

Abstract
Peripartum cardiomyopathy (PPCM) is a life-threatening pregnancy-associated cardiomyopathy in previously healthy women. Although PPCM is driven in part by the 16-kDa N-terminal prolactin fragment (16K PRL), the underlying molecular mechanisms are poorly understood. We found that 16K PRL induced microRNA-146a (miR-146a) expression in ECs, which attenuated angiogenesis through downregulation of NRAS. 16K PRL stimulated the release of miR-146a–loaded exosomes from ECs. The exosomes were absorbed by cardiomyocytes, increasing miR-146a levels, which resulted in a subsequent decrease in metabolic activity and decreased expression of Erbb4, Notch1, and Irak1. Mice with cardiomyocyte-restricted Stat3 knockout (CKO mice) exhibited a PPCM-like phenotype and displayed increased cardiac miR-146a expression with coincident downregulation of Erbb4, Nras, Notch1, and Irak1. Blocking miR-146a with locked nucleic acids or antago-miRs attenuated PPCM in CKO mice without interrupting full-length prolactin signaling, as indicated by normal nursing activities. Finally, miR-146a was elevated in the plasma and hearts of PPCM patients, but not in patients with dilated cardiomyopathy. These results demonstrate that miR-146a is a downstream-mediator of 16K PRL that could potentially serve as a biomarker and therapeutic target for PPCM.
Introduction
Peripartum cardiomyopathy (PPCM) is a life-threatening heart disease of unknown etiology characterized by a sudden onset of heart failure in the last month of pregnancy and/or in the first months postpartum (1). Recently, the antiangiogenic 16-kDa N-terminal prolactin fragment (16K PRL), produced by cleavage of the full-length nursing hormone prolactin (PRL) by cathepsin D, has been discovered as a potential factor initiating and driving PPCM (2). We previously showed that female mice with cardiomyocyte-specific Stat3 knockout (referred to herein as CKO mice) develop PPCM. In these mice, increased oxidative stress activates cardiac cathepsin D, which promotes generation of 16K PRL in the peripartum heart, leading to decreased cardiac capillary density and reduced cardiac function, a feature that could be prevented by treating mice with the PRL blocker bromocriptine (2). Moreover, oxidative stress, cathepsin D, and 16K PRL are elevated in plasma probes of PPCM patients (2). Treatment with bromocriptine yielded promising results in preventing PPCM in at-risk patients and in promoting recovery in PPCM patients (3, 4). The antiangiogenic properties of 16K PRL are widely documented, as it reduces abnormal retinal neovascularization in a model of oxygen-induced retinopathy (5) and tumor growth in xenograft mouse models (6, 7). 16K PRL acts specifically on ECs and inhibits migration (8), blocks cell cycle progression (9, 10), and induces apoptosis (11). Activation of the transcription factor NF-κB by 16K PRL in ECs appears central to these antiangiogenic properties (11). However, the precise mechanisms by which 16K PRL exerts its angiostatic effect have not been elucidated, nor has the precise way by which 16K PRL induces PPCM and impairs cardiac function.
MicroRNAs (miRNAs) are short (19–24 nt), noncoding small RNAs that repress gene expression posttranscriptionally by targeting the 3′-untranslated regions (3′UTRs) of specific mRNAs (12). miRNAs that stimulate or repress angiogenesis have opened up novel therapeutic options in pathophysiologies associated with deregulated angiogenesis (13) and vascular and cardiovascular diseases (14–16). Recently, miRNA detection in the blood, protected from endogenous RNase activity by exosomes, has emerged as a promising diagnostic tool (17, 18). Various cell types, including ECs, release exosomes loaded with miRNAs into the surrounding environment and blood, where they may work as systemic and intercellular communicators (19). Here, we discovered that 16K PRL mediated a large part of its effects via induction of microRNA-146a (miR-146a) in ECs. miR-146a promoted endothelial injury and, in a paracrine manner via miR-146a–loaded endothelial exosomes, reduced cardiomyocyte metabolic activities. This 16K PRL–miR-146a circuit appears to play a major role in the development of PPCM, since blocking miR-146a in CKO mice by the use of locked nucleic acid–modified (LNA-modified) antisense oligonucleotides or antago-miRs largely prevented the development of the PPCM phenotype. In contrast to complete PRL blockade with bromocriptine, neutralizing miR-146a left full-length PRL signaling intact, allowing CKO mice to nurse their offspring. miR-146a was elevated in plasma of PPCM patients, but not in pregnancy-matched healthy controls, bromocriptine-treated PPCM patients, or dilated cardiomyopathy (DCM) patients with a similar degree of heart failure. These data suggest that miR-146a may serve as a specific biomarker for the diagnosis of PPCM in patients and that miR-146a may represent a novel therapeutic target to treat PPCM.
Results
16K PRL–induced NF-κB signaling upregulates miR-146a in ECs.
We aimed to identify miRNAs involved in 16K PRL–mediated antiangiogenic effects. Since NF-κB activation is essential for the antiangiogenic effects of 16K PRL (10, 11), we assayed the expression of different miRNAs described as being NF-κB dependent — miR-125b, miR-132, miR-146a, miR-155, and miR-221 — in HUVECs stimulated with 16K PRL (20–22). 16K PRL increased expression of miR-146a, but did not alter miR-125b, miR-132, miR-155, and miR-221 expression (Figure 1A). Time course analyses of 16K PRL–induced miR-146a and the expression of the miR-146a primary transcript (pri-miR-146a) are shown in Supplemental Figure 1, A and B (supplemental material available online with this article; doi: 10.1172/JCI64365DS1). 16K PRL–mediated induction of miR-146a was abolished by the NF-κB pathway blocker BAY 11-7082 and attenuated by siRNA knockdown of the NF-κB subunit p65 (Figure 1, B and C, and Supplemental Figure 1, C and D). The involvement of NF-κB in 16K PRL–induced miR-146a expression was confirmed by the luciferase activity of the luciferase reporter vector consisting of the 600-bp segment of the WT promoter of miR-146a, with 2 NF-κB binding sites (21), compared with a vector containing mismatches in the 2 NF-κB binding sites (Figure 1, D and E).
Figure 1. 16K PRL mediates antiangiogenic effects in ECs via miR-146a.

