

Abstract
The use of water as a reagent in redox-driven reactions is advantageous because it is abundant and environmentally compatible. The conversion of water to dioxygen in photosynthesis illustrates one example, in which a redox-inactive CaII ion and four manganese ions are required for function. In this report we describe the stepwise formation of two new heterobimetallic complexes containing CoII/III and CaII ions, and either hydroxo or aquo ligands. The preparation of a 4-coordinate CoII synthon was achieved with the tripodal ligand, N,N′,N″-[2,2′,2″-nitrilotris(ethane-2,1-diyl)]tris(2,4,6-trimethylbenzenesulfonamido, [MST]3−. Water binds to [CoIIMST]− to form the 5-coordinate [CoIIMST(OH2)]− complex that was used to prepare the CoII/CaII complex [CoIIMST(μ-OH2)CaII⊂15-crown-5(OH2)]+ ([CoII(μ-OH2)CaIIOH2]+). [CoII(μ-OH2)CaOH2]+ contained two aquo ligands, one bonded to the CaII ion and one bridging between the two metal ions and thus represents an unusual example of a heterobimetallic complex containing 2 aquo ligands spanning different metal ions. Both aquo ligands formed intramolecular hydrogen bonds with the [MST]3− ligand. [CoIIMST(OH2)]− was oxidized to form [CoIIIMST(OH2)] that was further converted to [CoIIIMST(μ-OH)CaII⊂15-crown-5]+ ([CoIII(μ-OH)CaII]+) in the presence of base and CaIIOTf2/15-crown-5. [CoIII(μ-OH)CaII]+ was also synthesized from the oxidation of [CoIIMST]− with PhIO in the presence of CaIIOTf2/15-crown-5. Allowing [CoIII(μ-OH)CaII]+ to react with diphenylhydrazine afforded [CoII(μ-OH2)CaIIOH2]+ and azobenzene. Additionally, the characterization of [CoIII(μ-OH)CaII]+ provides another formulation for the previously reported CoIV–oxo complex, [(TMG3tren)CoIV(μ-O)ScIII(OTf)3]2+ to one that instead could contain a CoIII–OH unit.
INTRODUCTION
A significant challenge in synthetic chemistry is regulating the secondary coordination sphere of metal ion(s) to affect function.1 These types of regulatory interactions are found within the active sites of metalloproteins and aid in promoting efficient and selective transformations. Non-covalent interactions are the major forces that control the properties of the secondary coordination spheres in proteins. For instance, intramolecular hydrogen bonds (H-bonds) involving metal bound water ligands are often found within the structures of active sites and are instrumental in dictating subsequent function. Lipoxygenase is a non-heme iron enzyme that catalyzes the cleavage of C–H bonds by cycling between FeIII–OH and FeII–OH2 states.2 In both states, it is proposed that H-bonding networks involving the hydroxo/aquo ligands are necessary for catalysis. Additionally, recent structural evidence on the oxygen-evolving complex (OEC) within Photosystem II has shown that the active site contains an extensive H-bonding network surrounding the terminal aquo ligands on the Mn4CaO5 cluster that most likely influences function.3,4 The OEC also illustrates that calcium ions have an essential role in the oxidation of water to dioxygen.5 The presence of a CaII ion within the OEC cluster suggests that group 2 metal ions can influence other redox processes performed by transition metal complexes. In fact, synthetic molecular water oxidation catalysts often contain a mixture of transition metal ions and group 1 or 2 ions,6a such as in the cobalt/K3PO4 system developed by Kanan and Nocera.6b,c Recent examples of MnCaO clusters have also been reported; in particular, the Mn3CaO4 system of Agapie demonstrated the role of the CaII ion in modulating redox potentials.7,8 Nevertheless, the limited number of discrete molecular examples makes it difficult to assess the effects of H-bonds and group 2 metal ions on redox processes at transition metal ions. Moreover, there are few synthetic examples of heterometallic complexes containing aquo ligands bound similarly to those found in the OEC.
In this report we describe the stepwise assembly of a series of CoII/III–OH2 and CoIII–OH complexes from water that contain intramolecular H-bonds and CaII ions (Chart 1). The complexes are formed using the tripodal ligand N,N′,N″-[2,2′,2″-nitrilotris(ethane-2,1-diyl)]tris-(2,4,6-tri-methylbenzenesulfonamido) ([MST]3−) that contains three sulfonamido units. We have shown that this design supports formation of complexes with (CaII-(μ-OH)-MnIII) cores.9 The formation of the heterobimetallic Ca/Co complexes in the present study establishes the importance of CaII ions in stabilizing water binding and is crucial in activating the cobalt center for redox chemistry. Moreover, our findings suggest that the recently reported heterobimetallic [(TMG3tren)CoIV(μ-O)ScIII(OTf)3]2+ complex could be formulated to have a CoIII–OH unit rather than the more unusual CoIV–oxo moiety.10
Chart 1.
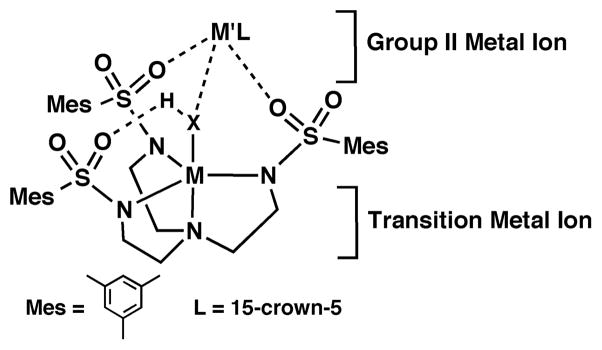
Design Aspects of the Heterometallic Complexes.
EXPERIMENTAL SECTION
General Methods
All reagents were purchased from commercial sources and used as received, unless otherwise noted. Solvents were sparged with argon and purified using a JC Meyer Co. solvent system. Potassium hydride (KH) as a 30% dispersion in mineral oil was filtered with a medium porosity glass frit and washed 5 times each with pentane and Et2O. Solid KH was dried under vacuum and stored under an inert atmosphere. The syntheses of metal complexes were conducted in a Vacuum Atmospheres, Co. drybox under an argon atmosphere. The ligand H3MST,9 Ca(OTf)2/15-crown-5,9 [FeCp2]OTf,11a acetyl ferrocenium tetrafluoroborate,11b and CaII[N(TMS)2]2THF2,11c and PhIO11d were synthesized according to literature methods. Tetrabutylammonium hexafluorophosphate (TBAP) was recrystallized from hot ethanol 3 times and dried at 150°C for several days under reduced pressure.
Preparative Methods
Preparation of Me4N[CoIIMST]
H3MST (900 mg, 1.3 mmol) was dissolved in 45 mL of DMA, and KH (160 mg, 3.9 mmol) was added and allowed to stir until gas evolution ceased (~30 min). This solution was allowed to react with CoII(OAc)2 (230 mg, 1.3 mmol) and Me4N(OAc) (260 mg, 1.9 mmol), and the mixture was allowed to stir overnight. The resulting purple solution was filtered to remove insoluble white material (KOAc, 330 mg, 3.4 mmol, 94% yield), the purple filtrate was concentrated (~5 mL), and Et2O was added to form a precipitate. The resulting blue solid was isolated on a fritted-glass funnel, washed several times with Et2O, and dried under vacuum for several hours (990 mg, 93%). Further purification of the crude material was possible by dissolving the blue solid in DCM and layering under pentane (660 mg, 62%). Dissolving the crude blue salt in a DMA and MeCN mixture (1:1) and allowing for slow vapor diffusion of Et2O afforded crystals suitable for X-ray diffraction analysis. FTIR: (KBr, cm−1) selected vibrations 3039, 3021, 2975, 2935, 2874, 2854, 2838, 1655, 1604, 1563, 1488, 1467,1446, 1405, 1379, 1346, 1279, 1237, 1221, 1141, 1102, 1073, 1055, 1039, 975, 960, 943, 932, 847, 826. UV–vis: λmax (DCM, nm (ε, M−1cm−1)) 420 (shoulder), 410 (23), 568 (72), 752 (14). Solid suitable for elemental analysis was recrystallized from DCM/pentane. Anal. Calcd (found) for Me4N[CoIIMST]·0.25 DCM (C37.25H57.5Cl0.5CoN5O6S3): C, 52.99 (52.68); H, 6.86 (6.91); N, 8.30 (8.33). An alternative method was also utilized: crystalline solid of this salt could be obtained by slow diffusion of Et2O into the initial DMA filtrate, which after 4 days yielded sky blue crystals (780 mg, 73%).
