

Summary
The enzyme Arylsulfatase B (ARSB; N-acetylgalactosamine-4-sulfatase) removes 4-sulfate groups from chondroitin-4-sulfate and dermatan sulfate and is required for the degradation of these sulfated glycosaminoglycans (GAGs). Since these GAGs accumulate in patients with Cystic Fibrosis (CF), we investigated the activity of ARSB in leukocytes of patients with CF, to consider if reduced activity of ARSB might contribute to the pathophysiology of CF. Previous cell-based experiments had demonstrated that when the deficiency of the cystic fibrosis transmembrane regulator (CFTR) was corrected in bronchial epithelial cells, the ARSB activity increased significantly. De-identified, citrated blood samples were collected from 16 children with cystic fibrosis and 31 control subjects, seen in the Pediatric Clinic at Rush University Medical Center. Polymorphonuclear (PMN) and mononuclear cell (MC) populations were separated by density gradient, and blinded determinations of ARSB activity were performed using the exogenous substrate 4-methylumbilliferyl sulfate. Interleukin-6 was measured in the plasma samples by ELISA. ARSB activity was significantly less in the PMN and MC from the CF patients than controls (p<0.0001, unpaired t-test, two-tailed). Interleukin-6 levels in plasma were significantly greater in the CF population (p<0.001). Mean age, age range, and male:female ratio of CF patients and controls were similar, and no association of ARSB activity with age, gender, or CFTR genotype was evident. Since recombinant human ARSB is used successfully for replacement therapy in Mucopolysaccharidosis VI, it may be useful to restore ARSB activity to normal levels and increase degradation of sulfated GAGs in CF patients.
Introduction
In patients with cystic fibrosis (CF), defective cystic fibrosis transmembrane conductance regulator (CFTR) impairs chloride transport across the apical surface of epithelial cells. Since the discovery of the CF gene in 1989, more than 1875 mutations have been reported in CFTR, but CFTR gene replacement therapy has not emerged as a viable therapeutic option [1,2]. Prior to determination of CFTR mutations as the cause of CF, CF was often considered in relation to the Mucopolysaccharidosis (lysosomal storage diseases), due to consistent observations of increased sulfated glycoconjugates in cells and secretions of CF patients [3,4]. Findings included accumulation of dermatan sulfate and chondroitin sulfate in cultured skin fibroblasts of CF patients [5]. Both dermatan sulfate (DS) and chondroitin-4-sulfate (C4S) are sulfated glycosaminoglycans (sGAG) that are metabolized by Arylsulfatase B (ARSB; N-acetylgalactosamine 4-sulfatase), which removes the sulfate group from C4 of the N-acetylgalactosamine-4-sulfate residue at the non-reducing end and is required for GAG degradation [6]. Previously, we hypothesized that ARSB deficiency had a critical role in the clinical manifestations of CF [7], and demonstrated that correction of defective CFTR leads to increased ARSB activity in cultured bronchial epithelial cells [8], and that reduced secretion of IL-8 and impaired neutrophil migration follow ARSB silencing in human bronchial epithelial cells [9].
ARSB is a member of the highly conserved family of sulfatase enzymes and can regulate the degradation of C4S and DS [10,11]. Experiments with CF cell lines demonstrated that ARSB activity was ~40% higher when defective CFTR was repaired by successful transfection [8]. Following silencing of ARSB in bronchial epithelial cells, the cellular content of sGAG and of chondroitin-4-sulfate was significantly increased, consistent with profound impact of ARSB on the sGAG abundance [9,12]. Other imaging and activity studies have demonstrated that ARSB is not only a lysosomal enzyme, but also localizes to the cell membrane [9,13-17]. ARSB requires post-translational modification for activity, with a conserved cysteine residue (C91) converting to a formylglycine (oxo-alanine) derivative to properly configure the active site [18]. Mucopolysaccharidosis VI or Maroteaux-Lamy-Syndrome, which arises from congenital deficiency of functional ARSB is being successfully treated with ARSB replacement therapy [19-21].
ARSB activity was observed to be deficient in lymphoid cell lines from two patients with CF in the 1970s [22], and investigators in Italy reported that ARSB activity was reduced in whole blood samples of 57 children with CF, compared to 181 control samples [23]. In this report, we present precise measurements of ARSB activity in the polymorphonuclear leukocytes and mononuclear cells of children with cystic fibrosis, compared to control subjects, including healthy controls, controls with asthma, and controls with other conditions.
Methods
Recruitment of study participants
Study participants were enrolled from the Out-Patient Pediatric clinics at Rush University Medical Center. Study enrollment procedures were approved by the Institutional Review Boards of Rush University Medical Center (RUMC) and the University of Illinois at Chicago (UIC). Cystic fibrosis patients were 2-17 years of age and were followed in the Rush Cystic Fibrosis Center Out-Patient clinic at Rush University. Patients 8 years and above signed assent forms, and parents of all subjects signed consent forms, permitting collection of peripheral blood samples. Control subjects included children with known underlying diagnoses and children with no known illness. Patients with acute asthmatic episodes or acute inflammatory conditions were excluded from participation. Venous blood was collected in citrated tubes and processed within two hours of collection. Blood samples were labeled with a numerical code that did not distinguish CF patients from control subjects. A registry linking patient identifiers with blood sample codes was developed, and relevant clinic information was collected from the patient charts, on-line CF database (PortCF), and incorporated into the study database.
