

Pharmacogenomic biomarkers can optimize an individual’s therapy; however, the overall role of genetic factors in drug response remains uncertain. The majority of genetic variants currently used as clinical pharmacogenomic biomarkers affect drug metabolism and transport, while fewer biomarkers accurately predict drug response (pharmacodynamics). This article evaluates heritable aspects of drug therapy, highlighting gene-gene-environment interactions, in light of recent human evolution. These concepts are critical to the development of new drug therapies and biomarkers tests.
Nature versus nurture – a futile discussion
Estimates as to what extent heritable and environmental factors contribute to complex traits often depend upon the study design, and the questions asked. For example, ability to taste phenylthiocarbamide (PTC), the bitter ingredient in broccoli, entirely depends on the status of PTC receptors, TAS2R38, with large portions of the population carrying null alleles unable to sense PTC bitterness – they like broccoli. One might say that this trait is 100% heritable. On the other hand, the bitter sensation of broccoli entirely depends on the presence of PTC, hence it is 100% environmental – both statements are correct in this gene-environment interaction. Similarly, heritability of drug addiction is typically pegged at 50–90%, while exposure and other environmental factors also play critical roles. We consider all drug therapy as a gene-environment interaction – a seemingly trivial insight that nevertheless can help in the search for multi-factorial conditions under which a drug has optimal efficacy.
The ‘missing heritability’
Despite massive studies on a genome-wide scale, we still understand only a small portion of the genetic factors contributing to complex disorders, with multiple genes implicated but each conveying only low risk. This gap has been termed the ‘missing heritability’. We can expect most penetrant risk variants to be under negative selection and hence relatively rare, leaving open the question of what contributes to the large heritability estimates in a population. Zuk et al. have proposed that considering dynamic interactions between genetic factors, or epistasis, could substantially narrow the gap (1), whereas additional large-scale sequencing may reveal many more rare risk variants but will fall short of bridging the gap. Since drugs target select genes and disease pathways, fewer gene variants with greater effect size might be involved in determining treatment outcomes. Indeed recent GWAS results on drug effects have revealed a few single variants with exceptional penetrance, but much of drug-heritability also remains hidden.
Role of evolution and gene regulation
Vast stretches of the human genome bear signatures of selection pressures, with regulation of gene expression a prevalent path towards high allele frequencies under positive selection. The impact of a regulatory variant typically depends on the cellular environment, thereby, enabling selection of favorable traits tailored to target organs, each with distinct optimal requirements. Because such variants may have no overt deleterious impact, GWA studies designed to detect deleterious risk factors are poorly suited for their discovery. By searching for genes under positive selection pressure that also double as drug targets, we have discovered a host of frequent regulatory variants in genes relevant to drug therapy (e.g., CYP3A4, NAT1, DRD2, DAT, MAOA, TPH2, CETP, ACE) (2), at least some carrying a signature of positive selection implying impact on a human trait. While associations with disease were marginal or absent, strong effects can emerge under exposure to drugs (gene-environment interactions). In addition, epistatic gene-gene interactions involving frequent variants can turn into risk factors under certain conditions (gene-gene-environment interactions) (Fig. 1).
Figure 1.
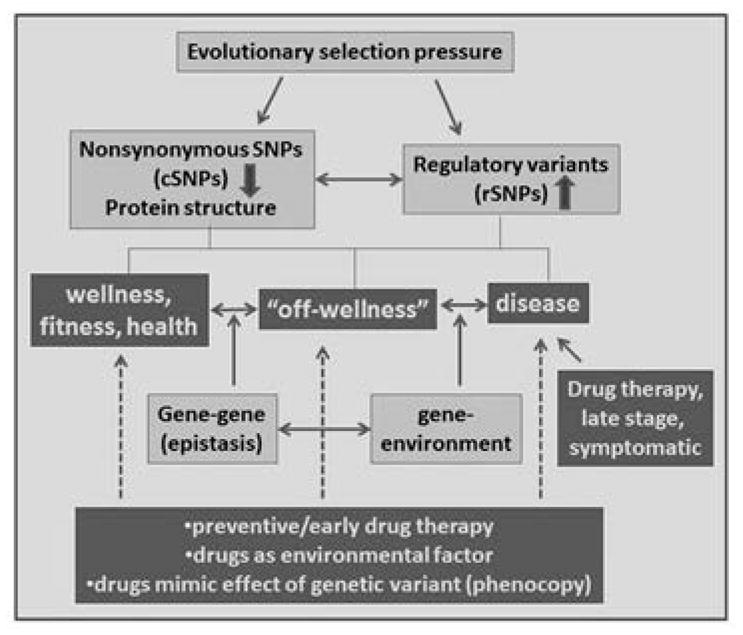
Concepts in pharmacogenomics with potential to guide successful therapies when applied in concerted fashion. nsSNPs also stand for other sequence variants affecting protein structure (e.g., indels). Size of the arrows (up and down) estimates main influence of evolutionary selection pressure, with effects in opposite directions possible. Requiring stable biomarker targets, early therapy and prevention tend to rely more strongly on genetic factors. A drug-induced phenocopy mimicking a genetic mutation can replace this mutation in a gene-gene epistatic interaction.