Preparation of Me4N[CoIIMST(OH2)]
Me4N[CoIIMST] (99 mg, 0.12 mmol) was dissolved in DCM (~5 mL) and treated with water (20 μL, 1.11 mmol), and the sky blue solution turned pink. Layering the pink solution under pentane produced a crystalline solid of the salt. The pink micro-crystals were isolated by decanting and then dried by flowing Ar over the solid for a few seconds (prolonged drying or use of vacuum resulted in loss of the aquo ligand). FTIR: (Nujol, cm−1) ν(OH) 3494, 3186; (KBr, cm−1) ν(OH) 3602, 3497, 3273; selected vibrations (KBr, cm−1) 3022, 2960, 2913, 2856, 1653, 1603, 1563, 1489, 1467, 1451, 1405, 1377, 1384, 1256, 1231, 1133, 1120, 1093, 1055, 1038, 978, 957, 932, 846, 826, 813. UV–Vis: λmax (DCM, nm (ε, M−1cm−1)) 530 (63), 712 (26). Anal. Calcd (found) for Me4N[CoIIMST(OH2)]·2H2O (C37H63CoN5O9S3): C, 50.67 (50.73); H, 7.24 (7.18); N, 7.99 (7.79)
Preparation of [CoIIMST(μ-OH2)CaII⊂15-crown-5-(OH2)]OTf
Me4N[CoIIMST] (100 mg, 0.13 mmol) was dissolved in 4 mL of DCM and CaIIOTf2/15-crown-5 (73 mg, 0.13 mmol) was added. After stirring for 5 min, the homogenous blue solution was treated with water (12 mg, 0.67 mmol) and the solution turned pink. Crystals suitable for diffraction were obtained by layering the pink solution under pentane. Crystallized compound was separated from Me4NOTf by vigorously stirring with pentane and decanting; the pink salt was isolated on a fritted-glass funnel and dried under reduced pressure (111 mg, 63%). FTIR: (Nujol, cm−1) ν(OH) 3430 and 3218; selected vibrations 1604, 1564, 1151, 1124, 1096, 1033, 992, 980, 955, 892, 875, 819. UV–vis: λmax (DCM, nm (ε, M−1cm−1)) 530 (49), 698 (28). Anal. Calcd (found) for [CoIIMST(μ-OH2)Ca⊂15-crown-5(OH2)]OTf·0.5DCM (C44.5H70CaClCoF3N4O16S4): C, 43.22 (43.01); H, 5.70 (5.87); N, 4.53 (4.49)
Preparation of [CoIIIMST(μ-OH)Ca⊂15-crown-5]OTf from H2O
Me4N[CoIIMST] (100 mg, 0.12 mmol) was suspended in 5 mL of THF and H2O (12 mg, 0.67 mmol) was added, causing the mixture to become homogenous and pink. The solution was then treated with [FeCp2]OTf (40 mg, 0.12 mmol) to produce a dark red mixture that was stirred for at least 30 min. The mixture was then treated with a 1 mL THF solution containing the following species: CaII[N(TMS)2]2(THF)2 (45 mg, 0.089 mmol), CaIIOTf2/15-crown-5 (50 mg, 0.090 mmol), and 15-crown-5 (20 mg, 0.091 mmol). This addition resulted in an immediate color change from dark red to yellow-brown. The solution was stirred for 1 h, after which it was filtered and the THF solvent was removed under reduced pressure. The brown solid was triturated with pentane (3×5 mL portions) until the washings were colorless, affording a yellow brown solid that was dried under reduced pressure. The solid was re-dissolved in a minimal amount of DCM and stored at −35°C overnight to produce a white solid (Me4NOTf) and a small amount of pink crystals ([CoIIMST(μ-OH2)Ca⊂15-crown-5(OH2)]OTf) that were removed from the solution via filtration. The resulting yellow-brown filtrate was layered under pentane and stored at −35°C to produce yellow brown crystals (44 mg, 31%). Crystals suitable for diffraction were obtained by dissolving the crystalline salt in DCM and layering under pentane at rt. FTIR: (KBr, cm−1) ν(OH) 3430; selected vibrations 3023, 2935, 2858, 1604, 1471, 1405, 1380, 1355, 1265, 1229, 1133, 1090, 1053, 1031, 978, 954, 872, 852, 822. UV–vis: λmax (DCM, nm (ε, M−1cm−1)) 390 (4800), 430 (shoulder), 730 (730). ESI-MS(+) Cald (m/z) (found) for [CoIIIMST(μ-OH)Ca⊂15-crown-5]+(C43H66CaCoN4O12S3): 1025.2798 (1025.2771)
Preparation of [CoIIIMST(μ-OH)Ca⊂15-crow-n5]OTf from PhIO
Me4N[CoIIMST] (99 mg, 0.12 mmol) was dissolved in 5 mL of DCM, CaIIOTf2/15-crown-5 (68 mg, 0.12 mmol) was added and the mixture was stirred until homogenous. The blue solution was then treated with PhIO (51 mg, 0.23 mmol), causing the blue solution to slowly turn yellow-brown (30 min). Unreacted PhIO was removed by filtration and the resulting yellow brown filtrate was concentrated to dryness to afford a solid that was washed with pentane (3×10 mL). The solid was extracted with benzene (3×10 mL). The benzene solution was reduced to dryness in vacuo. The resulting fine yellow-brown solid was isolated using a fritted-glass funnel, washed with pentane, and dried to afford 120 mg (85%) of the desired salt. The spectroscopic data match those obtained for samples of the salt made by the previously described method.
Preparation of [CoIIIMST(OH2)]
Me4N[CoIIMST] (50 mg, 0.061 mmol) was dissolved in 5 mL of DCM and treated with water (5.0 mg, 0.28 mmol), causing a color change from blue to pink. The homogenous solution was treated with [FeCp2]OTf (42 mg, 0.13 mmol) resulting in an immediate color change to deep red. After stirring for 30 min, the reaction mixture was filtered and the DCM filtrate was layered under pentane. A brown-red solid mixture of [CoIIIMST(OH2)], unreacted [FeCp2]OTf, and Me4NOTf was obtained, which was isolated on a fritted-glass funnel and washed with pentane until the washings were colorless. The solid was dried (45 mg) and suspended in ~10 mL of toluene, stirred for at least 1 h, and filtered to afford a red filtrate. Pentane (10 mL) was then added to the filtrate and the mixture was stored at −35°C, causing the precipitation of pure [CoIIIMST(OH2)]. The complex was isolated on a fritted-glass funnel, washed with pentane, and dried (13 mg, 28 %). Crystals suitable for diffraction were obtained by saturating a DCM:pentane solution with pure [CoIIIMST(OH2)] and storing at −35°C for an extended period of time. FTIR: (KBr, cm−1) ν(OH) 3308; selected vibrations 2970, 2936, 2867, 1603, 1564, 1469 1442, 1405, 1388, 1306, 1280, 1144, 1118, 1073, 1051, 1033, 983. UV–vis: λmax (DCM, nm (ε, M−1cm−1)) 403 (5700), 494 (5100), 845 (750). Anal. Calcd (found) for [CoIIIMST(OH2)]·0.75 DCM (C33.75H48.5Cl1.5CoN4O7S3): C, 48.80 (49.07); H, 5.89 (6.32); N, 6.75 (6.72)
Physical Methods
Elemental analyses were performed on a Perkin-Elmer 2400 CHNS analyzer. Electronic absorbance spectra were recorded with a Cary 50 UV-vis spectrophotometer and an 8453 Agilent UV-vis spectrophotometer equipped with an Unisoku Unispeks cryostat. Fourier transform infrared spectra were collected on a Varian 800 Scimitar Series FTIR spectrometer. 1H NMR spectra were recorded on a Bruker DRX500 spectrometer. High resolution mass spectra were collected using Waters Micromass LCT Premier Mass Spectrometer. Cyclic volt-ammetric experiments were conducted using a CHI600C electrochemical analyzer. A 2.0 mm glassy carbon electrode was used as the working electrode at scan velocities 0.1 V·s−1. A cobaltocenium/cobaltocene couple (−1.33 V vs. [FeCp2]+/0) was used as an internal reference to monitor the reference electrode (Ag+/Ag). Perpendicular-mode X-band electron paramagnetic resonance (EPR) spectra were collected using a Bruker EMX spectrometer equipped with an ER041XG microwave bridge, an ER4116DM dual-mode cavity, and an Oxford Instrument liquid He quartz cryostat. All EPR spectra were recorded at 10 K with the following experimental parameters: frequency 9.64 GHz, power 0.20 mW, modulation amplitude 9.02 G, time constant 20.48 seconds, conversion time 40.96 seconds. EPR spectral simulations were done using SpinCount 3.0.12 Crystals for X-ray diffraction were mounted on a Bruker SMART APEX II diffractometer and the APEX2 program package was used to determine the unit-cell parameters and for data collection.12 X-ray absorption spectra were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) on beamlines 7–3 at an electron energy of 3.0 GeV with an average current of 300 mA The radiation was monochromatized by a Si(220) double-crystal monochromator. The intensity of the incident X-ray (I0) was monitored by an N2-filled ion chamber in front of the sample. The data were collected as fluorescence excitation spectra with a Ge 30 element detector (Canberra). Energy was calibrated by the rising edge position of Co foil (7709.0 eV).
