53 compared with cells that contain nonfunctional p53. Second, DDX3 was found to have a regulatory effect in controlling the extent of functional wild-type p53 accumulation after DNA damage. These findings reveal that DDX3 has previously unrecognized actions that regulate these key cellular responses to DNA damage.
Although DDX3 is well-established to modulate the extrinsic apoptotic signaling pathway (14, 15), we are unaware of any study testing if it also has a regulatory role in the intrinsic apoptotic pathway, two pathways that share in common many components. There was no effect of DDX3 depletion on caspase-3 activation induced by the kinase inhibitor staurosporine or the ER stressor thapsigargin in MCF-7 cells, demonstrating that common components of the two apoptotic signaling pathways are not targets of DDX3 for its regulation of death receptor-mediated apoptosis. This further supports the previously reported conclusion that in the extrinsic apoptotic pathway, DDX3 inhibits caspase activation by an anti-apoptotic action as part of a protein complex associated with death receptors (15). Thus, the components of apoptotic signaling that are common to both the extrinsic and intrinsic apoptotic signaling pathways appear not to be influenced by DDX3.
In contrast to the finding that caspase activation induced by staurosporine and thapsigargin was independent of DDX3, caspase-7 activation following DNA damage induced by camptothecin was clearly impeded by DDX3 depletion in MCF-7 cells, an effect opposite to the promotion of death receptor-induced apoptosis caused by DDX3 depletion. However, upon testing if this regulation of the response to DNA damage was an effect of DDX3 common to other cell types, we encountered the paradoxical finding that camptothecin-induced caspase-3 activation was enhanced by DDX3 depletion in both HeLa and MDA-MB-231 cells. Thus, DDX3 depletion had opposite effects in MCF-7 cells compared with HeLa and MDA-MB-231 cells on DNA damage-induced executioner caspase-3/7 activation. This paradox appears to be due to the differences in p53 in the cells. Whereas MCF-7 cells express functional wild-type p53, which has a key role in the induction of apoptosis following DNA damage, HeLa cells, MDA-MB-231 cells and BJAB cells contain nonfunctional p53, and only with functional wild-type p53 did DDX3 depletion reduce caspase activation, whereas with nonfunctional p53 DDX3 depletion increased caspase-3 activation in response to DNA damage. The latter effect, increased caspase activation associated with DDX3 depletion, appears to be partially due to the engagement of the extrinsic apoptotic pathway in these cells following DNA damage, as activation of the extrinsic apoptotic signaling-associated caspase-8 was also increased in cells with DDX3 depletion, in accordance with the previous finding that DDX3 is inhibitory towards extrinsic apoptotic signaling (15). Thus, knocking down DDX3 level in cells with mutant or non-functional p53 permits greater activation of the extrinsic apoptosis pathway after DNA damage than otherwise occurs.
Oppositely to results in cells with non-functional p53, in cells that contain functional wild-type p53, DDX3 depletion impaired both caspase activation and p53 accumulation. The mechanism for DDX3 promoting DNA damage-induced caspase activation in cells containing functional wild-type p53 appeared to be due to DDX3 participating in the regulation of the accumulation of p53 following DNA damage, as knocking down DDX3 reduced p53 levels, and DDX3 overexpression increased p53 levels following DNA damage. However, accumulation of p53 following inhibition of the proteasome with MG132 was independent of DDX3. Since DDX3 co-immunoprecipitated with p53, together these findings suggest that DDX3 binding to p53 promotes the accumulation of p53, but not the expression of p53 based on the unchanged p53 levels in MG132-treated cells with or without knockdown of DDX3. In support of this, overexpression of DDX3 was capable of increasing p53 levels even after p53 was stabilized by signals emanating from the DNA damage cascade. These findings add to recent reports indicating that DEAD box proteins have a variety of actions related to cell survival as well as to modulation of mRNA [8, 9, 15, 37-39]. Furthermore, another prominent DEAD box protein DDX5/p68 also has been shown to be involved in regulating cell proliferation, apoptosis, and p53 [40-44]. Thus, the repertoire of actions of DEAD box proteins extends beyond their classical actions of regulating mRNA to also include functions important in regulating signaling events important in cell survival and apoptosis.
