

Abstract
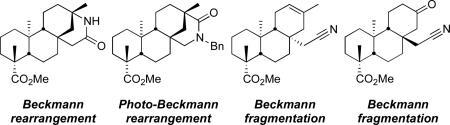
We have applied a diversity-oriented approach for the synthesis of skeletally diverse and stereochemically complex templates for small-molecule library production by performing Beckmann rearrangement and Beckmann fragmentation reactions on the bicyclo[3.2.1]octane rings of steviol and isosteviol, aglycones derived from the diterpene natural product stevioside. The optimization of these two reaction pathways is presented along with the successful application of a photo-Beckmann rearrangement. This work also led to the discovery of cyano-Prins-type and Thorpe-Ziegler-type cyclization reactions.
Major issues associated with the development of high-value information-rich small molecule libraries are achieving skeletal diversity, stereochemical complexity,1 and mining areas of biologically relevant chemical space.2 Natural products provide a solid platform for the discovery of biologically active small molecules.3 It has been suggested that natural product-derived libraries should provide high screening hit rates because natural products have been evolutionarily molded by protein domains, and are therefore likely to engage in interactions with conserved protein folds across protein families.4 To date, the systematic exploration of many regions of natural product chemical space has not been possible due to the scarcity of accessible material. Steviol (1, Figure 1), however, is readily available from the natural sweetener stevioside (5, Scheme 1)5 and an attractive template because stevioside (5) and its aglycones steviol (1) and isosteviol (6, Scheme 1) have shown diverse pharmacological activities.6 Potentially, this scaffold could also provide access to templates representative of the large and diverse family of diterpenes derived from the methylerythritol 4-phosphate pathway,7 and subsequent metabolic processes. Representative diterpenes from this family include gibberellic acid derivative GA-13315 (2), oridonin (3), and cafestol (4) with antiangiogenic,8 antitumor,9 and neuroprotective properties,10 respectively (Figure 1). These compounds have attracted much attention from the synthetic organic chemistry community and therefore, a large amount of literature has been produced over the last eight decades pertaining to the synthesis11 and structural modification of stevioside (5)12 and structurally related diterpenes.13
Figure 1.

Representative examples of diterpenes.
Scheme 1.

Access to Steviol (1) and Isosteviol (6)
We decided to employ the Beckmann rearrangement for ring expansion chemistry and the Beckmann fragmentation for ring cleavage reactions on the stevioside aglycones steviol (1) and isosteviol (6) for efficient generation of templates for library production.
Steviol (1) was obtained through a well-precedented enzyme mediated hydrolysis of stevioside (5).14,15 The D-ring isomer isosteviol (6) was obtained directly through a modification of existing methods and proceeds via a Wagner-Meerwein rearrangement (Scheme 1) of steviol (1).16 Initially we sought to access diverse heterocyclic intermediates through manipulation of the D-ring ketone of isosteviol (6, Scheme 2).
Scheme 2.

Beckmann Fragmentation and Rearrangement
Although the Beckmann rearrangement has previously been reported17 regarding an analogous substrate (the N-methyloxime), the lactam was sufficiently attractive to warrant further investigation. Methylation of the carboxylic acid moiety of isosteviol (6) under standard conditions delivered methyl ester 7. The ketone function of 7 was converted to the corresponding oxime 8 in 93% yield on treatment with hydroxylamine and potassium acetate. Reaction with thionyl chloride as reported17 then delivered nitrile 9, the corresponding Beckmann fragmentation product, in 65% yield and lactam 10 in 27% yield. The Beckmann fragmentation is well precedented18 in systems with a quaternary α-carbon. This pathway significantly retarded the yield of lactam 10 if fresh thionyl chloride was not used. Use of fresh thionyl chloride provided nitrile 9 in 11% yield and lactam 10 in 55% yield. A more robust two-step procedure is outlined in Scheme 3. The conversion of the oxime hydroxyl group in 8 to the mesylate followed by treatment with acid in methanol exclusively led to the desired lactam 10 in 87% yield. It is of interest that this procedure appears to shut down the Beckmann fragmentation. We propose that this reaction proceeds via a tetrahedral intermediate in a similar fashion to that described by White et al., in which the less sterically crowded anti-stereoisomer favors the migration of the bridgehead carbon in lactam formation.19 Lactam 10 was subsequently alkylated with methyl iodide and benzyl bromide to furnish compounds 12 and 13, respectively. Despite being relatively hindered, this amide lends itself well to alkylation and should therefore prove useful in the development of small-molecule libraries of lactam derivatives.
Scheme 3.