(A) miRNA level evaluated by qRT-PCR in HUVECs after 16K PRL treatment (50 nM, 8 hours). (B and C) miR-146a in HUVECs treated with 16K PRL (50 nM, 8 hours), (B) with or without BAY 11-7082 pretreatment (10 μM, 1 hour) or (C) with 72 hours transfection with p65 NF-κB subunit or control siRNA. (D) WT and mutated (Mut) miR-146a promoter luciferase vectors. (E) Luciferase activity after 16K PRL treatment (50 nM, 8 hours). (F) Proliferation status, reflected by BrdU incorporation, in HUVECs transfected with pre- or anti-miR-146a or -miR-control. (G) BrdU incorporation in HUVECs stimulated with 16K PRL (50 nM, 8 hours) with or without anti-miR-146a transfection (48 hours). (H) Apoptotic index, reflected by caspase-3 activity, in HUVECs transfected with pre- or anti-miR-146a or -miR-control. (I) Representative images of aortic rings 9 days after transfection with anti-miR-146a or -miR-control. Scale bars: 0.5 mm. (J) Quantification of sprout length in I (n = 8–10 aortic rings per condition). (K) Representative images of laser-induced choroidal neovascularization 7 days after transfection with pre-miR-control and -miR-146a injected intravitreously (n ≥ 8 eyes/condition; 4 lesions/eyes). Dashed outlines denote lesion area. Scale bars: 100 μm. (L) Quantification of the green signal present in lesion area. Data are mean ± SD (n ≥ 3) or mean ± SEM (J and L). *P < 0.05 vs. respective control. #P < 0.05 vs. 16K PRL–treated control. See also Supplemental Figure 1.
16K PRL mediates antiangiogenic effects in ECs through miR-146a.
Next we evaluated whether miR-146a mediates antiangiogenic effects of 16K PRL in HUVECs. miR-146a levels increased by pre-miR-146a transfection reduced HUVEC proliferation, whereas miR-146a inhibition by anti-miR-146a enhanced it (Figure 1F and Supplemental Figure 1E). 16K PRL treatment reduced HUVEC proliferation, an effect that was prevented by anti-miR-146a transfection (Figure 1G).
Furthermore, as reflected by caspase-3 activity, overexpression of miR-146a increased HUVEC apoptosis, whereas its inhibition reduced it (Figure 1H), a feature confirmed by annexin V–PI staining and by DNA fragmentation assays (Supplemental Figure 1, F and G). No effect of miR-146a overexpression or silencing was observed in HUVEC migration or tube formation ability (Supplemental Figure 1, H and I). In an ex vivo aortic ring assay, pre-miR-146a–mediated overexpression of miR-146a led to shorter sprouts, whereas anti-miR-146a–mediated reduction of miR-146a led to longer sprouts, compared with aortic rings transfected with respective controls (Figure 1, I and J). In a model of choroidal neovascularization (CNV) induced by laser injury, intravitreal injections of pre-miR-146a and the pre-miR-control, performed just after laser burns, showed that miR-146a overexpression decreased neoformation of blood vessels in choroidal lesions, as assessed by epifluorescence microscopy after FITC-dextran injection (Figure 1, K and L, and Supplemental Figure 1K).
Downregulation of NRAS, a novel target gene of miR-146a, is responsible for antiangiogenic effects of 16K PRL in ECs.
Potential target genes of miR-146a in ECs were analyzed by expression profiling on HUVECs transfected with pre-miR-146a. Overexpression of miR-146a significantly regulated 441 transcripts. Ingenuity Pathway Analysis software identified inflammation, proliferation, and apoptosis as the main functions regulated by miR-146a in ECs. The bioinformatic algorithm Sylamer (23) showed a strong enrichment (positive y axis) for the miR-146a seed match (sequence complementary to the seed) near the most downregulated genes (left x axis) (Supplemental Figure 2A), confirming that the global shift of RNA levels observed after miR-146a overexpression correlates with the expected miR-146a regulation of target transcripts. Among all miRNAs tested by Sylamer, miR-146a was the one for which the seed match was the most enriched in the downregulated genes. To identify targets regulated by miR-146a, the 164 downregulated transcripts observed in the microarray experiments were compared with the 130 different computationally predicted target transcripts for miR-146a using the TargetScan5.1 algorithm, which confirmed the already validated targets of miR-146a: TNF receptor–associated factor–6 (TRAF6) and IL-1 receptor–associated kinase 1 (IRAK1) (Supplemental Figure 2, B and C). In addition, neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) emerged as a new putative direct target of miR-146a conserved in human and mouse (Supplemental Figure 2D). NRAS mRNA and NRAS protein levels were decreased by miR-146a overexpression in HUVECs (Figure 2, A and B). A luciferase reporter vector encoding the complete 3′UTR of human NRAS and a mutated vector containing mismatches in the predicted miR-146a binding site were constructed. Cotransfection of the NRAS 3′UTR plasmid and pre-miR-146a in HEK293T cells resulted in a significant decrease in luciferase activity compared with cells cotransfected with pre-miR-control or the mutated 3′UTR target sequence (Figure 2C), which suggests that NRAS is a target of miR-146a. Downregulation of NRAS by miR-146a was also observed in rat heart ECs (RheA cells) (Supplemental Figure 2E). HUVECs with siRNA-mediated reduction of NRAS (Supplemental Figure 2, F and G) showed reduced proliferation rate and increased apoptosis (Figure 2, D–F). Likewise, in the model of CNV, intravitreal injections of NRAS siRNA decreased blood vessel density compared with the control siRNA (Figure 2, G and H). NRAS expression was downregulated in HUVECs treated with 16K PRL, but restored when HUVECs were transfected with anti-miR-146a (Figure 2I).
Figure 2. NRAS is a target gene of miR-146a in HUVECs.

(A) NRAS mRNA (qRT-PCR) and (B) protein levels (Western blot) in HUVECs transfected with pre-miR-146a and pre-miR-control. (C) Luciferase activity from NRAS 3′UTR WT reporter plasmid and mutated NRAS 3′UTR cotransfected into HEK293T cells with pre-miR-146a or pre-miR-control 48 hours after transfection. (D) BrdU incorporation, (E) FACS analysis for apoptosis by annexin V–PI staining, and (F) DNA fragmentation analysis in HUVECs transfected with NRAS or control siRNA for 48 hours. (G) Representative images of laser-induced choroidal neovascularization 7 days after transfection with NRAS or control siRNA injected intravitreously (n ≥ 8 eyes/condition; 4 lesions/eyes). Dashed outlines denote lesion area. Scale bars: 100 μm. (H) Quantification (by ImageJ) of the green signal present in lesion area. CNV, choroidal neovascularization. (I) NRAS mRNA level in HUVECs stimulated with 16K PRL (50 nM, 8 hours), with pretransfection (48 hours) with anti-miR-control or anti-miR-146a. All data are mean ± SD (n ≥ 3) or mean ± SEM (H). *P < 0.05 vs. respective control. See also Supplemental Figure 2.
16K PRL promotes release of miR-146a–loaded exosomes from ECs.
Analysis of HUVEC conditioned medium by dynamic light scattering analysis revealed that HUVECs secreted vesicles with a peak distribution around 100 nm (Figure 3A). These microparticles were positive for the exosomal marker CD63 (Figure 3B). miR-146a was detected in exosomes purified from HUVEC conditioned medium (Ct, 31.2) and upregulated in exosomes isolated from HUVECs stimulated by 16K PRL, but the amount of exosomes produced by HUVECs was not affected by 16K PRL treatment (Figure 3, C and D).
Figure 3. miR-146a can be exported from ECs in exosomes that can be transferred to cardiomyocytes and impair their metabolism.