Separation of leukocytes on gradient
Polymorphonuclear (PMN) and mononuclear (MC) white blood cells were separated from whole blood using the Polymorphprep™ kit (AXIS-SHIELD, Oslo, Norway). Each 2 ml citrated whole blood sample was carefully layered over 2 ml of Polymorphprep solution in a 5 ml centrifuge tube. Tubes were centrifuged at 500g at 25°C for 40 minutes. The blood components separated by density, with plasma at the top, then the mononuclear cells (MC), including monocytes and lymphocytes, then polymorphonuclear leukocytes (PMN), and with red blood cells at the bottom of the tube. The MC and PMN fractions were collected separately from the two distinct bands between plasma and RBCs. In four of the CF patients, distinct bands were not identified, so a single leukocyte sample was analyzed. Hepes-buffered saline (0.85% NaCl 10 mM pH 7.4) was diluted 50% with water and added to an equal volume of PMN and of MC, and centrifuged at 500g at 25°C for 20 minutes. The pellets of PMN and MC were then resuspended in 3 ml of RBC lysis buffer at 37°C for 7 minutes with occasional shaking to remove residual erythrocyte contamination. The purified PMN and MC were harvested and stored at -80°C until further analysis.
Measurement of Arylsulfatase B Activity
Blinded analysis of arylsulfatase B (ARSB) activity was determined by a fluorometric assay, following a standard protocol, using 20 μl of cell homogenate, 80 μl of assay buffer (0.05 M Na-acetate buffer, pH 5.6), and 100 μl of substrate [5 mM 4-methylumbilliferyl sulfate (MUS)] in assay buffer) combined in a microplate well, as reported previously [8,24]. The microplate was incubated for 30 min at 37°C, and the reaction was stopped by adding 150 μl of stop buffer (glycine–carbonate buffer, pH 10.7). Fluorescence was measured at 360 nm (excitation) and 465 nm (emission). Activity (expressed as nmol/mg protein/hour) was derived from a standard curve for prepared with known quantities of 4-methylumbilliferyl at pH 5.6.
Measurement of Interleukin-6 by ELISA
The concentration of Interleukin (IL)-6 in the plasma samples was measured by using the Quantikine ELISA kit for human IL-6 (R&D Systems, Minneapolis, MN). The IL-6 in the samples was captured into the wells of a microtiter plate pre-coated with specific anti-IL-6 monoclonal antibody. Immobilized IL-6 was then detected by a biotinylated second antibody and streptavidin-horseradish peroxidase (HRP)-conjugate with the use of the chromogenic substrate hydrogen peroxide/tetramethylbenzidine (TMB). Color intensity was read at 450 nm with a reference filter of 570 nm in an ELISA plate reader (FLUOstar, BMG LABTECH, Inc., Cary, NC). The IL-6 concentrations were extrapolated from a standard curve plotted by using known concentrations of IL-6, and are expressed as picograms per milliliter plasma (pg/ml).
Experiments in normal human bronchial/tracheal epithelial cells
Primary, normal human bronchial/tracheal epithelial cells were obtained [Clonetics® NHBE with retinoic acid, Lonza, Walkersville, MD] and grown in the recommended bronchial epithelial cell growth media (BEGM®, Lonza) following established procedures [9,13]. When cells reached 80% confluence, they were harvested with ReagentPack Subculture Reagents™ (Lonza) and sub-cultured. Cells were transfected with small interfering (si) RNA for ARSB (150 ng; 0.6 μl; Qiagen, Valencia, CA) or by control siRNA in 100 μl serum-free culture medium and 12 μl of HiPerfect Transfection Reagent (Qiagen) for 24 hours, as previously detailed [9,12]. Effective knockdown was confirmed by Western blot and by ARSB activity assay which indicated decline of ~81%. For ARSB silencing, the siRNA sequences were:
Sense: 5′- GGGUAUGGUCUCUAGGCAtt - 3′
Antisense: 5′- UUGCCUAGAGACCAUACCCtt - 3′
For the negative control, the siRNA sequences were:
Sense: 5′- UUCUCCGAACGUGUCACGUtt - 3′
Antisense: 5′- ACGUGACACGUUCGGAGAAtt -3′
Spent media were collected for IL-6 determinations to be performed by ELISA, as described above. Chondroitin-4-sulfate (C4S) was measured in control, control siRNA- and ARSB siRNA-treated cell lysates by sulfated GAG assay (Blyscan™, Biocolor Ltd, Newtownabbey, Northern Ireland), using labeled 1,9-dimethylmethylene blue, as previously reported [9,12,13,25]. Antibody to native chondroitin-4-sulfate (Abnova, Walnut, CA) at a concentration of 1 μg/mg cell protein was added to the cell lysates in tubes, and the tubes were rotated overnight in a shaker at 4°C, and processed as previously reported [9,12,13]. Pre-washed Protein L-Agarose beads (100 μl; SCBT, Santa Cruz, CA) were added to each tube, and the tubes were incubated overnight at 4°C. The beads were washed three times with PBS containing Protease Inhibitor Cocktail, and the precipitate was eluted with dye-free elution buffer and subjected to the Blyscan assay in the presence of excess unbound dye. The cationic dye and GAG at acid pH produced an insoluble dye-GAG complex, and the GAG content was determined by the amount of dye that is recovered from the test sample following exposure to Blyscan Dissociation Reagent. Absorbance maximum of 1,9-dimethylmethylene blue is 656 nm. Concentration is expressed as μg / mg protein of cell lysate. Dye-GAG complex is proportional (1:1) to the sulfate content for C4S.
