

Conditionally expressed progesterone receptor isoforms PRA and PRB enhance breast cancer cell migration through interaction with focal adhesion kinase (FAK) and differential regulation of FAK phosphorylation and turnover. PRB-stimulated migration is reduced by progestins, which is prevented by PR antagonists or agonist-bound PRA.
Abstract
Progesterone receptor (PR) and progestins affect mammary tumorigenesis; however, the relative contributions of PR isoforms A and B (PRA and PRB, respectively) in cancer cell migration remains elusive. By using a bi-inducible MDA-MB-231 breast cancer cell line expressing PRA and/or PRB, we analyzed the effect of conditional PR isoform expression. Surprisingly, unliganded PRB but not PRA strongly enhanced cell migration as compared with PR(–) cells. 17,21-Dimethyl-19-norpregna-4,9-dien-3,20-dione (R5020) progestin limited this effect and was counteracted by the antagonist 11β-(4-dimethylamino)phenyl-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one (RU486). Of importance, PRA coexpression potentiated PRB-mediated migration, whereas PRA alone was ineffective. PR isoforms differentially regulated expressions of major players of cell migration, such as urokinase plasminogen activator (uPA), its inhibitor plasminogen activator inhibitor type 1, uPA receptor (uPAR), and β1-integrin, which affect focal adhesion kinase (FAK) signaling. Moreover, unliganded PRB but not PRA enhanced FAK Tyr397 phosphorylation and colocalized with activated FAK in cell protrusions. Because PRB, as well as PRA, coimmunoprecipitated with FAK, both isoforms can interact with FAK complexes, depending on their respective nucleocytoplasmic trafficking. In addition, FAK degradation was coupled to R5020-dependent turnovers of PRA and PRB. Such an effect of PRB/PRA expression on FAK signaling might thus affect adhesion/motility, underscoring the implication of PR isoforms in breast cancer invasiveness and metastatic evolution with underlying therapeutic outcomes.
INTRODUCTION
Human progesterone receptor (PR) is a crucial transcription factor involved in development and differentiation of female reproductive tissue. It is expressed from a single gene as two isoforms, PRA (94 kDa) and PRB (116 kDa), at similar level, PRA being truncated for the 164 N-terminal amino acids of PRB. On hormone binding, PRA and PRB homodimers or heterodimers exhibit distinct transcriptional regulatory functions by targeting various subsets of genes (Graham et al., 2005; Jacobsen et al., 2005; Leo et al., 2005; Khan et al., 2012). However, unliganded PR is transcriptionally activated by growth factor stimuli irrespective of hormone status (Labriola et al., 2003). PRA is mainly localized in the nucleus, whereas PRB continuously shuttles between nuclear and cytoplasmic compartments (Guiochon-Mantel et al., 1994; Boonyaratanakornkit et al., 2008). Thus PRB can mediate either direct transcriptional events or rapid cytoplasmic changes by interacting with nonnuclear signaling pathways, such as mitogen-activated protein kinase (MAPK), c-Src, PI3K/Akt, and Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3; Migliaccio et al., 1998; Proietti et al., 2005; Boonyaratanakornkit et al., 2007; Hammes and Levin, 2007; Fu et al., 2008b). Therefore it is likely that both isoforms elicit distinct and coordinated functions in the two compartments through dynamic processes.
Association of estrogens and progestins has been shown to increase breast cancer risk factor in long-term hormone replacement therapy patients (Rossouw et al., 2002; Chlebowski et al., 2003, 2010). Moreover, alteration of PRA/PRB expression ratio is frequently observed in breast cancer cells (Graham et al., 1995; Mote et al., 2002). Elevated PRA expression (McGowan and Clarke, 1999; Bagheri-Yarmand et al., 2004), as well as loss of PR expression (Bogina et al., 2011), is generally associated with poor prognosis. PR also has been implicated in breast cancer metastatic progression through unclear molecular mechanisms (Weigelt et al., 2005). Although most breast cancer metastases lack estrogen receptor (ER) and PR expression, PR has been reported to facilitate metastasis evolution by increasing invasiveness of primary cancer cells through transcriptional regulation of key proteins involved in cellular migration and adhesion, such as matrix metalloproteases (MMPs), vascular endothelial growth factor, the plasminogen activator (PA) system (Kato et al., 2005; Carnevale et al., 2007), and focal adhesion kinase (FAK; Fu et al., 2010).
