

Abstract
We report a 2.3-year-old female patient with global developmental delay, infantile spasms, hypotonia, microcephaly, flat face, full cheeks, macroglossia, highly arched palate, retro-gnathia, narrow ear orifices, and café-au-lait spots. Molecular karyotyping revealed approximately a 1-Mb interstitial deletion of the long arm of one chromosome 12, del(12)(q24.31). The same deletion was identified in her father who presents insulin-dependent diabetes mellitus (IDDM) diagnosed at 14 years. Only one other patient with a similar de novo deletion has been reported previously [Mol Syndromol 2010;1:42-45]. A phenotype-genotype correlation is discussed, and the description of a novel rare microdeletion entity is raised.
Key Words : Chromosome 12q, Developmental delay, Haploinsufficient, HNF1A, Microdeletion 12q24.31
There have been only limited reports of patients with distal interstitial deletion of chromosome 12q [Meinecke and Meinecke, 1987; Watson et al., 1989; Sathya et al., 1999; Plotner et al., 2003; Baple et al., 2010; Qiao et al., 2012]. Among these, only 2 patients with a de novo deletion limited to the band 12q24.31 have been described [Baple et al., 2010; Qiao et al., 2012]. The patient described by Baple et al. [2010] has a more proximal deletion than the one described by Qiao et al. [2012] with a small overlapping region.
We report herein a female patient with an inherited proximal interstitial deletion of band 12q24.31 overlying the microdeletion described by Baple et al. [2010]. The microdeletion in the proband was inherited from her father, who had only an insulin-dependent diabetes mellitus explained by the fact that this deletion involves the entire coding sequence of the HNF1A gene. Finally, we offer a comparison to the rare described cases with deletions affecting this same region, raising the need to study a larger set of patients in order to better understand this new microdeletion.
Material and Methods
Clinical Report
The proband (fig. 1) was the first child of nonconsanguineous parents. Family history was unremarkable, except the fact that the father presented an insulin-dependent diabetes at age 14.
Fig. 1.

Proband at 2.3 years old.
The patient is the product of an uncomplicated pregnancy. A routine fetal ultrasound, during the whole pregnancy, was within normal limits. The patient was born at term by normal vaginal delivery. Her birth weight was 2.9 kg (10th percentile), birth length was 50 cm (median) and birth head circumference was 32 cm (10th percentile). The neonatal period was unremarkable.
At 4 months, she developed refractory infantile spasms and an electroencephalogram (EEG) confirmed hypsarrhythmia. She showed partial response to adrenocorticotropic hormone therapy, and her development was profoundly delayed. At 18 months of age, she was unable to sit without support, and she did not crawl, babble or coo. Her social interaction skills were lacking, and she rarely smiled in response. She was capable of transferring objects and walking a few steps with support. The girl had a flat face, full cheeks, highly arched palate, retrognathia, narrow ear orifices, and 4 café-au-lait spots: 1 on the back, 1 on the thigh and 2 on the buttocks.
At 18 months of age, her weight was 12.5 kg (90th percentile); she was considered obese. Between 8 and 18 months, she presented significant constipation.
Re-examination at 20 months of age showed moderate global psychomotor delay with a head circumference of 42.5 cm (<<3rd percentile). Her weight was 11.3 kg (50th percentile), and her height was 86 cm (50th percentile), within the normal ranges. No constipation was noted due to the fact that corticosteroid therapy was stopped, 3 months before.
Cytogenetic analysis was normal. Magnetic resonance imaging of the brain showed mild diffuse atrophy. She had no apparent problems with hearing or vision.
DNA Sequencing
The coding sequences of the following genes were sequenced in the patient after her DNA was amplified by PCR: MECP2 (GenBank accession number: NM_004992.3) and ACADS (NM_000017.2). Primers were designed using Primer 3 (http://frodo.wi.mit.edu) and OLIGOS v.9.3, and checked for specificity using BLAST (http://www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi). DNA sequences were obtained from UCSC and Genbank databases. PCR reactions were performed using Taq DNA polymerase (Invitrogen Life Technologies, Carlsbad, Calif., USA). The amplification conditions for each PCR were 95°C for 5 min, followed by 35 cycles of: 95°C for 30 s, the specific annealing temperature for 30 s, and 72°C for 30 s, with a post-cycling final extension of 10 min at 72°C.