RESULTS AND DISCUSSION
Sulfonamido groups have been used by others to prepare multidentate ligands. For instance, the work of Walsh illustrated that C2 symmetric sulfonamido ligands could be used to synthesize a series of metal complexes.13 Structural studies on these complexes showed that in some cases both the deprotonated nitrogen donors and the oxygen atoms could bind metal ions to form bridged dinuclear complexes. Sulfonamido groups also have been incorporated into tripodal compounds, but only those ligands of Mountford were used to prepare metal complexes.14 We have extended the use of sulfonamido-based tripods by preparing [MST]3−, which has allowed us to synthesize heterobimetallic complexes.9 Formation of this type of complex takes advantage of the two structurally distinct metal ion-binding sites on the ligand (Chart 1): one that is composed of four nitrogen atoms and a second site that incorporates oxygen atoms of the sulfonamido moieties. The nitrogen-based site can readily bind a transition metal ion, whereas the oxygen-based site is able to bind main-group metal ions. Furthermore, the sulfonamido groups are positioned to promote the formation of intramolecular H-bonds. We have applied these concepts to assemble a series of CoII–aquo and CoIII–OH complexes.
Preparation and Structure of [CoIIMST]−
Treating a DMA solution of H3MST with 3 equiv of KH followed by CoII(OAc)2 and metathesis with Me4N(OAc) resulted in the formation of Me4N[CoIIMST], which was purified via crystallization. The crystal structure of Me4N[CoIIMST] revealed that the anion contained a four-coordinate cobalt center having trigonal monopyramidal (TMP) coordination geometry (Figure 1). The average Co-Neq bond length and Neq-Co-Neq angle were 1.967(3) Å and 119.1(2)°, and the Co1–N1 bond distance was 2.114(1) Å (Table 1). The CoII center is displaced 0.207 Å from the plane formed by N2, N3, and N4 toward the vacant axial coordination site. These metrical parameters are similar to those of other CoII complexes with TMP geometry.15a,b;16 The structure of [CoIIMST]− also showed that the SO2R groups of the [MST]3− ligand formed a cavity around the vacant coordination site on the cobalt center. The UV-vis spectrum of Me4N[CoIIMST] in DCM had absorbance peaks at λmax = 410 nm, 572 nm and 712 nm (Figure S1)—these features are similar to the optical properties found for previously reported TMP CoII complexes.15a,b;16 Moreover, EPR spectra on frozen DCM solutions of Me4N[CoIIMST] showed that the complex has an S = 3/2 spin ground state (Table 2), similar to what is observed in other C3-symmetric, four-coordinate CoII complexes.15b,17 Based on these data we concluded that [CoIIMST]− maintained a TMP structure in solution and could serve as a synthon for the preparation of CoII/III–OH2 and CoIII–OH complexes.
Figure 1.
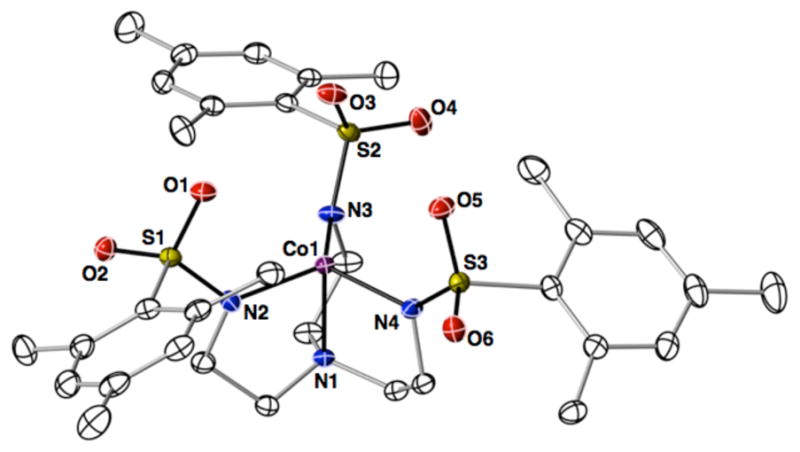
Thermal ellipsoid diagram depicting the molecular structure of [CoIIMST]−. The thermal ellipsoids are drawn at the 50% probability level and the hydrogen atoms are removed for clarity.
Table 1.
Selected metrical data for [CoIIMST]−, [CoIIMST(OH2)]−, and [CoIIIMST(OH2)].
| [CoIIMST]− | [CoIIMST(OH2)]− | [CoIIIMST(OH2)] | |
|---|---|---|---|
| Bond Distances (Å) | |||
| Co1–O7 | NA | 2.135(1) | 1.934(2) |
| Co1–N1 | 2.114(1) | 2.199(1) | 1.954(2) |
| Co1–N2 | 1.973(1) | 2.020(1) | 1.950(2) |
| Co1–N3 | 1.957(1) | 2.028(1) | 1.927(2) |
| Co1–N4 | 1.971(1) | 2.054(1) | 1.961(2) |
| d[Co-Neq] | 0.207 | 0.310 | 0.127 |
| Bond Angles (°) | |||
| N1-Co1-O7 | 175.52(5) | 178.20(7) | |
| N3-Co1-N4 | 118.90(5) | 119.83(5) | 119.48(8) |
| N4-Co1-N2 | 117.52(5) | 119.28(5) | 119.43(7) |
| N3-Co1-N2 | 120.31(5) | 114.04(5) | 118.78(7) |
Table 2.
EPR and Redox Properties of the CoII Complexes.
| Complex | g-values | Az (cm−1) | E1/2 (V)a |
|---|---|---|---|
| [CoIIMST]− | 4.36, 2.00 | 96×10−4 | 0.41 |
| [CoIIMST]−+CaII | 4.31, 2.00 | 94×10−4 | 0.62 |
| [CoIIMST(OH2)]− | 4.33, 2.04 | 90×10−4 | 0.068 |
| [CoII(μ-OH2)CaIIOH2]+ | 4.35,2.02 | 98×10−4 | 0.29 |
versus [FeCp2]+/0
Preparation and Structures of CoII–Aquo Complexes
The synthesis of [CoIIMST]− allowed us to examine the step-wise binding of external species to the complex. This process is illustrated with the preparation of the mono-aquo CoII complex [CoIIMST(OH2)]−, which was accomplished by treating a DCM solution of [CoIIMST]− with excess H2O causing a color change from sky blue to pink (Scheme 1). The UV-vis spectrum of [CoIIMST(OH2)]− is similar to that found for the CoII-OH complex [CoIIH3buea(OH)]2−, which has a TBP coordination geometry.15c The molecular structure of the complex was determined by X-ray diffraction methods and confirmed the trigonal bipyramidal coordination geometry of the cobalt center (Figure 2). The binding of the water molecule occurs at the open site within the sulfonamido cavity, having a Co1–O7 bond distance of 2.135(1) Å and an O7-Co1-N1 bond angle of 175.52(5)°. Minor structural differences in bond lengths of less than 0.085 Å were found between [CoIIMST]− and [CoIIMST(OH2)]−. In addition, the CoII ion is displaced 0.310 Å from the plane formed by N2, N3, and N4 atom of [MST]3−, a change of greater than 0.1 Å from that observed in [CoIIMST]−. The aquo ligand also forms two intramolecular H-bonds to the O3 and O 5 atoms of [MST]3− as indicated by the O7···O3 and O7···O5 distances of 2.677(2) Å.
Scheme 1.

Preparative routes to the cobalt complexes. Conditions: a) 5–10 equiv H2O, rt, DCM; b) Ca(OTf)2/15-crown-5, 5 equiv H2O, rt, DCM; c) [FeIIICp2]OTf, rt, DCM (or THF); d) Ca(OTf)2/15-crown-5, PhIO, rt, DCM; e) 0.5 equiv Ca[N(TMS)2]2·THF2, 0.5 equiv 15-crown-5, and equiv 0.5 Ca(OTf)2/15-crown-5, rt, THF; f) 10 equiv DPH, rt, DCM.
Figure 2.