Statistical analysis
Study data were compiled using coded identifiers, then stratified by case or control following linkage to the clinical data file, while maintaining confidentiality of subject identity. Measurements are presented as mean ± standard deviation (S.D.). Statistical significance of differences between CF patients and controls was determined using unpaired t-tests, two-tail (Instat3, GraphPad Software, San Diego, CA), unless stated otherwise. In figures and tables, **** indicates p<0.0001, *** indicates p≤0.001, ** p≤0.01 and * p≤0.05.
Results
Patient characteristics
Sixteen patients with cystic fibrosis (CF), ranging in ages from 6.5 -17.5 years with mean age 12.4 (± 4.0) were enrolled. Thirty-one control subjects, including 11 healthy controls, 10 patients with bronchial asthma, and 10 non-asthmatic patients with other medical diagnoses, and ranging in age from 3.5 -17 years with mean age of 10.8 (± 4.3), were enrolled. Summary data for age and gender are presented in Table 1. Differences in mean age were not significant.
Table 1.
Summary of Patient Characteristics - Age and Gender
| Cystic Fibrosis | All Controls | Controls: No disease | Controls: Asthma | Controls: Other Diagnosis | |
|---|---|---|---|---|---|
| Number | 16 | 31 | 11 | 10 | 10 |
| Age range | 6.5 - 17.5 | 3.5 - 17 | 5 - 16 | 3.5 - 16.5 | 3.5 - 17 |
| Mean Age | 12.4 (4.0) | 10.8 (4.3) | 11.4 (3.8) | 9.6 (3.7) | 11.2 (5.6) |
| Gender - M:F | 8:8 | 19:12 | 5:6 | 8:2 | 6:4 |
Arylsulfatase B activity in CF patients and controls
Arylsulfatase B (ARSB) activity was determined in the polymorphonuclear cells (PMN) and the mononuclear cells (MC) of the CF patients and the control subjects. In four CF patients, separate determinations of PMN and MC ARSB activity could not be performed, and a single WBC value is reported. Table 2 presents the ARSB data, the CFTR genotypes of the CF patients, and the status of the control subjects (no diagnosis, diagnosis of asthma, or diagnosis of other medical condition).
Table 2.
Subject Data
| Gender | Age (yrs) | PMN ARSBa (nmol/mg protein/h) | MC ARSBb (nmol/mg protein/h) | WBC ARSBc (nmol/mg protein/h) | IL-6d (pg/ml) | Other |
|---|---|---|---|---|---|---|
| CYSTIC FIBROSIS | ||||||
| Male | 10.5 | 52.3 | 44.7 | 63.9 | ΔF508/ΔF508 | |
| Female | 6.5 | 48.4 | 63.8 | ΔF508/ΔF508 | ||
| Male | 14.5 | 52.0 | 46.2 | 66.6 | ΔF508/G551D | |
| Female | 9 | 44.1 | 50.9 | 60.8 | 2307insA/3120+1G>A | |
| Female | 17.5 | 49.8 | 53.7 | 73.8 | NA | |
| Male | 17.5 | 57.1 | 53.0 | 73.8 | ΔF508/ΔF508 | |
| Female | 15.5 | 46.0 | 61.3 | ΔF508/1717-1G>A | ||
| Male | 6.5 | 52.1 | 68.6 | ΔF508/ΔF508 | ||
| Male | 16.5 | 53.6 | 52.2 | 66.2 | ΔF508/ΔF508 | |
| Female | 9 | 60.2 | 53.7 | 66.8 | S1255x(2)/11203V(2) | |
| Female | 15 | 42.9 | 69.4 | ΔF508/ΔF508 | ||
| Male | 17 | 50.1 | 58.1 | 64.3 | R74W/D1270N/(TG)11-5T/(TG)11-7T | |
| Female | 9 | 44.4 | 48.0 | 58.2 | ΔF508/ΔF508 | |
| Male | 8.5 | 59.7 | 48.7 | 65.2 | ΔF508/312011G>A | |
| Female | 10.5 | 53.8 | 58.3 | 75.1 | ΔF508/312011G>A | |
| Male | 15 | 45.5 | 48.2 | 50.0 | F508del/P74W/D1270N/P798S/G921E | |
| CONTROLS | ||||||
| Female | 5 | 67.4 | 66.4 | 4.8 | Neuromuscular | |
| Female | 4.5 | 72.7 | 64.8 | 5.1 | Down Syndrome | |
| Female | 17 | 71.7 | 60.2 | 4.3 | Down Syndrome; renal | |
| Male | 3.5 | 83.8 | 63.6 | 4.4 | Neuromuscular | |
| Male | 12 | 71.0 | 67.6 | 4.6 | Trauma | |
| Male | 16 | 68.8 | 59.4 | 5.8 | Neuromuscular | |
| Male | 7 | 82.8 | 59.4 | - | Heme/Onc | |
| Male | 17 | 78.7 | 55.8 | 5.3 | Renal | |
| Female | 16.5 | 76.9 | 65.2 | 4.1 | Renal | |