Optimized Beckmann Rearrangement of 11
Next, we decided to access the regioisomeric lactam through the photolysis of an oxaziridine (Scheme 4).20 Ketone 7 was converted to imines 14 through heating in the presence of benzylamine under dehydrating conditions.21 The imines were formed in a 7:1 ratio (determined by 1H NMR) in favor of the expected E-isomer – vide infra. The imines 14 were subsequently epoxidized to furnish the oxaziridines 15 in 85% over the two steps. Photolysis of the oxaziridines (254 nm, Hg lamp) then delivered lactam 16 in 56% yield as well as the regioisomer 13 in 7% yield. As with the Beckmann rearrangement, where the bond that migrates is that which is anti to the oxygen on nitrogen, the outcome of the photo-Beckmann is also stereoelectronically defined. As a general rule, the bond that migrates is the one anti to the lone pair on the nitrogen.22 Therefore, the isolation of the regioisomeric lactam 16 confirms the assignment of the E-imine 14 as the major isomer.
Scheme 4.

Formation of Lactam 16
Having established synthetic routes to the N-alkylated isomeric lactams 13 and 16, we recognized that nitrile 9 is also an attractive template for library design. However, in order to generate useful quantities of 9, the Beckmann fragmentation pathway needed to be optimized (Scheme 5).
Scheme 5.

Optimized Beckmann Fragmentation of Isosteviol Oxime 8
A similar fragmentation was recently reported by the Coates group for a closely related substrate employing TsCl in DMF as the reagent, but these reaction conditions delivered a 2:1 mixture of the alkenes, which required a difficult separation.23 Modification of reaction conditions through conversion of the oxime hydroxyl group in 8 to the corresponding acetate followed by treatment with pTsOH in acetonitrile at 90 °C cleanly delivered nitriles 9 and 17 in 84% overall yield and in an 8:1 ratio of alkenes (determined by 1H NMR). Through a single crystallization from dichloromethane and ethyl acetate, this mixture could be enriched to 20:1 in favor of the Δ6-alkene. Interestingly, treating oxime 8 with Ac2O, followed by reaction with pTsOH in benzene as the solvent at reflux delivered a 2:0.2:1 ratio of nitriles 9/17 to lactam 10. This suggests that the solvent plays an important role in the stabilization of the intermediates leading to either the lactam or nitrile. Attempts to drive the equilibrium to further favor the Δ6-alkene (Scheme 5) unexpectedly led to the formation of bicyclo[2.2.2]octane 19 (36%) and lactone 18 (37%). The former most likely proceeds through a cyano-Prins-type cyclization, while the latter is derived through hydration of 17 followed by cyclization (Figure 2).
Figure 2.

Proposed mechanisms for the formation of 18, 19, and 23.
After having established the chemistry of the isosteviol system, we turned our attention to the steviol (1) scaffold since it seemed reasonable that a similar fragmentation would occur in this ring system (Scheme 6). Again, methylation of the acid function under standard conditions delivered the methyl ester (96%), which was subsequently treated with Ac2O to provide acetate 20 (85%). The exo-methylene group in 20 was then ozonized to deliver the corresponding ketone in 67% yield. The ketone was converted to the oxime 21 in 90% yield and then treated with Ac2O and pTsOH in acetonitrile at 90 °C to deliver the expected nitrile 22 in 67% yield (Scheme 6). More vigorous heating in toluene initiated a Thorpe-Ziegler-type cyclization delivering the bicyclo[2.2.2]octane 23.24 The formation of 23 proceeds presumably in an analogous fashion to 19 (Figure 2).
Scheme 6.