The metastatic process mainly results from alterations in cell migration and invasiveness through multiple signaling cross-talks such as extracellular matrix (ECM) degradation and cell adhesion/deadhesion dynamics (Lauffenburger and Horwitz, 1996; Chiang and Massague, 2008; May et al., 2011; Rosenthal et al., 2011). In this regard, the PA system is strongly activated in aggressive tumor cells and is frequently involved in the development of metastatic phenotype (Chapman, 1997; Bernard-Trifilo et al., 2006; Mitra et al., 2006; Wei et al., 2007; Michael et al., 2009; Smith and Marshall, 2010). Urokinase plasminogen activator (uPA), the major actor of the PA system, is a serine protease targeting plasminogen and several MMPs, leading to the remodeling of ECM and activating cell migration. In addition, uPA binds to the membrane-anchored uPA receptor (uPAR), which exhibits multiple functions in proliferation, migration, and adhesion-dependent signal transduction. This receptor is coupled to signaling factors such as integrins, c-Src kinases, FAK, platelet-derived growth factor receptor, G protein–coupled receptor, and JAK/STAT (Busso et al., 1994; Stahl and Mueller, 1994; Wei et al., 1996; Chapman, 1997; Bernard-Trifilo et al., 2006; Mitra et al., 2006; Mitra and Schlaepfer, 2006; Wei et al., 2007; Michael et al., 2009; Smith and Marshall, 2010). Moreover, after binding to uPA, the plasminogen activator inhibitor type 1 (PAI-1) inhibits uPA serine protease activity and thus ECM proteolysis. Despite this function, PAI-1 is also able to promote cell migration (Chazaud et al., 2002; Czekay and Loskutoff, 2004; Providence and Higgins, 2004; Wilkins-Port et al., 2007; Fabre-Guillevin et al., 2008), as well as proliferation processes (Olson et al., 1992; Schneider et al., 2000; McMahon et al., 2001; Jo et al., 2005), via multiple interactions with ECM components such as low-density lipoprotein receptor–related protein (LRP) and integrins. Both PAI-1 and uPA have been validated as major prognostic factors for breast cancer evolution (Janicke et al., 2001; Harbeck et al., 2004; Sakakibara et al., 2004; Biermann et al., 2008).
Interaction of uPA-bound uPAR with β1-integrin triggers clustering of the complex, leading to autophosphorylation of FAK (Tang et al., 1998; Bernard-Trifilo et al., 2006; Mitra and Schlaepfer, 2006; Lim et al., 2008). This tyrosine kinase functions as an integrator of multiple signaling pathways, promoting formation and turnover of focal adhesion points, membrane ruffling, and cell shape and cytoskeletal reorganization involved in cell motility processes (Luo and Guan, 2010; Schaller, 2010). Under the control of extracellular signaling mediated by integrins and other cell surface receptors, FAK autophosphorylation on the Tyr-397 residue (Y397) creates a binding site for SH-2–containing proteins such as Src kinase. This association leads to subsequent phosphorylations of FAK, maximizing its activation (Luo and Guan, 2010). FAK Y397–dependent activation is strongly enhanced in metastatic cancer cells as compared with primary breast tumors (Sood et al., 2004). Moreover, knockdown of FAK (Ilic et al., 1995) or mutation on several phosphorylation sites results in a strong decrease in cancer cell motility (Luo and Guan, 2010).
Various studies highlighted the possible effect of progestins and PR on such signaling pathways regulating cell migration (Marbaix et al., 1992; Lin et al., 2000, 2001; Vincent et al., 2002; Fu et al., 2008a, 2010; Hiscox et al., 2010). PR inhibits cell growth and induces cell spreading and focal adhesion in association with modifications of FAK activity and β1-integrin signaling in metastatic MDA-MB-231 cells stably expressing both PRA and PRB recombinants (Lin et al., 2000). In contrast, progesterone (P4) enhances in vitro cell migration through matrix-coated membranes, although it strongly inhibits uPA mRNA synthesis (Lin et al., 2001). In nonmetastatic T47D cells that endogenously express PRA and PRB, PR-dependent cellular proliferation involves the activation of the c-Src/p21ras/MAPK signaling pathway (Migliaccio et al., 1998; Carnevale et al., 2007). In such cells, P4 enhances motility via activation of FAK signaling, relying on c-Src tyrosine kinase activity (Fu et al., 2010). This function was also recently reported to be effective in vascular endothelial cells (Zheng et al., 2012). However, owing to a lack of relevant models, the relative contributions of PR isoforms PRA and PRB have not been clearly elucidated.
Here we address these controversial questions using our newly established cellular model conditionally expressing PRA and/or PRB in MDA-MB-231 metastatic breast cancer cells (MDA-iPRAB; Khan et al., 2012). This model allows us to determine the differential effects of PR isoform induction on cell migration as a function of ligand status independent of estrogen signaling. We correlate the global promigratory effect of PRA and PRB coexpression with PR-responsive transcriptional modulation of factors involved in cell migration mechanism, as well as with nongenomic regulations directly targeting FAK complexes.