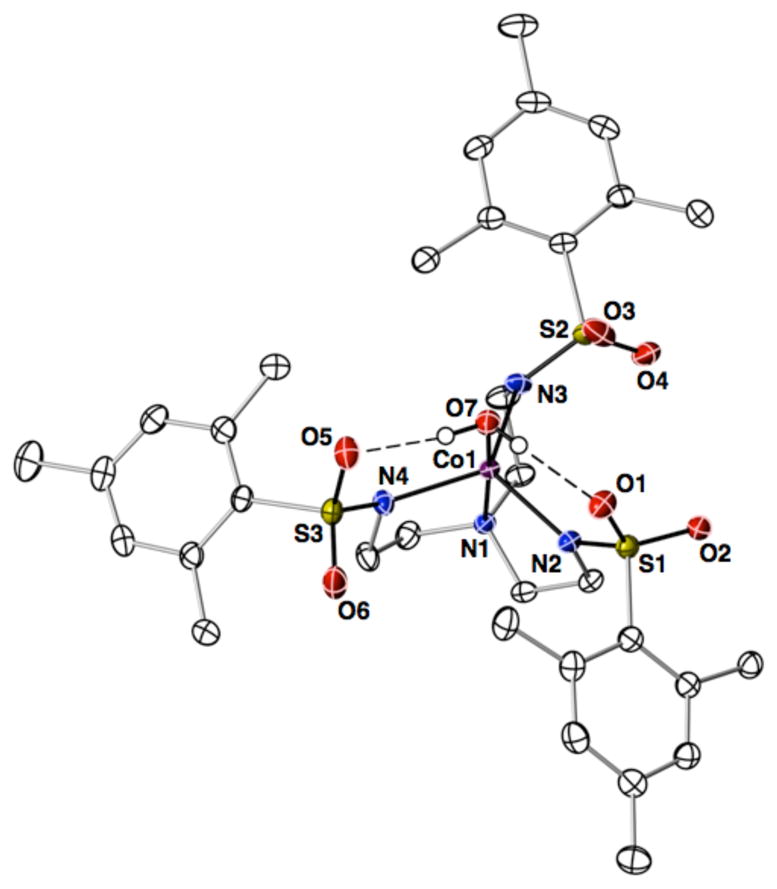
Thermal ellipsoid diagram depicting the molecular structure of [CoIIMST(OH2)]−. The thermal ellipsoids are drawn at the 50% probability level and only the hydrogen atoms of the water ligand are shown for clarity.
The binding of water to [CoIIMST]− also occurred when CaII ions were added to the complex. In the presence of CaOTf2/15-crown-5 and 5 equiv H2O (Scheme 1), a new species was isolated whose analytical and vibrational properties suggested the formation of a heterobimetallic complex containing a CoII–(μ-OH2)–CaII core. The visible spectrum of this species measured in DCM was similar to [CoIIMST(OH2)]−, a finding that is consistent with the complex maintaining a CoII–aquo unit with trigonal bipyramidal coordination geometry (Figure S1).18
Determination of the molecular structure of this new species revealed the heterobimetallic complex [CoIIMST(μ-OH2)CaII⊂15-crown-5(OH2)]+ ([CoII(μ-OH2)CaIIOH2]+) (Figure 3). As in [CoIIMST(OH2)]−, the CoII ion accommodates a single aquo ligand; however, the aquo ligand bridges the CoII and CaII ions to form the CoII–(μ-OH2)–CaII core. There are several small changes in the metrical parameters around the CoII ion as compared to those in [CoIIMST(OH2)]−. For instance, the Co1–O7 bond length of 2.221(2) Å in [CoII(μ-OH2)CaIIOH2]+ is elongated by 0.086 Å. In addition, the CoII ion in [CoII(μ-OH2)CaIIOH2]+ is slightly more displaced out of the trigonal plane by 0.015 Å. The bridging aquo ligand also forms two intramolecular H-bonds as indicated by the O7···O3 and O7···O5 distances of 2.656(2) Å and 2.693(2) Å. These changes suggested that the CaII ion does not perturb the primary coordination sphere of the CoII ion to a large extent, in agreement with the optical spectrum.
Figure 3.

Thermal ellipsoid diagram depicting the molecular structure of [CoII(μ-OH2)CaIIOH2]+. The thermal ellipsoids are drawn at the 50% probability level and only the hydrogen atoms of the water ligands are shown for clarity.
In addition to the small changes in the primary coordination sphere of the CoII center, there are several new structural features within the secondary coordination sphere. The [CoIIMST(OH2)]− moiety acts as a bidentate ligand to the CaII ion through the bridging aquo O7 atom and the O1 atom from a sulfonamido group with a Ca1–O1 bond distance of 2.399(1) Å and a longer Ca1–O7 length of 2.531(2) Å. The structure of [CoII(μ-OH2)CaIIOH2]+ also showed that the complex had a second terminal aquo ligand bonded to the CaII ion. The Ca1–O13 bond length of 2.369(2) Å is significantly shorter than the distance observed for the Ca1–O7 bond by 0.16 Å. The terminal aquo also forms a single intramolecular H-bond with an O13···O2 distance of 2.758(2) Å. The oxygen atoms of the 15-crown-5 ligand fill the remaining coordination sites on the CaII ion. The structure of [CoII(μ-OH2)CaIIOH2]+ also showed that the two aquo ligands are relatively close to one another, having an O7···O13 interatomic distance of 3.762(3) Å.
Even though the structural properties of the primary coordination sphere of the CoII center in [CoIIMST(OH2)]− and [CoII(μ-OH2)CaIIOH2]+ are nearly the same, the addition of CaII ion affects the lability of Co–OH2 unit in the solid state. Conversion of [CoIIMST(OH2)]− to [CoIIMST]− was accomplished by simply applying a small vacuum (P ~ 10−3 torr). However, attempts to remove either the terminal or bridging aquo ligands in [CoII(μ-OH2)CaIIOH2]+ were unsuccessful, even after prolonged exposure to vacuum, determined by comparison of FTIR spectra (Figure S2B) and elemental analyses to those obtained from independently pre-pared samples of [CoII(μ-OH2)-Ca2+⊂15-crown-5-(OH2)]OTf.
Electrochemical and EPR Properties
The redox properties of the CoII complexes were explored using cyclic voltammetry in DCM (Figures 4A and S3, Table 2). For [CoIIMST]−, a reversible one-electron process was observed at 0.41 V vs [FeCp2]+/0 that is assigned to the CoII/III couple. The potential shifted to more positive values when CaII ions were introduced to the electrochemical cell. Treating [CoIIMST]− with 1 equiv Ca(OTf)2/15-crown-5 produced a new quasi-reversible couple at 0.62 V vs [FeCp2]+/0, suggesting that CaII ions can interact with [CoIIMST]− in the absence of an external ligand within the cavity (Figure S3).
Figure 4.

Cyclic voltammograms (A) and ⊥-mode EPR spectra (B) of [CoIIMST(OH2)]− (black line) and [CoII(μ-OH2)CaIIOH2]+ (gray line). Cobaltocenium (*) was used as an internal standard in the CV experiments. The voltammograms were recorded at room temperature in DCM (with 0.1 M TBAPF6) and the EPR data were collected at 10 K on frozen DCM solution (6 mM). Inset: comparison of the hyperfine interactions for the two CoII–aquo complexes. See Table 2 for E1/2, g- and Az-values.
The electrochemical properties of the CoII–OH2 complexes were substantially different from [CoIIMST]−. For [CoIIMST(OH2)]−, a reversible couple was found at 0.068 V, a shift of nearly 0.4 V from that of [CoIIMST]−. This shift is consistent with a more facile oxidation of a five-coordinate CoII center than one that is four-coordinate. The CoII/III potential for [CoII(μ-OH2)CaIIOH2]+ appeared at 0.29 V vs. [FeCp2]+/0, a shift of more than 0.2 V compared to [CoIIMST(OH2)]−. The increase in redox potential reflects that change in overall positive charge of the complex upon the binding of the CaII ion.
The ⊥-mode EPR spectra measured on frozen solutions of the CoII complexes all have features with g-values centered near 4.3 and 2.0, which is indicative of S = 3/2 spin ground states.17 However, there are distinguishable differences in the g-values (Figures 4B and S4, Table 2) that reflect the changes in the coordination environments of the complexes. In addition, the complexes have similar eight-line hyperfine patterns on the features at g ~ 2.0 (Table 2), which is consistent with monomeric CoII species. Notice that the largest difference in the hyperfine splitting (ΔAz = 8 × 10−4 cm−1) is observed between the two CoII–OH2 species, [CoIIMST(OH2)]− and [CoII(μ-OH2)CaIIOH2]+.
