

Abstract
Background and Purpose
Brain ischemia causes immediate and delayed cell death that is exacerbated by inflammation. Recent studies show that hypocretin-1/orexin-A (Hcrt-1) reduces ischemic brain injury, and Hcrt-positive neurons modulate infection-induced inflammation. Here we tested the hypothesis that Hcrt plays a protective role against ischemia by modulating inflammation.
Methods
Orexin/ataxin-3 (AT) mice, a transgenic strain in which Hcrt-producing neurons degenerate in early adulthood, and wild type (WT) mice were subjected to transient middle cerebral artery occlusion (MCAO). Infarct volume, neurological score, and spontaneous home cage activity were assessed. Inflammation was measured using immunohistochemistry, ELISA and assessment of cytokine mRNA levels.
Results
Infarct volumes 24 and 48 hours after MCAO were significantly larger, neurological score was worse, and spontaneous activity decreased in AT compared to WT mice.
Macrophage/microglial infiltration and myeloperoxidase-positive cells were higher in AT compared to WT mice. Pre-MCAO intracerebroventricular injection of Hcrt-1 significantly reduced infarct volume and macrophage/microglial infiltration in both genotypes, and improved neurological score in AT mice. Post-MCAO treatment decreased infarct size in both WT and AT mice, but had no effect on neurological score in either genotype. Microglia express the Hcrt-1 receptor following MCAO. TNFα production by LPS-stimulated microglial BV2 cells was significantly reduced by Hcrt-1 pretreatment. Sham AT mice exhibit increased brain TNFα and IL-6 mRNA, suggesting chronic inflammation.
Conclusion
Loss of Hcrt neurons in AT mice resulted in worsened stroke outcomes, which were reversed by administration of exogenous Hcrt-1. The mechanism underlying Hcrt-mediated neuroprotection includes attenuation of inflammatory responses following ischemic insult.
Keywords: Hypocretin, orexin, brain ischemia, inflammation, neurobehavior
Introduction
The hypocretin/orexin (Hcrt) neurons which produce Hcrt neuropeptides (Hcrt-1 and 2, i.e., orexin A and B) are localized in the hypothalamus.1,2 They project broadly throughout the brain and mediate many physiological functions, including wakefulness and sleep, energy homeostasis, glucose metabolism, autonomic function,2-11 and stress-adaptive responses such as stress-induced analgesia.12, 13 Loss of Hcrt neurons or dysfunction in the Hcrt system has been observed in several disorders, including narcolepsy14, 15 and subarachnoid hemorrhage.16 Recently, a few studies have indicated that the Hcrt system may be involved in cerebral ischemic injury. Increased expression of the Hcrt-1 receptor on neurons, astrocytes, and oligodendrocytes was observed 48 hours following mouse global ischemia,17 and in neurons 4-24 hours following permanent middle cerebral artery occlusion (MCAO) in rat.18 Moreover, intracerebroventricular administration of Hcrt-1 before MCAO in rat19, 20 and mouse21 decreased the infarct size. Separately, recent work has found that lipopolysaccharide-induced lethargy may be due in part to damage of Hcrt-positive neurons,22 suggesting that the Hcrt system may play a role in inflammatory processes.
Brain ischemia causes both immediate and delayed cell death and is accompanied by a robust inflammatory response that can exacerbate injury during reperfusion. In the present study we tested whether endogenous Hcrt-producing neurons promote neuroprotection following MCAO, and whether Hcrt-mediated protection is associated with modulation of the inflammatory response. Using the orexin/ataxin-3 mice (AT) in which the hypocretin/orexin neurons degenerate during early adulthood, we performed transient focal ischemia on wild type (WT) and AT mice, and found that AT mice had larger infarcts, greater behavioral deficits, and increased microglial activation compared to WT mice. mRNA analysis revealed higher levels of both TNFα and IL-6 in AT mice, suggesting a chronic inflammatory state in this genotype. Importantly, administration to the brain of Hcrt-1 pre- or post-MCAO decreased infarct size in both WT and AT mice, suggesting effects in AT mice are likely a direct result of loss of Hcrt neuropeptides. In vitro experiments support an anti-inflammatory effect of Hcrt that may contribute to its neuroprotection.
