

Abstract
Mitochondrial dysfunction and associated oxidant stress have been linked with numerous complex diseases and aging largely by in vitro determination of mitochondria oxidant production and scavenging. We applied targeted in vivo fluorescence analyses of mitochondria-dense pharyngeal tissue in C. elegans to better understand relative mitochondrial effects, particularly on matrix oxidant burden, of respiratory chain complex, MnSOD, and insulin receptor mutants displaying variable longevity. The data demonstrate significantly elevated in vivo matrix oxidant burden in the short-lived complex I mutant, gas-1(fc21), which was associated with limited superoxide scavenging capacity despite robust MnSOD induction, as well as decreased mitochondria content and membrane potential. Significantly increased MnSOD activity was associated with in vivo matrix oxidant levels similar to wild-type in the long-lived respiratory chain complex III mutant, isp-1(qm150). Yet, despite greater superoxide scavenging capacity in the complex III mutant than in the significantly longer-lived insulin receptor mutant, daf-2(e1368), only the former showed modest oxidative stress sensitivity. Furthermore, increased longevity was seen in MnSOD knockout mutants (sod-2(ok1030) and sod-2(gk257)) that had decreased MnSOD scavenging capacity and increased in vivo matrix oxidant burden. Thus, factors beside oxidant stress must underlie RC mutant longevity in C. elegans. This work highlights the utility of the C. elegans model as a tractable means to non-invasively monitor multi-dimensional in vivo consequences of primary mitochondrial dysfunction.
Keywords: Complexes I, II, and III; MnSOD; membrane potential; MitoSOX; TMRE; fluorescence microscopy
1. INTRODUCTION
Oxidative stress resulting from increased production of reactive species and/or concomitant decline in antioxidant scavenging capacity may damage proteins, lipids, nucleic acids and other cellular structures (Valko et al. 2006). Such multi-faceted cellular damage may contribute to many sporadic (Schapira 2006) and inherited (DiMauro 2004) mitochondrial diseases and aging (Rea et al. 2007). Up to 2% of total oxygen consumed by the mitochondrial respiratory chain (RC) in in vitro studies, and 0.2% under more physiologic conditions (Balaban et al. 2005), has been shown to be improperly reduced to generate superoxide, which is then released into the matrix primarily from complex I (Murphy 2009) or intermembrane space exclusively from complex III (Chen et al. 2003; Murphy 2009). Superoxide dismutase (SOD) offers a primary oxidant defense through rapid conversion of superoxide radicals into hydrogen peroxide and oxygen. Three human SOD genes localize to different subcellular compartments, where SOD2 encodes the manganese SOD that functions within the mitochondria matrix.
Assessing the relative balance of oxidant production and scavenging in vivo remains a significant challenge (Yang et al. 2007). The development of small, lipophilic molecules that emit fluorescence only in the oxidized form has permitted a method of semi-quantitative evaluation of oxidant burden in cellular model systems. In particular, mitochondria-specific oxidant levels can be assessed using the fluorescent probe, MitoSOX Red, a lipophilic hydroethidine (HE) derivative that accumulates 100- to 1000-fold within mitochondria due to charge attraction of its triphenylphosphonium cation through the mitochondria membrane bilayers into the negatively-charged mitochondria matrix (Robinson et al. 2008). Mitochondria-generated oxidants react with MitoSOX to yield two primary fluorescent products, a 2-hydroxyethidium derivative (2-OH-Mito-E+) resulting from superoxide oxidation, and Mito-E+, a non-specific oxidized product (Zielonka et al. 2008). Reliable interpretation of fluorescence analyses that rely upon mitochondria membrane potential (DeltaPsi(m) or ΔΨm) for dye distribution may be subject to diminished or enhanced dye uptake, although several fluorescent dyes are now commonly used for in vitro ΔΨm assessment (O’Reilly et al. 2003). Membrane potential indicator dyes have recently been applied in vivo in the model animal, C. elegans (Gaskova et al. 2007; Zuryn et al. 2008).
C. elegans offers a facile model in which to study a host of in vivo mitochondria functions. Nematodes incorporate ingested fluorescent dyes into their cells and are optically transparent, which permits precise assessment of tissue and cellular dye localization. Furthermore, several well-characterized C. elegans strains harboring mutations in nuclear genes encoding mitochondrial proteins are easily accessible (www.wormbase.org). However, assessing mitochondria functions by quantitative analysis of whole animal fluorescence may be limited by non-specific binding of lipophilic, fluorescence-based reagents, particularly within the lipid-rich granules of the gastrointestinal tract (Clokey and Jacobson 1986). Thus, we sought to identify C. elegans tissues having high metabolic activity that might offer a focus for the further study of mitochondria physiology. C. elegans actively transports and grinds food (i.e., bacteria) between its mouth and intestines through a muscle-tube like pharynx, which consists largely of anterior and terminal pharyngeal bulbs (PB) joined by an isthmus (Altun and Hall 2008). The pharynx shares several similarities with the mammalian heart including near-continuous and intrinsic myogenic electrical activity (up to 250 beats per minute) (Avery and Horvitz 1989), electrical coupling between muscle cells (Starich et al. 1996), calcium-based action potentials (Shtonda and Avery 2005), and high mitochondria density (Altun and Hall 2008). Capitalizing on these anatomical features and the existence of well-characterized C. elegans mutants, we evaluated the relative in vivo mitochondria oxidant production and mitochondria membrane potential (ΔΨm) in RC and insulin receptor mutants displaying variable longevity, as well as in MnSOD mutants with primary impairment of mitochondria superoxide scavenging capacity. We demonstrate that C. elegans offers a powerful model in which to assess the in vivo, multi-dimensional consequences of mitochondrial dysfunction, as well as to explore the complex relationship between oxidant stress and longevity under varying genetic and environmental conditions.
2. METHODS
2.1 Strains growth and maintenance
C. elegans worm strains were obtained from the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN) (Table 1). Strains studied included wild-type (N2 Bristol), three mitochondrial RC mutants in complexes I, II, and III [gas-1(fc21), mev-1(kn1), isp-1(qm150), respectively], and the insulin receptor mutant (daf-2 (e1368)). All superoxide scavenging (MnSOD) mutant strains studied harbored loss-of-function mutations, including two sod-2 alleles [sod-2(gk257) is homozygous for a complex mutation characterized by an 18 basepair insertion and 159 basepair deletion; sod-2(ok1030) has a 900 basepair homozygous deletion] and a single allele of sod-3 [sod-3(gk235) is homozygous for a complex rearrangement characterized by a 6 basepair insertion and 390 basepair deletion] (www.wormbase.org). Nematodes were grown at 20°C on nematode growth media (NGM) plates spread with OP50 E. coli. For all fluorescent studies, synchronous young adult populations were obtained by bleaching gravid adults on NGM plates (Hope 1999), plating the recovered eggs onto unspread NGM plates overnight, and then transferring L1-arrested animals the next day to NGM plates spread with OP50 E. coli. Upon reaching adulthood (defined by the presence of eggs laid on the plate), nematodes were washed off in S. basal (3.4 g KH2PO4, 4.4 g K2HPO4, 5.85 g NaCl, 1 liter water, pH 7.0) and adults were separated from eggs by gravity. For mass spectrometry study of whole worm aliquots (see below) as well as for mitochondria isolation, nematodes were grown at 20°C on 20 large NGM plates spread with OP50 E. coli for two generations, and then grown in 1 liter liquid culture flasks with K12 E. coli at 200 rpm in shaking 20°C incubators (I26/R Series, New Brunswick Scientific, Edison, NJ). Worms were harvested from liquid culture, as previously described (Falk 2006, Hope 1999).
Table 1. C. elegans strain description, homology, and lifespan.