| Male | 12.5 | 69.7 | 66.6 | 6.3 | Renal | |
| Male | 8 | 78.0 | 56.2 | 5.0 | None | |
| Female | 8 | 69.6 | 67.9 | 5.6 | None | |
| Female | 5 | 66.8 | 64.0 | 4.8 | None | |
| Female | 11 | 77.9 | 68.6 | 5.8 | None | |
| Male | 14 | 85.5 | 61.2 | 5.0 | None | |
| Female | 9 | 77.6 | 65.3 | 5.0 | None | |
| Female | 16 | 67.4 | 64.0 | 5.0 | None | |
| Female | 9 | 75.9 | 57.4 | 4.7 | None | |
| Male | 16 | 81.9 | 58.8 | 5.4 | None | |
| Male | 14 | 81.3 | 69.1 | 6.2 | None | |
| Male | 15.5 | 73.0 | 62.9 | 3.6 | None | |
| Male | 7.5 | 73.9 | 57.2 | 13.5 | Asthma | |
| Male | 11 | 62.5 | 69.9 | 18.2 | Asthma | |
| Female | 11 | 70.1 | 65.3 | 13.8 | Asthma | |
| Female | 8.5 | 81.3 | 56.8 | 11.4 | Asthma | |
| Male | 9 | 53.1 | 55.6 | 12.2 | Asthma | |
| Male | 3.5 | 60.0 | 47.6 | 17.8 | Asthma | |
| Male | 5.5 | 68.6 | 39.0 | 13.0 | Asthma | |
| Male | 12 | 84.4 | 45.8 | 24.9 | Asthma | |
| Male | 12 | 68.1 | 51.4 | 26.6 | Asthma | |
| Male | 16.5 | 56.3 | 47.8 | 26.6 | Asthma |
No differences in ARSB activity for the PMN or the MC were present in relation to age (Fig. 1A, 1B) or gender (Fig. 1C, 1D), for the study participants, including the CF patients and the control subjects. No association between ARSB activity and specific genotype was apparent for the CF patients.
Figure 1. No association between ARSB activity and age or gender.

A. Distribution of ARSB activity in PMN vs. age of all subjects indicates no relationship between ARSB activity and age.
B. Distribution of ARSB activity in MC vs. age of all subjects indicates no relationship between ARSB activity and age.
C. Distribution of ARSB activity in PMN vs. gender indicates no relationship between ARSB activity and gender.
D. Distribution of ARSB activity in MC vs. gender indicates no relationship between ARSB activity and gender. [ARSB=arylsulfatase B; PMN=polymorphonuclear leukocytes; MC=mononuclear cells]
Mean ARSB activity was significantly less in the CF patients than in the total control subjects for both PMN and MC (p<0.001) (Table 3). In the controls, including the no disease controls (n=11), the asthmatic controls (n=10), and the other disease controls (n=10), ARSB activity in the PMNs was significantly higher than in the CF patients (p<0.001). The CF patients all had PMN ARSB activity less than 61 nmol/mg protein/hr, and the non-asthmatic controls all had PMN ARSB activity greater than 66 nmol/mg protein/hr (Fig. 2A).
Table 3.
Summary of ARSB Activity
| Cystic Fibrosis | All Controls | Controls: No Disease | Controls: Asthma | Controls: Other Diagnosis | |
|---|---|---|---|---|---|
| ARSB Activity ± (S.D.) [nmol/mg protein/h] | |||||
| Neutrophils | |||||
| Mean | 51.9 (5.5)**** | 73.0 (8.2) | 75.9 (6.1) | 67.8 (10.2)* | 74.3 (5.8) |
| Range | 44.1 - 60.2 | 53.1 - 85.5 | 66.8 - 85.5 | 53.1 - 84.4 | 67.4 - 83.8 |
| Mononuclear Cells | |||||
| Mean | 51.3 (4.3)#### | 60.0 (7.6) | 63.2 (4.4) | 53.6 (9.3)## | 62.9 (3.9) |
| Range | 44.7 - 58.3 | 39.0 - 69.9 | 56.2 - 69.1 | 39.0 - 69.9 | 55.8 - 67.6 |
for p<0.0001, unpaired t-test, two-tail, cystic fibrosis (n=12) compared to total controls, no disease, and other disease, and p=0.0002 compared to asthma
for p<0.05, unpaired t-test, two-tail, asthma compared to no disease
for p<0.0001, unpaired t-test, two-tail, cystic fibrosis (n=12) compared to no disease and other disease, and p=0.0006 compared to total controls. NS between cystic fibrosis and asthma.
for p<0.01, unpaired t-test, two-tail, for asthma compared to no disease and to other disease.
Figure 2. Differences between ARSB Activity in PMN and MC in Control and CF Subjects.