Beckmann Fragmentation of Steviol Derivative 21
In conclusion, we devised practical methods to access a number of diverse chemotypes, possessing high stereochemical complexity, and as single enantiomers from stevioside, which is readily available in kg quantities. We have shown a different approach toward the optimization of the Beckmann rearrangement of isosteviol to form lactam derivative 10 as the exclusive reaction product. Lactam 10 provides a classical point for diversification via N-alkylation. The regioisomeric lactams of type 16 can be obtained from ketone 7 as shown in Scheme 4 by reaction with diverse amines. Alternatively, a library can be prepared by removal of the N-benzyl group of 16, followed by N-alkylation. Additionally, we demonstrated that the Beckmann fragmentation of isosteviol- and steviol-derived compounds form nitrile derivatives. The nitriles 9 and 22 will allow double functionalization via the ester and nitrile functional groups. The formation of 19 via a cyano-Prins-type cyclization and of 23 by a Thorpe-Ziegler-type reaction have not been previously reported. These types of reactions are underrepresented in the literature.
Supplementary Material
Acknowledgement
We gratefully acknowledge financial support from the National Institutes of Health Grant GM081267 and the University of Minnesota through the Vince and McKnight Endowed Chairs.
Footnotes
Supporting Information. Experimental details and copies of 1H and 13C NMR spectra for compounds 6 - 10, 12 – 13, 15, 16, and 18 - 23. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Burke MD, Berger EM, Schreiber SL. Science. 2003;302:613. doi: 10.1126/science.1089946.Thomas GL, Wyatt EE, Spring DR. Curr. Opin. Drug. Discovery Dev. 2006;9:700.Mao S, Probst D, Werner S, Chen J, Xie X, Brummond KM. J. Comb. Chem. 2008;10:235. doi: 10.1021/cc7001843. For a recent related approach see: Huigens RW, III, Morrison KC, Hicklin RW, Flood TA, Jr., Richter MF, Hergenrother PJ. Nat. Chem. 2013;5:195. doi: 10.1038/nchem.1549.
- 2.Feher M, Schmidt JM. J. Chem. Inf. Comput. Sci. 2003;43:218. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 3.Newman DJ. J. Med. Chem. 2008;51:2589. doi: 10.1021/jm0704090. [DOI] [PubMed] [Google Scholar]
- 4.Breinbauer R, Vetter IR, Waldmann H. Angew. Chem. Int. Ed. 2002;41:2878. doi: 10.1002/1521-3773(20020816)41:16<2878::AID-ANIE2878>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 5.Hanson JR, De Oliveira BH. Nat. Prod. Rep. 1993;10:301. doi: 10.1039/np9931000301. [DOI] [PubMed] [Google Scholar]
- 6.Brahmachari G, Mandal LC, Roy R, Mondal S, Brahmachari AK. Arch. Pharm. Chem. Life Sci. 2011;1:5. doi: 10.1002/ardp.201000181. [DOI] [PubMed] [Google Scholar]