Materials and methods
Animals
Adult male (wildtype C57/BC6 and orexin/ataxin-3 mice, 3-5 months old, 25-35g) were used. While AT mice are normal during early development, the strain has Hcrt-specific expression of ataxin-3, a disease protein that results in gradual degeneration of Hcrt-expressing neurons that is completed by 3 months of age.23 Details of strain production and animal care can be found in Supplemental Materials and Methods.
Power analysis for a 30% change in infarct size showed that 11 animals/group were needed for p<0.05, power 80%. Mice that died before perfusion, exhibited brain hemorrhage, or were surgical failures indicated by a fully normal neuroscore of 0 were excluded from analysis. Mice were randomized by being chosen by a blinded experimenter. Numbers of mice excluded and exclusion reasons for each experiment are indicated in Supplemental Table 3.
Focal cerebral ischemia
Anesthesia was induced with 4% isoflurane and maintained by 1.5-2% isoflurane in 70% air and balanced oxygen by a facemask. Rectal temperature was maintained at 37 ± 0.5° C with a heating pad (Harvard Apparatus, Hollister, MA). Transient focal ischemia was induced by middle cerebral artery occlusion (MCAO) for 60 min, which generates infarction in both cortex and striatum, as previously described.24-26 Details of surgery protocol can be found in Supplemental Materials and Methods. The surgeon was blinded to genotype and experimental treatment.
Behavioral Testing
Neurological score was evaluated 24 and 48 hours after MCAO according to a neurological grading score,25, 26 from 0 (no observable neurological deficit) to 4 (unable to walk spontaneously and a depressed level of consciousness). The evaluator was blinded to genotypes and experimental treatment. The SmartCage™ system (AfaSci, Inc., Redwood City, CA) was used for automated analysis of spontaneous activity as described previously.26, 27 The homecage activity variables (locomotion, travel distance, velocity, and rear-ups) were determined by photo-beam breaks and automatically analyzed using CageScore™ software (AfaSci, Inc.). Mice were assessed continuously for 30 min during the light phase, 24 and 48 hours following reperfusion.
Measurement of cerebral infarction area
Twenty-four or 48 hours after MCAO and immediately following neuroscore assessment, mice were anesthetized with isoflurane and decapitated. Brains were removed and sectioned coronally with a rodent brain slicer matrix (Zivic Instruments, Pittsburgh, PA). Sections were incubated in 2% 2,3,5-triphenyletrazolium chloride (TTC, #T8877, Sigma-Aldrich, St. Louis, MO) and infarction core volume as defined by an absence of TTC staining (percent of hemispheric volume) was determined by a blinded observer using 4 sections per brain and corrected for edema using the NIH ImageJ program (Image J 1.37v, Wayne Rasband, NIH) as described previously.25, 26
Immunofluorescence
Ischemic or sham-operated mice were euthanized with an overdose of isoflurane and perfused with ice-cold phosphate-buffered saline (PBS; pH 7.4) 48 hours after MCAO, followed by 4% paraformaldehyde in PBS as previously described.28 Brains were removed and post-fixed for 72 hours in 4% paraformaldehyde in PBS and cut into 50 μm coronal sections. Details of the immunofluorescence protocol, including antibodies used and cell counting protocol, can be found in Supplemental Materials and Methods.
Reverse Transcription Quantitative real-time Polymerase Chain Reaction (RT-qPCR) for mRNA quantitation
Total RNA was isolated with TRIzol® (Invitrogen) from the ischemic hemisphere (from +0.8 to -1.2mm relative to bregma) of WT or AT mice 4 hours following MCAO. Reverse transcription was performed using the TaqMan MicroRNA Reverse Transcription Kit according to manufacturer's instructions (Applied Biosystems). Predesigned primer/probes (Applied Biosystems) for mRNAs and GAPDH were also from Applied Biosystems. The expression of mRNAs was normalized using GAPDH as the internal control. Measurements were normalized to GAPDH (ΔCt) and the comparison calculated as the inverse log of ΔΔCt to give relative fold change value.
Treatment with recombinant Hcrt-1 in vivo
Hcrt-1 was injected intracerebroventricularly (icv) as previously described.26 Two μl of either vehicle (0.1% bovine serum albumin in 0.9% PBS) or containing 2nmol of Hcrt-1 dissolved in the vehicle was infused over 10 min into the left lateral ventricle 30 min before or after MCAO. After 48 hours of reperfusion neurological score was determined, animals were sacrificed, and brains removed for TTC staining, as described above.