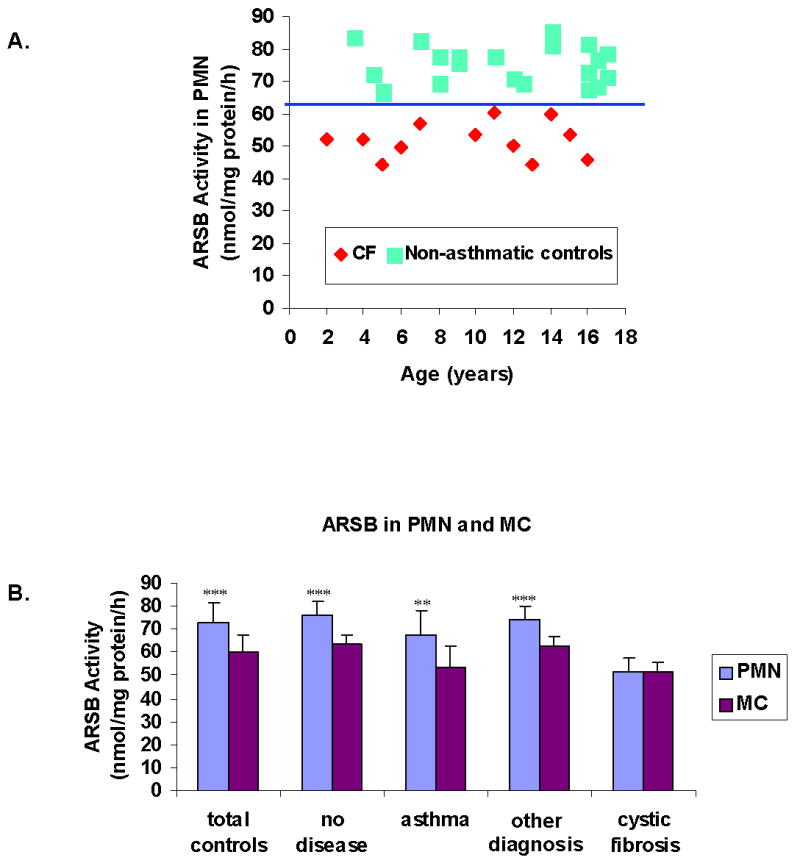
A. ARSB activity in the PMN of the CF patients is less in all cases than in the non-asthmatic control subjects, with a break at 61 nmol/mg protein/hr. [ARSB=arylsulfatase B; PMN=polymorphonuclear leukocytes; CF=cystic fibrosis]
B. In the total controls, the no disease controls, the other disease controls, and the asthma controls, mean ARSB activity is significantly higher in the PMN than in the MC (p<0.001, p<0.01 for asthma). In contrast in the CF patients, no significant difference is demonstrated between the mean PMN and mean MC ARSB activity. [ARSB=arylsulfatase B; PMN=polymorphonuclear leukocytes; MC=mononuclear cells; CF=cystic fibrosis]
In the control subjects, mean ARSB activity in the PMN was significantly higher than in the MC (p<0.001 all controls, no disease, and other disease, and p<0.01 for asthma controls) (Fig. 2B). In contrast, in the CF patients, there was no difference between the mean ARSB activity in PMN (51.9 ± 5.5 nmol/mg protein/h) vs. the MC (51.3 ± 4.3 nmol/mg protein/h) in the twelve samples in which distinct fractions were obtained. For the non-asthmatic controls, PMN ARSB activity was higher than MC ARSB activity for each of the subjects (Table 2).
Arylsulfatase B activity in asthma controls
The ARSB activity in the MC of the asthmatic patients was significantly reduced in comparison to the no disease and other disease controls (Table 3) (p<0.01). Also, in contrast to the finding in the non-asthmatic controls, in some of the asthmatic patients, the ARSB activity in the PMN was not greater than in the MC, although the mean PMN ARSB activity in the group of asthmatic patients was significantly higher than the MC ARSB activity (Fig. 2B) (p<0.01).
Plasma Interleukin-6 Activity in CF patients and controls
Plasma Interleukin (IL)-6 activity was markedly increased in the CF patients, compared to both the non-asthmatic and asthmatic controls (p<0.001) (Fig. 3A, Table 4). The IL-6 activity in the asthmatic subjects was also increased, relative to the non-asthmatic controls.
Figure 3. IL-6 in CF patients and control subjects.
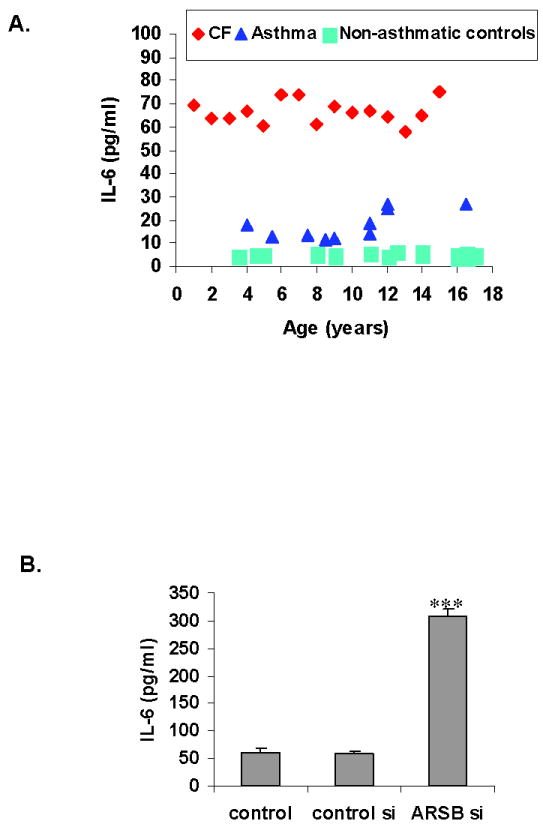
A. IL-6 activity is significantly greater in the CF patients than in the asthmatic and non-asthmatic control populations (p<0.001) with a mean value of 65.6 ± 6.3 pg/ml. In the asthmatic patients, mean IL-6 was 17.8 ± 6.1 pg/ml, significantly greater than in the non-asthmatic control population (p<0.001)
B. When ARSB was silenced by siRNA in bronchial epithelial cells in cell culture, marked increase in the IL-6 secreted into the spent media was observed (p<0.001, one-way ANOVA with Tukey-Kramer post-test), consistent with the observed increases in IL-6 in (A) when ARSB activity is less.