(A) Dynamic light scattering analysis of conditioned medium of 16K PRL–treated HUVECs (50 nM, 24 hours). (B) Flow cytometry analysis of exosomes purified from HUVEC medium and labeled with CD63. (C) miR-146a level in exosomes from HUVECs treated with 16K PRL or not treated (NT). (D) Exosome production by HUVECs treated or not with 16K PRL (50 nM, 48 hours). (E) miR-146a level in NRCMs treated or not with 16K PRL. (F) Fluorescence microscopy detecting fusion of miR-146a–loaded endothelial exosomes labeled with the green fluorescent PHK67 membrane linker with NRCMs (α-actinin, red; DAPI, blue). Scale bars: 50 μm. (G) Higher-magnification views of boxed regions in F. Scale bars: 15 μm. (H) Electron micrographs of NRCM sections showing vesicles (arrows); after a 16-hour incubation with HUVEC exosomes, NRCMs showed larger multivesicular vesicles containing the exosomes (inset; enlarged ×2-fold). Scale bars: 500 nm. (I) miR-146a level in NRCMs exposed to miR-146a-exosomes or control-exosomes or transfected with pre-miR-146a or pre-miR-control. (J) Expression level of pri-miR-146a in NRCMs exposed to miR-146a- or control-exosomes. (K) miR-146a expression level, (L) metabolic activity, assessed by MTS assay, and (M) Erbb4 level in NRCMs exposed to control exosomes, miR-146a exosomes, and miR-146a exosomes cotransfected with anti-miR-control or anti-miR-146a. All data are mean ± SD (n ≥ 3). *P < 0.05 vs. respective control; #P < 0.05 vs. miR-146a-exosomes with anti-miR-control. See also Supplemental Figure 3.
Cardiomyocytes absorb miR-146a–loaded endothelial exosomes.
Compared with ECs, the expression of miR-146a in cultured neonatal rat cardiomyocytes (NRCMs) was low and could not be induced by direct stimulation with 16K PRL (Figure 3E and Supplemental Figure 3, A and B). In contrast, 16K PRL stimulated miR-146a expression in primary cardiac fibroblasts (Supplemental Figure 3C). Fluorescence microscopy revealed efficient uptake by NRCMs of exosomes purified from HUVECs overexpressing miR-146a and labeled with the green fluorescent PKH67 membrane linker (Figure 3, F and G). Confirming this result, NRCMs incubated with exosomes for 16 hours showed large intracellular vesicles loaded with numerous vesicular bodies (exosomes), and those structures were not observed in cells not incubated with exosomes (Figure 3H). The level of miR-146a in NRCMs was substantially increased after fusion with exosomes derived from pre-miR-146a–transfected ECs (miR-146a-exosomes) compared with NRCMs incubated with exosomes from pre-miR-control–transfected ECs (control-exosomes; Figure 3I). The efficiency of miR-146a transfer via exosomes in NRCMs was comparable to the effect of direct pre-miR-146a transfection using lipofection (Figure 3I). Addition of miR-146-exosomes did not induce pri-miR-146a expression in NRCMs (Figure 3J). Increased miR-146a after transfer via exosomes in NRCMs was completely abolished by cotransfection with anti-miR-146a (Figure 3K).
miR-146a–loaded endothelial exosomes or pre-miR-146a transfection reduces metabolic activity and decreases expression of target genes Erbb4, Notch1, and Irak1 in cardiomyocytes. miR-146a-exosomes and pre-miR-146a transfection substantially decreased the metabolic activity of NRCMs compared with respective control transfection, a feature that was normalized by cotransfection with anti-miR-146a (Figure 3L and Supplemental Figure 3, D and E).
Reduced metabolic activity was not accompanied by enhanced cell death, neither in NRCMs treated with miR-146a-exosomes nor upon transfection with pre-miR-146a, compared with their respective controls (Supplemental Figure 3F). We confirmed previous findings (24) that Erbb4 was a target of miR-146a in NRCMs (Figure 3M and Supplemental Figure 3, G and H). miR-146a target sites were confirmed in human ERBB4 and mouse Erbb4 genes (Supplemental Figure 3, I–K). Ribonucleoprotein immunoprecipitation with Ago2 (25) proved that Erbb4 and 2 other miR-146a target genes, Notch1 (26) and Irak1 (21), were direct physical targets of miR-146a in NRCMs (Supplemental Figure 3, L and M). ERBB4 appears to be involved in the regulation of metabolic activities in cardiomyocytes by promoting glucose uptake upon stimulation by its ligand, neuregulin-1 (NRG1) (27). This increase in NRG1-mediated metabolic activity was abolished in NRCMs overexpressing miR-146a (miR-146a, –1.3%; miR-control, +6.2%; P < 0.05).
Cardiac tissue of CKO mice with PPCM displays increased levels of miR-146a associated with reduced expression of Nras, Erbb4, Notch1, and Irak1.
Previously, we presented evidence that 16K PRL is a major factor responsible for PPCM in CKO mice (2). In line with in vitro results in ECs and NRCMs, elevated levels of miR-146a were detected in hearts of CKO mice with PPCM compared with pregnancy-matched postpartum WT siblings (Figure 4A). In situ hybridization localized upregulated miR-146a predominantly to nonmyocyte cells, including ECs (Figure 4B and Supplemental Figure 4A). In line with 16K PRL as an inducer of miR-146a, nullipara WT mice systemically injected with a 16K PRL–expressing adenovirus displayed increased cardiac miR-146a expression that was associated with decreased cardiac function (Supplemental Figure 4B and Supplemental Table 1). Increased cardiac miR-146a levels in CKO mice with PPCM were associated with reduced mRNA levels of Erbb4, Notch1, and Irak1 (Figure 4C). Western blot confirmed reduced protein levels of ERBB4 and NRAS compared with postpartum WT mice (Figure 4, D and E). Blocking PRL with bromocriptine in CKO mice with PPCM, which prevented PPCM onset (2), significantly reduced cardiac miR-146a levels and elevated Erbb4 mRNA levels (Supplemental Figure 4, C–E). As observed in in vitro experiments, miR-carrying exosomes produced by ECs were taken up by cardiomyocytes in vivo after intracardiac injection (Figure 4F).
Figure 4. Role and regulation of miR-146a in postpartum (PP) WT mice and in postpartum CKO mice treated or not with LNA-miR-control, LNA-miR-146a, or recombinant NRG1.