Synthesis & Structure of [CoIIIMST(OH2)]
The reversible CoII/III couple measured for [CoIIMST(OH2)]− prompted our investigation into isolating this oxidized product by chemical methods. We found that treating [CoIIMST(OH2)]− with [FeIIICp2]OTf in DCM (or THF) resulted in an immediate color change from pink to deep red (Scheme 1, Figure S5). The optical spectrum in DCM of the new species revealed intense features at λmax (εM) = 403 (5700), 494 (5100), 845 (750) nm that are characteristic of CoIII complexes with TBP geometry.15c Additional spectroscopic and analytical measurements suggested that this new red species was the expected CoIII–OH2 complex, [CoIIIMST(OH2)]. We also investigated this oxidative process in the presence of Ca(OTf)2/15-crown-5 and found that the reaction was incomplete when using [FeIIICp2]OTf as the oxidant, which agrees with our electrochemical observations. Complete conversion to an oxidized species was accomplished using the stronger oxidant acetyl ferrocenium tetrafluoroborate (E1/2 = 0.27 V vs. [FeCp2]+/0);11b this complex had optical properties nearly identical to those of [CoIIIMST(OH2)] but we were unable to obtain the complex in sufficient purity to determine its formulation.
The molecular structure of [CoIIIMST(OH2)] showed that the primary coordination sphere of the cobalt center had the expected contraction compared to its CoII–OH2 analog (Table 1 and Figure S6). The Co1–N1 bond distance in [CoIIIMST(OH2)] is 1.954(2) Å and its average Co1–Neq bond distance is 1.946 Å, which are shorter by 0.245 Å and 0.088 Å, respectively compared to those in [CoIIMST(OH2)]−. In addition, the Co1–O7 bond distance in [CoIIIMST(OH2)] decreased by 0.201 Å upon oxidation to 1.934(2) Å. There are also intramolecular H-bonds between the aquo ligand and [MST]3− in [CoIIIMST(OH2)], yet the O7···O1 and O7···O5 distances of 2.589 Å and 2.576 Å are significantly shorter than those found in [CoIIMST(OH2]−. The shortening of these distances corresponds to a strengthening of the H-bonds in [CoIIIMST(OH2)], which is expected because the acidity of the aquo ligand will increase upon coordination to the CoIII center. This premise is further supported by FTIR studies: the ν(O-H) bands in [CoIIIMST(OH2)] are broadened and at lower energies than those found in [CoIIMST(OH2)]−.
Oxidation Chemistry: Preparation and Structure of [CoIIIMST(μ-OH)-Ca⊂15-crown-5]+
[CoIIMST]− did not react with dioxygen even in the presence of CaII ions, in agreement with our electrochemical measurements. The reactivity of [CoIIMST]− with PhIO was also explored and we found no evidence for a reaction, even after several days. However, when [CoIIMST]− was treated with PhIO in the presence of Ca(OTf)2/15-crown-5, a new dark yellow-brown species was formed within 1 h (Scheme 1). The new species had an absorption spectrum in DCM with features at λmax (εM) = 390 (4800), 430 (sh), 730 (730) nm, which are similar to another monomeric TBP CoIII–OH complex (Figure S5).15c ESI-HRMS results showed a species with a strong ion peak with mass-to-charge ratio (m/z) of 1025.2771 (calcd, 1025.2798) corresponding to a species having a formulation of [CoIIIMST(μ-OH)-Ca⊂15-crown-5]+ ([CoIII(μ-OH)CaII]+). Furthermore, FTIR analysis revealed a sharp peak at 3430 cm−1 supporting the presence of a hydroxo ligand (Figure S7).
Repeated attempts to crystallize [CoIII(μ-OH)CaII]+ synthesized from PhIO were unsuccessful and thus a new synthetic route was sought. We reasoned that deprotonation of [CoIIIMST(OH2)] with an appropriate base could lead to a putative CoIII-OH complex which could then be treated with Ca(OTf)2/15-crown-5 to form [CoIII(μ-OH)CaII]+. This approach was realized by treating a THF solution of [CoIIIMST(OH2)] with 0.5 equiv Ca[N(TMS)2]2·THF2, 0.5 equiv 15 -crown-5, and 0.5 equiv Ca(OTf)2/15-crown-5 that produced a species that had identical spectroscopic properties to [CoIII(μ-OH)CaII]+ (Scheme 1). Moreover, this approach yielded single crystals that were suitable for X-ray structural determination, thereby confirming the mass spectral formulation of the yellow-brown species as [CoIIIMST(μ-OH)-Ca⊂15-crown-5]+ (Figure 5). The molecular structure of [CoIII(μ-OH)CaII]+ contained a hydroxo ligand bridging the CoIII center and the CaII ion, in a manner similar to the manganese analog.9 The CoIII center has TBP coordination geometry provided by the nitrogen donors of the [MST]3− ligand and the hydroxo ligand. The Co1–N1 and Co1–Neq bond distances are comparable to those found for [CoIIIMST(OH2)]. However, the Co1–O7 bond distance in [CoIII(μ-OH)CaII]+ is 1.854(1) Å,19 a decrease in length of 0.080 Å from [CoIIIMST(OH2)] and 0.367 Å from [CoII(μ-OH2)CaIIOH2]+. The N1-Co1-O7 bond angle in [CoIII(μ-OH)CaII]+ is statistically equivalent to that in [CoII(μ-OH2)CaIIOH2]+, whereas the Ca1–O7 bond distance in [CoIII(μ-OH)CaII]+ is 0.265 Å shorter than that in [CoII(μ-OH2)CaIIOH2]+. In addition, there is a large contraction of the Co1-Ca1 interatomic distance of over 0.5 Å between [CoII(μ-OH2)CaIIOH2]+ and [CoIII(μ-OH)CaII]+.
Figure 5.
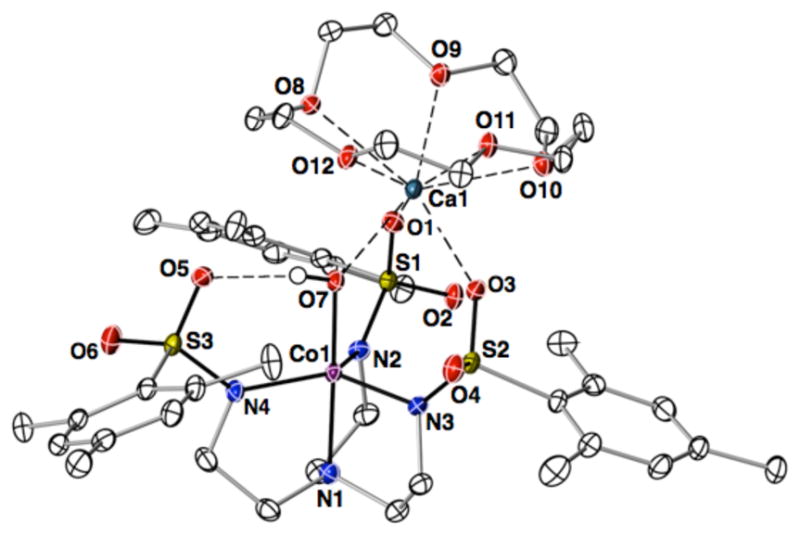
Thermal ellipsoid diagram depicting the molecular structure of [CoIII(μ-OH)CaII]+. The thermal ellipsoids are drawn at the 50% probability level and only the hydrogen atom of the hydroxo ligand is shown for clarity.
A comparison of the molecular structures of [CoII(μ-OH2)CaIIOH2]+ and [CoIII(μ-OH)CaII]+ (Table 3) illustrate the versatility of the [MST]3− ligand to accommodate a second metal ion and form intramolecular H-bonds. In [CoIII(μ-OH)CaII]+ a single intramolecular H-bond between the hydroxo ligand and O5 of the [MST]3− ligand was present. Because of this lone H-bond, the remaining sulfonamido groups, along with the hydroxo ligand, formed a triden-tate-binding site for the calcium ion: binding to the CaII ion occurred through O7 and the O1 and O3 atoms of the sulfonamido groups. In [CoII(μ-OH2)CaIIOH2]+, the bridging aquo ligand (the one containing O7) is involved in two intramolecular H-bonds with the [MST]3− ligand. Thus, the O1 atom of the remaining sulfonamido group and O7 act as a bidentate ligand to the calcium center. These results suggest that [MST]3− can regulate the mode of coordination to a second metal ion depending on the type of intramolecular H-bonding network in the complex.
Table 3.