Table 4.
Summary of IL-6 Values
| Cystic Fibrosis | All Controls | Controls: No Disease | Controls: Asthma | Controls: Other Diagnosis | |
|---|---|---|---|---|---|
| Interleukin-6 pg/ml ± (S.D.) | |||||
| Mean | 65.5 (6.3)*** | 9.3 (7.0) | 5.1 (0.7) | 17.8 (6.1)### | 5.0 (0.7) |
| Range | 50.0 - 75.1 | 3.6 - 26.6 | 3.6 - 6.2 | 11.4 - 26.6 | 4.1 - 6.3 |
for p<0.001, unpaired t-test, two-tail, cystic fibrosis compared to each of the other groups.
for p<0.001, unpaired t-test, two-tail, asthma compared to no disease and other disease.
Consistent with this finding, in experiments with primary human bronchial epithelial cells in cell culture, when ARSB was silenced by small interfering (si) RNA, the IL-6 that was released into the media increased significantly (Fig. 3B), compared to media from untreated cells or cells treated with control siRNA. In these cell-based experiments, ARSB activity declined ~80%, with an associated increase to ~2.3 times the baseline level of chondroitin-4-sulfate, as previously reported [9,25].
Discussion
The enzyme ARSB is emerging as an important enzyme in regulation of vital cellular functions, due to its regulation of sulfation and degradation of chondroitin-4-sulfate (C4S). By increasing the sulfate content of the C4S and inhibiting C4S degradation, ARSB deficiency can affect the C4S binding with Interleukin-8, and thereby the secretion of IL-8 [9]. IL-8 establishes a chemotactic gradient for neutrophils, so increased cellular sequestration and reduced secretion of IL-8 when ARSB activity is reduced, may lead to increased neutrophil adherence to the bronchial epithelium, such as associated with biofilm formation [26,27]. Hence, ARSB deficiency may contribute to both the accumulation of sulfated GAGs in the tissues and secretions of patients with CF and to impaired neutrophil responsiveness due to reduced IL-8 secretion, consistent with an important role in mediating the pathophysiology of CF. Also, since Pseudomonas has sulfatase activity [28], the increase in sulfated GAGs due to ARSB deficiency may provide an enriched environment for Pseudomonas growth, consistent with the proclivity of CF patients to have persistent Pseudomonas colonization.
The findings of this report present clear evidence that ARSB activity is reduced in the PMN and MC of patients with CF, compared to controls. The decline of ARSB activity in the PMN is greater than in the MC, and the mean PMN ARSB activity is equivalent to the mean MC ARSB activity in the CF patients. In contrast, in the controls, the mean PMN ARSB activity is significantly greater than the mean MC ARSB activity. Unexpectedly, study results also demonstrated reduced ARSB activity in the leukocytes of patients with asthma, compared to the no disease and other diagnosis controls. The decline in ARSB activity in the asthmatic subjects is more significant in their MC than in the PMNs. Also, plasma IL-6 level was increased in the CF patients to seven times the level of the controls, and in the asthmatic patients to 3.6 times the level in the non-asthmatic control subjects. The finding of increased IL-6 in cultured primary bronchial/tracheal epithelial cells when ARSB is silenced supports this observed clinical association between reduced ARSB activity and increased IL-6.
Since chondroitin-4-sulfation impacts upon the attachment of plasmodia in the placenta and in the microcirculation [29,30], and ARSB is required for removal of sulfate groups and the degradation of C4S, we hypothesize that other cellular mechanisms may have evolved to regulate the critically important ARSB activity. Mutations in the CFTR gene affect the regulation of chloride secretion, and the CFTR mutations associated with CF generally lead to a non-functional CFTR in which the mutated protein is either unable to be translocated to its correct position on the cell membrane or when localized at the cell membrane is unable to properly regulate the chloride channel [31,32]. It is pertinent that the sulfatase modifying factor (SUMF) [also known as the formylglycine modifying enzyme (FGE)] is an endoplasmic reticulum enzyme that is required for the formylglycine (oxo-alanine) modification of ARSB, and of other sulfatases, and is essential for their biological activity. The SUMF has a halide binding cleft that appears to influence its reactivity [33]. This suggests a potential site of interaction between CFTR and ARSB activation, since the CFTR effects on chloride may inhibit ARSB post-translational modification. Anionic effects on ARSB activity are well-known, and chloride, sulfate, sulfite, and phosphate have been reported to inhibit ARSB activity [8,34,35]. Since the etiologic association between mutations of the CFTR and the clinical manifestations of CF is well-established by genetic analysis, but the functional impact of defective CFTR on the specific pathophysiology that is characteristic of CF is not well-explained, elucidation of the molecular interactions between CFTR and SUMF and ARSB may be highly informative to our understanding of CF pathogenesis.
