

Abstract
Williams-Beuren syndrome is a rare multisystem neurodevelopmental disorder caused by a 1.55-1.84-Mb hemizygous deletion on chromosome 7q11.23. The classical phenotype consists of characteristic facial features, supravalvular aortic stenosis, intellectual disability, overfriendliness, and visuospatial impairment. So far, 26-28 genes have been shown to contribute to the multisystem phenotype associated with Williams-Beuren syndrome. Among them, haploinsufficiency of the ELN gene has been shown to cause the cardiovascular anomalies. Identification of patients with atypical deletions has provided valuable information for genotype-phenotype correlation, in which other genes such as LIMK1,CLIP2, GTF2IRD1, or GTF2I have been correlated with specific cognitive profiles or craniofacial features. Here, we report the clinical and molecular characteristics of a patient with an atypical deletion that does not include the GTF2I gene and only partially includes the GTF2IRD1 gene.
Key Words : Deletion 7q11.23, Genomic disorders, GTF2I, GTF2IRD1 , Segmental duplications, Williams-Beuren syndrome, Williams syndrome
Williams-Beuren syndrome (WBS; OMIM 194050) is a complex multisystem neurodevelopmental disorder characterized by diverse physical and neurological abnormalities. Children with WBS manifest characteristic facial features, including bitemporal narrowing, depressed nasal root, bulbous nasal tip, periorbital fullness, stellate irides, full cheeks, long philtrum, full lips, wide mouth, and dental malocclusion [Morris et al., 1988; Morris, 2010]. The most common cardiovascular abnormalities include supravalvular aortic and peripheral pulmonary stenosis. Patients with WBS have developmental delay, intellectual disability commonly ranging from a mild to a moderate degree, visuospatial construction deficit, and motor functions impairment [Morris, 2010].
This distinctive spectrum of symptoms and features arises from a 1.5-1.8-Mb hemizygous deletion of 26-28 genes on chromosome 7q11.23. Individuals with atypical deletions smaller than 1.5 Mb (1-2%) present variable subsets of WBS signs and symptoms. The phenotype-genotype correlation relies mainly on evidence obtained through mouse models and analysis of individuals with atypical deletions. Certain genes within the WBS deletion region, such as LIMK1, CLIP2, GTF2IRD1, and GTF2I, have been associated to the specific cognitive profile and craniofacial features [Tassabehji et al., 2005; Antonell et al., 2010].
Here, we report the clinical and molecular characterization of a girl with a ∼1.3 Mb deletion that does not include the GTF2I gene and comprises partially the GTF2IRD1 gene.
Methods
A cohort of 37 patients referred to the Centro Nacional de Genética Médica (CENAGEM) because of a suspected diagnosis of WBS has been studied. Diagnostic confirmation by FISH was performed with a commercial Williams-Beuren syndrome region probe kit. Genomic DNA from the probands and their parents was extracted using standard methods. The current work has been approved by the Ethics Committee of CENAGEM. Informed consent was obtained from all families.
All DNA samples were tested for 7q11.23 microdeletion size using 2 different techniques: multiplex ligation-dependent probe amplification (MLPA) and real-time quantitative PCR (qPCR). The MLPA assay was performed using SALSA kit P029 (MRC Holland, http://www.mlpa.com), which contains 12 probes from the WBS chromosomal region (fig. 1a) and 20 control probes. For the MLPA analysis, 50 ng of DNA was used as instructed by the manufacturer's protocol. DNA from 3 healthy individuals was used as control. The PCR products were analyzed by capillary electrophoresis, and data normalization was performed by Coffalyser software (MRC Holland). Power SYBR Green PCR Master Mix (Applied Biosystems) was used for qPCR, as instructed by the manufacturer's protocol. Primers were designed for the amplification of DNA fragments located within and flanking the WBS chromosomal region and qPCR reactions were carried out as previously described [Howald et al., 2006]. Control DNA fragments were located on alternate chromosomes (fig. 1a). Samples were run in triplicate using standard conditions. Calculations were carried out by the comparative Ct method.
Fig. 1.