Gene-environment interactions
From an evolutionary vantage point, exposure to countless drugs has occurred largely over the immediate past, whereas nutrients and toxins have a long history of shaping the human genome. Therefore, drugs inadvertently encounter genetic variants molded by environmental pressures, with strong effects on efficacy and toxicity. We expect hepatic drug metabolizing enzymes (DMEs) to be under strong environmental selection pressure, as primary lines of defense against toxins. Among the DMEs, the cytochrome P450 gene family plays an eminent role, recognizing ~65% of drugs as substrates (Fig. 2). Not only are there multiple CYP450 genes, but many also carry frequent mutations, mostly abrogating enzyme activity, strongly affecting drug metabolism and response. But how might P450 loss-of-function mutations rise to high frequency in response to environmental toxins? The answer may lie in competition between CYP isoforms. The liver is loaded with CYP enzymes, contributing to its dark red-brown color, possibly saturating the liver’s expression capacity and cofactor supply. Hence, reduced expression of major P450 enzymes allows other subtypes to be expressed more robustly, suggesting an indirect evolutionary selection process in response to toxins. The most abundant P450, i.e., CYP3A4, shows large variability but few if any polymorphisms affecting expression, even though genetic factors in basal and induced expression have been suggested to account for >60% of the variability. Certainly, transcription factors such as PXR, CAR, GR, HNF4A, VDR, and FXR coordinately sense environmental cues, fine-tune CYP3A4 content, and carry genetic variants themselves, resulting in substantial variation in CYP3A4 activity. On the other hand, CYP3A4 was thought to be largely devoid of mutations. We have discovered an intronic SNP (CYP3A4*22) reducing expression 2–6 fold in the liver but not in the intestines (3), such tissue specificity a hallmark of regulatory variants. CYP3A4*22 is located in a large ancestral haplotype, one possible indication of recent positive selection. But even with considerations of genetic variants in transcription factors, part of the overall genetic influence remains unresolved.
Figure 2.
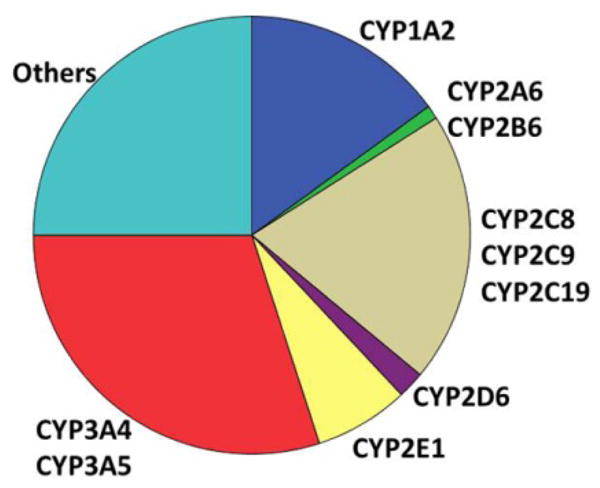
Distribution of the main cytochrome P450 drug-metabolizing enzymes in the liver. Assuming saturation of the ability of the liver to produce the high CYP protein load and energy demand of P450-mediated oxidative metabolism, reduced expression of main P450 enzymes enables lesser P450 subtypes to increase activity. (Graph by D. Wang)
Gene-environment interactions also play a role in pharmacodynamics. For example, we have found frequent splicing polymorphisms in the dopamine receptor DRD2 to modulate cognitive processing, but also to increase risk of cocaine-induced death threefold, the cocaine exposure revealing a strong genetic influence. Generalizing these findings, genes under possible positive selection pressures can contain one or more frequent functional variants with little effect on disease risk but strong response to drug exposure – the external challenge in a gene-environment interaction.