Replacement therapy of deficient ARSB in CF may provide a new opportunity for effective intervention and for improved health status of patients with CF. In MPS VI patients treated with recombinant human ARSB, improved FEV1.0 and FVC were reported [21], suggesting that these parameters, as well as other measures of improved health, might also improve in patients with CF, and possibly other respiratory disorders, including asthma. Few adverse effects of ARSB replacement therapy have occurred, encouraging our consideration and hope that replacement therapy with ARSB will emerge as a new therapeutic option for CF.
References
- 1.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC. Identification of the Cystic Fibrosis gene: Cloning and characterization of complementary DNA. Science. 1090;245(4922):1066–73. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Mueller C, Flotte TR. Gene therapy for cystic fibrosis. Clin Rev Allergy Immunol. 2008;35(3):164–178. doi: 10.1007/s12016-008-8080-3. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Doranz B, Yankaskas JR, Engelhardt JF. Genotypic analysis of respiratory mucous sulfation defects in cystic fibrosis. J Clin Invest. 1995;96:2997–3004. doi: 10.1172/JCI118372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng PW, Boat TF, Cranfill K, Yankaskas JR, Boucher RC. Increased sulfation of glycoconjugates by cultured nasal epithelial cells from patients with cystic fibrosis. J Clin Invest. 1989;84:68–72. doi: 10.1172/JCI114171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matalon R, Dorfman A. Acid mucopolysaccharides in cultured fibroblasts of cystic fibrosis of the pancreas. Biochem Biophys Res Commun. 1968;33:954–958. doi: 10.1016/0006-291x(68)90405-1. [DOI] [PubMed] [Google Scholar]
- 6.Litjens T, Hopwood JJ. Mucopolysaccharidosis type VI: Structural and clinical implications of mutations in N-acetylgalactosamine-4-sulfatase. Hum Mutat. 2001;18(4):282–295. doi: 10.1002/humu.1190. [DOI] [PubMed] [Google Scholar]
- 7.Tobacman JK. Does deficiency of arylsulfatase B have a role in cystic fibrosis? Chest. 2003;123:2130–2139. doi: 10.1378/chest.123.6.2130. [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharyya S, Look D, Tobacman JK. Increased arylsulfatase B activity in cystic fibrosis cells following correction of CFTR. Clin Chim Acta. 2007;380:122–127. doi: 10.1016/j.cca.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 9.Bhattacharyya S, Solakyildirim K, Zhang Z, Linhardt RJ, Tobacman JK. Cell-bound IL-8 increases in bronchial epithelial cells following arylsulfatase B silencing. Am J Respir Cell Mol Biol. 2010;42(1):51–61. doi: 10.1165/rcmb.2008-0482OC. [DOI] [PubMed] [Google Scholar]
- 10.Glaser JH, Conrad HE. Chondroitin SO4 catabolism in chick embryo chondrocytes. J Biol Chem. 1979;254(7):2316–2325. [PubMed] [Google Scholar]
- 11.deSousa JF, Nader HB, Dietrich CP. Sequential degradation of chondroitin sulfate in molluscs. J Biol Chem. 1990;265(33):20150–20155. [PubMed] [Google Scholar]
- 12.Bhattacharyya S, Kotlo K, Shukla S, Danziger RS, Tobacman JK. Distinct effects of N-acetylgalactosamine-4-sulfatase and galactose-6-sulfatase expression on chondroitin sulfate. J Biol Chem. 2008;283(15):9523–9530. doi: 10.1074/jbc.M707967200. [DOI] [PubMed] [Google Scholar]
- 13.Bhattacharyya S, Solakyildirim K, Zhang Z, Linhardt RJ, Tobacman JK. Chloroquine reduces arylsulfatase B activity and increases chondroitin 4-sulfate: Implications for mechanisms of action and resistance. Malaria J. 2009;8(1):303. doi: 10.1186/1475-2875-8-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharyya S, Tobacman JK. Arylsulfatase B regulates colonic epithelial cell migration by effects on MMP9 expression and RhoA activation. Clin Exp Metastasis. 2009;26(6):535–545. doi: 10.1007/s10585-009-9253-z. [DOI] [PubMed] [Google Scholar]
- 15.Prabhu S, Bhattacharyya S, Guzman G, Macias V, Kajdacsy-Balla A, Tobacman JK. Arylsulfatase B in normal and malignant colonic epithelium. J Histochem Cytochem. 2011;59(3):328–335. doi: 10.1369/0022155410395511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharyya S, Kotlo K, Danziger RS, Tobacman JK. Role of arylsulfatase B regulation of chondroitin-4-sulfate and kininogen interaction: Implications for blood pressure regulation. Biochim Biophys Acta. 2010;1802(5):472–477. doi: 10.1016/j.bbadis.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 17.Mitsunaga-Nakatsubo K, Kusunoki S, Kawakami H, Akasaka K, Akimoto Y. Cell-surface arylsulfatase A and B on sinusoidal endothelial cells, hepatocytes, and Kupffer cells in mammalian livers. Med Mol Morphol. 2009;42(2):63–69. doi: 10.1007/s00795-009-0447-x. [DOI] [PubMed] [Google Scholar]