Measurement of TNFα production by BV2 cells
BV2 microglial cells were treated with 10 ng/ml lipopolysaccharide (LPS, Sigma) for 24 hours and fixed with 4% paraformaldehyde. Details of immunocytochemistry protocol, including quantification, can be found in Supplemental Materials and Methods.
BV2 cells were treated with control media or media containing Hcrt-1 (100 nM) for 1 hour prior to treatment with LPS (10 ng/ml). Four hours following LPS treatment, supernatant was collected and TNFα measured using the TNFα Mouse ELISA Kit (Life Technologies). Cell number was assessed by DAPI staining and counting with NIH ImageJ. Fluorescence images were acquired at 2.5× magnification. TNFα measurements were normalized to cell number.
Statistical Analyses
Data is expressed as mean ± SEM. Differences were considered statistically significant for p <0.05. Student's t-tests were used when two groups were compared. Two-way ANOVAs were used when both genotype and treatment were taken into account, followed by Bonferroni post-tests using Prism 5 (GraphPAD Software for Science, San Diego, CA). All assessments were by blinded observers. Power analysis was completed using the POWER procedure in SAS 9.3 (Cary, NC).
Results
Infarction volume and neurological deficits are increased in AT mice
Infarct volumes at 24 and 48 hours post-MCAO were significantly larger (Figure 1A,B) and neurological score was significantly worse at 24 hours in AT compared to WT mice (Figure 1C). Physiological variables were not significantly different between WT and AT mice before, during MCAO, or 10 min after reperfusion (Supplemental Table 1).
Fig. 1.
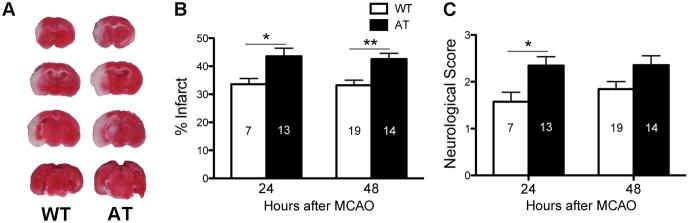
Infarct volume and neurological score are increased in AT mice. (A) Representative TTC-stained coronal sections showing infarcts in WT (left) and AT (right) mice. (B) Quantification of infarct volume expressed as a percent of hemispheric volume at 24 and 48 hours. (C) Neuroscore was assessed 24 and 48 hours after MCAO. Numbers in bars represent n/group. Two-way ANOVA revealed a significant genotype effect for both infarct size and neuroscore. Post-hoc tests: *P <0.05, **P<0.01 compared to WT.
Spontaneous locomotor activity is reduced in AT mice after MCAO
Twenty-four and 48 hours after surgery, spontaneous activity was monitored using the SmartCage™ system. AT mice exhibited decreased activity during the dark phase, but did not differ from WT during light phase (our unpublished data). Consistent with this, light phase activity measurements showed no difference between sham WT and AT mice. Following MCAO AT mice exhibited more profound and significant reductions in active time (Figure 2A) and distance traveled (Figure 2B) when compared to WT mice 24 and 48 hours post MCAO. AT mice also exhibited a significant decrease in rearing activity, indicative of reduced exploration, compared to WT mice (Figure 2C). AT and WT mice had similar average velocities before and after MCAO (Figure 2D). Together, these results are consistent with the differences in neurological scores and infarct volumes observed between the genotypes (Figure 1).
Fig. 2.