PCR products from genomic DNA were purified using the illustraTM GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, Buckinghamshire, UK), and both strands of the resultant products were sequenced using the BigDye® Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, Calif., USA) under standard conditions. The labeled products were subjected to electrophoresis on an ABI 3130 Genetic Analyzer sequencing system (Applied Biosystems). Electropherograms were analyzed using Sequence Analysis Software version 5.2 (Applied Biosystems) and compared to reference sequences using ChromasPro version 1.5 (Technelysium, South Brisbane, Qld., Australia). Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequences.
Genome-Wide Microarray Analysis
Genomic DNA from whole blood was extracted, amplified and purified. Following denaturation of probe DNA, hybridization used the Affymetrix Cytogenetics Whole-Genome 2.7 M Array at 50°C for 16-18 h, using the Human Mapping Cytogenetic 2.7 M assay kit and following the manufacturer's standard protocol. This higher resolution array contains approximately 400,000 SNP markers and 2.3 million nonpolymorphic markers, with high density coverage across cytogenetically significant regions. Data was collected using GeneChip® Scanner 3000 7G, and CEL files were analyzed using Affymetrix Chromosome Analysis Suite software (ChAS v.1.0.1). Initial analysis and quality assessment of the array data were performed with a Genotyping Console (Affymetrix). The median absolute pair-wise difference of each chip was used as a quality assessment of the array data. The median absolute pair-wise difference of this array was 0.27, which meets the QC criteria by Affymetrix. To minimize potential false positives from the signal-to-noise ratio, only copy number variants (CNVs) that involved at least 10 consecutive probe sets were considered, thus providing a median resolution of 7 kb.
The annotation file used in our analysis can be found on the Affymetrix website, listed as ArrayNA30.1 (hg18). Using previously described criteria for defining common variants [Qiao et al., 2010] additional CNVs detected with the high resolution array were compared to the Database of Genomic Variants (http://pro-jects.tcag.ca/variation) for overlap with CNVs in controls.
Real-Time PCR (RQ-PCR) Confirmation
Reactions were conducted in a 96-well plate with the ABI 7500 Sequence Detection System (Applied Biosystems). Amplicons were designed against the putative altered locus (HNF1A: forward primer: 5′-GTGTGGCGAAGATGGTCAAGT-3′, reverse primer: 5′-CTGGTTGAGGCCAGTGGTATC-3′) and against a control locus (β-actin) to control differences in genomic-DNA purity and concentration. PCR was performed in a 20-μl reaction volume containing 10 μl Power SYBR-GreenPCR Master Mix (Applied Biosystems), 10 pmol forward and reverse primers and 10 ng of genomic DNA. The reaction cycling conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and at 60°C for 1 min. Sequence Detection Software was used for exporting the threshold cycle data and further analyzing differences in threshold cycle values (ΔCt) between the test locus and the control locus.
Results
Fluorescent sequencing of the coding region of the patient's MECP2 gene showed no mutation, thus excluding Rett syndrome as the disease responsible for the symptoms.
Genome-wide microarray analysis showed a heterozygous 980-kb cryptic interstitial deletion of the long arm of chromosome 12 (fig. 2). The breakpoints of the deletion were within band 12q24.31, between 119,633,574 Mb and 120,613,673 Mb, and encompassed 820 SNP and CNV markers. Annotated genes in the deleted region are shown in figure 4.
Fig. 2.
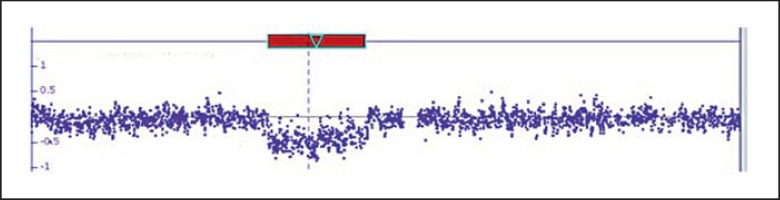
Schematic diagram of the sub-microscopic deletion of chromosome 12q24.31 detected by Affymetrix cytogenetic 2.7M arrays. DNA copy number within 12q24.31 region offset down from normal baseline indicates deletion detected by array.
Fig. 4.

Comparison between the genes deleted in the proband (ind3, red track) and the genes deleted in the patients described by Baple et al. [2010] (ind1, blue track) and Qiao et al. [2012] (ind2, brown track).