In summary, the results reported here reveal that DDX3 regulates apoptotic signaling after DNA damage and contributes to the stabilization of functional wild-type p53. Since DDX3 associates with p53, the stabilization and nuclear accumulation of p53 following DNA damage, the effect of DDX3 is likely at least in part due to promoting nuclear retention of p53. Overall, these results further demonstrate that DDX3 plays important roles in regulating processes that are associated with carcinogenesis and chemotherapy.
Conclusion
This study found that DDX3 associates with p53, increases p53 accumulation, and positively regulates camptothecin-induced apoptotic signaling in cells expressing functional wild-type p53, whereas in cells expressing mutant p53 DDX3 inhibits activation of the extrinsic apoptotic pathway to reduce caspase activation. These results demonstrate that DDX3 not only regulates extrinsic apoptotic signaling, as previously reported, but also selectively regulates intrinsic apoptotic signaling following DNA damage.
Methods
Cell culture and materials
HeLa and MCF-7 cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 100 U/ml penicillin, 100 μg/ml streptomycin, and 15 mM HEPES, in humidified, 37°C chambers with 5% CO2. BJAB cells were grown in RPMI 1640 medium (Cellgro, Herndon, VA) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium-pyruvate, 2.5 g/L glucose, 100 U/ml penicillin and 100 μg/ml streptomycin. Human neuroblastoma SH-SY5Y cells were grown in 50% Minimum Essential Medium Eagle (MEM) (Cellgro, Herndon, VA) and 50% Kaighn’s Modification of Ham’s F-12 (ATCC) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Nuclei were extracted from cells with a nuclear extraction kit according to the manufacturer’s instructions (Active Motif). Sources of chemicals were: lithium chloride, camptothecin (Sigma, St. Louis, MO), thapsigargin, etoposide, staurosporine (Alexis Biochemicals, San Diego, CA), MG132 (Calbiochem, San Diego, CA). The following sources provided antibodies: caspase-8, cleaved, active caspase-3/7, cleaved 85 kDa fragment of poly(ADP-ribose) polymerase (PARP), p38, p-p38 and p-JNK (Cell Signaling Technology, Beverly, MA), β-actin (Sigma), p53 (Transduction Laboratories, Lexington, KY). DDX3 antibodies and DDX3 adenovirus were prepared in the laboratory of Dr. T. Zhou [12]. All horseradish peroxidase (HRP)-conjugated secondary reagents were purchased from Southern Biotechnology Associates (Birmingham, AL).
shRNA and expression
Lentiviral mediated shRNA was performed using shRNA lentiviral (pLKO.1-puro) plasmids (Sigma). The oligonucleotides containing the DDX3 target sequence that were used are: sequence #1, 5′-CCGGCCCTGCCAAACAAGCTAATATCTCGAGATATTAGCTTGTTTGGCAGGGTTTT and sequence #4, 5′-CCGGCGCTTGGAACAGGAACTCTTTCTCGAGAAAGAGTTCCTGTTCCAAGCGTTTT One 100 mm dish of 293FT cells (Invitrogen) was co-transfected with 3 μg of the pLKO.1-puro plasmids plus 3 μg each of the packaging vectors pLP1, pLP2, and pLP/VSVG (Invitrogen) using Lipofectamine 2000. The media was changed approximately 16 hr after transfection, and the cells were cultured an additional 48–72 hr. The media was then collected, centrifuged at 3000rpm for 5 min, and filtered through a 0.45 μm filter. Experimental cells were incubated with the virus-containing medium overnight in 6-well plates, the media was changed, and cells incubated for 24 hr. Cells were transferred to 100 mm dishes and infected cells were selected by incubation in puromycin (1 μg/ml). To overexpress DDX3, cells were infected with adenovirus in serum-free media for 30 min, the media was changed and cells grown for 48 hr, followed by experiment assays.