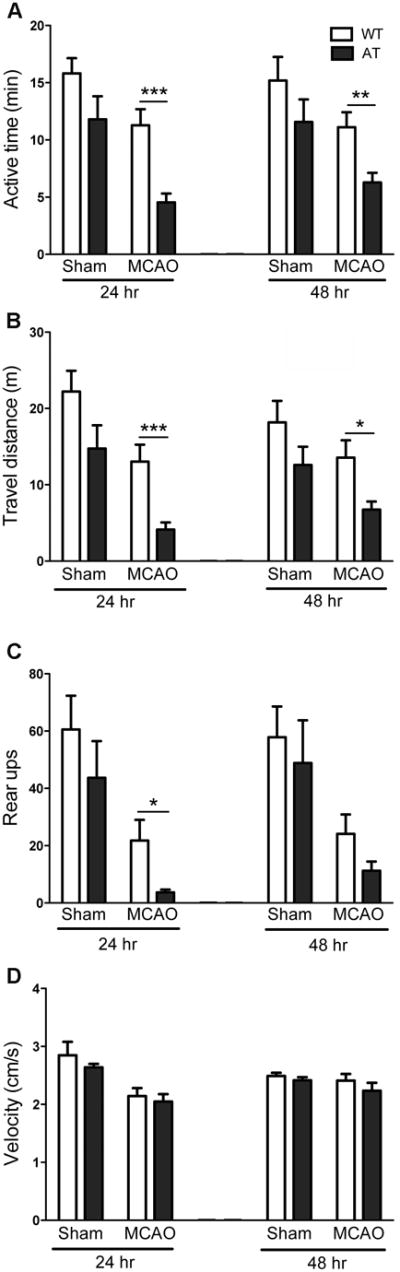
AT mice have reduced light-phase spontaneous activity after MCAO. AT mice showed significantly greater reductions in active time (A), travel distance (B), and rear up counts (C), but no significant difference in average velocity compared to WT (D). For all panels, Sham n=5-7/group, MCAO n=18-22/group. Two-way ANOVA revealed significant genotype and surgery (Sham vs. MCAO) differences for active time (24 and 48 hours), travel distance (24 and 48 hours), and rear ups (24 hours, 48 hours only surgery effect). Two-way ANOVA revealed a significant surgery effect in average velocity at 24h. Post hoc tests: *P < 0.05, **P < 0.01, ***P < 0.001 compared to WT.
AT mice exhibit increased macrophage/microglia and neutrophil infiltration after MCAO
Infiltration of macrophages and neutrophils is prominent following MCAO.29 Morphometric analysis revealed that the total number of activated macrophages/microglia significantly increased in the ischemic core (IC) of AT compared to WT mice (Figure 3A,B). However, no significant differences were observed in the cortical penumbra (WT = 36.8 ± 4.3 vs AT = 39.0 ± 2.0, P = 0.65). The increased number of activated macrophages/microglia in the IC was associated with significantly increased infiltration of leukocytes, detected as MPO-positive cells (Figure 3A,C). MPO positive cells were restricted to the IC.
Fig. 3.
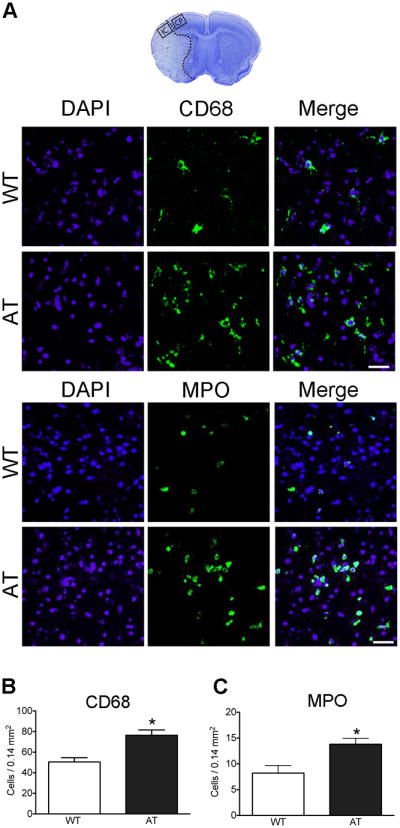
AT mice exhibit increased numbers of activated macrophages/microglia and MPO-positive cells after MCAO. (A) The top image shows a representative coronal brain section with cresyl violet staining on which the squares represent the area where pictures of immunostaining were taken and cells were counted (IC = ischemic core, CP = cortical penumbra). The middle panels are representative immunofluorescence images of CD68-stained, and the bottom panels of MPO-stained, sections counterstained with DAPI 48 hours after stroke in the ischemic core. (B-D) Quantification of CD68- (B) and MPO- (C) positive cells in IC. n=5/group. Bar=50μm. * P<0.05 vs. WT.