Sequencing of the coding region of ACADS gene, included in the deletion, in the patient showed no mutation.
The parents were also tested by genome-wide microarray analysis. The mother was normal, and the father presented the same 980-kb deletion with the same breakpoints found in the patient.
This 980-kb deletion was confirmed in the patient and parents by RQ-PCR (fig. 3). The mother gave normal results. The father and the affected child showed loss of 1 copy of HNF1A gene, indicating that the deletion in the patient was inherited from the father, as expected from the genome-wide microarray analysis.
Fig. 3.

Confirmation test results for HNF1A gene by RQ-PCR. The bars represent the relative quantification of each sample. F (father) and A (affected) are patients with HNF1A gene deletion, C1-6 are normal controls. There is a significant reduction in relative quantification by the patients in comparison with the normal controls (p < 0.05, by t test).
Discussion
We report herein on a 2.3-year-old girl with a microdeletion affecting the band 12q24.31. To date, only one patient with a similar deletion (fig. 4) has been described [Baple et al., 2010]. Both patients share a number of features: hypotonia, developmental delay, full cheeks, macroglossia, high-arched palate, and café-au-lait spots (table 1). Microcephaly is absent in the patient reported by Baple et al. [2010], while overgrowth and toe and finger anomalies are absent in the present case. Anxiety and autistic behavior have not been observed, but may appear later. Two other patients with larger and more distal deletions of 12q24.31 have been described [Sathya et al., 1999; Plotner et al., 2003]. They present additional heart and genital abnormalities (table 1), and one can speculate that genes responsible for these features may be located more distally from the deleted region described here. Exploring the presumed region in databases has not yet revealed any candidate genes related to these 2 defects.
Table 1.
Clinical comparison between patients with 12q deletions
| Baple et al., 2010 | Present patient | Qiao et al., 2012 | Sathya et al., 1999 | Plotner et al., 2003 | |
|---|---|---|---|---|---|
| Deletion | 12q24.31 (119,647,954–120,984,333) | 12q24.31 (119,633,574–120,613,673) | 12q24.31 (120,742,688–122,308,925) | 12q24.31q24.33 | 12q24.31q24.32 |
| Size of the deletion | between 1.58 and 2.53 Mb | 980 kb | 1566 kb | ||
| De novo deletion | yes | inherited from the father | yes | – | |
| Age at examination | 11 years | 27 months | 8 years 4 months | 9 months | 20 months |
| Healthy parents | yes | father: insulin-dependent diabetes at 14 years | yes | ||
| Mother pregnancy | sudden unexplained weight gain at 36 weeks | – | polyhydramnios at 31 weeks | cephalopelvic disproportion | |
| Weight at birth | 4.1 kg (90–95th centile) | 2.9 kg | 2.6 kg (45th centile) | 2.84 kg (10th centile) | |
| Weight | 11.8 kg at 1 year | 12.5 kg at 1.5 years (90th centile) | 5.25 kg at 3 months (25th centile) | ||
| Overgrowth despite dietary regimens | ++ at 11 years | 11.3 kg at 2.3 years (50th centile) | |||
| Microcephaly | – | ++ (42.5 cm at 2.3 years, <<3rd centile) | + (30.5 cm, <5th centile) | + (37 cm at 3 months, <5th centile) | |
| Hypoglycaemia | ++ immediately after birth | – | – | ||
| Glucose stabilisation | 4 years | ||||
| Hypotonia | + | + | + | ||
| Macroglossia | ++ | +/– | + | – | |
| Overgrowth | ++ | – | disproportionate growth failure | ||
| Developmental delay | ++ | ++ | ++ | ++ | ++ |
| Behavioural problems | + | ||||
| Developmental progress | very slow | very slow | slow | ||
| Walking | 18 months | few steps with support | few steps with support | ||
| Speaking | few words at 2 years | – | few words | ||
| Anxiety | ++ | – | |||
| Autistic spectrum disorder | + at 4 years | – | simplex | ||
| Urinary continence | + | – | |||
| Epilepsy (West syndrome) | – | + at birth | – | ||
| Constipation | ++ | ++ until 2 years | – | ||
| Upslanted palpebral fissures | + | – | + synophrys | short palpebral fissures, ptosis left eye | |
| Nose | broad nasal base | bulbous nasal tip | high nasal root with thick alae nasi and a square tip | short nose, anteverted nares | large bulbous nose, anteverted nares, smooth philtrum |