(A) miR-146a level in LV tissue from CKO and WT mice. (B) miR-146a in situ hybridization (green) in LV sections of CKO and WT mice counterstained with isolectin B4 (blood vessels, red). Higher-magnification image (top right) and digitized image illustrating miR-146a and isolectin B4 (yellow) colocalization (bottom right) are also shown. Scale bars: 100 μm (left and middle); 25 μm (right). (C) mRNA (qRT-PCR) and (D) protein levels (Western blot) in LVs of CKO and WT mice. (E) Quantification of results in D. (F) Fluorescence microscopy detecting human endothelial exosomes (anti-human CD63, red, arrows) in mouse hearts (α-actinin, green) 24 hours after intracardiac injection in WT mice. Higher-magnification views of boxed regions are shown at far right. Control with corresponding IgG is shown below. Scale bars: 20 μm; 5 μm (far right). (G) H&E staining of LV sections. Scale bars: 100 μm. (H) Quantification of fibrosis. (I) LV sections stained with isolectin B4 (blood vessels, yellow), WGA (cell membranes, red), and nuclei (DAPI, blue). Scale bars: 20 μm. (J) Capillary/cardiomyocyte (CM) ratio. (K) mRNA (qRT-PCR on mRNA pools of n = 5 mice/group). (L) Protein levels (Western blot), (M) quantified in bar graph in LVs of CKO mice treated with LNA-miR-control and LNA-miR-146a. (N) Fractional shortening in CKO mice injected with recombinant NRG1 (1.25 μg/d i.p.) or NaCl (control). *P < 0.05 vs. respective control. n = 3–6 per group. Data are mean ± SEM. See also Supplemental Figure 4.
LNA-modified antisense oligonucleotide– or antago-miR–mediated inhibition of miR-146a in vivo attenuates PPCM in CKO mice.
To assess whether miR-146a is causally linked to PPCM in CKO mice, we used LNA-modified antisense oligonucleotides to silence miR-146a in vivo (referred to herein as LNA-miR-146a). LNA-miR-146a substantially attenuated a decrease in systolic function compared with the respective LNA-miR-control, but did not attenuate LV dilatation compared with CKO females with no pregnancies (Table 1). Cardiac fibrosis was markedly reduced, and postpartum cardiac capillary density was higher, in LNA-miR-146a– versus LNA-miR-control–treated CKO mice (Figure 4, G–J). The expression of Erbb4 mRNA (confirmed in n = 5 individuals), as well as Notch1 and Irak1 mRNA (determined in mRNA pools of n = 5 hearts per treatment), was higher in the LV of LNA-miR-146a– versus LNA-miR-control–treated CKO mice, whereas Nras mRNA was not different (Figure 4K). Protein levels of ERBB4 and NRAS were significantly higher in LNA-miR-146a– versus LNA-miR-control–treated CKO mice (Figure 4, L and M).
Table 1.
Cardiac function in CKO mice with no pregnancy and after 2 subsequent pregnancies with systemic inhibition of miR-146a by injection of LNA-modified antisense oligonucleotides or cholesterol-modified antago-miRs
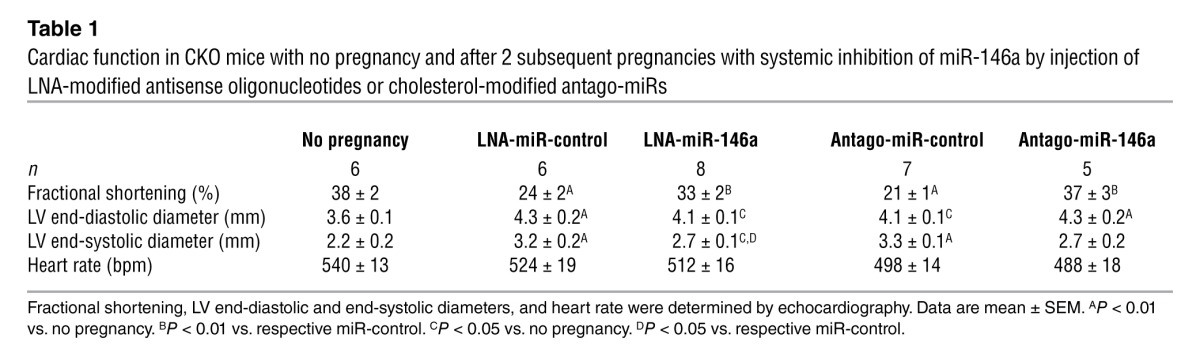
Importantly, LNA-miR-146a treatment of CKO mice did not interfere with lactation, as LNA-miR-146a–treated mice were able to raise offspring with similar success as LNA-miR-control–treated or nontreated CKO mice (LNA-miR-146a, 11 ± 5 offspring weaned after 2 subsequent pregnancies; LNA-miR-control, 11 ± 5 offspring weaned; untreated, 11 ± 3 offspring weaned; P = NS). Similarly, cholesterol-modified antago-miRs (28) used to block miR-146a attenuated cardiac dysfunction, reduced fibrosis, and increased Erbb4, Nras, Notch1, and Irak1 expression in CKO mice (Table 1 and Supplemental Figure 4, F–M). Nursing ability also remained intact (antago-miR-control, 10 ± 4 offspring weaned after 2 subsequent pregnancies, n = 5; antago-miR-146a, 8 ± 4 offspring weaned, n = 6; P = NS). Daily treatment with recombinant NRG1 applied for 2 subsequent nursing periods attenuated cardiac dysfunction compared with NaCl-treated CKO mice (Figure 4N, Supplemental Data, and Supplemental Figure 4N).
miR-146a is specifically elevated in plasma from patients with acute PPCM and normalized after recovery associated with PRL blockade by bromocriptine treatment.
We analyzed miR-146a levels in plasma probes from patients with acute PPCM (n = 38), from healthy postpartum controls (n = 18), and from healthy age-matched nonpregnant women (n = 5). Plasma levels of miR-146a were significantly higher in patients with acute PPCM compared with healthy postpartum controls (P < 0.001; Figure 5A). Levels of miR-146a were not significantly different between healthy postpartum controls and healthy nonpregnant women. Compared with 30 patients with a similar degree of heart failure due to dilated cardiomyopathy (DCM) and with no history of PPCM (ejection fraction [EF], PPCM, 28% ± 9%; DCM, 29% ± 9%; P = NS), miR-146a levels were significantly higher in the PPCM group (P < 0.001; Figure 5A). NT-proBNP, a biomarker for heart failure, was elevated to a similar degree (PPCM, 5,106 ± 4,326 fmol/ml; DCM, 3,346 ± 11,516 fmol/ml; P = NS), whereas total PRL was higher in the PPCM group (PPCM, 36 ± 48 μg/l; DCM, 8 ± 4 μg/l; P < 0.01).
Figure 5. miR-146a is elevated in plasma and LV tissue of patients with acute PPCM compared with controls and DCM and is reduced after recovery.

(A) Ratio between miR-146a and 2 spike-in miRNAs (cel-39 and cel-238) in plasma from patients with acute PPCM (n = 38), postpartum-matched (PP-matched) healthy controls (n = 18), age-matched healthy controls (n = 5), DCM patients (n = 30), and recovered PPCM patients treated with standard heart failure therapy and bromocriptine (n = 12). **P < 0.01, ***P < 0.001 vs. PPCM. (B) Follow-up ratio between miR-146a and 2 spike-in miRNAs in plasma from 7 PPCM patients, at baseline and after treatment with standard therapy for heart failure and bromocriptine. **P < 0.01 vs. PPCM. (C) qRT-PCR of miR-146a in LVs from PPCM patients (n = 3), DCM patients (n = 8), and nonfailing organ donors (NF; n = 4). (D) Level of ERBB4 mRNA in LVs from PPCM patients (n = 3) and nonfailing organ donors (n = 4). *P < 0.05 vs. nonfailing; #P < 0.005 vs. PPCM. Data are mean ± SEM.