Immunoprecipitation and immunoblotting
After washing with PBS twice, cells were harvested in lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM sodium orthovanadate, 100 μM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 50 mM NaF, 1 nM okadaic acid, 1% Triton X-100, and 10% glycerol) and centrifuged at 20,800xg for 15 min. For immunoprecipitation of p53, cell lysates (500 μg protein) were incubated with anti-p53 antibody overnight, followed by incubation with protein G sepharose beads (GE Healthcare, Piscataway, NJ) for 2 hr. Beads were washed three times with 400 μl lysis buffer, resuspended in 40 μl of Laemmli buffer, and the denatured samples were run on 8-15% Tris/Glycine gels to detect the indicated proteins.
For immunoblotting, cells were washed twice with PBS, and lysed in lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM sodium orthovanadate, 100 μM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 50 mM NaF, 1 nM okadaic acid, 1% Triton X-100, and 10% glycerol). The lysates were incubated for 30 min on ice, centrifuged at 20,800 g for 15 min, and supernatants were collected. Protein concentrations were determined using the bicinchoninic method (Pierce, Rockford, IL). Cell lysates were mixed with Laemmli sample buffer and placed in a boiling water bath for 5 min. Proteins were resolved in SDS-polyacrylamide gels, and transferred to nitrocellulose. Blots were probed with the indicated antibodies, and were developed using horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence.
Highlights.
DDX3 promotes camptothecin-induced apoptosis in cells expressing wild-type p53.
DDX3 impedes apoptosis in cells with nonfunctional p53.
DDX3 associates with p53.
DDX3 promotes nuclear wild-type p53 accumulation and activates p21 expression.
Acknowledgements
The authors thank Dr. Ling Song for excellent technical support. This research was supported by grant MH038752 from the National Institutes of Health.
List of Abbreviations
- DDX3
DEAD box protein 3
- shRNA
short hairpin RNA
Footnotes
Competing interests The authors have no potential conflicts of interest
Author’s contributions SM carried out the experimental work, SM, TZ, EJ and RSJ contributed to the design, data analysis, and manuscript preparation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Abdelhaleem M, Maltais L, Wain H. The human DDX and DHX gene families of putative RNA helicases. Genomics. 2003;81:618–622. doi: 10.1016/s0888-7543(03)00049-1. [DOI] [PubMed] [Google Scholar]
- [2].Rocak S, Linder P. DEAD-box proteins: the driving forces behind RNA metabolism. Nat. Rev. Mol. Cell. Biol. 2004;5:232–241. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- [3].Tarn WY, Chang TH. The current understanding of Ded1p/DDX3 homologs from yeast to human. RNA Biol. 2009 Jan-Mar;6(1):17–20. doi: 10.4161/rna.6.1.7440. [DOI] [PubMed] [Google Scholar]
- [4].Owsianka AM, Patel AH. Hepatitis c virus core protein interacts with a human dead box protein DDX3. Virology. 1999;257:330–340. doi: 10.1006/viro.1999.9659. [DOI] [PubMed] [Google Scholar]
- [5].Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- [6].Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T, Kato N. DDX3 DEAD-Box RNA helicase is required for hepatitis C virus RNA replication. J. Virol. 2007;81:13922–13926. doi: 10.1128/JVI.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sekiguchi T, Iida H, Fukumura J, Nishimoto T. Human DDX3Y, the Y-encoded isoform of RNA helicase DDX3, rescues a hamster temperature-sensitive ET24 mutant cell line with a DDX3X mutation. Exp. Cell. Res. 2004;300:213–222. doi: 10.1016/j.yexcr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- [8].Chang PC, Chi CW, Chau GY, Li FY, Tsai YH, Wu JC, Lee YH. DDX3, a DEAD box RNA helicase, is deregulated in hepatitis virus-associated hepatocellular carcinoma and is involved in cell growth control. Oncogene. 2006;25:1991–2003. doi: 10.1038/sj.onc.1209239. [DOI] [PubMed] [Google Scholar]