TNFα and IL-6 mRNA are increased in sham AT compared to WT mice
To assess levels of inflammatory cytokines acutely after sham and MCAO surgery (4 hours),30 RT-qPCR was used to measure Ccl2, Ccl3, IL-10, IL-1α, IL-1β, IL-6, and TNFα. Following MCAO, these cytokines all markedly increased compared to sham, but there were no significant differences between genotypes (Supplemental Table 2). Both IL-6 and TNFα were found to be significantly higher in sham AT compared to sham WT (Table 1).
Table 1.
TNFα and IL-6 mRNA values in sham and MCAO WT and AT mice 4 hours following surgery.
| Sham TNFα | MCAO TNFα | Sham IL-6 | MCAO IL-6 | |
|---|---|---|---|---|
| WT | 1.10 ± 0.25 | 42.97 ± 13.99 | 1.04 ± 0.14 | 10.04 ± 2.36 |
| AT | 2.19 ± 0.35* | 34.81 ± 11.90 | 1.58 ± 0.13* | 18.67 ± 6.58 |
Values shown are mean ± SEM, normalized to sham WT levels. n=3-5/group.
P < 0.05 compared to WT.
Hcrt-1 decreases infarct volume and inflammation
Hcrt-1 administered either 30 min prior to or 30 min after MCAO significantly reduced infarct volume in WT and AT mice 48 hours after reperfusion (Figure 4A,B). While Hcrt-1 pretreated AT mice showed a significantly improved neurological score, pre- or post-treatment had no effect on neurological score of WT mice (Figure 4A,B). Hcrt-1 administration also decreased CD68-positive cells in the IC but did not change the number of CD68+ cells in the cortical penumbra (Figure 4C).
Fig. 4.

Administration of Hcrt-1 before or after MCAO protects both WT and AT mice. (A) Infarct volumes in WT mice decreased with Hcrt-1 treatment 30 min prior or after MCAO compared to vehicle groups, n=6-15/group, but there was no improvement in neuroscore at 48 hr. (B) Infarct volumes in AT mice decreased with Hcrt-1 treatment 30 min or after MCAO compared to vehicle groups, neuroscore was only decreased by Hcrt-1 pretreatment compared to vehicle in AT mice. (C) Hcrt-1 decreased CD68+ cells in the ischemic core in WT and AT mice compared to vehicle, but did not change counts in the penumbra in either genotype, n=3-6/group. *P < 0.05, **P < 0.01, ***P < 0.001 compared to Vehicle (Veh).
Hcrt-1 attenuates microglial TNFα production
Immunostaining of brains 48 hours after MCAO demonstrated that the only cells expressing Hcrt-1R were CD68+ microglia in the ischemic penumbra (Figure 5A) with little to no expression in the infarct core. GFAP+ astrocytes (Figure 5A) and neurons (data not shown) exhibited no detectable expression. To further investigate effects of Hcrt-1 on microglial response, we measured TNFα levels in response to LPS, an inducer of inflammation. LPS exposure significantly increased expression of Hcrt-1R on BV2 microglial cells (Figure 5B,C). Untreated BV2 cells express TNFα about the detection limit of our method, 2-5 pg/ml, while LPS treatment induced a very large increase in TNFα production, approximately 300-fold. When the cells were treated with Hcrt-1 1 hour prior to LPS stimulation TNFα production was significantly reduced approximately 15% (Figure 5D).
Fig. 5.
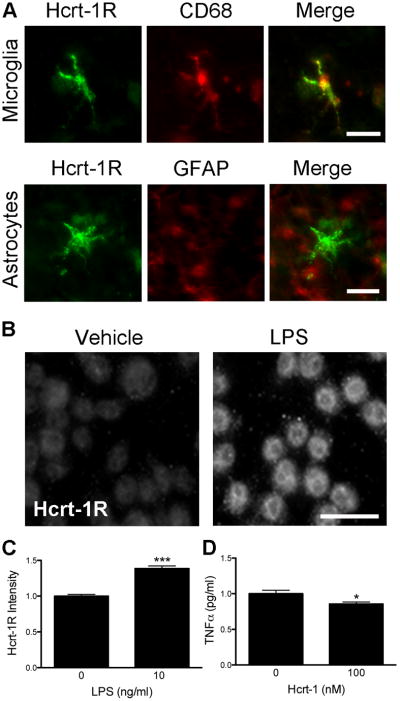
The Hcrt system affects inflammation. (A) The Hcrt-1R receptor is expressed on CD68-positive microglia (top panel), but not on GFAP-positive astrocytes (bottom panel) 48 hours after stroke. Bar=25μm. (B-C) Treatment of BV2 cells with LPS increased Hcrt receptor expression. (B) Fluorescence micrographs representative of cells treated with vehicle (left) or LPS (right) then immunolabeled for Hcrt-1R. Bar=25μm. (C) Quantification of Hcrt-1R immunostaining shows a significant increase following LPS treatment. n=5/group. (D) Hcrt-1 treatment (100 nM) significantly reduced the production of TNFα in BV2 cells stimulated by LPS. Values normalized to LPS treatment without Hcrt-1. n=16/group. * P < 0.05, *** P < 0.001.