In 12 PPCM patients who recovered after being treated with standard therapy for heart failure (beta blocker and ACE inhibitor) and 2.5–5 mg/d bromocriptine for 6–8 weeks (baseline EF, 23% ± 11%; follow-up EF, 51% ± 13%; P < 0.01; baseline LV end-diastolic diameter, 59 ± 6 mm; follow-up LV end-diastolic diameter, 53 ± 6 mm; P = NS), miR-146a plasma levels were within normal range (Figure 5A). Paired follow-up clinical data sets with corresponding blood samples were available from 7 of these PPCM patients (mean follow-up, 5 ± 2.5 months). miR-146a plasma levels decreased compared with the acute phase of PPCM (P < 0.01, paired t test; Figure 5B), which was associated with substantially improved cardiac function in all 7 patients (baseline EF, 22% ± 13%; follow-up EF, 53% ± 15%; P < 0.01; Supplemental Table 2).
Analysis from LV tissue revealed that miR-146a levels were significantly higher in patients with PPCM (n = 3) than in nonfailing organ donors (n = 4; P < 0.05) or in patients with end-stage heart failure due to DCM (n = 8; P < 0.005; Figure 5C). LV tissue levels of ERBB4 mRNA were reduced in PPCM patients compared with nonfailing organ donors (Figure 5D).
Discussion
Our experimental findings and these first clinical data suggest that 16K PRL generated by the coincidence of unbalanced oxidative stress, subsequent activation of PRL cleaving enzymes (cathepsin D), and high PRL levels (pituitary and cardiac) appear to be causative for PPCM. This hypothesis was supported by the prior observations that the PRL blocker bromocriptine showed beneficial effects in first clinical trials in PPCM patients (3, 4), completely prevented PPCM in CKO mice (2), and attenuated PPCM in mice with a cardiomyocyte-restricted deficiency of PPARγ coactivator 1α (PGC1α-KO mice) (29). To date, a specific receptor for 16K PRL has not been identified, but its downstream signaling cascade involving NF-κB in ECs is well documented (11). Our screen for NF-κB–activated miRNAs identified miR-146a as being specifically induced by 16K PRL. Furthermore, in vitro studies showed that miR-146a mediated a large part of the known antiangiogenic effects of 16K PRL in ECs. Moreover, after EC-exosomal transfer in cardiomyocytes, miR-146a impaired their metabolic activity. In fact, the 16K PRL–miR-146a cascade appeared to be responsible for a large portion of 16K PRL–induced pathological changes in PPCM, since targeting miR-146a in CKO mice by 2 different pharmacological approaches, LNA-miR-146a and antago-miR-146a, largely prevented PPCM symptoms. The finding that LNA-miR-146a– and antago-miR-146a–treated CKO mice were able to nurse their offspring confirmed that these strategies only affected 16K PRL–mediated effects, leaving the normal full-length PRL signaling system intact. Indeed, beside its role in nursing, full-length PRL has multiple beneficial effects, including promotion of angiogenesis and bonding between mother and child (30).
Although miR-146a appears to be a major downstream mediator of 16K PRL, we did not obtain full rescue of the PPCM phenotype with LNA-miR-146a or antago-miR-146a treatment, which was different from the complete rescue observed with the PRL blocker bromocriptine (2). One reason for the incomplete rescue of the PPCM phenotype after LNA treatment could be that blocking miR-146a had no effect on the pathomechanisms upstream of 16K PRL, including reduced manganese superoxide dismutase expression and enhanced oxidative stress (2). Another reason could be the presence of additional miR-146a–independent 16K PRL–mediated pathophysiological mechanisms.
Our previous studies in CKO and in PGC1α-KO mice suggest a major role for a systemically damaged vasculature in PPCM onset, in which 16K PRL plays a key role (2, 29). We discovered that 16K PRL exerted part of its negative effects in ECs by upregulating miR-146a, which in turn impaired EC proliferation and survival. To date, miR-146a has been mainly described for its implication in innate immunity, since, by targeting TRAF6 and IRAK1, it prevents constitutive activation of NF-κB during the inflammatory response (21). Our present findings highlight a new role of miR-146a in angiogenesis: reducing EC proliferation and viability. A suggested mechanism in these processes involves the downregulation of NRAS, previously described as a key gene in cell survival and proliferation (31), which we identified as a novel target of miR-146a that strongly attenuated EC proliferation and survival in vitro and in vivo. The observation that miR-146a was upregulated and Nras downregulated in hearts from CKO mice with PPCM suggests that miR-146a is at least in part responsible for angiogenic impairment in PPCM. This idea is further supported by the observations that a systemically injected adenovirus expressing 16K PRL led to higher miR-146a levels in hearts of WT mice, that pharmacological blockade of miR-146a normalized cardiac Nras expression and capillary density in postpartum CKO mice, and that blocking PRL in CKO mice by bromocriptine treatment prevented miR-146a upregulation and capillary loss (2). Taken together, the induction of miR-146a by 16K PRL and the subsequent reduction of Nras seems to be one mechanism by which 16K PRL impairs endothelial homeostasis in the postpartum maternal heart. Sylamer analyses suggested that, besides Nras, additional miR-146a targets may also be involved in this process, a feature that will be explored in future projects.
Besides its direct miR-146a–mediated effects on ECs, 16K PRL promoted shedding of exosomes from ECs containing elevated levels of miR-146a. Exosomes appear to protect miRNAs from degradation (32) and to act as vehicles to transport and transfer miRNAs to recipient cells (19). In vitro analyses showed that 16K PRL was not inducing miR-146a expression in NRCMs, but that NRCMs efficiently took up miR-146a–loaded endothelial exosomes, which led to increased intracellular levels of the mature form — but not the pre-form — of miR-146a. This transfer also seems to occur in vivo, as injected endothelial exosomes were detected in cardiomyocytes in the mouse heart. Moreover, in situ hybridization in postpartum CKO hearts mainly detected pre-mRNA and mature miR-146a in ECs and other nonmyocyte cardiac cells, such as cardiac fibroblasts, which suggests that in addition to ECs, fibroblasts may also be a source for miR-146a in hearts exposed to 16K PRL. These observations illustrate how 16K PRL can increase miR-146a levels in cells that do not directly respond to 16K PRL stimulation.
Erbb4 is constitutively expressed in cardiomyocytes forming homo- and heterodimers with ERBB2, and a functional ERBB signaling system is essential to the physiological status in the adult heart, since postnatal conditional knockout of cardiac Erbb2 in mice leads to DCM (33, 34).