- [9].Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, Lee YH. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res. 2006;66:6579–6588. doi: 10.1158/0008-5472.CAN-05-2415. [DOI] [PubMed] [Google Scholar]
- [10].Lai MC, Chang WC, Shieh SY, Tarn WY. DDX3 regulates cell growth through translational control of cyclin E1. Mol Cell Biol. 2010 Nov;30(22):5444–53. doi: 10.1128/MCB.00560-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wu DW, Liu WS, Wang J, Chen CY, Cheng YW, Lee H. Reduced p21(WAF1/CIP1) via alteration of p53-DDX3 pathway is associated with poor relapse-free survival in early-stage human papillomavirus-associated lung cancer. Clin Cancer Res. 2011 Apr 1;17(7):1895–905. doi: 10.1158/1078-0432.CCR-10-2316. [DOI] [PubMed] [Google Scholar]
- [12].Huang JS, Chao CC, Su TL, Yeh SH, Chen DS, Chen CT, Chen PJ, Jou YS. Diverse cellular transformation capability of overexpressed genes in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2004;315:950–958. doi: 10.1016/j.bbrc.2004.01.151. [DOI] [PubMed] [Google Scholar]
- [13].Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard P, Jr., Mukadam S, Van Diest P, Chen JH, Farabaugh P, Patel AH, Raman V. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene. 2008;27:3912–3922. doi: 10.1038/onc.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li Y, Wang H, Wang Z, Makhija S, Buchsbaum D, Lobuglio A, Kimberly R, Zhou T. Inducible resistance of tumor cells to tumor necrosis factor-related apoptosis-inducing ligand receptor 2-mediated apoptosis by generation of a blockade at the death domain function. Cancer Res. 2006;66:8520–8528. doi: 10.1158/0008-5472.CAN-05-4364. [DOI] [PubMed] [Google Scholar]
- [15].Sun M, Song L, Li Y, Zhou T, Jope RS. Identification of an anti-apoptotic protein complex at death receptors. Cell Death Differ. 2008;15:1887–1900. doi: 10.1038/cdd.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun M, Song L, Zhou T, Gillespie GY, Jope RS. The role of DDX3 in regulating Snail. Biochim Biophys Acta. 2011;1813(3):438–47. doi: 10.1016/j.bbamcr.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71:875–886. doi: 10.1016/0092-8674(92)90562-q. [DOI] [PubMed] [Google Scholar]
- [18].Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- [19].Bieging KT, Attardi LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012 Feb;22(2):97–106. doi: 10.1016/j.tcb.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kubbutat MH, Ludwig RL, Ashcroft M, Vousden KH. Regulation of Mdm2-directed degradation by the C terminus of p53. Mol. Cell Biol. 1998;18:5690–5698. doi: 10.1128/mcb.18.10.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- [22].Haupt Y, Robles AI, Prives C, Rotter V. Deconstruction of p53 functions and regulation. Oncogene. 2002;21:8223–8231. doi: 10.1038/sj.onc.1206137. [DOI] [PubMed] [Google Scholar]
- [23].Rüegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol. Sci. 1989;10:218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- [24].Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu. Rev. Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- [26].Wu GS. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther. 2004;3(2):156–161. doi: 10.4161/cbt.3.2.614. [DOI] [PubMed] [Google Scholar]
- [27].Yoon CH, Kim MJ, Park MT, Byun JY, Choi YH, Yoo HS, Lee YM, Hyun JW, Lee SJ. Activation of p38 mitogen-activated protein kinase is required for death receptor-independent caspase-8 activation and cell death in response to sphingosine. Mol Cancer Res. 2009;7(3):361–70. doi: 10.1158/1541-7786.MCR-08-0069. [DOI] [PubMed] [Google Scholar]