The role of in vivo oxidant stress on longevity was studied in three RC subunit missense mutants, three MnSOD knockout mutants, and an insulin receptor missense mutant. FUDR-based median lifespan assessment was largely consistent with previously reported lifespan findings in these strains. * indicates concurrent wild-type (N2 Bristol) control for MnSOD mutant lifespan comparison. All other strains were concurrently studied with the unmarked wild-type (N2 Bristol) control. RC, respiratory chain. Lifespan plots and animal number studied for each mutant are provided in Supplementary Figure 4.
| C. elegans Strain Name | Gene Function and Human Homologue | Mutation Type | Human-Worm Protein Similarity | Lifespan (20°C) | |||
|---|---|---|---|---|---|---|---|
| Interpretation | Median | Mean | Maximum | ||||
| N2 Bristol | - | wild-type | - | vs. RC Mutants vs. MnSOD mutants* |
14 17 |
16.9 19.9 |
21 23 |
| gas-1 (fc21) | Complex I subunit (NDUFS2) | missense | 83.4% | short-lived | 12 | 15.6 | 20 |
| mev-1 (kn1) | Complex II subunit (SDH-C) | missense | 75.3% | unchanged | 14 | 16.4 | 22 |
| isp-1 (qm150) | Complex III subunit (UQCRFS1) | missense | 97.5% | long-lived | 20 | 22.4 | 31 |
| daf-2 (e1368) | Insulin receptor (IR) | missense | 62.0% | long-lived | 31 | 31.1 | 38 |
| sod-2 (gk257) | Mitochondrial MnSOD (SOD2) - Cel chr. X | knockout | 97.3% | long-lived* | 19 | 20.7 | 24 |
| sod-2 (ok1030) | Mitochondrial MnSOD (SOD2) - Cel chr. X | knockout | 97.3% | long-lived* | 20 | 21.6 | 25 |
| sod-3 (gk235) | Mitochondrial MnSOD (SOD2) - Cel chr. I | knockout | 97.2% | long-lived* | 18.5 | 20.0 | 23 |
2.2 Lifespan assessment
Animals were maintained at 20°C throughout the experiment. Synchronized nematode cultures were initiated by bleaching young adults to obtain eggs. Collected eggs were allowed to hatch overnight on 10 cm unspread NGM plates, after which L1-arrested larvae were transferred to 10 cm NGM plates spread with OP50 E. coli. Upon reaching the first day of egg laying, synchronous young adults were moved to fresh 3.5 cm NGM plates seeded with OP50 E. coli (lifespan experiment “Day 0”). For purposes of higher-throughput lifespan screening by preventing offspring from nematodes under study from reaching adulthood, fluorodeoxyuridine (FUDR) was added to OP50-spread NGM plates to a final concentration of 100 ug/ml (Hamilton et al. 2005). 40 to 60 nematodes were studied per strain, divided on two 3.5 cm NGM plates. Mortality was confirmed by stimulating nematodes lightly with a platinum wire; nematodes that did not move after stimulation were scored as dead and removed from the plate. Worms that died of protruding/bursting vulva, bagging, or crawling off the agar were censored. All lifespan studies were performed by a single technician (J.O.). Median, mean, and maximal lifespan were determined for each strain relative to concurrently studied wild-type worms.
2.3 Relative quantitation of mitochondria superoxide levels by fluorescence microscopy
All fluorescent studies were performed at 20°C in dim light. An initial time course experiment was carried out in wild-type (N2 Bristol) worms to assess the optimal duration of dye exposure in living animals. Terminal PB mean fluorescence intensity was determined in synchronous young adult nematodes following 12, 36, and 60 hours of 10 uM MitoSOX exposure with or without paraquat (either at a low dose of 200 uM or a high dose of 20 mM). The relative contribution of E. coli to observed fluorescence was assessed through comparison of nematodes fed OP50 that was live or killed by UV exposure for 30 minutes. Fluorescence microscopy analysis and quantitation was performed on an Olympus IX70 fluorescence microscope with a HQ500/30x Q530LP HQ610/75m cube set (CHROMA Technology Group, Rockingham, VT), using the Metamorph image acquisition and analysis software package (Molecular Devices Corp, Downingtown, PA). Two additional time-course experiments were subsequently performed in N2 Bristol nematodes fed 10 uM MitoSOX Red using the same microscope and software detailed below to confirm that similar peak fluorescence occur following 24 hour exposures regardless of analysis platform.
Synchronous young adult stage worm populations of each strain were grown on NGM plates spread with OP50 E. coli and either 10 uM MitoSOX Red (Molecular Probes, Eugene, OR) alone or with 1 mM paraquat (Sigma, St. Louis, MO) to induce oxidant stress (Bus and Gibson 1984; Cocheme and Murphy 2009). Following 24-hour incubation, nematodes were transferred to fresh NGM plates spread with OP50 E. coli for one hour to clear their guts of residual dye. Living nematodes were paralyzed in situ by directly adding 1 mg/ml levamisole to NGM agar plates. Photographs were taken immediately in a dark room at 160X magnification and 2 second exposure time using a CY3 fluorescence cube set (MZFLIII, Leica, Bannockburn, IL) with a Cool Snap cf2 camera (Nikon, Melville, NY). Subsequently, the terminal PB was manually circled to quantify the region’s mean intensity using NIS Elements BR imaging software (Nikon, Melville, NY). A baseline CY3 intensity value near 0 represents the minimum quantifiable terminal bulb intensity in animals not previously exposed to MitoSOX Red. All MitoSOX analyses were performed by two technicians (S.D. and R.L.); observer influence on mean strain fluorescence intensity was negligible as assessed by ANOVA (see Section 2.12).
2.4 Mass spectrometric validation of 2-OH-Mito-E+ production in C. elegans
C. elegans were grown in liquid culture and washed clear of remaining bacteria, as above (Section 2.1). Worm number was determined by pelleting washed worms, resuspending them in 50 ml S. basal in Falcon tubes, inverting several times to mix, and counting worm number in triplicate 20 ul aliquots under 5X magnification. Approximately 10,000 young adult stage nematodes were fed 10 uM MitoSOX in liquid culture in 20°C incubators (I26/R Series, New Brunswick Scientific, Edison, NJ) shaking at 220 rpm for 24 hours, washed three times in 5 ml S. basal, centrifuged at 300g for 5 minutes at 4°C to pellet the worms, and stored at −80°C until use. Three 500 ul worm pellets were combined, freeze-thawed twice, and then ground in a homogenizer with a Teflon pestle for 2 minutes on ice. Following the addition of 2 volumes of 100% ethanol, the mixture was further homogenized until approximately 90% of the worms were broken open and the cellular material was freed from the cuticle. Homogenates were incubated on ice in the dark for 1 hour with agitation every 15 minutes, and then centrifuged at 20,000 × G at 4°C for 20 minutes.
Mass spectrometric analysis was performed on worm extract supernatants using an Agilent 1100 series LC-MS instrument equipped with an electrospray (ESI) source and quadrupole mass filter. Synthesis of 2-OH-Mito-E+ standard was performed as previously described (Zielonka et al. 2008). LC-MS analysis of MitoSOX Red (Mito-HE) and its oxidation products from whole worm extracts was performed as previously published (Zielonka et al. 2008), with slight modification. Reverse phase chromatography was conducted using a Phenomenex Synergi Polar RP column (250mm × 2.0 mm, 4 uM), which was directly coupled to an ESI source operating in positive ion mode. Buffers A and B were 0.1 % formic acid/0.01% trifluoroacetic acid in water and 0.1% formic acid/0.01% trifluoroacetic acid in acetonitrile, respectively. Samples were subjected to a linear gradient at a flow rate of 0.1 ml per min for 35 min from 50 – 75% B. Mito-HE and 2-OH-Mito-E+ standards (10 pmol) were used to optimize LC-MS single ion monitoring parameters. Initially, full scan spectra (m/z = 200 – 800) were repeatedly acquired over the linear gradient to determine retention time, charge state(s), and m/z value(s). The following m/z values were used during SIM analyses: Mito-HE (Group 1): 632.2 and 316.6 for the singly and doubly-charged molecular ions, respectively; 2-OH-Mito-E+ (Group 2): 323.6 (2+) for the doubly-charged molecular ion. Aliquots of ethanol extracts (40 ul) were analyzed by LC-SIM-MS with a cycle time of 1.20 sec and peak width of 0.2.
2.5 Quantitation of mitochondria membrane potential and mitochondria density by fluorescence microscopy
Young adult worms were incubated for 24 hours at 20°C on NGM plates spread with OP50 E. coli that were pre-treated for relative quantitation of mitochondria membrane potential with 1 uM tetramethylrhodamine ethyl ester perchlorate (TMRE) and of mitochondria density with 10 uM MitoTracker Green FM (Invitrogen, Carlsbad, CA) (Scaduto and Grotyohann 1999). Worms were subsequently transferred to fresh NGM plates containing only OP50 E. coli for one hour to clear their intestinal tract of residual dye, after which they were paralyzed in situ on NGM plates by adding 10 mg/ml levamisole (Sigma, St. Louis, MO). Photographs were immediately taken (MZFLIII, Leica, Bannockburn, IL). Terminal PB mean fluorescence was quantified using NIS Elements Imaging software (Nikon, Melville, NY), as described above, except that an exposure time of 320 milliseconds and CY3 filter were used. All TMRE analyses were performed by a single technician (S.D.).
2.6 Confocal microscopy
Confocal microscopy (TCS SP2, Leica, Bannockburn, IL) was used to confirm subcellular fluorescence localization within the terminal PB of wild-type and mutant worms (with the assistance of Andrea Stout, Ph. D in the Cell Biology Core Facility, University of Pennsylvania, Philadelphia, PA). Worms were exposed for 24 hours individually or in combination to a MitoTracker Dye (Green, Red, or Deep Red, Invitrogen, Molecular Probes, Carlsbad, CA), MitoSOX (Mito-HE), or TMRE. Following one hour intestinal clearing on unlabelled E. coli, nematodes were paralyzed onto glass slides in 20 ul of 10 mg/ml levamisole. Double labeling of was not reproducibly achieved due to extensive dye bleed-through at multiple concentrations attempted. Optical stacks were obtained at 1 um resolution, and recombined into movie files using Image J (http://rsbweb.nih.gov/ij/).