Comparison of metrical parameters for [CoIII(μ-OH)CaII]+ and [CoII(μ-OH2)CaIIOH2]+.
| [CoIII(μ-OH)CaII]+ | [CoII(μ-OH2)CaIIOH2]+ | |
|---|---|---|
| Bond Distances (Å) | ||
| Co1–O7 | 1.854(1) | 2.221(2) |
| Co1–N1 | 1.997(1) | 2.126(2) |
| Co1–N2 | 1.971(2) | 2.037(2) |
| Co1–N3 | 1.950(2) | 2.031(2) |
| Co1–N4 | 1.962(2) | 2.013(2) |
| Ca1–O1 | 2.361(2) | 2.399(1) |
| Ca1–O3 | 2.379(1) | – |
| Ca1–O7 | 2.266(1) | 2.531(2) |
| Ca1–O13 | – | 2.369(2) |
| Ca1···Co1 | 3.804(1) | 4.315(1) |
| d[Co-Neq] | 0.212 | 0.325 |
| Bond Angles (°) | ||
| N1-Co1-O7 | 177.38(6) | 177.75(7) |
| Co1-O7-Ca1 | 134.56(7) | 130.41(7) |
| O7-Ca1-O13 | – | 100.29(6) |
| N3-Co1-N4 | 124.35(7) | 113.87(8) |
| N4-Co1-N2 | 123.14(7) | 11414(8) |
| N3-Co1-N2 | 109.01(7) | 12440(8) |
The reactivity of [CoIII(μ-OH)CaII]+ with DPH was also explored. [CoIII(μ-OH)CaII]+ reacts slowly with 0.5 equiv of DPH at room temperature. However, when 10 equiv DPH were employed, [CoIII(μ-OH)CaII]+ was converted to a new species in less than 20 min, whose EPR spectrum was identical to that of [CoII(μ-OH2)CaIIOH2]+ (Figures S8 and S9), along with the formation of azobenzene in a yield of 70%.
Comparison of [CoIII(μ-OH)CaII]+ to a Proposed [CoIV(μ-O)ScIII]2+ Complex
We compared the properties of the CoIII/II complexes with [MST]3− to other cobalt complexes with C3-symmetric ligands.10,15 In particular, we found noticeable similarities in the reactivity and physical properties to complexes synthesized by Ray derived from [CoIITMG3tren(OTf)]+, a five-coordinate CoII complex with a tripodal ligand that is comparable to [MST]3−.10 Both [CoIIMST]− and [CoIITMG3tren(OTf)]+ are only oxidized with oxygen-atom transfer reagents in the presence of redox-inactive metal ions. Ray reported that treating [CoIITMG3tren(OTf)]+ with sPhIO and Sc(OTf)3 produced a new species formulated as [(TMG3tren)CoIV(μ-O)ScIII(OTf)3]2+, whose EPR features were assigned to a CoIV(μ-O)ScIII core having an S = 3/2 spin ground state. This presumed CoIV product had a visible spectrum with features at λmax = 489 and 810 nm that are strikingly similar to those found for [CoIII(μ-OH)CaII]+ (Figure S5) and other CoIII–O(H) species.15c,d Using EXAFS methods, a Co–O bond length of 1.85 Å was found in [(TMG3tren)CoIV(μ-O)ScIII(OTf)3]2+, which was used as evidence for a CoIV–oxo unit; yet this bond length is identical to the Co1–O7 bond distance of 1.854(2) Å found by X-ray diffraction methods for [CoIII(μ-OH)CaII]+.19 Moreover, data from X-ray absorption near-edge spectra (XANES) showed a 1.25 eV shift in edge energies between [CoIITMG3tren]2+ and the [(TMG3tren)CoIV(μ-O)ScIII(OTf)3]2+ complex. The XANES spectra for both [CoII(μ-OH2)CaIIOH2]+ and [CoIII(μ-OH)CaII]+ have also been measured20 (Figure 6) and we found an edge energy shift of 1.42 eV even though the cobalt centers in our complexes differ by only one oxidation state. From these results we propose that treating [CoIITMG3tren(OTf)]+ with sPhIO in the presence of Sc3+ ions produced a meta-stable [(TMG3tren)CoIII(μ-OH)ScIII(OTf)3]2+ complex rather than the CoIV–oxo analog proposed previously. Our premise is supported by the similarities in the electronic and structural properties of the oxidized product of [CoIITMG3tren(OTf)]+ with those found in other CoIII–hydroxo complexes, including [CoIII(μ-OH)CaII]+. A [(TMG3tren)CoIII(μ-OH)ScIII(OTf)3]2+ complex should also have analogous types of reactivity as we observed for [CoIII(μ-OH)CaII]+; namely it can react with X–H bonds to produce a CoII–containing species. Our proposal suggests that both CoIII and CoII complexes could be present at the same time21 and accounts for the observation of an S = 3/2 species (i.e., these EPR features are derived from a CoII center, rather than CoIV). Note that the EPR parameters, specifically the hyperfine interactions, between the putative Ray’s CoIV–oxo complex and our [CoII(μ-OH2)CaIIOH2]+ are nearly identical (Az = 100 × 10−4 and 98 × 10−4 cm−1, respectively).
Figure 6.

XANES spectra for [CoII(μ-OH2)CaIIOH2]+ (gray line) and [CoIII(μ-OH)CaII]+ (black line) collected on solid samples (10% by weight) in boron nitride.20
SUMMARY
We have demonstrated for the first time the stepwise generation of Co–aquo complexes that led to the isolation of a system containing a CoII(μ-OH2)CaIIOH2 core, a motif resembling structural elements found in the OEC. The active site includes both transition metal ions (Mn) and a CaII ion, in addition to an extensive H-bonding network, and all these features are critical for water oxidation.3,4 The presence of a CaII ion in the OEC has prompted several synthetic efforts to prepare transition metal clusters that include calcium ions.5–9 In addition, intramolecular H-bonds have been proposed to be involved in some synthetic water oxidation systems22 and have been used to isolate metal-aquo complexes.23 However, incorporation of a CaII ion, transition metal ions, at least two water molecules, and an intramolecular H-bond network into a single molecular species has been difficult to replicate in synthetic systems. We accomplished the synthesis of this type of heterobimetallic complex using the multifunctional tripodal ligand [MST]3−, which coordinated both CaII and CoII ions, as it positioned two aquo ligands within 3 Å of each other. Its design took advantage of non-covalent interactions that linked the two metal centers and stabilized the aquo ligands through intramolecular H-bonds. Utilizing non-covalent interactions produced versatile systems in which the cobalt and CaII centers could readily adopt different primary and secondary coordination spheres.
Changes in redox properties of the CoII complexes correlated with the coordination of CaII ions. For example, the redox potential of the [CoIIMST]− shifted over 200 mV in the presence of CaII ions. A similar positive shift was observed when CaII ions were added to [CoIIMST(OH2)]−. Moreover, [CoIIMST]− does not react with dioxygen but only stronger oxidants, such as PhIO, in the presence of CaII ions to afford the heterobimetallic complex [CoIII(μ-OH)CaII]+. The dependency of this reaction on CaII ions suggests an inner sphere process, whereby the CaII ion is essential for binding and oxidation. The need for CaII ions to promote reactivity is similar to what we observed in the synthesis of the analogous [MnIII(μ-OH)CaII]+ species.9,24
The conversion of [CoIII(μ-OH)CaII]+ to [CoII(μ-OH2)CaIIOH2]+ when treated with DPH illustrated that CoIII–OH complexes can homolytically cleave X–H bonds. This type of reactivity is observed in the enzyme lipoxygenase which utilizes FeIIIOH species to cleave C–H bonds.2 Similar X–H bond cleavage reactivity by synthetic MIII–OH complexes are known,25 but to our knowledge [CoIII(μ-OH)CaII]+ is the first example of a CoIII–OH species exhibiting this kind of chemistry. This finding, along with the structural and physical properties of [CoIII(μ-OH)CaII]+ and [CoII(μ-OH2)CaIIOH2]+, indicated that the recently reported CoIV–oxo species10 is actually a complex containing a CoIII center. Theoretical studies using DFT methods on in silico-generated CoIV–oxo species suggest that the oxo ligand can have appreciable spin density,10,26 leading to the possibility of CoIII–oxyl radical complexes. However, the existence of this type of species has yet to be validated experimentally. We think it is an unlikely assignment based on the data presented here, which is consistent with the more plausible formulation of a CoIII(μ-OH)ScIII core.
Supplementary Material
Scheme 2.

Comparison of possible heterobimetallic complex formed when treating [CoIITMGtren(OTf)]+ with sPhIO and Sc(OTf)3 as proposed in the literature10 (A) and reformulated based on the results obtained herein (B).