a Physical map of the WBS region at 7q11.23 showing the relative location of the MLPA probes (green vertical lines) and qPCR amplicons (blue vertical lines). Ac, Bc, Cm, Bm, and Am indicate segmental duplication blocks flanking the common deletion interval. b Summary of the mapping of WBS atypical deletions reported in the literature and WBS269-023A1 patient. Deleted fragments are denoted by gray segments, not deleted ones by black segments. Dotted segments represent undefined deletion status. c Profiles of qPCR assays for detection of deletion size. Relative DNA amplification of several markers was quantified for the WBS269-023A1 (green triangles) and 2 patients with classical WBS: WBS268-022A1 (red squares) and WBS270-024A1 (purple circles). WT is a reference sample (blue diamonds). d Profiles of qPCR assay to narrow the distal deletion breakpoint within the GTF2IRD1 gene. WBS269-023A1 (green large squares), WBS160 (purple triangles) carrying the classical deletion, WT2 (turquoise small squares), and WT (blue diamonds) are controls. e Mapping of atypical deletion of patient WBS269-023A1. The distal breakpoint is schematically represented about 19-50 kb downstream of exon 1 of the GTF2IRD1 gene. 4.12_GTF2IRD1 was the more distal deleted marker and 4.11_GTF2IRD1 was the first not deleted marker.
To refine the distal breakpoint, we used several additional primer pairs to amplify the region between the more distal deleted fragment and the adjacent nondeleted segment (primer sequences are listed in online supplementary table 1, see www.k-arger.com/doi/10.1159/000347167).
Results and Discussion
The WBS269-023A1 patient is the first daughter of healthy, nonconsanguineous parents and was 5 months old at the time of diagnosis. Family history showed ventricular septal defect in the father and atrial septal defect in a paternal uncle. The pregnancy was controlled and delivered by caesarean at 39 weeks of gestational age due to fetal distress.
Birth weight, length, and OFC were: 2.650 kg (∼3rd centile), 50 cm (∼50th centile) and 35 cm (50-75th centile), respectively. She was diagnosed with peripheral pulmonary stenosis and ventricular septal defect, and had a flat midface, depressed nasal bridge, anteverted nostrils, bitemporal narrowing, periorbital fullness, long philtrum, thick lips, and mild micrognathia (fig. 2). When she was 2 years old, these facial features became accentuated as in typical WBS. She showed a mildly delayed development profile for her chronological age, based on the Gesell Developmental Schedules [Ball, 1977]. At 2 years of age, she also showed delays mainly in the adaptive fine motor development. Patterns of development for children between 15 months and 2 years old for motor, adaptive, language and personal-social areas were evaluated. The patient complied with the development guidelines envisaged by Gesell Schedules for the age of 15-18 months, except for some guidelines in the adaptive and fine motor development areas. For example, she was not able to turn the pages of a book and inserted no more than one cube on the form board. She also had speech delay with the incapacity to say 5 words, which is what we would expect for a child her age by the Gesell's standards. Unfortunately, she was too young to unambiguously determine her social behavior.
Fig. 2.
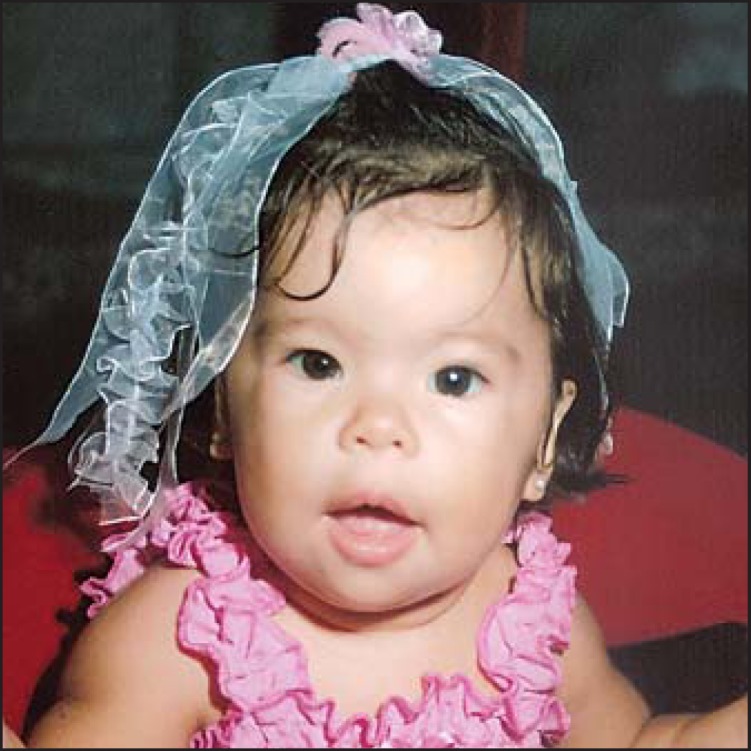
Facial appearance of WBS269-023A1. Picture of patient WBS269-023A1 shows the classical WBS facial appearance, including bitemporal narrowing, periorbital fullness, flat nasal bridge, short nose, anteversion of nares, wide mouth, and long philtrum.