During pregnancy, expression of Erbb4 increases in the maternal heart and plays a modulatory role during physiological hemodynamic overload associated with pregnancy, as pharmacological inhibition of ERBB2 leads to cardiomyopathy in this condition (35). The ligand of ERBB2/4 receptors is NRG1, a factor that is mainly produced by ECs; therefore, the NRG1/ERBB signaling system is a way through which cardiomyocytes and ECs communicate in the heart (27, 36, 37). Recently, Horie et al. described that part of the cardiotoxic effect of the chemotherapeutic agent doxorubicin is derived from the upregulation of miR-146a and the subsequent downregulation of Erbb4 in cardiomyocytes (24). We confirmed by ribonucleoprotein immunoprecipitation assay that Erbb4, but also Notch1 and Irak1, were direct physical targets of miR-146a in cardiomyocytes.
Pharmacological blockade of miR-146a or blocking PRL with bromocriptine in postpartum CKO mice attenuated Erbb4 downregulation and improved cardiac function, which supports the idea that a 16K PRL–miR-146a-exosome–mediated circuit impairs Erbb4 in hearts from CKO mice with PPCM (Figure 6). Our finding of increased miR-146a and decreased Erbb4 expression in LV tissue from PPCM patients compared with nonfailing hearts suggests that this mechanism may be also active in human PPCM.
Figure 6. Role of miR-146a in PPCM and proposed alternative treatment that maintains normal PRL functions.

(A) In PPCM patients, PRL is cleaved into 16K PRL, which (via NF-κB) increases miR-146a expression in ECs. By targeting NRAS, miR-146a reduces proliferation and viability in ECs and contributes to destruction of the cardiac microvasculature. miR-146a is released from ECs protected by the exosomes that fuse with cardiomyocytes, where miR-146a targets ERBB4 and impairs metabolism. (B and C) Proposed therapeutic options for PPCM management. (B) Blocking PRL completely by use of bromocriptine eliminates pathophysiological 16K PRL, but also nursing ability. (C) Use of anti-miR-146a in less severely affected patients may improve PPCM recovery, while keeping normal nursing functions.
Our previous work pointed to a metabolic impairment in PPCM, in part as a consequence of insufficient nutrition and oxygen supply due to reduced vasculature, but potentially also due to direct impairment of cardiomyocyte metabolism (2, 29). Indeed, 16K PRL via miR-146a impaired angiogenesis by downregulating NRAS in ECs. Since NRAS was barely expressed in cardiomyocytes, downregulation of this target gene may not affect cardiomyocytes directly. In turn, exosome-mediated transfer of miR-146a reduced the metabolic activity of cardiomyocytes, in part through downregulation of Erbb4, given that metabolic impairment was improved by treatment with antago-miR-146a (which increased Erbb4 expression) and activation of ERBB signaling by NRG1 was not able to increase metabolic activity in cardiomyocytes overexpressing miR-146a. Moreover, enhancing ERBB signaling in postpartum CKO mice with NRG1 only moderately attenuated cardiac dysfunction, probably due to downregulated Erbb4 expression. Thus, our results showed for the first time that 16K PRL impairs the crosstalk between ECs and cardiomyocytes by promoting the release of miR-146a–loaded exosomes from ECs, thereby also altering cardiomyocyte metabolism, which could — in addition to the impaired vasculature — contribute to heart failure in PPCM. However, our experimental design in the CKO PPCM model could not clearly distinguish between the contribution of decreased nutrition and oxygen supply due to the insufficient vasculature and the direct metabolic impairment of the cardiomyocytes caused by the downregulated ERBB signaling system in vivo.
PPCM and DCM patients with similar degrees of heart failure displayed similar increases in NT-proBNP, an unspecific marker for heart failure, while miR-146a plasma levels were only upregulated in PPCM patients. miR-146a levels were also higher in cardiac tissue from PPCM patients compared with cardiac tissue from nonfailing organ donors or DCM patients, further supporting the idea of a specific pathophysiological condition in PPCM, related to high 16K PRL, that is not present in other forms of heart failure. Therefore, the plasma level of miR-146a may emerge as a specific biomarker to distinguish PPCM patients who have a good potential for recovery from those PPCM patients with preexisting heart disease (i.e., DCM or genetic forms of heart failure) who have a much lower recovery potential and are more likely to require heart transplantation or assist device support. Such information would be of great importance for risk stratification and clinical management of patients. In addition, controlling miR-146a plasma levels in PPCM patients may be used to monitor recovery or progression of the disease.
In conclusion, our findings suggest a novel pathophysiologic circuit in PPCM involving enhanced local and systemic generation of 16K PRL, upregulated endothelial miR-146a expression with antiangiogenic properties, and impaired metabolism and function of cardiomyocytes. Within this circuit, we propose a miRNA-based intercellular communication system between ECs and cardiomyocytes via exosomes (Figure 6). Our discovery that miR-146a represents an important downstream effector of 16K PRL in PPCM suggests therapeutic strategies that could specifically remove adverse downstream effects of 16K PRL while leaving normal PRL signaling intact, which — in combination with bromocriptine — may help to speed recovery in critically ill PPCM patients (Figure 6, B and C). In addition, miR-146a may serve as a highly specific blood biomarker useful for diagnosis and risk stratification of patients with peripartum heart failure.
Methods
Further information can be found in Supplemental Methods.
Accession number.
Microarray data have been deposited in GEO (accession no. GSE43016).
Cell culture and reagents.
Isolation and culture of HUVECs (passages 6–11) were described previously (38). NRCMs and fibroblasts were isolated by enzymatic disassociation of neonatal rat hearts, as described previously (39). NRCMs were cultivated in a 4:1 mixture of 4.5 g/l DMEM high-glucose/M199, and fibroblasts were cultivated in 4.5 g/l DMEM high-glucose. RheA cells (40) and HEK293T cells were cultured in DMEM 4500 supplemented with 10% FBS. Recombinant 16K PRL (0.14 μg/ml) was produced and purified as described previously (41). BAY 11-7082 was purchased from Calbiochem. siRNAs were transfected using DharmaFECT-4 (Dharmacon Research Inc.) according to the manufacturer’s protocol.
Patients.
Plasma samples were obtained from patients with PPCM at their first presentation (n = 38; diagnosis based on criteria defined by Sliwa et al.; ref. 4), from healthy nursing women (n = 18), from age-matched healthy controls (n = 5), from patients with heart failure due to DCM (n = 30), and from patients with PPCM receiving bromocriptine treatment (see Supplemental Data). LV tissue was obtained from 1 PPCM patient undergoing heart transplantation, from 2 PPCM patients obtaining an assist device, from 8 DCM patients at the time of heart transplantation, and from donor hearts not suited for transplantation.
Animal experiments.
Generation of CKO mice, with cardiomyocyte-restricted knockout of Stat3 (αMHC-CreTg/+;Stat3fl/fl); WT littermates (Stat3fl/fl); and their PPCM phenotype has been described previously (2). LNA-miR-146a or LNA-miR-control (Exiqon) was given by 3 i.v. injections (20 mg/kg/injection; 3 days before and 1 and 3 days after delivery; ref. 42) for 2 consecutive pregnancies. Antago-miR-146a and antago-miR-control (VBC Biotech) was given by 4 i.v. injections (8 mg/kg/injection; 2 days before and 1, 3, and 7 days after delivery; ref. 28) for 2 consecutive pregnancies. NRG1 (50 μg/kg/d; Immunotools) was injected daily i.p. for 2 consecutive pregnancies and nursing periods (starting 3 days before delivery until 3 weeks after delivery).