2.7 Mitochondria-fraction isolation from C. elegans
When a majority of animals had reached adult stage following growth for a single generation in liquid culture, worms were washed extensively in S. basal with sucrose gradient centrifugation protocol to separate the adult population, as previously described (Falk et al. 2006). The adult-enriched nematode fraction was immediately subject to a mitochondria isolation protocol on ice involving homogenization, proteinase degradation of the outer nematode cuticle, and differential centrifugation, as previously described (Falk et al. 2006), exception an additional slow centrifugation spin (300 × G, 10 minutes, 4°C) was performed to remove remaining intact worms from the mitochondria fraction prior to the first high-speed centrifugation spin (7000 × G, 10 minutes, 4°C).
2.8 Quantitation of sod-2 and sod-3 RNA expression in nematodes at baseline and following paraquat exposure
Worms of each strain were grown to high density on two 10 cm NGM agar plates seeded with OP50 E. coli. Synchronous first-day young adult worms of each strain were incubated at 20°C for 24 hours on 10 cm NGM plates with and without 1 mM paraquat. Total RNA was extracted from populations of 1,000 nematodes (estimated by dilution, as per Section 2.4) using modified Trizol protocol (Invitrogen Corporation, Carlsbad, CA) and purified in RNeasy spin columns (Qiagen, Inc., Valencia, Ca), as previously described (Falk et al. 2008). Total RNA concentration and dissolution was determined spectrophotometrically at 230 nm, 260 nm, and 280 nm wavelengths (Nanodrop ND-100 Spectrophotometer v3.1.2, NanoDrop Technologies, Inc., Wilmington, DE). 2–10 ug of total RNA was DNase-treated using the TURBO DNA-free kit (Ambion Inc., Austin, TX). 400 ng–1.4 ug of DNase-treated RNA was reverse transcribed in a 20μl reaction mixture to generate cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). 40 ng of cDNA was used per quantitative PCR (qPCR) reaction containing Taqman gene expression assays (Applied Biosystems, Foster City, CA) for endogenous housekeeping gene drs-1 (Ce02451127_g1), as well as the target genes sod-2 (Ce02410777_g1) or sod-3 (Ce02404515_g1). drs-1 was selected based on analysis of genes having unchanging expression in C. elegans mitochondria mutants relative to N2 in previously reported expression microarray datasets (Falk et al. 2008) that are publicly accessible (GEO series accession number GSE9967) at NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). Taqman Universal PCR Master Mix (Applied Biosystems, manufactured by Roche, Branchburg, NJ) was used per Applied Biosystems standard protocol for Taqman Gene Expression Assay (Taqman MGB Probes, FAM dye-labeled). Real-time qPCR was performed on a SDS-7500 qPCR machine (Applied Biosystems, Foster City, CA) that was available in the Nucleic Acid and Protein Core Facility at The Children’s Hospital of Philadelphia (Philadelphia, PA). Sequence Detection Software 1.2.3 version was used to perform relative quantitation analysis of gene expression (Applied Biosystems, Foster City, CA). Nematode growth and RNA extraction was performed by the same technician (M.R). All qPCR studies were performed by a single individual (E.P.).
2.9 MnSOD enzyme activity assay in C. elegans isolated mitochondria
Mitochondria MnSOD-specific activity was measured by slight modification of the assay described by McCord and Fridovich (McCord and Fridovich 1969). Intact mitochondria were isolated from C. elegans (Section 2.7), and frozen in −80°C until subsequently being thawed on ice for MnSOD activity analysis. The assay buffer contained 50 mM KH2PO4 (Fisher Scientific, Pittsburgh, PA) pH 7.8, 200 uM xanthine (Sigma, St. Louis, MO), 60 uM acetylated-cytochrome-C (Sigma) 0.02 % Triton-X100 (Sigma), 2 mM KCN (to inhibit any contaminating Cu/Zn SOD) and 0.02 U xanthine oxidase (Calbiochem, San Diego, CA). The rate of acetylated-cytochrome C reduction was measured spectrophotometrically at 550 nm using an Olis Modernized DW-2/2000 Spectrophotometer and Olis Global Works software (Mitochondria Research Core Facility, The Children’s Hospital of Philadelphia, Philadelphia, PA). In a single cuvette reaction, a baseline reduction rate was first observed without mitochondria present. Mitochondria protein was then added to the cuvette and the change in the rate of acetylated-cytochrome C reduction was recorded. The rate of reduction in the presence and absence of mitochondria sample was determined using Olis Global Works software. The amount of MnSOD presence in each mitochondria sample was calculated with the following formula (adapted from Kuthan et al. 1986): [SOD] (Unit) = [(V0/V1)−1]/mg protein, where V0 = the rate of reduction of acetylated-cytochrome C in the absence of protein, and V1= the rate of reduction of acetylated-cytochrome C in the presence of mitochondria protein. One unit of SOD was defined as the amount required to cause a 50% inhibition in the rate of reduction of acetylated-cytochrome C. As the reaction constant was not linear, values were only accepted for mitochondria samples that fell within a 40% to 60% rate reduction. Mean ± SEM MnSOD enzyme activity was determined by averaging a minimum of two technical replicates for each of at least two independent mitochondria preparations of each strain. All analyses were performed by a single individual (E.P.).
2.10 MnSOD protein quantitation by Western analysis of C. elegans isolated mitochondria
Mitochondria proteins were prepared and subjected to Western Blot analysis of MnSOD protein levels. Isolated mitochondria proteins that were prepared from different worm strains (as above, per Section 2.7), were first diluted to an appropriate concentration with MSM buffer pH 7.0 containing 220 mM mannitol, 70 mM sucrose, and 5 mM MOPS (SIGMA-Aldrich, St. Louis, MO). These samples were then further diluted with Laemmli sample buffer (BIO-RAD Laboratories, Hercules, CA) containing β-mercaptoethanol, in a 1:1 ratio and boiled at 98°C to 100°C for 4 minutes. Equal amounts of protein samples (50 ug) were separated on 12% SDS-PAGE (BIO-RAD Mini Protean 3 Cell, BIO-RAD Laboratories, Hercules, CA). Resolved proteins were transferred to nitrocellulose or PVDF membranes using Mini Trans-Blot Cell system (BIO-RAD) and labeled with rabbit polyclonal anti-SOD2 antibody (Abcam Inc., Cambridge, MA) in 1:1000 dilutions. This antibody was designed to detect amino acids 25–222 of human SOD2. Proteins were detected with Infrared IRDYE 680 conjugated goat anti-rabbit IgG in 1:20000 dilution and protein bands were visualized by scanning the membranes with Odyssey Infrared Imaging Systems (LI-COR Biosciences, Lincoln, NE). Intensity of the 22 kDa band in each sample was quantified to determine the relative amount of MnSOD protein in each mutant strain.
2.11 Analysis of 4-HNE protein adducts from isolated mitochondria
Frozen mitochondria protein was thawed on ice and subjected to Western blot analysis of 4-hydroxy-2-nonenal (4-HNE) protein adducts (Catala 2009), following similar preparation as described above (Section 2.10). Equal amounts of mitochondria protein (20 ug) were separated on 12% SDS-PAGE and transferred to nitrocellulose or PVDF membranes. Resolved proteins were labeled with rabbit anti-HNE (4-hydroxy-2-nonenal) antibody (Alpha Diagnostic International, San Antonio, TX) in 1:1000 dilutions. Proteins were detected with Infrared IRDYE 680 conjugated goat anti-rabbit IgG in 1:20000 dilution and protein bands were visualized by scanning the membranes with Odyssey Infrared Imaging Systems (LI-COR Biosciences, Lincoln, NE).
2.12 Statistical analyses
Two-sided, non-parametric ANOVA analyses were performed in SAS version 9.1 (SAS Institute Inc, Cary, NC) to compare mean intensity differences between strains both when compared to wild-type, and when compared between baseline and following oxidant stress for a given strain for all live worm fluorescence studies (Figures 2 and 3A). Statistical analysis was performed by student’s t-test in Excel (Microsoft) for other analyses as indicated, where * p<0.05, ** p<0.01, and *** p<0.001.
Figure 2. In vivo relative oxidant quantitation in C. elegans terminal pharyngeal bulbs.