Discussion
In the present study using the transgenic AT mice, which develop normally but exhibit degeneration of Hcrt neurons in young adulthood, we found worsened outcome following experimental stroke. Increased infarct size correlated with more severe neurobehavioral deficits in the AT mice, by both standard neurological scoring26 and automated quantitation of spontaneous activity. Because the velocity of the mice did not differ between genotypes, it is unlikely that the deficits in active time, distance traveled and rearing activity are due to physical impairment in the AT mice. Instead, this likely reflects reduced alertness or neuropsychological impairment in the AT mice. Administration of Hcrt-1 reduces infarct size in both AT and WT mice, consistent with previous reports.21,20 Importantly, we report here that Hcrt-1 treatment after MCAO effectively reduces infarct volumes. Improved neurobehavioral function was only seen in pretreated AT mice, suggesting that release of endogenous Hcrt during brain injury and reperfusion might reach a level to produce maximal functional improvement in WT mice under our experimental conditions, or that sensitivity to detect differences is reduced at 48 hours. Although our results are promising, lack of neuroscore improvement after post-treatment suggests that additional work is needed to optimize post-treatment and further assess whether Hcrt-1 treatment could be a potential therapy following stroke. A further limitation of this study is the lack of activity assessment after Hcrt-1 treatment.
Thus far, a few potential mechanisms of Hcrt-induced protection against ischemia have been proposed. Administration of Hcrt restored hepatic and skeletal insulin receptor levels close to sham, and decreased post-ischemic glucose intolerance that leads to neuronal death.21 In addition, Hcrt-1 was shown to increase levels of protective hypoxia-induced factor-1α.20 Moreover, a study of gastrointestinal ischemia/reperfusion found that exogenous Hcrt-1 resulted in decreased lipid peroxidation and MPO+ cells,31 in agreement with our results showing decreased neutrophils in the cortical ischemic core with Hcrt-1 treatment.
In our study CD68+ microglia in the cortical penumbra were the predominant cells expressing the Hcrt-1R, 48 hours after MCAO. This is in contrast to a previous study showing that neurons and some glial cells expressed the Hcrt-1R 24 hours after permanent MCAO in rat.18 These discrepancies may be due to differences in time course of expression, species, or antibodies used.
In light of our immunohistochemical findings, including a marked increase in activated microglia in the ischemic core in the AT mice, we hypothesized that endogenous Hcrt-1 may regulate acute inflammation, thereby contributing to its neuronal protective properties. Indeed, microglial BV2 cells pretreated with Hcrt-1 exhibited decreased LPS-induced TNFα production. These data, along with the reduction of MPO- and CD68-cell counts with Hcrt-1 treatment in vivo, suggest that Hcrt-1 can be anti-inflammatory, which may complement other postulated neuroprotective mechanisms mentioned above.20, 21
Currently, the mechanism by which Hcrt-1 affects inflammation is unknown. Although previous studies have shown that inflammatory agents, such as LPS, decrease the activity of Hcrt-positive neurons in the hypothalamus,22 TNFα-R-deficient mice have increased expression of Hcrt mRNA,32 and treatment of B35 neuroblastoma cells with TNFα decreases Hcrt precursor half-life.32 The present study is the first to directly assess the effects of Hcrt-1 on inflammatory responses following cerebral ischemia. One possible mechanism underlying Hcrt modulation of inflammation is via Hcrt's antioxidant effects.31 Recent work has suggested that reactive oxygen species can directly induce pro-inflammatory cytokine production.33 Also, Hcrt increases insulin receptor expression.21 Insulin has been identified as an anti-inflammatory mediator,34 suggesting that increases in insulin sensitivity by Hcrt may be partially responsible for Hcrt's anti-inflammatory actions. Although there is no previous documentation of Hcrt-regulated changes in toll-like receptors or pro-inflammatory signaling, these are potential future directions to explore.