- 7.Brandle JE, Telmer PG. Phytochemistry. 2007;68:1855. doi: 10.1016/j.phytochem.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Zhang H, Chen J, Zhao H, Zeng X, Zhang H, Qing C. Invest New Drugs. 2012;30:8. doi: 10.1007/s10637-010-9501-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhou GB, Kang H, Wang L, Gao L, Liu P, Xie J, Zhang FX, Weng XQ, Shen ZX, Chen J, Gu LJ, Yan M, Zhang DE, Chen SJ, Wang ZY, Chen Z. Blood. 2007;109:3441. doi: 10.1182/blood-2006-06-032250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trinh K, Andrews L, Krause J, Hanak T, Lee D, Gelb M, Pallanck L. J Neurosci. 2010;30:5525. doi: 10.1523/JNEUROSCI.4777-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a Cook IF, Knox JR. Tetrahedron Lett. 1970:4091. [Google Scholar]; b Mori K, Nakahara Y, Matsui M. Tetrahedron Lett. 1970:2411. [Google Scholar]; c Nakahara Y, Mori K, Matsui M. Agr. Biol. Chem. 1971;35:918. [Google Scholar]; d Mori K, Nakahara Y, Matsui M. Tetrahedron. 1972;28:3217. [Google Scholar]; e Ziegler FE, Kloek JA. Tetrahedron. 1977;33:373. [Google Scholar]; f Ogawa T, Nozaki M, Matsui M. Tetrahedron. 1980;36:2641. [Google Scholar]
- 12.a Coates RM, Bertram EF. Tetrahedron Lett. 1968:5145. [Google Scholar]; b Coates RM, Bertram EF. J. Org. Chem. 1971;36:2625. [Google Scholar]; c Terauchi T, Asai N, Doko T, Taguchi R, Takenaka O, Sakurai H, Yonaga M, Kimura T, Kajiwara A, Niidome T, Kume T, Akaike A, Sugimoto H. Bioorg. Med. Chem. 2007;15:7098. doi: 10.1016/j.bmc.2007.07.037. [DOI] [PubMed] [Google Scholar]
- 13.a Wang F-P, Liang X-T. Alkaloids Chem. Biol. 2002;59:1. doi: 10.1016/s0099-9598(02)59008-8. [DOI] [PubMed] [Google Scholar]; b Mander LN. Nat. Prod. Rep. 2003;20:49. doi: 10.1039/b007744p. [DOI] [PubMed] [Google Scholar]; c Hanson JR. Nat. Prod. Rep. 2007;24:1332. doi: 10.1039/b705951p. [DOI] [PubMed] [Google Scholar]
- 14.Stevioside can be obtained in kilogram quantities from several vendors.
- 15.Pezzuto JM, Compadre CM, Swanson SM, Nanayakkara NPD, Kinghorn AD. Proc. Natl. Acad. Sci. U.S.A. 1985;82:2478. doi: 10.1073/pnas.82.8.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avent AG, Hanson JR, De Oliviera BH. Phytochemistry. 1990;29:2712. [Google Scholar]
- 17.Militsina OI, Kovyljaeva GI, Bakaleynik GA, Strobykina IY, Kataev VE, Alfonsov VA, Musin RZ, Beskrovny DV, Litvinov IA. Mendeleev Commun. 2005:27. [Google Scholar]
- 18.Gawley RE. Org. React. 1988;35:1. [Google Scholar]
- 19.White JD, Hrncliar P, Stappenbeck F. J. Org. Chem. 1999;64:7871. [Google Scholar]
- 20.Judd WR, Katz CE, Aube J. Science of Synthesis. 2005;21:133. [Google Scholar]
- 21.Al'fonsov VA, Andreeva OV, Bakaleinik GA, Beskrovnyi DV, Gubaidullin AT, Kataev VE, Kovylyaeva GI, Konovalov AI, Korochkina MG, Litvinov IA, Militsina OI, Strobykina IY. Russ. J. Gen. Chem. 2003;73:1255. [Google Scholar]
- 22.Aube J. Chem. Soc. Rev. 1997;26:269. [Google Scholar]
- 23.Roy A, Roberts FG, Wilderman PR, Zhou K, Peters RJ, Coates RM. J. Am. Chem. Soc. 2007;129:12453. doi: 10.1021/ja072447e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Compound 23 has been prepared previously by a different route. Mori K, Nakahara Y, Matsui M. Tetrahedron. 1972;28:3217.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.