Steady-state oxidant levels in young adult nematodes at baseline as determined by mean terminal pharyngeal bulb (PB) fluorescence at 160X magnification at baseline and following 24 hour exposure to 10 uM MitoSOX Red (“S” = baseline, blue bars) ± 1 mM paraquat (“S+P” = oxidant stress, red bars) at 20°C. Steady-state oxidant levels were significantly increased only in the complex I and both MnSOD paralog knockout mutants. With paraquat exposure, in vivo relative oxidant levels rose significantly in all three RC mutants (gas-1, mev-1, isp-1). Statistical analyses were performed by ANOVA, where ** p<0.01, *** p<0.001). Box length represents 25th to 75th percentile inter-quartile range, interior horizontal line represents median, vertical lines issuing from the box extend to minimum and maximum values of the adjacent analysis variable [calculated as third quartile ± (1.5*interquartile range)], open red circles indicate outliers defined as values exceeding the upper adjacent level, and asterisks represent severe outliers (exceed 3 box lengths above the upper adjacent level). N, total animal number studied over three replicate experiments per strain, as indicated within boxes. sod-2 strain presented is sod-2(gk235).
Figure 3. In vivo quantitation of relative membrane potential and mitochondria density in C. elegans terminal pharyngeal bulbs.

A) Mitochondria membrane potential (ΔΨm) in living nematodes at baseline as determined by mean fluorescence at 160X magnification following 24 hour exposure to 1 uM TMRE at 20°C. ΔΨm was most dramatically reduced relative to wild-type (N2) in the complex I mutant (gas-1, mean PB fluorescence decreased by 63%). Mean TMRE PB fluorescence was also substantially reduced in the complex III mutant (isp-1) and both sod-2 mutants, but was only decreased by 12% in the complex II mutant (mev-1) and by 9% in the insulin receptor mutant (daf-2). Statistical analyses were performed by ANOVA, where *** p<0.001). Box plot components indicate values as detailed in Figure 2 legend. N, total animal numbers studied across three replicate experiments per strain, as indicated within boxes. B) Decreased mitochondria density in complex I mutant worms is suggested by a 48% decrease in mean PB fluorescence intensity in gas-1 relative to N2 following 24 hour exposure of synchronous young adult worms to 10 uM MitoTracker Green FM dye, whose mitochondria localization is largely independent of membrane potential. Statistical analysis by student’s t-test, where *** p<0.001. N, animal number studied per strain. PB, pharyngeal bulb.
3. RESULTS and DISCUSSION
3.1 Ingested MitoSOX dye predominantly localized to mitochondria-rich pharyngeal bulbs in C. elegans
Upon feeding cationic, mitochondria-targeted, fluorescent dyes to C. elegans, a consistent pattern of preferential labeling was observed within their mitochondria-dense PB (Figure 1). This mitochondria distribution closely approximates that observed in a previous report of an EGFP-fusion product of the promoter for the mitochondria complex I K09A9.5 (NDUFS2 homologue) subunit (Kayser et al. 2001), which we also observed upon confocal analysis of that same strain (data not shown; pKEK strain obtained from Philip Morgan, M.D. and Margaret Sedensky, M.D., University of Washington, Seattle, WA). Confocal microscopy demonstrated that MitoSOX-based fluorescence assessed 24 hours following dye ingestion localized to PB cytoplasmic organelles measuring approximately one micron in diameter (Figure 1I). MitoSOX fluorescence exhibited similar PB labeling patterns to other mitochondria-localizing dyes (eg. MitoTracker Green). In addition, large fluorescent foci were consistently observed cephalad to the grinder of the terminal PB in multiple strains following MitoSOX treatment, but not after MitoTracker treatment. Confocal microscopy reconstruction (Supplementary Figure 1) demonstrated that these fluorescent foci can be resolved to multiple mitochondrion-sized particles organized into a c-shaped, tripartite structure that appears bilaterally symmetric with two cephalad arms cradling the terminal PB. This fluorescent mitochondria cluster observed with MitoSOX treatment represents an apparently unique structure that is not characteristic of known pharyngeal, neuronal, or support cells (www.wormatlas.org and personal communication with Leon Avery, PhD) (Albertson and Thomson 1976). While the size of the component vesicles that comprise the c-shaped structure are individually consistent with mitochondria, our inability to co-label them with MitoTracker dyes leaves open several possibilities. Possible explanations include that these particular vesicles may represent viable mitochondria that simply fail to co-label due to co-operative loading of the two dyes, depolarized mitochondria in the process of being degraded, or lipophilic structures of non-mitochondria origin.
Figure 1. Nematode terminal pharyngeal bulb labeling with mitochondria-targeted dyes.

a) Light micrograph of young adult wild-type (N2) nematode, where cephalad portion (red circle) highlights the two pharyngeal bulbs (m, metacorpus and tb, terminal bulb) and initial portion of the gastrointestinal tract (g); b) MitoSOX fluorescence in N2 demonstrates consistent labeling within the C. elegans pharynx, as well as in lipid-rich gut granules that non-specifically bind lipophilic dyes. The top worm shows nuclear sparing of the mitochondria-localized dye both in the terminal pharyngeal bulb (tb) and in cells of the gastrointestinal tract (g); c) MitoSOX fluorescence of N2 pharynx overlay with DIC image demonstrates preferential labeling in terminal pharyngeal bulb; d) Threshholding permits focused analysis of terminal pharyngeal bulb (tb) fluorescence. Similar fluorescent patterns were observed by confocal microscopy maximal projection images whether animals are fed: e) MitoTracker Green in N2, f) MitoTracker Red in N2, g) Overlay of e and f, h) Typical MitoSOX terminal pharyngeal bulb fluorescent pattern in sod-3 knockout mutant, i) 1000x magnification image taken with CY3 filter of N2 fed MitoTracker Red demonstrated individual cytoplasmic vesicle labeling on the order of ~800 nm diameter fluorescent foci.
Optimal nematode exposure times to mitochondria-targeted dyes sufficient to achieve reproducible, steady-state levels of fluorescence within their terminal PB were determined by an initial feeding time-course experiment (Supplementary Figure 2A). In wild-type (N2 Bristol) synchronous young adult nematodes fed 10 uM MitoSOX, increased fluorescence was observed between 12 and 36 hours of dye ingestion, whereas exposure times under four hours proved inadequate to permit reproducible fluorescence quantitation. Repeat time-course analyses applying the same experimental conditions and analyses subsequently used in strain comparisons (Section 3.2) confirmed that within a single strain, N2 Bristol, 24 hour dye exposure produced highly reproducible results, with fluorescence intensity largely unchanged between 24 and 97 hours of dye exposure (Supplementary Figure 2B). 24 hour dye exposure time to synchronized nematode populations beginning on their first day of adulthood (defined by the first day of egg-laying) was selected for comparative studies between strains (Section 3.2), although variable rate of dye uptake between strains may be a potential confounding factor. Live bacteria were used for all experiments, since wild-type nematodes fed dye for prolonged periods (eg. 60 hours) on UV-killed bacteria had poor growth and early death that closely followed observed spikes in relative oxidant levels assessed by mean terminal PB MitoSOX fluorescence, most evident following high-dose (20 mM) paraquat treatment (Supplementary Figure 2A).
Under these feeding conditions, MitoSOX ingestion and subsequent oxidation by superoxide generates a specific product, 2-OH-Mito-E+, whose production within mitochondria can be detected by LC-MS in worm extracts from the complex I mutant, gas-1(fc21) (Supplementary Figure 3). MitoSOX can also be oxidized by other oxidants, albeit at lower yields, forming the non-superoxide oxidant product, Mito-E+, which can also be monitored by LC-MS. A major challenge to in vivo detection of superoxide has been the specificity of its detection (Zielonka et al. 2008). As monitoring of the 2-OH-Mito-E+ product offers a unique opportunity to perform comparative quantitative assessment of superoxide production, the LC-MS data provided here is one of the few examples in the existing literature that support in vivo production of superoxide. However, technical limitations of this method prohibited reliable relative quantification of 2-OH-Mito-E+ production between strains. Therefore, fluorescence microscopy was employed for relative quantification of in vivo oxidant levels following MitoSOX exposure in C. elegans (Section 3.2).
3.2 In vivo matrix oxidant burden was greatest in mutants for RC complex I and MnSOD
In comparison with wild-type (N2), significantly increased baseline MitoSOX fluorescence was observed in gas-1 (29%, p<0.001), sod-2(gk257) (21%, p<0.01), and sod-3 (29%, p<0.001) mutants (Figure 2, blue bars). The long-lived daf-2 mutant showed no significant difference in PB fluorescence from N2 (4% decrease, p=0.355). Upon 1 mM paraquat exposure, increased oxidant levels assessed by terminal PB MitoSOX fluorescence relative to N2 were observed in gas-1 (55%, p<0.001), mev-1 (21%, p<0.001), isp-1 (8%, p<0.01), and sod-3 (17%, p<0.001) (Figure 2, red bars). In vivo oxidant burden upon exposure to paraquat remained unchanged in both N2 and the insulin receptor mutant (daf-2), as is consistent with the well-documented oxidative stress resistance of daf-2 (Feng et al. 2001). Intrastrain comparison of mean terminal PB fluorescence following 1 mM paraquat exposure relative to the same mutant at baseline demonstrated significantly increased oxidant levels upon oxidant stress in all three RC mutants: gas-1 (59%, p<0.01), mev-1 (21%, p<0.001), and isp-1 (10%, p<0.001). Surprisingly, intrastrain comparison of two MnSOD mutants, sod-2(gk257) and sod-3(gk235), each showed decreased oxidant levels by 14% (p<0.001) following 1 mM paraquat treatment. This unanticipated decrease in fluorescence appeared to result from diminished dye uptake due to poor feeding and general activity in the MnSOD mutants, which implies the MnSOD mutants manifest excessive oxidative stress sensitivity rather than resistance. Indeed, all MnSOD mutants appeared sickly and died prematurely following paraquat exposure (data not shown), whereas mortality did not appear to be substantially increased following 24 hour paraquat exposure in the other strains.