Control or 16K PRL adenovirus vector (109 PFU; ref. 2) was injected i.v. into the lateral tail vein of C57BL/6J female mice (6–8 weeks of age; n = 5 per group). Echocardiography and Millar catheter analysis were performed as described previously (43, 44). Mice were sacrificed, and hearts and blood were harvested.
Exosome purification, labeling, and analysis.
Exosomes were purified from conditioned cell culture media as previously described (see Supplemental Methods and ref. 32).
HUVEC transfection and functional assays.
Pre- and anti-miRs (50 nM; Ambion) were transfected into HUVECs with Dharmafect-4 (Dharmacon Research Inc.) according to the manufacturer’s instructions. p65 and control siRNAs (20 nM) or NRAS and control siRNAs (50 nM) were transfected based on calcium phosphate transfection. Transfected cells were plated in SFM (Lonza) supplemented with 5 ng/ml bFGF or EGM-2 (Lonza) and 0.5% serum, respectively. After a 24-hour transfection, cells were washed and kept for additional 48 or 72 hours in plasma-free medium. Functional assays were performed as previously described (see Supplemental Methods and ref. 45).
Transfection, cell metabolism assay, and cell viability of NRCMs.
Pre-miR-146a and pre-miR-control (100 nM; Ambion) were transfected into NRCMs with Lipofectamine (Invitrogen) as previously described (40). After transfection for 24 hours, cells were washed and kept for an additional 96 hours in serum-free medium. Cell metabolism was determined by MTS assay (Promega) according to the manufacturer’s instructions. Cell viability was evaluated by trypan blue staining of dead cells and expressed as a percentage of total NRCMs. Cell culture experiments were performed on 4–5 independent cell isolations.
Ex vivo aortic ring assay.
Mouse aortic rings were cultured and quantified as previously described (46), transfected with 50 nM of anti- or pre-miR-control and -miR-146a with Dharmafect-4 (Dharmacon Research Inc.), and incubated at 37°C.
In vivo CNV experiment.
CNV was induced in mice (2-month-old C57BL/6) by laser burns on the Bruch membrane, as described previously (47). 2 μl pre-miR-control and pre-miR-146a (5 μM) or control or NRAS siRNA (50 μM) were injected intravitreally. Quantification was performed using ImageJ software on the green signal (fluorescein-labeled dextran) in the lesion area.
Statistics.
All values are expressed as mean ± SD (in vitro and in vivo experiments) or mean ± SEM (in vivo experiments and patient analyses). Comparisons between different conditions were assessed using 2-tailed Student’s t test. If the normality test failed, the Mann-Whitney test was performed. Differences between groups were analyzed by ANOVA followed by Bonferroni post-hoc analyses as appropriate (Figure 5A and Table 1). 2-tailed paired t test was used for patient follow-up (PPCM vs. bromocriptine-treated PPCM; Figure 5B). A P value less than 0.05 was considered significant.
Study approval.
All animal procedures were approved by the IACUC and performed in accordance with the guidelines of the local animal ethics commission. PPCM cases were reported to our registry from university hospitals, tertiary hospitals, or cardiologists in private practice. This PPCM registry, as well as patient serum and tissue analyses, were approved by the local ethics commission. All patients provided written informed consent.
Supplementary Material
Acknowledgments
We thank Michelle Lion, Karin Battmer, Birgit Brandt, Silvia Gutzke, Patricia Piscicelli, Lea Greune, and Sergej Erschow for technical assistance. We thank Eric Rozet for advice in statistical analysis and Khalid Bajou for expertise in animal models. We thank Igor Tudorache for contribution of human LV tissue. We acknowledge the GIGA-genomics (Benoît Hennuy), GIGA-mouse (Pierre Drion), GIGA-imaging, and GIGA-sequencing facilities. This study was supported by University of Liège; Fonds pour la Recherche Industrielle et Agricole, Belgium (FRIA); Fonds National de la Recherche Scientifique, Belgium (FNRS); Belgian Science Policy; grant IUAP06/30 from the federal government Belgium; Neoangio program no. 616476 of the “Service Public de Wallonie”; German Research Foundation (DFG); Foundation Leducq; Bundesministerium für Bildung und Forschung (BMBF); Belgian Foundation against Cancer; and European Commission project FP7-HEALTH-2007-A, proposal no. 201279 “MICROENVIMET.”
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2013;123(5):2143–2154. doi:10.1172/JCI64365.
See the related Commentary beginning on page 1925.
References
- 1.Hilfiker-Kleiner D, Sliwa K, Drexler H. Peripartum cardiomyopathy: recent insights in its pathophysiology. Trends Cardiovasc Med. 2008;18(5):173–179. doi: 10.1016/j.tcm.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Hilfiker-Kleiner D, et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. 2007;128(3):589–600. doi: 10.1016/j.cell.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 3.Hilfiker-Kleiner D, et al. Recovery from postpartum cardiomyopathy in 2 patients by blocking prolactin release with bromocriptine. J Am Coll Cardiol. 2007;50(24):2354–2355. doi: 10.1016/j.jacc.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Sliwa K, et al. Evaluation of bromocriptine in the treatment of acute severe peripartum cardiomyopathy: a proof-of-concept pilot study. Circulation. 2010;121(13):1465–1473. doi: 10.1161/CIRCULATIONAHA.109.901496. [DOI] [PubMed] [Google Scholar]
- 5.Pan H, et al. Molecular targeting of antiangiogenic factor 16K hPRL inhibits oxygen-induced retinopathy in mice. Invest Ophthalmol Vis Sci. 2004;45(7):2413–2419. doi: 10.1167/iovs.03-1001. [DOI] [PubMed] [Google Scholar]
- 6.Bentzien F, Struman I, Martini JF, Martial JA, Weiner RI. Expression of the antiangiogenic factor 16K hPRL in human HCT116 colon cancer cells inhibits tumor growth in Rag1(–/–) mice. Cancer Res. 2001;61(19):7356–7362. [PubMed] [Google Scholar]
- 7.Nguyen NQ, et al. Inhibition of tumor growth and metastasis establishment by adenovirus-mediated gene transfer delivery of the antiangiogenic factor 16K hPRL. Mol Ther. 2007;15(12):2094–2100. doi: 10.1038/sj.mt.6300294. [DOI] [PubMed] [Google Scholar]
- 8.Lee SH, Kunz J, Lin SH, Yu-Lee LY. 16-kDa prolactin inhibits endothelial cell migration by down-regulating the Ras-Tiam1-Rac1-Pak1 signaling pathway. Cancer Res. 2007;67(22):11045–11053. doi: 10.1158/0008-5472.CAN-07-0986. [DOI] [PubMed] [Google Scholar]
- 9.D’Angelo G, Struman I, Martial JA, Weiner RI. Activation of mitogen-activated protein kinases by vascular endothelial growth factor and basic fibroblast growth factor in capillary endothelial cells is inhibited by the antiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proc Natl Acad Sci U S A. 1995;92(14):6374–6378. doi: 10.1073/pnas.92.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabruyn SP, Nguyen NQ, Cornet AM, Martial JA, Struman I. The antiangiogenic factor, 16-kDa human prolactin, induces endothelial cell cycle arrest by acting at both the G0-G1 and the G2-M phases. Mol Endocrinol. 2005;19(7):1932–1942. doi: 10.1210/me.2004-0515. [DOI] [PubMed] [Google Scholar]