In conclusion, we have shown that the endogenous Hcrt system is protective against transient MCAO in mice, in part likely attributed to Hcrt-1-mediated anti-inflammatory actions. Our findings are consistent with previous studies suggesting that Hcrt can be a potentially useful therapeutic or element of a protective cocktail to reduce stroke-induced brain damage, even if given during reperfusion, and suggest further studies are needed to understand how Hcrt attenuates inflammation following ischemia.
Supplementary Material
Acknowledgments
We are grateful to Ms. T.S. Chen, Dr. S. Black and Dr. T. Kilduff, at SRI International for breeding and genotyping the AT and WT mice used in this study, and Dr. M. Zheng for help with statistics.
Sources of Funding: This work was supported by NIH grants R01MH078194, R43MH076309, R43NS065555, and R43 NS073311 to X Xie, R01 GM49831 to RGG and T32 GM089626 to REW.
Footnotes
Conflict of interest: X. Xie is the founder and a stockholder of AfaSci, Inc. All other coauthors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: A family of hypothalamic neuropeptides and g protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 2.de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, et al. The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakurai T. The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–181. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- 4.Shiuchi T, Haque MS, Okamoto S, Inoue T, Kageyama H, Lee S, et al. Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle via sympathetic nervous system. Cell Metab. 2009;10:466–480. doi: 10.1016/j.cmet.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Nishino S, Kanbayashi T. Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications in the hypothalamic hypocretin/orexin system. Sleep Med Rev. 2005;9:269–310. doi: 10.1016/j.smrv.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Culebras A. Cerebrovascular disease and sleep. Curr Neurol Neurosci Rep. 2004;4:164–169. doi: 10.1007/s11910-004-0032-6. [DOI] [PubMed] [Google Scholar]
- 7.Rousseaux M, Muller P, Gahide I, Mottin Y, Romon M. Disorders of smell, taste, and food intake in a patient with a dorsomedial thalamic infarct. Stroke. 1996;27:2328–2330. doi: 10.1161/01.str.27.12.2328. [DOI] [PubMed] [Google Scholar]
- 8.Espiner EA, Leikis R, Ferch RD, MacFarlane MR, Bonkowski JA, Frampton CM, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002;56:629–635. doi: 10.1046/j.1365-2265.2002.01285.x. [DOI] [PubMed] [Google Scholar]
- 9.Payne RS, Tseng MT, Schurr A. The glucose paradox of cerebral ischemia: Evidence for corticosterone involvement. Brain Res. 2003;971:9–17. doi: 10.1016/s0006-8993(03)02276-5. [DOI] [PubMed] [Google Scholar]
- 10.Ohno K, Sakurai T. Orexin neuronal circuitry: Role in the regulation of sleep and wakefulness. Front Neuroendocrinol. 2008;29:70–87. doi: 10.1016/j.yfrne.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Xie X, Crowder TL, Yamanaka A, Morairty SR, Lewinter RD, Sakurai T, et al. Gaba(b) receptor-mediated modulation of hypocretin/orexin neurones in mouse hypothalamus. J Physiol. 2006;574:399–414. doi: 10.1113/jphysiol.2006.108266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerashchenko D, Horvath TL, Xie XS. Direct inhibition of hypocretin/orexin neurons in the lateral hypothalamus by nociceptin/orphanin fq blocks stress-induced analgesia in rats. Neuropharmacology. 2011;60:543–549. doi: 10.1016/j.neuropharm.2010.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie X, Wisor JP, Hara J, Crowder TL, LeWinter R, Khroyan TV, et al. Hypocretin/orexin and nociceptin/orphanin fq coordinately regulate analgesia in a mouse model of stress-induced analgesia. J Clin Invest. 2008;118:2471–2481. doi: 10.1172/JCI35115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 16.Dohi K, Ripley B, Fujiki N, Ohtaki H, Shioda S, Aruga T, et al. Csf hypocretin-1/orexin-a concentrations in patients with subarachnoid hemorrhage (sah) Peptides. 