3.3 Complex I mutant had most substantially decreased in vivo mitochondria membrane potential
In vivo ΔΨm measured by TMRE fluorescence was significantly decreased (p<0.001) in all mitochondria mutant strains relative to N2, as is consistent with a recent report using a different fluorescent indicator dye (Lemire et al. 2009). The most dramatic differences were observed in strains harboring mutations in RC complexes that generate the proton gradient (mean fluorescence intensity decreased by 62.7% in gas-1(fc21) and by 24.2% in isp-1(qm150), respectively) as well as in all three MnSOD knockout alleles studied (decreased mean fluorescence by 20% in sod-2(gk257), 26% in sod-2(ok1030), and 17% in sod-3(gk235)) (Figure 3A). As gas-1 had a substantially less polarized ΔΨm than did N2 at baseline, increased oxidant levels assessed by MitoSOX fluorescence in this strain are not attributable to changes in mitochondria polarization. Furthermore, study of a reporter dye whose uptake into mitochondria is largely independent of membrane potential (MitoTracker Green FM) showed that gas-1 had a 48% decrease in mean PB fluorescence intensity, suggesting decreased PB mitochondria density in this complex I mutant (Figure 3B). Thus, the 29% relative increase in MitoSOX PB fluorescence in the complex I mutant was likely an underestimate of mitochondria oxidant burden, since their reduced ΔΨm and decreased mitochondria content would be predicted to limit MitoSOX uptake and retention.
3.4 Mitochondria superoxide scavenging capacity was most substantially enhanced in complex III dysfunction
C. elegans has two functional MnSOD paralogs (sod-2 and sod-3) that share extensive protein sequence similarity with human SOD2 (Table 1) (Hunter et al. 1997). Relative quantitation of sod-2 and sod-3 expression by qRT-PCR analysis of total RNA isolated from synchronous young adult nematode populations revealed substantially greater transcriptional upregulation of sod-3 than sod-2 in all mutants studied (Figure 4A). sod-2 expression was most upregulated at baseline relative to N2 in mev-1 (by 2.2-fold), and following paraquat-induced oxidant stress in gas-1 (by 1.8-fold). By comparison, sod-3 expression showed dramatically greater upregulation in all RC mutants (gas-1, mev-1, and isp-1) both at baseline relative to N2 (by 11.5-fold, 8.5-fold, and 15-fold, respectively) (Table 2A) and following oxidative stress relative to paraquat-treated N2 (7.5-fold, 5.5-fold, and 9-fold, respectively) (Table 2B). Intrastrain comparison upon paraquat challenge revealed sod-2 expression was most increased above baseline levels in gas-1 (25%), whereas sod-3 was substantially induced following oxidant stress in all three RC mutants and N2. These data suggest that sod-3 is the predominant MnSOD gene in C. elegans responding to mitochondria oxidative stress caused by primary RC dysfunction. However, determination of absolute sod-2 vs sod-3 transcript levels would be necessary to prove the functional significance of this upregulated response.
Figure 4. Whole worm MnSOD expression and protein levels in mitochondria mutants.

A) Whole worm MnSOD total RNA levels were measured by qPCR. sod-3 upregulation dramatically exceeded sod-2 upregulation in primary RC dysfunction, both at baseline and following oxidant stress. B) MnSOD western blot analysis in isolated mitochondria from RC mutant strains confirmed that total MnSOD protein is upregulated (22kDa band) in RC mutants (complex I, gas-1; complex II, mev-1, and complex III, isp-1). By contrast, MnSOD protein was not increased in the insulin receptor mutant (daf-2), despite its 4-fold increased sod-2 transcript level (Figure 4A, above). MnSOD protein levels were diminished relative to N2 in the sod-3 MnSOD knockout mutant, suggesting both sod-2 and sod-3 contribute to total MnSOD protein levels. C) MnSOD western blot analysis in isolated mitochondria from three distinct knockout alleles of two sod-2 paralogs again demonstrated decreased MnSOD in an independent mitochondria preparation of sod-3(gk235), mutant as well as decreased MnSOD in the sod-2(gk257) compound heterozygous deletion/insertion allele. However, MnSOD protein levels were increased relative to wild-type in the long-lived sod-2(ok1030) homozygous deletion allele. Relative changes are reported in Table 2A.
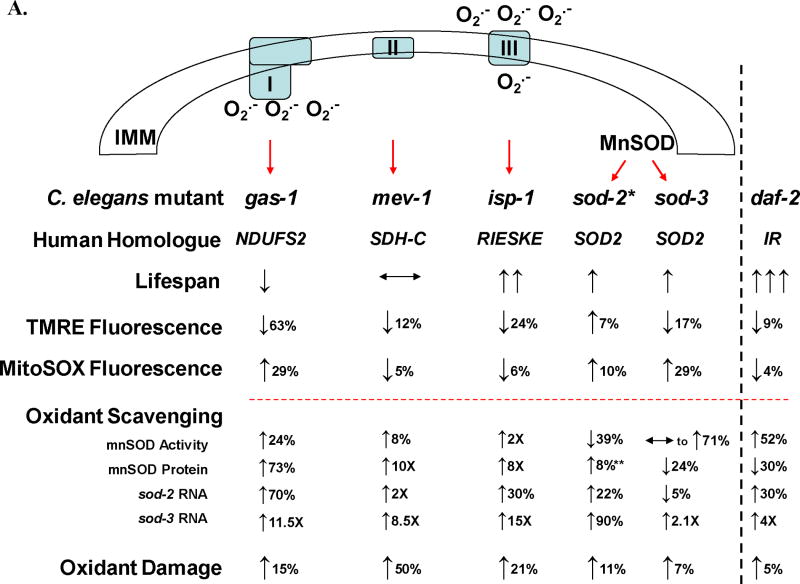
Table 2. Multi-dimensional summary of relative alterations in lifespan, oxidant levels, oxidant scavenging, and oxidant damage in C. elegans mitochondrial respiratory chain mutants.
A) Relative steady-state oxidant burden (assessed by MitoSOX PB fluorescence), mitochondria membrane potential (ΔΨm, assessed by TMRE PB fluorescence) and antioxidant defenses in synchronous young adult stage animals of each strain compared to wild-type nematodes under non-stressed conditions. Red dotted line separates in vivo (above line) from in vitro (below line) parameters. Black arrows indicate direction of change relative to wild-type, where % and X indicate percent change below and above two-fold, respectively. O• indicates oxidant levels primarily in the form of superoxide, as well as its site of production in the mitochondria matrix via complex I and predominantly into the intermembrane space via complex III. *, results are provided for sod-2(gk257), which was the main sod-2 allele analyzed in most assays. **, 8% increase (essentially no change) was observed by MnSOD Western for sod-2(gk257) mitochondria, while a 3-fold increased MnSOD protein level was observed for sod-2(ok1030) (Figure 4C). These allelic differences may relate to the binding site of the antibody within the protein, rather than reflecting increased intact protein. B) Relative oxidant levels and MnSOD transcriptional response in two paralagous MnSOD mutants following 24 hour oxidant stress (1 mM paraquat exposure) relative to wild-type in young adult stage animals. In vivo oxidant levels following oxidative stress increase to the greatest extent in the complex I mutant (gas-1, 55% increased MitoSOX PB fluorescence) and to a lesser extent in the complex II mutant (mev-1, 21% increased MitoSOX PB fluorescence) and complex III mutant (isp-1, 8.3% increased MitoSOX PB fluorescence). Increased in vivo oxidant burden occurred despite marginally increased sod-2 expression (below a 2-fold level of induction) and significant sod-3 induction (greater than 5-fold level of induction) in all three respiratory chain (RC) mutants.
|
| B. | |||
|---|---|---|---|
| C. elegans Strain | OXIDANT LEVEL | OXIDANT SCAVENGING | |
| PB Fluorescence (MitoSOX) | sod-2 Relative Expression | sod-3 Relative Expression | |
| gas-1(fc21) | ↑ (55%) | ↑ (84%) | ↑ (7.4 X) |
| mev-1(kn1) | ↑ (21%) | ↑ (30%) | ↑ (5.6 X) |
| isp-1(qm150) | ↑ (8.3%) | ↑ (40%) | ↑ (9.1 X) |
| daf-2(e1368) | ↓ (7.2%) | ↑ (23%) | ↑ (20%) |
| sod-2(gk257) | ↓ (1.2%) | ↓ (65%) | ↓ (7%) |
| sod-3(gk235) | ↑ (16%) | ↓ (30%) | ↓ (13%) |
To assess mitochondria superoxide removal capacity of each mutant strain regardless of individual transcript levels, total MnSOD protein levels (Figures 4B) and MnSOD enzymatic activity (Figure 5) were quantified in intact mitochondria isolated from each strain. MnSOD protein levels were variably increased in the RC mutants (73% in gas-1, 10-fold in mev-1, and 8-fold in isp-1) (Table 2B). However, MnSOD enzyme activity was only significantly increased in the complex III mutant, isp-1 (by 2-fold) relative to N2. MnSOD enzyme activity showed non-significant trends toward increase in gas-1, daf-2, and sod-3(gk235), and was essentially unchanged in mev-1. Thus, increased MnSOD transcript did not translate to similar magnitude increases in MnSOD protein or enzyme activity in all strains. We postulate that MnSOD is itself a target of oxidant damage in C. elegans.