| Fontanelles | – | large anterior fontanelle | |||
| Full cheeks | + | + | + | + | |
| Palate | higly arched | +/– higly arched | narrow palate | high narrow palate with thick gums | |
| Overcrowded teeth | + | – | prominent front incisors | ||
| Full and everted lower lip | + | thin lower lip | |||
| Ears | large and narrow ears with a thick helix | narrow ear orifice | small, low-set and posteriorly rotated ears | normal ears | large prominent ears |
| Inverted nipples | + | – | |||
| Café-au-lait spot | 1 truncal | 1 back, 1 thigh, 2 buttock | 1 lower back | mongolian spot on the lower back | |
| Large hands and feet | + | – | bilateral pes planovalgus | ||
| Tapering of the fingers | + | – | ++ | + | |
| Toes | short toes | – | bilateral hypoplastic nails on both halluces | great toes dorsiflexed | |
| Fingers | left 4th finger proximally implanted | – | prominent fingertip pads | long fingers, patulous finger tips, long thumb | 5th finger clinodactyly |
| Bone | shortening of the 4th metacarpal bone | ||||
| Cardiac defects | – | – | – | + (PFO-PDA) | + (ASD-VSD-PFO-PDA) |
| Tracheomalacia | – | – | + | – | |
| Genital abnormalities | – | – | bifid scrotum, grade 2 hypospadias, chordee | sacral pit, large pubic fat pad |
– = Absent
+ = present
++ = strongly noted
+/– = weakly noted
PFO = patent foramen ovale
PDA = patent ductus arteriosus
ASD = atrial septal defect
VSD = ventricular septal defect.
The deletion identified in the patient reported here was inherited from her father. He was 2 m tall and had an insulin-dependent diabetes mellitus that appeared at age 14 years. No dysmorphic features were noted. The diabetes might be explained by the deletion of the entire coding sequence of the HNF1A gene. Indeed, heterozygous loss-of-function mutations in the HNFA1 gene are a common cause of monogenic diabetes diagnosed in adolescence and early adulthood [Ellard et al., 2007]. The absence of any other features in the father may be due to the presence of maternally imprinted genes in the deleted region or to a reduce penetrance of this microdeletion. Nevertheless, according to the current Catalogue of Parent of Origin Effects (http://www.otago.ac.nz/IGC) there are no imprinted genes identified in the 12q24.32 deleted region. More research is needed in order to resolve the cause of this phenomenon.
The deleted region comprises the gene ACADS that might be responsible for some of the clinical findings in the patient, such as the developmental delay and the presence of infantile spasms. Indeed, patients with homozygous mutations in ACADS (OMIM 606885) present with variable features including hypoglycaemia, developmental delay, seizures, and behavioral difficulties. In some patients, these symptoms are transient [Van Maldegem et al., 2006]. Sequencing of the coding region of ACADS gene in the patient did not reveal any sequence variation.
Other genes, that are more or less likely haploinsufficient, are found in the deleted region: 37% UNC119B, 24% ANAPC5, 34% RNF34, 44% P2RX4, and 18% CAMKK2 (fig. 5, DECIPHER: http://decipher.sanger.ac.uk/). UNC119B has unknown function, ANAPC5 controls cell cycle progression by targeting a number of cell cycle regulators [Jin et al., 2008]. RNF34 presents an anti-apoptotic function [Sasaki et al., 2002]. The product of P2RX4 and P2RX7 belongs to the family of purinoceptors for ATP [Yamamoto et al., 2000]. CAMKK2 plays a role in the calcium/calmodulin-dependent (CaM) kinase cascade by phosphorylating the downstream kinases [Hsu et al., 1998]. Unfortunately, none of these genes have been identified as responsible for a specific disease so far to potentially explain the developmental delay and the presence of infantile spasms in the patient.
Fig. 5.

Classification of the genes deleted in the region according to their haploinsufficiency, from unlikely haploinsufficient to likely haploinsufficient, according to DECIPHER (http://decipher.sanger.ac.uk/). High ranks (e.g. 0-10%) indicate that a gene is more likely to exhibit haploinsufficiency, low ranks (e.g. 90-100%) indicate a gene is more likely to not exhibit haploinsufficiency.
On the other hand, a regular follow-up of the patient is needed since the deletion of several genes is known to be involved in cancer development [Delpu et al., 2011].