Acknowledgments
We thank the NIH (GM50781) for support of this work. Portions of this research were supported by the Department of Energy (DOE), Office of Science, Office of Basic Energy Science (OBES), Division of Chemical Sciences, Geosciences and Biosciences, under contract DE-AC02–05CH11231, and carried out at the Stanford Synchrotron Radiation Lightsource (SSRL). SSRL is operated by DOE and OBES. The SSRL SMB Program is supported through DOE, OBER, and by the NIH, the National Center for Research Resources (NCRR).
ABBREVIATIONS
- [H3buea]3−
tris(tert-butylureaylethylene)aminato
- TMP
trigonal monopyramidal
- TBP
trigonal bipyramidal
- TBAP
tetrabutylammonium hexafluorophosphate
- d[Co-Neq]
Co displacement from the equatorial plane formed by three deprotonated sulfonamido nitrogen donors of [MST]3−
- DPH
diphenylhydrazine
- PhIO
iodosylbenzene
- sPhIO
2-(tert-butylsulfonyl)iodosylbenzene
- ESI-HRMS
electrospray ionization high-resolution mass spectrometry
- N(TMS)2
bis(trimethylsilyl)amide
- TMG3tren
tris(tetramethylguanidino)tren
Footnotes
ASSOCIATED CONTENT
Crystallographic details for all the molecular structures with CIFs, and Figures S1–S9. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Shook RL, Borovik AS. Inorg Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shook RL, Borovik AS. Chem Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Borovik AS. Acc Chem Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]; (d) Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. Proc Acad Natl Sci. 2003;100:3629–3634. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lu Y. Inorg Chem. 2006;45:9930–9940. doi: 10.1021/ic052007t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lu Y, Berry SM, Pfister TD. Chem Rev. 2001;101:3047–f3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]; (h) Natale D, Mareque-Rivas JC. Chem Commun. 2008:425–437. doi: 10.1039/b709650j. [DOI] [PubMed] [Google Scholar]
- 2.(a) Schurmann K, Anton M, Ivanov I, Richter C, Kuhn H, Walther M. J Biol Chem. 2011;286:23920–23927. doi: 10.1074/jbc.M110.211821. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saam J, Ivanov I, Walther M, Hozhütter HG, Kuhn H. Proc Natl Acad Sci. 2007;104:13319–13324. doi: 10.1073/pnas.0702401104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schenk G, Neidig ML, Zhou J, Holman TR, Solomon EI. Biochemistry. 2003;42:7294–7302. doi: 10.1021/bi027380g. [DOI] [PubMed] [Google Scholar]
- 3.Umena Y, Kawakami K, Shen J, Kamiya N. Nature. 2011;473:55–61. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 4.(a) Barry BA, Chen J, Keough J, Jenson D, Offenbacher A, Pagba C. J Phys Chem Lett. 2012;3:543–554. doi: 10.1021/jz2014117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saito K, Shen JR, Ishida T, Ishikita H. Biochemistry. 2011;50:9836–9844. doi: 10.1021/bi201366j. [DOI] [PubMed] [Google Scholar]; (c) Haumann M, Junge W. Biochim Phys Acta. 1999;1411:121–133. doi: 10.1016/s0005-2728(99)00045-6. [DOI] [PubMed] [Google Scholar]; (d) Saito K, Shen JR, Ishida T, Ishikita H. Biochemistry. 2011;50:9836–9844. doi: 10.1021/bi201366j. [DOI] [PubMed] [Google Scholar]; (e) Hwang HJ, Dilbeck P, Debus RJ, Burnap RL. Biochemistry. 2007;46:11987–11997. doi: 10.1021/bi701387b. [DOI] [PubMed] [Google Scholar]; (f) Meyer TJ, Huynh MHV, Thorp H. H Angew Chem Int Ed. 2007;46:5284–5304. doi: 10.1002/anie.200600917. [DOI] [PubMed] [Google Scholar]; (g) Polander BC, Barry BA. Proc Natl Acad Sci. 2012;109:6112–6117. doi: 10.1073/pnas.1200093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Ghanotakis DF, Babcock GT, Yocum CF. FEBS Lett. 1984;167:120–130. [Google Scholar]; (b) Boussac A, Rutherford AW. Biochemistry. 1988;27:3476–3483. [Google Scholar]; (c) Ananyev GA, Zaltsman L, Vasko C, Dismukes GC. Biochim Biophys Acta. 2001;1503:52–68. doi: 10.1016/s0005-2728(00)00215-2. [DOI] [PubMed] [Google Scholar]; (d) Yocum CF. Coord Chem Rev. 2008;252:296–305. [Google Scholar]; e) Miqyass M, Gorkom HJ, van Yocum CF. Photosynth Res. 2007;92:275–287. doi: 10.1007/s11120-006-9124-2. [DOI] [PubMed] [Google Scholar]; (f) Miqyass M, Yocum CF, van Gorkom HJ. Calcium Requirements for S-State Transitions. In: Allan JF, Gantt E, Golbeck JH, Osmond B, editors. Photosynthesis Energy from the Sun. Springer; Netherlands: 2008. pp. 459–462. [Google Scholar]; (g) Pecoraro VL, Baldwin MJ, Caudle MT, Hsieh WY, Law NA. Pure Appl Chem. 1998;70:925–929. [Google Scholar]; (h) Cady CW, Crabtree RH, Brudvig GW. Coord Chem Rev. 2008;252:444–455. doi: 10.1016/j.ccr.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Brudvig GW. Philos Trans R Soc, B. 2008;8:1211–1219. doi: 10.1098/rstb.2007.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Mullins CS, Pecoraro VL. Coord Chem Rev. 2008;47:1849–1861. doi: 10.1016/j.ccr.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Betley TA, Wu Q, Van-Voorhis T, Nocera DG. Inorg Chem. 2008;47:1894–1861. doi: 10.1021/ic701972n. [DOI] [PubMed] [Google Scholar]
- 6.(a) Wiechen M, Zaharieva I, Dau H, Kurz P. Chem Sci. 2012 doi: 10.1039/C2SC20226C. [DOI] [Google Scholar]; (b) Kanan MW, Nocera DG. Science. 2008;321:1072–1075. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]; (c) Reece SY, Hamel JA, Sung K, Jarvi TD, Esswein AJ, Pijpers JJH, Nocera DG. Science. 2011;334:645–648. doi: 10.1126/science.1209816. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kotzabasaki V, Siczek M, Lis T, Milios CJ. Inorg Chem Commun. 2011;14:213–216. [Google Scholar]; (b) Nayak S, Nayek HP, Dehnen S, Powell AK, Reedijk J. Dalton Trans. 2011;40:2699–2702. doi: 10.1039/c0dt01463j. [DOI] [PubMed] [Google Scholar]; (c) Hewitt IJ, Tank J-K, Madu NT, Clérac R, Buth G, Anson CE, Powell AK. Chem Commun. 2006:2650–2652. doi: 10.1039/b518026k. [DOI] [PubMed] [Google Scholar]; (d) Mishra A, Wernsorfer W, Abboud KA, Christou G. Chem Commun. 2005:54–56. doi: 10.1039/b413680b. [DOI] [PubMed] [Google Scholar]
- 8.Kanady JS, Tsui EY, Day WM, Agapie T. Science. 2011;333:733–736. doi: 10.1126/science.1206036. [DOI] [PubMed] [Google Scholar]
- 9.Park YJ, Ziller JW, Borovik AS. J Am Chem Soc. 2011;133:9258–9261. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfaff FF, Kundu S, Risch M, Pandian S, Heims F, Pry-jomska-Ray I, Haack P, Metzinger R, Bill E, Dau H, Comba P, Ray K. Angew Chem Int Ed. 2011;50:1711–1715. doi: 10.1002/anie.201005869. [DOI] [PubMed] [Google Scholar]
- 11.(a) Margulieux GW, Weidermann N, Lacy DC, Moore CE, Rheingold AL, Figueroa JS. J Am Chem Soc. 2010;132:5033–5035. doi: 10.1021/ja1012382. [DOI] [PubMed] [Google Scholar]; (b) Connelly NG, Geiger WE. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]; (c) Panda TK, Hrib CG, Jones PG, Jenter J, Roesky PW, Tamm M. Eur J Inorg Chem. 2008:4270–4279. doi: 10.1021/ic9024052. [DOI] [PubMed] [Google Scholar]; (d) Dauban P, Saniere L, Tarrade A, Dodd R. H J Am Chem Soc. 2001;123:7707–7708. doi: 10.1021/ja010968a. [DOI] [PubMed] [Google Scholar]