Figure 5. MnSOD enzyme activity assessment in isolated mitochondria of RC mutants.

Only the complex III mutant (isp-1) had significantly elevated MnSOD enzyme activity in isolated mitochondria. Trends toward upregulation observed in gas-1, daf-2, and sod-3 did not reach statistical significance, perhaps because of small sample size. No discrimination was made by this assay between protein encoded by sod-2 or sod-3. Bars represent mean ± standard deviation of two to five biological replicates per strain. Single mitochondria preparations were assessed for each of the sod-2 knockout alleles, which demonstrated similar reductions in MnSOD activity relative to N2. *, p < 0.05. Percent change in each mutant strain relative to N2 is reported in Table 2A.
3.5 Altered longevity in mitochondria RC mutants correlated directly with oxidant scavenging capacity and indirectly with in vivo oxidant levels
High-throughput assessment of mutant lifespan in the presence of FUDR validated previous reports of decreased longevity in complex I RC mutants (Kayser et al. 2004), and increased longevity in complex III RC mutants (Feng et al. 2001) and insulin receptor mutants (Kenyon et al. 1993) (Supplementary Figure 4). In contrast to prior reports of shortened lifespan in mev-2(kn1) (Honda 2008), FUDR-based lifespan analysis of the complex II mutant was unaltered relative to N2. A modest increase in median lifespan was observed for the MnSOD mutants relative to N2, although this was largely limited to sod-2 (9% and 4% increased median lifespan in the ok1030 and gk257 knockout alleles, respectively) rather than sod-3 (0.5% increased median lifespan in the gk257 knockout allele). While these data were consistent with increased longevity recently reported in sod-2 (Van Raamsdonk and Hekimi 2009), we observed a less dramatic effect. Furthermore, the magnitude of altered longevity of any MnSOD mutant allele did not approximate that seen for the complex III RC mutant (43% increased median lifespan in isp-1(qm150)).
We observed that MnSOD induction is common to many RC mutants and does not alone predict lifespan. However, correlation of longevity with in vivo matrix oxidant burden in RC mutants demonstrated substantially increased in vivo oxidant levels and reduced oxidant scavenging capacity in the short-lived complex I mutant (gas-1(fc21)) relative to the long-lived complex III mutant (isp-1(qm150)) (Table 2). These data suggest that greater MnSOD enzyme scavenging capacity is associated with lower in vivo oxidant levels and increased longevity in the setting of primary RC dysfunction. Study of additional RC complexes and mutations will be necessary to determine the generalizability of this association in primary RC dysfunction.
Oxidant damage assessed by immunoreactive 4-HNE protein adducts in isolated mitochondria was modestly increased in all RC mutants (similarly as previously reported in gas-1(fc21) and mev-1(kn1) by Kayser et al. 2004) and MnSOD mutants, but not in the insulin receptor mutant (Supplementary Figure 5). The extent of 4-HNE adducts did not correlate with animal longevity (Table 2). Indeed, the complex II mutant having the least change in lifespan had the greatest increase in 4-HNE adducts (50% increase in mev-1 relative to N2).
3.6 Biological relevance
Understanding the complex relationship between oxidant stress and altered longevity in genetically-based mitochondrial RC dysfunction is facilitated by integrated analysis of the in vivo effects of individual mutations on oxidant levels, membrane potential, and lifespan, along with in vitro assessment of mitochondria superoxide scavenging capacity and lipid peroxidation damage in C. elegans. Application of novel methods to quantify mitochondria-specific fluorescence in living animals permits sensitive assessment of relative mitochondria membrane potential and matrix oxidant levels, both in individual genetic mutations and/or with oxidative stress. While MitoSOX PB fluorescence quantitation permits assessment of matrix oxidant burden, oxidant production from enzymes that release oxidants into the intermembrane space will not be measured by this approach. Quantifying terminal PB fluorescence is preferable to whole-worm fluorescence quantification for enhancing the sensitivity and specificity of functional mitochondria assessment owing to the high mitochondria density in the PB, while minimizing effects attributable to the lipophilic quality of the dyes that enables their binding to non-mitochondria organelles, such as lipid-rich gut granules. However, this approach necessitates manual analysis of a large number of individual animals to discern whether relative differences in mitochondrial function are attributable to a specific mutation, exposure, or stochastic variation. Development of computer algorithms to specifically quantify terminal PB fluorescence is a future goal to enhance the practical utility of this approach. A potential alternative approach to permit quantitation of whole animal fluorescence is to assess mitochondria-localized dyes in animals lacking lipophilic gut granules (such as the lysosomal mutant, glo-1) (Hermann et al. 2005). However, we did not pursue this approach to avoid potentially confounding effects of combined lysosomal and mitochondrial dysfunction.
The only mutants in our study that had significantly increased in vivo oxidant levels were the complex I mutant (gas-1) and two paralogous MnSOD mutants, sod-2(gk257) and sod-3(gk235) (Figure 2). Since complex I is the major site for matrix oxidant production (Hirst et al. 2008), it is reasonable to postulate that the gas-1 (NDUFS2) mutation would increase oxidant production due to its location in the complex I matrix arm along the pathway of electron transfer and near the presumed site of oxidant production. With oxidant stress caused by prolonged paraquat exposure, however, all three RC mutants showed increased in vivo matrix oxidant levels despite concordant robust MnSOD induction. Although absolute transcript quantitation was not performed, relative quantitation suggested sod-3 induction far exceeded that of sod-2 in all RC mutants. Regardless, greater than 11-fold upregulation of sod-3 in the complex I mutant, gas-1(fc21) proved inadequate to compensate for ongoing increased oxidant production by inherently mutant mitochondria. Similarly, 2-fold increased sod-3 transcript levels in sod-2(gk257) knockouts were insufficient to scavenge that mutant’s in vivo oxidant load despite its having presumably normal inherent mitochondrial RC composition. Both mutant MnSOD paralogs studied displayed significantly increased in vivo matrix oxidant levels, which was greater in sod-3(gk235) relative to sod-2(gk257). Interestingly, although lifespan of both of these MnSOD paralog knockouts were perhaps marginally increased relative to wild-type, we and others observed the extent to be less pronounced than for sod-2 (ok1030) (Van Raamsdonk and Hekimi 2009). The sod-2 knockout allele initially assessed by in vivo fluorescence studies of matrix oxidant burden was sod-2(gk257), although subsequent inclusion of the sod-2(ok1030) allele did not demonstrate substantial differences between the two sod-2 mutants in analyses of in vivo membrane potential or mitochondria MnSOD enzyme activity. Overall, both MnSOD paralogous genes clearly contribute to mitochondria oxidant scavenging capacity in C. elegans, as knockout of either increases in vivo oxidant levels and loss of sod-2 alone reduces in vitro superoxide scavenging activity. However, sod-3 appears to be the primary MnSOD transcript induced by primary RC dysfunction.
As fluorescence-based oxidant quantitation requires mitochondria uptake of positively charged dyes, we investigated whether impaired membrane potential and/or altered mitochondria density accounted for MitoSOX fluorescence differences in the RC mutants. We observed that in vivo membrane potential was significantly decreased in all RC mutants relative to wild-type, and most substantially reduced in the complex I mutant where mean TMRE fluorescence intensity was decreased by 63% in gas-1(fc21) (Figure 3A). Therefore, the observed 29% increase in MitoSOX fluorescence in the complex I mutant cannot be attributed to altered membrane potential, but likely represents an underestimate of the actual in vivo oxidant levels occurring consequent to complex I dysfunction. When also factoring in the nearly 50% decrease in MitoTracker Green FM fluorescence quantitation seen in gas-1 relative to N2 suggestive of decreased mitochondria density (Figure 3B), corrected MitoSOX fluorescence quantitation would exceed a 2-fold increase in the complex I mutant, although TMRE and MitoTracker dye uptake might not truly be independent. Similarly, relatively increased MitoSOX fluorescence levels identified in the other RC mutants also likely provide underestimations of their in vivo oxidant burden to an extent that is proportionate to their relative reduction in mitochondria membrane potential. In other words, a linear reduction in mitochondria membrane potential will result in an exponential reduction in the amount of MitoSOX loaded into the mitochondria and hence available for oxidation. Nonetheless, relative analysis of oxidant levels and mitochondria membrane potential in three primary RC mutants using fluorescence microscopy suggests that complex I function may make the largest contribution to in vivo membrane potential. An alternative possibility may be that observed fluorescence alterations are attributable to the specific RC subunit and amino acid residue affected in each of the three RC mutants studied, rather than generalizable to the normal function of their respective complexes. Since membrane potential follows a logarithmic scale, it is also possible that the millivolt differences in membrane potential in living nematodes are not as great between the RC mutants studied as would be suggested by linear-scale fluorescence analyses.