In conclusion, we have further delineated the clinical aspect of the 12q24.31 deletion characterized by developmental delay, hypotonia, microcephaly, full cheeks, macroglossia, and the presence of café-au-lait spots. Patients with same deletion must be properly followed to prevent the misdeclaration of diabetes or any psychiatric feature. The presence of this deletion in a healthy father indicates the need to study a larger set of patients and normal control individuals with similar deletions in order to better understand the etiology and pathogenesis of such defects and also to offer appropriate genetic counseling.
Acknowledgement
We are grateful to the family for making this research possible.
References
- Baple E, Palmer R, Hennekam RC. A microdeletion at 12q24.31 can mimic Beckwith-Wiedemann syndrome neonatally. Mol Syndromol. 2010;1:42–45. doi: 10.1159/000275671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpu Y, Hanoun N, Lulka H, Sicard F, Selves J, et al. Genetic and epigenetic alterations in pancreatic carcinogenesis. Curr Genomics. 2011;12:15–24. doi: 10.2174/138920211794520132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellard S, Thomas K, Edghill EL, Owens M, Ambye L, et al. Partial and whole gene deletion mutations of the GCK and HNF1A genes in maturity-onset diabetes of the young. Diabetologia. 2007;50:2313–2317. doi: 10.1007/s00125-007-0798-6. [DOI] [PubMed] [Google Scholar]
- Hsu LS, Tsou AP, Chi CW, Lee CH, Chen JY. Cloning, expression and chromosomal localization of human Ca2+/calmodulin-dependent protein kinase kinase. J Biomed Sci. 1998;5:141–149. doi: 10.1007/BF02258368. [DOI] [PubMed] [Google Scholar]
- Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653–665. doi: 10.1016/j.cell.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinecke P, Meinecke R. Multiple malformation syndrome including cleft lip and palate and cardiac abnormalities due to an interstitial deletion of chromosome 12q. J Med Genet. 1987;24:187. doi: 10.1136/jmg.24.3.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotner PL, Smith JL, Northrup H. Deletion 12q: a second patient with 12q24.31q24.32 deletion. Am J Med Genet A. 2003;118A:350–352. doi: 10.1002/ajmg.a.10232. [DOI] [PubMed] [Google Scholar]
- Qiao Y, Harvard C, Tyson C, Liu X, Fawcett C, et al. Outcome of array CGH analysis for 255 subjects with intellectual disability and search for candidate genes using bioinformatics. Hum Genet. 2010;128:179–194. doi: 10.1007/s00439-010-0837-0. [DOI] [PubMed] [Google Scholar]
- Qiao Y, Tyson C, Hrynchak M, Lopez-Rangel E, Hildebrand J, et al. : Clinical application of 2.7M Cytogenetics array for CNV detection in subjects with idiopathic autism and/or intellectual disability. Clin Genet (2012), E-pub ahead of print. [DOI] [PubMed]
- Sasaki S, Nakamura T, Arakawa H, Mori M, Watanabe T, et al. Isolation and characterization of a novel gene, hRFI, preferentially expressed in esophageal cancer. Oncogene. 2002;21:5024–5030. doi: 10.1038/sj.onc.1205627. [DOI] [PubMed] [Google Scholar]
- Sathya P, Tomkins DJ, Freeman V, Paes B, Nowaczyk MJ. De novo deletion 12q: report of a patient with 12q24.31q24.33 deletion. Am J Med Genet. 1999;84:116–119. doi: 10.1002/(sici)1096-8628(19990521)84:2<116::aid-ajmg6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- van Maldegem BT, Duran M, Wanders RJ, Niezen-Koning KE, Hogeveen M, et al. Clinical, biochemical, and genetic heterogeneity in short-chain acyl-coenzyme A dehydrogenase deficiency. JAMA. 2006;296:943–952. doi: 10.1001/jama.296.8.943. [DOI] [PubMed] [Google Scholar]
- Watson MS, McAllister-Barton L, Mahoney MJ, Breg WR. Deletion (12)(q15q21.2) J Med Genet. 1989;26:343–344. doi: 10.1136/jmg.26.5.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Korenaga R, Kamiya A, Ando J. Fluid shear stress activates Ca(2+) influx into human endothelial cells via P2X4 purinoceptors. Circ Res. 2000;87:385–391. doi: 10.1161/01.res.87.5.385. [DOI] [PubMed] [Google Scholar]