- 18.Roeser D, Preusser-Kunze A, Schmidt B, Gasow K, Wittmann JG, Dierks T, von Figura K, Rudolph MG. A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme. Proc Natl Acad Sci USA. 2006;103(1):81–86. doi: 10.1073/pnas.0507592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harmatz P, Whitley CB, Waber L, Pais R, Steiner R, Plecko B, Kaplan P, Simon J, Butensky E, Hopwood JJ. Enzyme replacement therapy in Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) J Pediatr. 2004;144(5):574–580. doi: 10.1016/j.jpeds.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 20.Harmatz P, Giugliani R, Schwartz IV, Guffon N, Teles EL, Miranda MC, Wraith JE, Beck M, Arash L, Scarpa M, Ketteridge D, Hopwood JJ, Plecko B, Steiner R, Whitley CB, Kaplan P, Yu ZF, Swiedler SJ, Decker C MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for Mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine ASB. Mol Genet Metab. 2008;95(4):469–475. doi: 10.1016/j.ymgme.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Harmatz P, Yu ZF, Giugliani R, Schwartz IV, Guffon N, Teles EL, Miranada MC, Wraith JE, Beck M, Arash L, Scarpa M, Ketteridge D, Hopwood JJ, Plecko B, Steiner R, Whitley CB, Kaplan P, Swiedler SJ, Hardy K, Berger KI, Decker C. Enzyme replacement therapy for Mucopolysaccharidosis VI: Evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis. 2010;33(1):51–60. doi: 10.1007/s10545-009-9007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beratis NG, Turner BM, Weiss R, Hirschhorn K. Arylsulfatase B deficiency in Maroteaux-Lamy Syndrome: Cellular studies and carrier identification. Pediatr Res. 1975;9:475–480. doi: 10.1203/00006450-197505000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Ferrero GB, Pagliardini S, Veljkovic A, Porta F, Bena C, Tardivo I, Restagno G, Silengo MC, Bignamini E. In vivo specific reduction of arylsulfatase B enzymatic activity in children with Cystic Fibrosis. Mol Genet Metab. 2008;94:39. doi: 10.1016/j.ymgme.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 24.Bhattacharyya S, Tobacman JK. Steroid sulfatase, arylsulfatases A and B, galactose 6-sulfatase, and iduronate sulfatase in mammary cells and effects of sulfated and non-sulfated estrogens on sulfatase activity. J Steroid Biochem Mol Biol. 2006;103(1):20–34. doi: 10.1016/j.jsbmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharyya S, Tobacman JK. Hypoxia reduces arylsulfatase B activity and silencing arylsulfatase B replicates and mediates the effects of hypoxia. PLoS One. 2012;7(3):e33250. doi: 10.1371/journal.pone.0033250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Medina R, Dunne WM, Singh PK, Brody SL. Pseudomonas aeruginosa acquires biofilm-like properties within airway epithelial cells. Infect Immun. 2005;73:8298–8305. doi: 10.1128/IAI.73.12.8298-8305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreau-Marquis S, Stanton BA, O’Toole GA. Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm Pharmacol Ther. 2008;21:595–599. doi: 10.1016/j.pupt.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hummerjohann J, Laudenbach S, Rétey J, Leisinger T, Kertesz MA. The sulfur-regulated arylsulfatase gene cluster of Pseudomonas aeruginosa, a new member of the cys regulon. J Bacteriol. 2000;182(7):2055–2058. doi: 10.1128/jb.182.7.2055-2058.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Achur RN, Valiyaveettil M, Gowda DC. The low sulfated chondroitin sulfate proteoglycans of human placenta have sulfate group-clustered domains that can efficiently bind Plasmodium falciparum-infected erythrocytes. J Biol Chem. 2003;278:11705–11713. doi: 10.1074/jbc.M211015200. [DOI] [PubMed] [Google Scholar]
- 30.Muthusamy A, Achur RN, Bhavanandan VP, Fouda GG, Taylor DW, Gowda DC. Plasmodium falciparum-infected erythrocytes adhere both in the intervillous space and on the villous surface of human placenta by binding to the low-sulfated chondroitin sulfate proteoglycan receptor. Am J Pathol. 2004;164:2013–2025. doi: 10.1016/S0002-9440(10)63761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuthbert AW. New horizons in the treatment of cystic fibrosis. Br J Pharmacol. 2011;163(1):173–183. doi: 10.1111/j.1476-5381.2010.01137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma M, Benharounga M, Hu W, Lukacs GL. Conformation and temperature-sensitive stability defects of the ΔF508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem. 2001;276(12):8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- 33.Roeser D, Schmidt B, Preusser-Kunze A, Rudolph MG. Probing the oxygen-binding site of the human formylglycine generating enzyme using halide ions. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 5):621–627. doi: 10.1107/S0907444907009961. [DOI] [PubMed] [Google Scholar]
- 34.Wojczyk B. Lysosomal arylsulfatases A and B from horse blood leukocytes: purification and physico-chemical properties. Biol Cell. 1986;57(2):147–152. doi: 10.1111/j.1768-322x.1986.tb00471.x. [DOI] [PubMed] [Google Scholar]
- 35.Rao GJ, Christe ME. Inhibition of rabbit liver arylsulfatase B by phosphate esters. Biochim Biophys Acta. 1984;788(1):58–61. doi: 10.1016/0167-4838(84)90297-8. [DOI] [PubMed] [Google Scholar]