Correlation of longevity with in vivo oxidant assessment in mitochondrial RC mutants demonstrated substantially greater oxidant stress based on increased matrix oxidant levels as well as reduced mitochondria oxidant scavenging capacity in the short-lived, complex I mutant (gas-1(fc21)) relative to the long-lived, complex III mutant (isp-1(qm150)). In contrast, upregulated MnSOD expression was common to all three RC mutants and did not predict lifespan, perhaps because of oxidant-induced impairment of MnSOD activity in some cases. These data suggest that greater MnSOD enzyme scavenging capacity is associated with lower in vivo oxidant levels and increased longevity in the setting of primary RC dysfunction. Study of additional RC complexes and mutations will be necessary to determine the generalizability of this association in primary RC dysfunction.
Our data further demonstrate that daf-2 mutants have a less robust increase in MnSOD expression and enzyme activity than do the complex III mutants. However, matrix scavenging capacity proved sufficient in daf-2 but insufficient in isp-1 to prevent increased in vivo matrix oxidant burden following oxidant stress. Furthermore, a greater than two-fold increased median lifespan in the insulin receptor mutant (daf-2(e1368)) relative to any RC mutant further supports that the mechanism of longevity due to altered insulin/FOXO signaling pathway is distinct from that caused by primary RC dysfunction and not solely attributable to oxidative stress, as is consistent with prior studies (Honda and Honda 1999).
4. CONCLUSION
C. elegans is a microscopically transparent, multi-cellular animal whose muscle tube-like, continually pumping pharynx is densely populated by mitochondria that can be readily and preferentially labeled with targeted fluorescent dyes. C. elegans terminal PB fluorescence quantitation thus offers a sensitive and specific means of non-invasively monitoring relative, steady-state, in vivo mitochondria oxidant burden and membrane potential. Targeted analyses of the seven-cell, terminal PB tissue in living C. elegans mutants for three different RC complexes, two MnSOD paralogous genes, and the insulin receptor demonstrated that significantly increased MitoSOX fluorescence and reduced TMRE fluorescence, indicative of increased matrix oxidant burden and decreased ΔΨm, respectively, occur only in mutants for RC complex I, gas-1(fc21), and two C. elegans paralogs of human SOD2 (MnSOD). Upon exposure to the oxidant generating agent, paraquat, in vivo oxidant levels rose most substantially in the short-lived complex I mutant, gas-1(fc21), and near normal-lived complex II mutant, mev-1(kn1), despite robust MnSOD induction at the level of expression (primarily sod-3) in both and at the protein level in mev-1(kn1). However, significantly increased MnSOD enzyme activity only occurred in the long-lived complex III mutant, isp-1(qm150), whose in vivo oxidant burden was notably not elevated. Thus, greater MnSOD scavenging capacity was associated with lower in vivo mitochondria oxidant burden and increased longevity in the setting of primary RC dysfunction, particularly when comparing the complex III (isp-1(qm150)) and complex I (gas-1(fc21)) mutations. Nevertheless, isp-1(qm150) manifested modest oxidative stress sensitivity and only half as prolonged median lifespan as the longer-lived insulin receptor, daf-2(e1368) mutant. Furthermore, modestly increased longevity was seen in a MnSOD knockout (sod-2) that had decreased MnSOD scavenging capacity and increased in vivo oxidant burden. Thus, factors beside oxidant stress must underlie RC mutant longevity in C. elegans.
Supplementary Material
This representative movie depicts fluorescence localization in the terminal pharyngeal bulb of wild-type C. elegans following 24 hour MitoSOX exposure. It demonstrates a unique, fluorescent, c-shaped, tripartite foci that appears bilaterally symmetric in the two most cephalad arms and cradles the mitochondria-dense terminal pharyngeal bulb. This fluorescence pattern was consistently observed in wild-type and mutant animals following MitoSOX-treatment, but not following exposure only to Mitotracker Green or Mitotracker Red.
A) Synchronous, young adult stage, N2 worms were fed on NGM agar plates containing either live or UV-killed OP50 E. coli in the presence of 10 uM Mitosox Red and “low” (200 uM) or “high” dose (20 mM) Paraquat for 12, 36, and 60 hours. MitoSOX fluorescence significantly increased between 12 and 36 hours. High dose paraquat fed to worms in the presence of UV-killed bacteria resulted in a spike in MitoSOX fluorescence intensity by 36 hours, with all animals dead by 60 hours. Of note, these initial analyses were performed using a different microscope and software than were subsequent fluorescent studies, prohibiting direct comparison of fluorescence intensity units. B) Repeat time-course analyses of MitoSOX dye fed to wild-type (N2) synchronous young adult nematodes performed using the same experimental conditional, microscope, and software as in subsequent studies (Figures 2 and 3) confirmed that fluorescence intensity values within a given strain (N2) were highly reproducible and increased substantially over the first 24 hours, but then largely leveled off at least through 96 hours of dye exposure.
In contrast to fluorescence microscopy, LC/MS can differentiate MitoSOX (hydroethidine, Mito-HE), its superoxide-specific oxidized product (2-hydroxy-ethidium, 2-OH-Mito-E+), and its non-superoxide converted product (ethidium, Mito-E+) in synchronous, young adult populations of 5,000 gas-1(fc21) worms labeled for 24 hours with MitoSOX. Worms that were not exposed to MitoSOX (top graph) do not contain these products. Unfortunately, LC/MS or isolated MS applied to C. elegans did not permit ready assessment of relative 2-OH-Mito-E+ levels among various RC mutants due to technical limitations.
A) Lifespan studies in RC mutants and insulin receptor mutant. N, number of animals studied per strain. All lifespan analyses were performed at 20°C by a single observer (J.O.) with 100 ug/ml FUDR from the first day of adulthood. B) Lifespan studies in two MnSOD paralog mutants (two alleles of sod-2 and a single allele of sod-3). Differences in wild-type (N2) strain lifespan may have resulted from variation in E. coli lawn thickness, as relative caloric restriction on a thinner bacterial lawn can prolong lifespan. For this reason, all mutant lifespans were interpreted relative to concurrently studied N2.
Qualitative assessment of 4-HNE protein adducts in isolated mitochondria suggested the greatest cumulative lipid peroxidation occurred in the RC mutants (particularly mev-1) and the MnSOD mutants. In contrast, mitochondria lipid peroxidation did not appear increased in the long-lived insulin receptor mutant relative to wild-type.
Acknowledgments
This work was funded by NIH grant K08-DK073545 and Ellison Medical Foundation New Scholar’s Award in Aging (AG-NS-0327-06) (MJF) as well as supported by the Gisela and Dennis Alter Chair in Pediatric Neonatology, and the Joseph Strokes Jr. Investigator program (HI). We thank Alexandra Gladstone for her assistance developing C. elegans fluorescence-based membrane potential analyses; Andrea Stout, PhD, of the Cell Biology Confocal Core Facility at The University of Pennsylvania School of Medicine for her expertise in confocal microscopy and movie generation; Todd Lamitina, PhD, for his helpful discussions and guidance in fluorescence microscopy; Evgueni Daikhin, MD, PhD and Marc Yudkoff, MD of the CHOP Mass Spectrometry Core Facility for their assistance with MS validation of MitoSOX specific products; Mary Selak, PhD, for her assistance in optimizing the MnSOD enzyme activity assay; Philip G. Morgan, M.D. and Margaret M. Sedensky, M.D. for sharing the pKEK EGFP mutant strain; as well as Abdul Salam, PhD of Westat Statistical Core and Zhe Zhang, PhD of the CHOP Bioinformatics Core for their assistance with statistical analyses and generation of Figures 2 and 3, respectively.
Abbreviations
- C. elegans
Caenorhabditis elegans
- MnSOD
manganese superoxide dismutase
- RC
respiratory chain
- PB
pharyngeal bulb
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Stephen Dingley, Email: stephen.dingley@gmail.com.
Erzsebet Polyak, Email: polyake@email.chop.edu.
Richard Lightfoot, Email: lightfoot@email.chop.edu.