- 12.See supporting information for experimental details
- 13.(a) Walsh PJ. Acc Chem Res. 2003;36:739–749. doi: 10.1021/ar0300219. [DOI] [PubMed] [Google Scholar]; (b) Walsh PJ, Lurain AE, Balsells J. Chem Rev. 2003;103:3297–3344. doi: 10.1021/cr0000630. [DOI] [PubMed] [Google Scholar]; (c) Pritchett S, Woodmansee DH, Gantzel P, Walsh PJ. J Am Chem Soc. 1998;120:6423–6424. [Google Scholar]; (d) Nanthakumar A, Miura J, Diltz S, Lee CK, Aguirre G, Ortega F, Ziller JW, Walsh PJ. Inorg Chem. 1999;38:3010–3013. doi: 10.1021/ic981093c. [DOI] [PubMed] [Google Scholar]
- 14.(a) Schwarz AD, Chu Z, Mountford P. Organometallics. 2010;29:1246–1260. [Google Scholar]; (b) Schwarz AD, Herbert KR, Paniagua C, Mountford P. Organometallics. 2010;29:4171–4188. [Google Scholar]
- 15.(a) Ray M, Hammes BS, Yap GPA, Rheingold AL, Borovik AS. Inorg Chem. 1998;37:1527–1532. [Google Scholar]; (b) Lucas RL, Zart MK, Murkerjee J, Sorrell TN, Powell DR, Borovik AS. J Am Chem Soc. 2006;128:15476–15489. doi: 10.1021/ja063935+. [DOI] [PubMed] [Google Scholar]; (c) Hammes BS, Young GV, Jr, Borovik AS. Angew Chem Int Ed. 1999;38:666–669. doi: 10.1002/(SICI)1521-3773(19990301)38:5<666::AID-ANIE666>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; (d) Larsen LP, Parolin TJ, Powell DR, Hendrich MP, Borovik AS. Angew Chem Int Ed. 2003;42:85–89. doi: 10.1002/anie.200390060. [DOI] [PubMed] [Google Scholar]
- 16.(a) Ciampolini M. Spectra of 3d Five-Coordinate Complexes. In: Jorgenen CK, Neilands JB, Nyholm RS, Reinen D, Williams RJP, editors. Structure and Bonding. Vol. 6. Springer-Verlag; New York: 1969. pp. 52–93. [Google Scholar]; (b) Ciampolini M, Paoletti P. Inorg Chem. 1967;6:1261–1262. [Google Scholar]; (c) Suh MP, Lee J, Han MY, Yoon TS. Inorg Chem. 1997;36:5651–5654. [Google Scholar]; (d) Vaira MD, Orioli PL. Inorg Chem. 1967;6:955–957. [Google Scholar]
- 17.(a) Banci L, Bencini A, Benelli C, Bohra R, Dance J-M, Gatteschi D, Jain V, Mehrotra R, Tressaud A, Woolley R, Zanchini C. In Structures Versus Special Properties, Structure and Bonding. Vol. 52. Springer Berlin; Heidelberg: 1982. Spectral-Structural Correlations in High-Spin Cobalt(II) Complexes; pp. 37–86. [Google Scholar]; (b) Jenkins DM, Bilio AJD, Allen MJ, Betley TA, Peters J. C J Am, Chem Soc. 2002;124:15336–15350. doi: 10.1021/ja026433e. [DOI] [PubMed] [Google Scholar]
- 18.We have spectrophotometrically monitored the formation of this CoII-aquo species and found changes in the optical spectrum until one equiv of water was added to [CoIIMST]− and CaII(OTf)2/15-crown-5; further addition of water caused no changes in the optical properties. Because we are only monitoring changes at the CoII center, these findings suggest that only one water molecule affect the electronic transitions of the CoII center (Figure S2), similar to what was observed for [CoIIMST(OH2]−. However, these spectral data do not allow us to determine the exact structure of the complex in solution, in particular, the coordination spheres around the CaII center.
- 19.This bond distance is comparable to the CoIII–O bond length of 1.894(2) Å reported for the monomeric [CoIIIH3buea(OH)]−.15c However, this bond distance is longer than the average CoIII–O bond length found in a homobimetallic CoIII complex with a CoIII2(μ-O)2core.15d
- 20.(a) The edge energies determined from the 2nd derivative of the XANES spectra are 7716.40 eV for [CoII(μ-OH2)CaIIOH2]+ and 7717.82 eV for [CoIII(μ-OH)CaII]+Bonnitcha PD, Hall MD, Underwood CK, Foran GJ, Zhang M, Beale PJ, Hambley T. W J Inorg Biochem. 2006;100:963–971. doi: 10.1016/j.jinorgbio.2006.02.015.Kanan MW, Yano J, Surendranath Y, Dincâ M, Yachandra VK, Nocera D. G J Am Chem Soc. 2010;132:13692–13701. doi: 10.1021/ja1023767.
- 21.The kinetics of this reaction would allow for both the CoIII– OH and CoII–OH2 species to be present at the same time.
- 22.(a) Dogutan DK, McGuire R, Jr, Nocera D. J Am Chem Society. 2011;133:9178–9180. doi: 10.1021/ja202138m. [DOI] [PubMed] [Google Scholar]; (b) Dogutan DK, Stoian SA, McGuire R, Jr, Schwalbe M, Teets TS, Nocera D. G J Am Chem Soc. 2011;133:131–140. doi: 10.1021/ja108904s. [DOI] [PubMed] [Google Scholar]; (c) Boyer JL, Polyansky DE, Szalda DJ, Zong R, Thummel RP, Fujita E. Angew Chem Int Ed. 2011;50:12600–12604. doi: 10.1002/anie.201102648. [DOI] [PubMed] [Google Scholar]
- 23.Mareque-Rivas JC, Prabaharan R, Rosales RTM. Chem Commun. 2004:76–77. doi: 10.1039/b310956a. [DOI] [PubMed] [Google Scholar]
- 24.Selected examples of the importance of redox-inactive metal ions on processes involving transition metal complexes: Fukuzumi S, Ohkubo K. Chem Eur J. 2000;6:4532–4535. doi: 10.1002/1521-3765(20001215)6:24<4532::aid-chem4532>3.0.co;2-9.Ohkubi K, Menon SC, Orita A, Otera J, Fukuzumi S. J Org Chem. 2003;68:4720–4726. doi: 10.1021/jo034258u.Darensbourg MY, Darensbourg DJ, Burns D, Drew D. A J Am Chem Soc. 1976;98:3127–3136.Gambarotta, Francesco A, Floriani C, Zanazzi PF. J Am Chem Soc. 1982;104:5082–5092.Lee Y, Mankad NP, Peters JC. Nature Chem. 2010;2:558–565. doi: 10.1038/nchem.660.Betley TA, Peters J. C J Am Chem Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f.Ding K, Brennessel WW, Holland PL. J Am Chem Soc. 2009;131:10804–10805. doi: 10.1021/ja902812y.Ding K, Pierpont AW, Brennessel WW, Lukat-Rodgers G, Rodgers KR, Cundari TR, Bill E, Holland P. L J Am Chem Soc. 2009;131:9471–9472. doi: 10.1021/ja808783u.Morimoto Y, Kotani H, Park J, Lee Y-M, Nam W, Fukuzumi S. J Am Chem Soc. 2011;133:403–405. doi: 10.1021/ja109056x.Fukuzumi S, Morimoto Y, Kotani H, Naumov P, Lee YM, Nam W. Nature Chem. 2010;2:756–759. doi: 10.1038/nchem.731.Karlin KD. Nature Chem. 2010;2:711–712. doi: 10.1038/nchem.813.
- 25.(a) Goldsmith CR, Cole AP, Stack TDP. J Am Chem Soc. 2005;127:9904–9912. doi: 10.1021/ja039283w. [DOI] [PubMed] [Google Scholar]; (b) Goldsmith CR, Stack TDP. Inorg Chem. 2006;45:6048–6055. doi: 10.1021/ic060621e. [DOI] [PubMed] [Google Scholar]; (c) Donoghue PJ, Tehranchi J, Cramer CJ, Sarangi R, Solomon EI, Tolman WB. J Am Chem Soc. 2011;133:17602–17605. doi: 10.1021/ja207882h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ogo S, Wada S, Watanabe Y, Iwase M, Wada A, Harata M, Jitsukawa K, Masuda H, Einaga H. Angew Chem Int Ed. 1998;37:2102–2104. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2102::AID-ANIE2102>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]; (e) Ogo S, Yamahara R, Roach M, Suenobu T, Aki M, Ogura T, Kitagawa T, Masuda H, Fukuzumi S, Watanabe Y. Inorg Chem. 2002;41:5513–5520. doi: 10.1021/ic0200040. [DOI] [PubMed] [Google Scholar]
- 26.Michel C, Baerends EJ. Inorg Chem. 2009;48:3628–3638. doi: 10.1021/ic802095m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.