Gene-gene (epistatsis) and gene-gene-environment interactions
The study of epistasis in disease is hampered by low frequency of risk alleles. Evidence that epistasis may be common stems from the literature on evolution, while epistatic models in drug therapy by definition encompass gene-gene-environment interactions. An example taken from nematode behavior – variable response to nutrient levels via a gene- gene interaction in catecholamine/peptide signaling (4) – demonstrates that evolutionary forces have long been at play in neuronal signaling -presaging possible insights into the therapy of mental disorders. We have found evidence of strong epistasis between DRD2 and the dopamine transporter DAT in affecting risk of lethal cocaine exposure (unpublished). Yet, very few drug studies have explicitly addressed gene-gene-environment interactions, and clinical applications have yet to emerge.
Exploiting epistasis in drug design and therapy
In cancer chemotherapy, epistasis has already led to a novel strategy termed synthetic lethality. This idea implies the presence of a non-lethal mutation critical to tumor development, for instance conveying a mutator phenotype. BRCA mutations lead to breast cancer associated with defective homologous recombination, affecting repair of double-strand breaks (5). Adding a second non-lethal defect, for example in PARP contributing to excision repair, effectively kills the cancer cell. This insight has led to therapies of BRCA-deficient tumors with PARP inhibitors, generating a phenocopy of PARP mutations, with potent antitumor potency but low overall toxicity in normal cells that retain BRCA functions (5). Additional gene-gene pairs in cancer chemotherapy are being tested, but epistasis might also offer novel treatments of other complex disorders. In “epistasis-guided therapy”, a strong epistatic interaction is exploited by generating a drug-induced phenocopy at one gene that interacts with a genetic variant in another gene. If the epistatic effect is desirable, a drug choice can be made, requiring genetic companion tests (Fig. 1). If the effect is detrimental, the test would recommend alternative therapy. Owing to the unique evolution of the human brain, and a growing number of genes with frequent variants affecting CNS functions, I predict that epistasis-guided therapy might prove fruitful in treating mental disorders.
Where to go from here? The concepts depicted in Figure 1 are well supported but have yet to systematically influence drug design and therapy in a concerted way. The following approaches may prove useful. First, genetic effects on drug therapy fall into the category of gene-environment interactions, requiring study in the context of human evolution. Second, consideration of dynamic gene-gene interactions (epistasis) has potential for developing biomarker tests with utility in clinical practice, guiding drug therapy, early intervention, and prevention. Understanding epistatic and environmental interactions can further lead to novel options in drug therapy, as illustrated with ‘synthetic lethality’. Third, we must unravel the molecular genetic mechanism of variants affecting drug response, to focus on relevant gene-gene interactions. Frequent variants with phenotypic impact are likely to be regulatory, possibly under positive evolutionary selection, and tissue selective, features that suggest affected phenotype, and hence type of therapy. Fourth, many rare variants with strong phenotypic penetrance will emerge as equally important factors, but they should be considered in the context of the frequent variants discussed here. Taken together, these approaches could shed light on the ‘missing heritability’ and guide optimal drug therapy.
Acknowledgments
Studies reported here were in part supported by NIH grant U01 GM092655.
Footnotes
The author does not have any conflict of interest with respect to the content of this paper.
References
- 1.Zuk O, et al. The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci USA. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadee W, et al. Pharmacogenomics of the RNA world: structural RNA polymorphisms in drug therapy. Clin Pharmacol Ther. 2011;89:355–365. doi: 10.1038/clpt.2010.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang D, et al. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 2011;11:274–286. doi: 10.1038/tpj.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bendesky A, et al. Catecholamine receptor polymorphisms affect decision-making in C. elegans. Nature. 2011;472:313–318. doi: 10.1038/nature09821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]