- [28].Houghton JA, Harwood FG, Tillman DM. Thymineless death in colon carcinoma cells is mediated via fas signaling. Proc. Natl. Acad. Sci. U.S.A. 1997;94:8144–8149. doi: 10.1073/pnas.94.15.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-κB and AP-1. Mol. Cell. 1998;1:543–551. doi: 10.1016/s1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
- [30].Tillman DM, Petak I, Houghton JA. A Fas-dependent component in 5-fluorouracil/leucovorin-induced cytotoxicity in colon carcinoma cells. Clin. Cancer Res. 1999;5:425–430. [PubMed] [Google Scholar]
- [31].Wen J, Ramadevi N, Nguyen D, Perkins C, Worthington E, Bhalla K. Antileukemic drugs increase death receptor 5 levels and enhance Apo-2L-induced apoptosis of human acute leukemia cells. Blood. 2000;96:3900–3906. [PubMed] [Google Scholar]
- [32].Nagane M, Pan G, Weddle JJ, Dixit VM, Cavenee WK, J. H. Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo. Cancer Res. 2000;60:847–853. [PubMed] [Google Scholar]
- [33].Poulaki V, Mitsiades CS, Mitsiades N. The role of Fas and FasL as mediators of anticancer chemotherapy. Drug Resist. Update. 2001;4:233–242. doi: 10.1054/drup.2001.0210. [DOI] [PubMed] [Google Scholar]
- [34].Peták I, Houghton JA. Shared pathways: death receptors and cytotoxic drugs in cancer therapy. Pathol. Oncol. Res. 2001;7:95–106. doi: 10.1007/BF03032574. [DOI] [PubMed] [Google Scholar]
- [35].Beurel E, Kornprobst M, Blivet-Van Eggelpoël MJ, Ruiz-Ruiz C, Cadoret A, Capeau J, Desbois-Mouthon C. GSK-3β inhibition by lithium confers resistance to chemotherapy-induced apoptosis through the repression of CD95 (Fas/APO-1) expression. Exp. Cell Res. 2004;300:354–364. doi: 10.1016/j.yexcr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- [36].Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GVW, Jope RS. Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fuller-Pace FV. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006;34:4206–4215. doi: 10.1093/nar/gkl460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rosner A, Rinkevich B. The DDX3 subfamily of the DEAD box helicases: divergent roles as unveiled by studying different organisms and in vitro assays. Curr. Med. Chem. 2007;14:2517–2525. doi: 10.2174/092986707782023677. [DOI] [PubMed] [Google Scholar]
- [39].Lee CS, Dias AP, Jedrychowski M, Patel AH, Hsu JL, Reed R. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res. 2008;36:4708–4718. doi: 10.1093/nar/gkn454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kahlina K, Goren I, Pfeilschifter J, Frank S. p68 DEAD box RNA helicase expression in keratinocytes. Regulation, nucleolar localization, and functional connection to proliferation and vascular endothelial growth factor gene expression. J. Biol. Chem. 2004;279:44872–44882. doi: 10.1074/jbc.M402467200. [DOI] [PubMed] [Google Scholar]
- [41].Bates GJ, Nicol SM, Wilson BJ, Jacobs AM, Bourdon JC, Wardrop J, Gregory DJ, Lane DP, Perkins ND, Fuller-Pace FV. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J. 2005;24:543–553. doi: 10.1038/sj.emboj.7600550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 2007;67:7572–7578. doi: 10.1158/0008-5472.CAN-06-4652. [DOI] [PubMed] [Google Scholar]
- [43].Yang L, Lin C, Zhao S, Wang H, Liu ZR. Phosphorylation of p68 RNA helicase plays a role in platelet-derived growth factor-induced cell proliferation by up-regulating cyclin D1 and c-Myc expression. J. Biol. Chem. 2007;282:16811–16819. doi: 10.1074/jbc.M610488200. [DOI] [PubMed] [Google Scholar]
- [44].Yang L, Lin C, Sun SY, Zhao S, Liu ZR. A double tyrosine phosphorylation of P68 RNA helicase confers resistance to TRAIL-induced apoptosis. Oncogene. 2007;26:6082–6092. doi: 10.1038/sj.onc.1210427. [DOI] [PubMed] [Google Scholar]