The molecular assessment of this patient showed an atypical short 7q11.23 deletion that partially included the GTF2IRD1 gene, but not GTF2I gene (fig. 1c). The qPCR assay showed a deletion of the marker 4.12_GTF2IRD1, located 19 kb downstream of exon 1 of the GTF2IRD1 gene, but not of 4.1_GTF2IRD1 marker, 80 kb from exon 1.
To narrow the telomeric breakpoint, we performed a new qPCR assay using 3 additional markers located in between 4.12_GTF2IRD1 and 4.1_GTF2IRD1 (fig. 1d). In this new qPCR assay, 4.12_GTF2IRD1 remained the most distal deleted marker, while 4.11_GTF2IRD1, located about 50 kb downstream exon 1, was the more proximal not deleted marker. This new assay showed that the distal breakpoint of the deletion was between 19-50 kb downstream of exon 1 of GTF2IRD1 gene, indicating that this patient had a deletion of ∼1.3 Mb (fig. 1d, e). The MLPA assay did not detect the atypical deletion in this patient because the commercial kit does not provide probes located at the distal breakpoints.
Several reports have shown that the distal breakpoints of many atypical WBS deletions are clustered around the GTF2IRD1 gene [Gagliardi et al., 2003; Hirota et al., 2003; Tassabehji et al., 2005; Dai et al., 2009; Ferrero et al., 2010] (fig. 1b). Such patients suggest that CLIP2, GTF2IRD1 and GTF2I are associated to the specific cognitive profile and craniofacial features [Tassabehji et al., 2005; Ferrero et al., 2010], although such evidences are debated and need further investigations [Vandeweyer et al., 2012]. A number of studies have established mice models of the more telomerically positioned WBS genes. Analysis of these mice has shown a series of phenotypes that share some intriguing parallels with WBS. A mouse model harboring a deletion between Clip2 and Gtf2ird1 showed craniofacial and dental abnormalities [Tassabehji et al., 2005]. Mice haploinsufficient for both Gtf2ird1 and Gtf2i are often growth retarded, and show hyploplasia of the mandible as well as other craniofacial defects resembling the anomalies and dental problems of WBS individuals [Enkhmandakh et al., 2009]. Finally, haploinsufficiency of GTF2IRD1 protein contributes to abnormalities of facial development, motor function and specific behavioral disorders that accompany WBS disease [Young et al., 2008; Howard et al., 2012; Schneider et al., 2012].
The phenotype of the patient reported here is in accordance with Dai et al. [2009], who described a patient carrying a full deletion of GTF2IRD1 gene but preserving the GTF2I gene. Of note, these clinical manifestations do not completely fit with the data reported by other authors which showed that patients with WBS and smaller deletions preserving both GTF2IRD1 and GTF2I genes had mild craniofacial features [Gagliardi et al., 2003; Hirota et al., 2003; Tassabehji et al., 2005] (table 1).
Table 1.