- 11.Tabruyn SP, et al. The antiangiogenic factor 16K human prolactin induces caspase-dependent apoptosis by a mechanism that requires activation of nuclear factor-kappaB. Mol Endocrinol. 2003;17(9):1815–1823. doi: 10.1210/me.2003-0132. [DOI] [PubMed] [Google Scholar]
- 12.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Olson EN. AngiomiRs — key regulators of angiogenesis. Curr Opin Genet Dev. 2009;19(3):205–211. doi: 10.1016/j.gde.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuehbacher A, Urbich C, Dimmeler S. Targeting microRNA expression to regulate angiogenesis. Trends Pharmacol Sci. 2008;29(1):12–15. doi: 10.1016/j.tips.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 15.van Rooij E, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103(48):18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thum T, Catalucci D, Bauersachs J. MicroRNAs: novel regulators in cardiac development and disease. Cardiovasc Res. 2008;79(4):562–570. doi: 10.1093/cvr/cvn137. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell PS, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008;110(1):13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 19.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 20.Mayoral RJ, Pipkin ME, Pachkov M, van Nimwegen E, Rao A, Monticelli S. MicroRNA-221-222 regulate the cell cycle in mast cells. J Immunol. 2009;182(1):433–445. doi: 10.4049/jimmunol.182.1.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tili E, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179(8):5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 23.van Dongen S, Abreu-Goodger C, Enright AJ. Detecting microRNA binding and siRNA off-target effects from expression data. Nat Methods. 2008;5(12):1023–1025. doi: 10.1038/nmeth.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horie T, et al. Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc Res. 2010;87(4):656–664. doi: 10.1093/cvr/cvq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keene JD, Komisarow JM, Friedersdorf MB. RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc. 2006;1(1):302–307. doi: 10.1038/nprot.2006.47. [DOI] [PubMed] [Google Scholar]
- 26.Mei J, Bachoo R, Zhang CL. MicroRNA-146a inhibits glioma development by targeting Notch1. Mol Cell Biol. 2011;31(17):3584–3592. doi: 10.1128/MCB.05821-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Odiete O, Hill MF, Sawyer DB. Neuregulin in cardiovascular development and disease. Circ Res. 2012;111(10):1376–1385. doi: 10.1161/CIRCRESAHA.112.267286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonauer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324(5935):1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 29.Patten IS, et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012;485(7398):333–338. doi: 10.1038/nature11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corbacho AM, Martinez De La Escalera G, Clapp C. Roles of prolactin and related members of the prolactin/growth hormone/placental lactogen family in angiogenesis. J Endocrinol. 2002;173(2):219–238. doi: 10.1677/joe.0.1730219. [DOI] [PubMed] [Google Scholar]
- 31.Shapiro P. Ras-MAP kinase signaling pathways and control of cell proliferation: relevance to cancer therapy. Crit Rev Clin Lab Sci. 2002;39(4–5):285–330. doi: 10.1080/10408360290795538. [DOI] [PubMed] [Google Scholar]
- 32.Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010;101(10):2087–2092. doi: 10.1111/j.1349-7006.2010.01650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozcelik C, et al. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc Natl Acad Sci U S A. 2002;99(13):8880–8885. doi: 10.1073/pnas.122249299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pentassuglia L, Sawyer DB. The role of Neuregulin-1beta/ErbB signaling in the heart. Exp Cell Res. 2009;315(4):627–637. doi: 10.1016/j.yexcr.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemmens K, Doggen K, De Keulenaer GW. Activation of the neuregulin/ErbB system during physiological ventricular remodeling in pregnancy. Am J Physiol Heart Circ Physiol. 2011;300(3):H931–H942. doi: 10.1152/ajpheart.00385.2010. [DOI] [PubMed] [Google Scholar]
- 36.Lemmens K, Segers VF, Demolder M, De Keulenaer GW. Role of neuregulin-1/ErbB2 signaling in endothelium-cardiomyocyte cross-talk. J Biol Chem. 2006;281(28):19469–19477. doi: 10.1074/jbc.M600399200. [DOI] [PubMed] [Google Scholar]
- 37.Hedhli N, et al. Endothelium-derived neuregulin protects the heart against ischemic injury. Circulation. 2011;123(20):2254–2262. doi: 10.1161/CIRCULATIONAHA.110.991125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tabruyn SP, et al. NF-kappaB activation in endothelial cells is critical for the activity of angiostatic agents. Mol Cancer Ther. 2009;8(9):2645–2654. doi: 10.1158/1535-7163.MCT-09-0383. [DOI] [PubMed] [Google Scholar]
- 39.Hilfiker-Kleiner D, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95(2):187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 40.Haghikia A, et al. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-conjugating enzymes. Eur Heart J. 2011;32(10):1287–1297. doi: 10.1093/eurheartj/ehq369. [DOI] [PubMed] [Google Scholar]
- 41.Struman I, et al. Opposing actions of intact and N-terminal fragments of the human prolactin/growth hormone family members on angiogenesis: an efficient mechanism for the regulation of angiogenesis. Proc Natl Acad Sci U S A. 1999;96(4):1246–1251. doi: 10.1073/pnas.96.4.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boon RA, et al. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res. 2011;109(10):1115–1119. doi: 10.1161/CIRCRESAHA.111.255737. [DOI] [PubMed] [Google Scholar]
- 43.Hilfiker-Kleiner D, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122(2):145–155. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 44.Hilfiker-Kleiner D, et al. Lack of JunD promotes pressure overload-induced apoptosis, hypertrophic growth, and angiogenesis in the heart. Circulation. 2005;112(10):1470–1477. doi: 10.1161/CIRCULATIONAHA.104.518472. [DOI] [PubMed] [Google Scholar]
- 45.Sabatel C, et al. MicroRNA-21 exhibits antiangiogenic function by targeting RhoB expression in endothelial cells. PLoS One. 2011;6(2):e16979. doi: 10.1371/journal.pone.0016979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berndt S, et al. Angiogenic activity of human chorionic gonadotropin through LH receptor activation on endothelial and epithelial cells of the endometrium. FASEB J. 2006;20(14):2630–2632. doi: 10.1096/fj.06-5885fje. [DOI] [PubMed] [Google Scholar]
- 47.Lambert V, et al. Influence of plasminogen activator inhibitor type 1 on choroidal neovascularization. FASEB J. 2001;15(6):1021–1027. doi: 10.1096/fj.00-0393com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.