Julian Ostrovsky, Email: ostrovskyj@email.chop.edu.
Meera Rao, Email: meerao@sas.upenn.edu.
Todd Greco, Email: tgreco@mail.med.upenn.edu.
Harry Ischiropoulos, Email: ischirop@mail.med.upenn.edu.
References
- Albertson DG, Thomson JN. The pharynx of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1976;275(938):299–325. doi: 10.1098/rstb.1976.0085. [DOI] [PubMed] [Google Scholar]
- Altun ZF, Hall DH. Alimentary System: Pharynx. WormAtlas. 2008. http://www.wormatlas.org/hermaphrodite/pharynx/mainframe.htm.
- Avery L, Horvitz HR. Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans. Neuron. 1989;3(4):473–85. doi: 10.1016/0896-6273(89)90206-7. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bus JS, Gibson JE. Paraquat: model for oxidant-initiated toxicity. Environ Health Perspect. 1984;55:37–46. doi: 10.1289/ehp.845537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catala A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem Phys Lipids. 2009;157(1):1–11. doi: 10.1016/j.chemphyslip.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Chen Q, et al. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278(38):36027–31. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- Clokey GV, Jacobson LA. The autofluorescent “lipofuscin granules” in the intestinal cells of Caenorhabditis elegans are secondary lysosomes. Mech Ageing Dev. 1986;35(1):79–94. doi: 10.1016/0047-6374(86)90068-0. [DOI] [PubMed] [Google Scholar]
- Cocheme HM, Murphy MP. Chapter 22 The uptake and interactions of the redox cycler paraquat with mitochondria. Methods Enzymol. 2009;456:395–417. doi: 10.1016/S0076-6879(08)04422-4. [DOI] [PubMed] [Google Scholar]
- DiMauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004;1658(1–2):80–8. doi: 10.1016/j.bbabio.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Falk MJ, et al. Mitochondrial complex I function modulates volatile anesthetic sensitivity in C. elegans. Curr Biol. 2006;16(16):1641–5. doi: 10.1016/j.cub.2006.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, et al. Metabolic pathway profiling of mitochondrial respiratory chain mutants in C. elegans. Mol Genet Metab. 2008;93(4):388–97. doi: 10.1016/j.ymgme.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bussiere F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1(5):633–44. doi: 10.1016/s1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- Gaskova D, DeCorby A, Lemire BD. DiS-C3(3) monitoring of in vivo mitochondrial membrane potential in C. elegans. Biochem Biophys Res Commun. 2007;354(3):814–9. doi: 10.1016/j.bbrc.2007.01.073. [DOI] [PubMed] [Google Scholar]
- Hamilton B, et al. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19(13):1544–55. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GJ, et al. Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol Biol Cell. 2005;16(7):3273–88. doi: 10.1091/mbc.E05-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. 2008;36(Pt 5):976–80. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13(11):1385–93. [PubMed] [Google Scholar]
- Honda Y, Tanaka M, Honda S. Modulation of longevity and diapause by redox regulation mechanisms under the insulin-like signaling control in Caenorhabditis elegans. Exp Gerontol. 2008;43(6):520–529. doi: 10.1016/j.exger.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Hope IA. C. elegans A practical approach. Oxford, UK: Oxford University Press; 1999. [Google Scholar]
- Hunter T, Bannister WH, Hunter GJ. Cloning, expression, and characterization of two manganese superoxide dismutases from Caenorhabditis elegans. J Biol Chem. 1997;272(45):28652–9. doi: 10.1074/jbc.272.45.28652. [DOI] [PubMed] [Google Scholar]
- Kayser EB, et al. Mitochondrial expression and function of GAS-1 in Caenorhabditis elegans. J Biol Chem. 2001;276(23):20551–8. doi: 10.1074/jbc.M011066200. [DOI] [PubMed] [Google Scholar]
- Kayser EB, Sedensky MM, Morgan PG. The effects of complex I function and oxidative damage on lifespan and anesthetic sensitivity in Caenorhabditis elegans. Mech Ageing Dev. 2004;125(6):455–64. doi: 10.1016/j.mad.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Kenyon C, et al. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366(6454):461–4. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kuthan H, Haussmann HJ, Werringloer J. A spectrophotometric assay for superoxide dismutase activities in crude tissue fractions. Biochem J. 1986;237(1):175–80. doi: 10.1042/bj2370175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire BD, et al. C. elegans longevity pathways converge to decrease mitochondrial membrane potential. Mech Ageing Dev. 2009;130(7):461–5. doi: 10.1016/j.mad.2009.05.001. [DOI] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. An enymatic function for erythrocuprein (Hemocuprein) J of Bio Chem. 1969;244:6049–55. [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly CM, et al. Quantitative analysis of spontaneous mitochondrial depolarizations. Biophys J. 2003;85(5):3350–7. doi: 10.1016/S0006-3495(03)74754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007;5(10):e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3(6):941–7. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- Scaduto RC, Jr, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. 1999;76(1 Pt 1):469–77. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial disease. Lancet. 2006;368(9529):70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- Shtonda B, Avery L. CCA-1, EGL-19 and EXP-2 currents shape action potentials in the Caenorhabditis elegans pharynx. J Exp Biol. 2005;208(Pt 11):2177–90. doi: 10.1242/jeb.01615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starich TA, et al. eat-5 and unc-7 represent a multigene family in Caenorhabditis elegans involved in cell-cell coupling. J Cell Biol. 1996;134(2):537–48. doi: 10.1083/jcb.134.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, et al. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5(2):e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Li J, Hekimi S. A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics. 2007;177(4):2063–74. doi: 10.1534/genetics.107.080788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc. 2008;3(1):8–21. doi: 10.1038/nprot.2007.473. [DOI] [PubMed] [Google Scholar]
- Zuryn S, Kuang J, Ebert P. Mitochondrial modulation of phosphine toxicity and resistance in Caenorhabditis elegans. Toxicol Sci. 2008;102(1):179–86. doi: 10.1093/toxsci/kfm278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This representative movie depicts fluorescence localization in the terminal pharyngeal bulb of wild-type C. elegans following 24 hour MitoSOX exposure. It demonstrates a unique, fluorescent, c-shaped, tripartite foci that appears bilaterally symmetric in the two most cephalad arms and cradles the mitochondria-dense terminal pharyngeal bulb. This fluorescence pattern was consistently observed in wild-type and mutant animals following MitoSOX-treatment, but not following exposure only to Mitotracker Green or Mitotracker Red.
A) Synchronous, young adult stage, N2 worms were fed on NGM agar plates containing either live or UV-killed OP50 E. coli in the presence of 10 uM Mitosox Red and “low” (200 uM) or “high” dose (20 mM) Paraquat for 12, 36, and 60 hours. MitoSOX fluorescence significantly increased between 12 and 36 hours. High dose paraquat fed to worms in the presence of UV-killed bacteria resulted in a spike in MitoSOX fluorescence intensity by 36 hours, with all animals dead by 60 hours. Of note, these initial analyses were performed using a different microscope and software than were subsequent fluorescent studies, prohibiting direct comparison of fluorescence intensity units. B) Repeat time-course analyses of MitoSOX dye fed to wild-type (N2) synchronous young adult nematodes performed using the same experimental conditional, microscope, and software as in subsequent studies (Figures 2 and 3) confirmed that fluorescence intensity values within a given strain (N2) were highly reproducible and increased substantially over the first 24 hours, but then largely leveled off at least through 96 hours of dye exposure.
In contrast to fluorescence microscopy, LC/MS can differentiate MitoSOX (hydroethidine, Mito-HE), its superoxide-specific oxidized product (2-hydroxy-ethidium, 2-OH-Mito-E+), and its non-superoxide converted product (ethidium, Mito-E+) in synchronous, young adult populations of 5,000 gas-1(fc21) worms labeled for 24 hours with MitoSOX. Worms that were not exposed to MitoSOX (top graph) do not contain these products. Unfortunately, LC/MS or isolated MS applied to C. elegans did not permit ready assessment of relative 2-OH-Mito-E+ levels among various RC mutants due to technical limitations.
A) Lifespan studies in RC mutants and insulin receptor mutant. N, number of animals studied per strain. All lifespan analyses were performed at 20°C by a single observer (J.O.) with 100 ug/ml FUDR from the first day of adulthood. B) Lifespan studies in two MnSOD paralog mutants (two alleles of sod-2 and a single allele of sod-3). Differences in wild-type (N2) strain lifespan may have resulted from variation in E. coli lawn thickness, as relative caloric restriction on a thinner bacterial lawn can prolong lifespan. For this reason, all mutant lifespans were interpreted relative to concurrently studied N2.
Qualitative assessment of 4-HNE protein adducts in isolated mitochondria suggested the greatest cumulative lipid peroxidation occurred in the RC mutants (particularly mev-1) and the MnSOD mutants. In contrast, mitochondria lipid peroxidation did not appear increased in the long-lived insulin receptor mutant relative to wild-type.