Molecular and clinical characteristics of patient WBS269-023A1 compared to the classical WBS phenotype and other patients with similar atypical deletions
| Clinical and molecular characteristics | Classical deletion (%) Morris et al., (1988) | Comparable atypical deletions |
|||||
|---|---|---|---|---|---|---|---|
| Gagliardi et al., (2003) | Hirota et al., (2003); case 1 | Tassabehji et al., (2005); HR | Dai et al., (2009); 5889 | Ferrero et al., (2010) | Patient WBS269-023A1 | ||
| Molecular characteristics | |||||||
| Deletion size, Mb | 1.5–1.8 | 1.1–1.2 | ~1.2 | ~1.1 | ~1.4 | ~0.9 | ~1.3 |
| CLIP2 deletion | + | + | + | + | + | + | + |
| GTF2IRD1 deletion | + | – | – | +/– | + | – | +/– |
| GTF2I deletion | + | – | – | – | – | – | – |
| Clinical features | |||||||
| Age | 5 years | 23 years | 3 years | 7 years | 11 years | 5 months | |
| Craniofacial | |||||||
| Dolicocephaly | + | – | – | – | + | – | + |
| Bitemporal narrowing | + | + | – | + | + | + | + |
| Periorbital fullness | + | – | – | + | + | + | + |
| Epicanthal folds | + | – | – | + | + | + | + |
| Strabismus | 50 | – | + | – | + | + | – |
| Short upturned nose | + | + | – | + | + | + | + |
| Full cheeks | + | + | – | + | + | + | + |
| Long philtrum | + | – | – | + | + | + | + |
| Thick lips | + | – | – | – | + | + | + |
| Dental malocclusion | 85 | – | + | – | + | + | NA |
| Cardiovascular | |||||||
| Supravalvular aortic stenosis | 75 | + | + | – | + | + | NA |
| Peripheral pulmonary stenosis | 50 | – | + | + | + | – | + |
| Ventricular septal defect | 10 | – | – | – | – | – | + |
| Hypertension | 50 | NR | – | NR | + | – | NA |
| Musculoskeletal | |||||||
| Joint hypermobility | 90 | – | – | – | – | – | + |
| Joint contractures | 50 | – | – | – | – | – | NA |
| Kyphosis/lordosis | 20/40 | – | – | – | – | – | NA |
| Hernias | 50 | – | – | – | – | + | – |
| Neuropsychological | |||||||
| Developmental delay | 95 | +/– | NR | – | – | – | NA |
| Intellectual disability | 75 | +/– | + | + | NR | – | NA |
| Overfriendliness | + | + | NR | – | – | – | NA |
| Visuospatial construction deficit | + | +/– | – | + | – | +/– | NA |
| Others | |||||||
| Genitourinary malformation | 20 | + | – | NR | NR | – | – |
| Hyperacusis | 80 | NR | – | – | – | – | + |
| Hypercalcemia | 15 | NR | NR | NR | – | – | NA |
+ = Present
– = not present
+/– = partially present
NR = not registered
NA = not available at clinical examination.
Although the patient reported here does not have a complete deletion of GTF2IRD1 gene, it is possible that, as consequence of the rearrangement, the gene expression level is affected, as it has been demonstrated for the WBS flanking region genes [Merla et al., 2006]. Nevertheless, as we do not have access neither to cell lines nor to fresh blood from the patient, this hypothesis has not been investigated in this work.
The identification of more atypical patients would enable better understanding of the individual role of each gene in the development of clinical features and the neuropsychological profiles. This is important because it gives us information about the pathogenic mechanisms of disease, which could contribute to the pursuit of new therapeutic treatments and eventually allow more precise prediction of the natural history in affected individuals. Because WBS is a rare disease with atypical deletions occurring only in 2% of patients, it is necessary to study a large population to obtain significant data, which would allow a more precise genotype-phenotype correlation and achieve more accurate conclusions. We encourage that larger consortia being developed to achieve this aim and the development of a set of quantitative measurements for clinical, cognitive and behavioral features in order to provide a far better mean of optimizing genotype-phenotype correlation.
Acknowledgements
We thank the families who participated in this project and made this study possible. This work was supported in part by grants from the Italian Ministry of Health (Ricerca Corrente 2008-10), the Fondazione Banca del Monte di Foggia ‘Domenico Siniscalco Ceci’, and received support from Ministero degli Affari Esteri, Direzione Generale per la Promozione e la Cooperazione Culturale (2009-2010) to G.M.