2005;26:2339–2343. doi: 10.1016/j.peptides.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Nakamachi T, Endo S, Ohtaki H, Yin L, Kenji D, Kudo Y, et al. Orexin-1 receptor expression after global ischemia in mice. Regul Pept. 2005;126:49–54. doi: 10.1016/j.regpep.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 18.Irving EA, Harrison DC, Babbs AJ, Mayes AC, Campbell CA, Hunter AJ, et al. Increased cortical expression of the orexin-1 receptor following permanent middle cerebral artery occlusion in the rat. Neurosci Lett. 2002;324:53–56. doi: 10.1016/s0304-3940(02)00176-3. [DOI] [PubMed] [Google Scholar]
- 19.Kitamura E, Hamada J, Kanazawa N, Yonekura J, Masuda R, Sakai F, et al. The effect of orexin-a on the pathological mechanism in the rat focal cerebral ischemia. Neurosci Res. 2010;68:154–157. doi: 10.1016/j.neures.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 20.Yuan LB, Dong HL, Zhang HP, Zhao RN, Gong G, Chen XM, et al. Neuroprotective effect of orexin-a is mediated by an increase of hypoxia-inducible factor-1 activity in rat. Anesthesiology. 2011;114:340–354. doi: 10.1097/ALN.0b013e318206ff6f. [DOI] [PubMed] [Google Scholar]
- 21.Harada S, Fujita-Hamabe W, Tokuyama S. Effect of orexin-a on post-ischemic glucose intolerance and neuronal damage. J Pharmacol Sci. 2011;115:155–163. doi: 10.1254/jphs.10264fp. [DOI] [PubMed] [Google Scholar]
- 22.Grossberg AJ, Zhu X, Leinninger GM, Levasseur PR, Braun TP, Myers MG, Jr, et al. Inflammation-induced lethargy is mediated by suppression of orexin neuron activity. J Neurosci. 2011;31:11376–11386. doi: 10.1523/JNEUROSCI.2311-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- 24.Xu L, Dayal M, Ouyang YB, Sun Y, Yang CF, Frydman J, et al. Chaperonin groel and its mutant d87k protect from ischemia in vivo and in vitro. Neurobiol Aging. 2006;27:562–569. doi: 10.1016/j.neurobiolaging.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 25.Han RQ, Ouyang YB, Xu L, Agrawal R, Patterson AJ, Giffard RG. Postischemic brain injury is attenuated in mice lacking the beta2-adrenergic receptor. Anesth Analg. 2009;108:280–287. doi: 10.1213/ane.0b013e318187ba6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42:2026–2032. doi: 10.1161/STROKEAHA.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khroyan TV, Zhang J, Yang L, Zou B, Xie J, Pascual C, et al. Rodent motor and neuropsychological behavior measured in home cages using the integrated modular platform - smartcage(tm) Clin Exp Pharmacol Physiol. 2012;39:614–622. doi: 10.1111/j.1440-1681.2012.05719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG. Overexpression of mitochondrial hsp70/hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab. 2009;29:365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998;56:149–171. doi: 10.1016/s0301-0082(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 30.Sieber MW, Claus RA, Witte OW, Frahm C. Attenuated inflammatory response in aged mice brains following stroke. PLoS One. 2011;6:e26288. doi: 10.1371/journal.pone.0026288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulbul M, Tan R, Gemici B, Ongut G, Izgut-Uysal VN. Effect of orexin-a on ischemia-reperfusion-induced gastric damage in rats. J Gastroenterol. 2008;43:202–207. doi: 10.1007/s00535-007-2148-3. [DOI] [PubMed] [Google Scholar]
- 32.Kapas L, Bohnet SG, Traynor TR, Majde JA, Szentirmai E, Magrath P, et al. Spontaneous and influenza virus-induced sleep are altered in tnf-alpha double-receptor deficient mice. J Appl Physiol. 2008;105:1187–1198. doi: 10.1152/japplphysiol.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyun E, Ramachandran R, Hollenberg MD, Vergnolle N. Mechanisms behind the anti-inflammatory actions of insulin. Crit Rev Immunol. 2011;31:307–340. doi: 10.1615/critrevimmunol.v31.i4.30. [DOI] [PubMed] [Google Scholar]
- 35.Barreto GE, Sun X, Xu L, Giffard RG. Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS One. 2011;6:e27881. doi: 10.1371/journal.pone.0027881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.