We thank Lilian Furforo for her helpful review of the text and María Cecilia Luna for her cooperation in this work.
References
- Antonell A, Del Campo M, Magano LF, Kaufmann L, de la Iglesia JM, et al. Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile. J Med Genet. 2010;47:312–320. doi: 10.1136/jmg.2009.071712. [DOI] [PubMed] [Google Scholar]
- Ball RS. The Gesell developmental schedules. J Abn Child Psychol. 1977;5:233–239. doi: 10.1007/BF00913694. [DOI] [PubMed] [Google Scholar]
- Dai L, Bellugi U, Chen XN, Pulst-Korenberg AM, Järvinen-Pasley A, et al. Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am J Med Genet Part A. 2009;149A:302–314. doi: 10.1002/ajmg.a.32652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkhmandakh B, Makeyev AV, Erdenechimeg L, Ruddle FH, Chimge NO, et al. Essential functions of the Williams-Beuren syndrome-associated TFII-I genes in embryonic development. Proc Natl Acad Sci USA. 2009;106:181–186. doi: 10.1073/pnas.0811531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero GB, Howald C, Micale L, Biamino E, Augello B, et al. An atypical 7q11.23 deletion in a normal IQ Williams syndrome patient. Eur J Hum Genet. 2010;18:33–38. doi: 10.1038/ejhg.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi C, Bonaglia MC, Selicorni A, Borgatti R, Giorda R. Unusual cognitive and behavioural profile in a Williams syndrome patient with atypical 7q11.23 deletion. J Med Genet. 2003;40:526–530. doi: 10.1136/jmg.40.7.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota H, Matsuoka R, Chen XN, Salandanan LS, Lincoln A, et al. Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on chromosome 7q11.23. Genet Med. 2003;5:311–321. doi: 10.1097/01.GIM.0000076975.10224.67. [DOI] [PubMed] [Google Scholar]
- Howald C, Merla G, Digilio MC, Amenta S, Lyle R, et al. Two high throughput technologies to detect segmental aneuploidies identify new Williams-Beuren syndrome patients with atypical deletions. J Med Genet. 2006;43:266–273. doi: 10.1136/jmg.2005.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard ML, Palmer SJ, Taylor KM, Arthurson GJ, Spitzer MW, et al. Mutation of Gtf2ird1 from the Williams-Beuren syndrome critical region results in facial dysplasia, motor dysfunction, and altered vocalisations. Neurobiol Dis. 2012;45:913–922. doi: 10.1016/j.nbd.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Merla G, Howald C, Henrichsen CN, Lyle R, Wyss C, et al. Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am J Hum Genet. 2006;79:332–341. doi: 10.1086/506371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CA. Introduction: Williams syndrome. Am J Med Genet Part C Semin Med Genet. 2010;154C:203–208. doi: 10.1002/ajmg.c.30266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL. Natural history of Williams syndrome: physical characteristics. J Pediatr. 1988;113:318–326. doi: 10.1016/s0022-3476(88)80272-5. [DOI] [PubMed] [Google Scholar]
- Schneider T, Skitt Z, Liu Y, Deacon RM, Flint J, et al. Anxious, hypoactive phenotype combined with motor deficits in Gtf2ird1 null mouse model relevant to Williams syndrome. Behav Brain Res. 2012;233:458–473. doi: 10.1016/j.bbr.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, et al. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005;310:1184–1187. doi: 10.1126/science.1116142. [DOI] [PubMed] [Google Scholar]
- Vandeweyer G, Van der Aa N, Reyniers E, Kooy RF. The contribution of CLIP2 haploinsufficiency to the clinical manifestations of the Williams-Beuren syndrome. Am J Hum Genet. 2012;90:1071–1078. doi: 10.1016/j.ajhg.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young EJ, Lipina T, Tam E, Mandel A, Clapcote SJ, et al. Reduced fear and aggression and altered serotonin metabolism in Gtf2ird1-targeted mice. Genes Brain Behav. 2008;7:224–234. doi: 10.1111/j.1601-183X.2007.00343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]