RESULTS
PRA and PRB differentially enhance migration of MDA-MB-231 breast cancer cells
We previously established a bi-inducible cell line derived from metastatic breast cancer MDA-MB-231 (PR−, ERα−) cells (Khan et al., 2012) conditionally expressing PRA and/or PRB. Addition of diacylhydrazine (RSL1) and/or doxycycline (Dox) as nonsteroidal inducers to MDA-iPRAB cells triggers expression of PRA and/or PRB, respectively (Figure 1, top). This model led us to investigate in a single cell line the differential effects of PR isoform expression on cell motility in the absence, as well as in the presence, of ligand. As shown in Figure 1, the experimental conditions for PR isoform expression were fixed to a PRA/PRB ratio of 2 after 24 h of treatment, depending on the relative concentrations of inducers. We then performed wound-healing repair assays on cycle-arrested cells by incubating them with 10−8 M 17,21-dimethyl-19-norpregna-4,9-dien-3,20-dione (R5020) or vehicle at various time in the absence of inducers for up to 24 h. We previously showed that inducer withdrawal did not markedly change expression of PRA and PRB for 12 h and reduced it to 50% after 24 h (Khan et al., 2012). Surprisingly, as presented in Figure 1 for time 10 h, in the absence of ligand, induction of PRB expression led to strong increase of MDA-iPRAB cell migration, whereas induction of PRA alone had no significant effect. When cells were treated with R5020, cell migration was still significantly enhanced in PRB-induced cells for a prolonged time until 24 h, although to a limited extent (2.5-fold) as compared with untreated PRB cells (5-fold). Cells coexpressing both isoforms (PRAB) migrated independently of the ligand at a rate similar to that observed for untreated PRB cells. We further determined that neither Dox nor RSL1 inducer provoked any change in adherent cell number, thus excluding experimental bias in wound-healing assays (data not shown). Next we determined whether the 11β-(4-dimethylamino)phenyl-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one (RU486) antagonist affected R5020-dependent cell migration. As shown in Supplemental Figure S1, RU486 completely abolished the antimigratory action of R5020, showing that this antagonist specifically restored the effect of unliganded PRB expression. Therefore, compared with the high basal migration rate of MDA-MB-231 cells lacking PR, coinduction of PRA and PRB expression provoked a global promigratory change in MDA-MB-231 cell behavior, which can be partially but specifically counteracted by R5020 only when PRA coexpression is low.
FIGURE 1:

PRA and PRB differentially enhance migration of MDA-MB-231 breast cancer cells. (A) MDA-iPRAB cells were incubated with vehicle (PR−), RSL1 (inducing PRA) or Dox (inducing PRB), or both (inducing PRA and PRB) for 24 h. Immunoblot analysis was performed from whole-cell extracts using anti-PR antibody recognizing both PR isoforms and anti-tubulin antibody for loading control. (B) MDA-iPRAB cells were induced as in A for 24 h and treated by mitomycin C for 1 h. At zero time point wound-healing repair assays were performed in the presence of either 10−8 M R5020 or vehicle for various time until 24 h. Photographs of each wounded area were taken at regular time and distance intervals (left). Results from three independent experiments are presented (right) as the random distance covered by cells after 10 h treatment (mean ± SEM, n = 3). Statistical analyses were done using Mann–Whitney tests (stars).
PRB regulates key genes of the PA system, enhancing cell migration
Movement of cancer cells results in part from ECM degradation at the leading edge of cell progression by different proteases and alteration of cell adhesion/deadhesion processes, both regulated by the PA system. To determine whether the effect of PRB on cell migration might be associated with relevant transcriptional regulations, we quantified uPA and uPAR transcripts in iPRAB cells by real-time quantitative reverse transcription PCR (qRT-PCR; Figure 2). Unliganded PRB but not PRA increased urokinase (uPA) transcripts twofold compared with the basal expression level observed in PR− cells. Moreover, induction of both PRA and PRB enhanced threefold uPA mRNA in the absence of hormone. In contrast, R5020 down-regulated uPA and uPAR mRNAs in PRB and PRAB cells compared with ligand-free conditions. Of note, the hormone counteracted the constitutive effect of PRB on uPA transcripts, in agreement with cell migration observed in wound-healing assays. We also analyzed expression of β1-integrin, which is required for matrix-dependent signaling, in particular by interacting with uPAR and promoting FAK autophosphorylation. As shown in Figure 2, unliganded PRB but not PRA significantly enhanced β1-integrin expression, whereas R5020 did not provoke any variation in this effect. Thus β1-integrin synthesis might be constitutively enhanced in PRB-expressing cells, potentially favoring cell progression.
FIGURE 2:
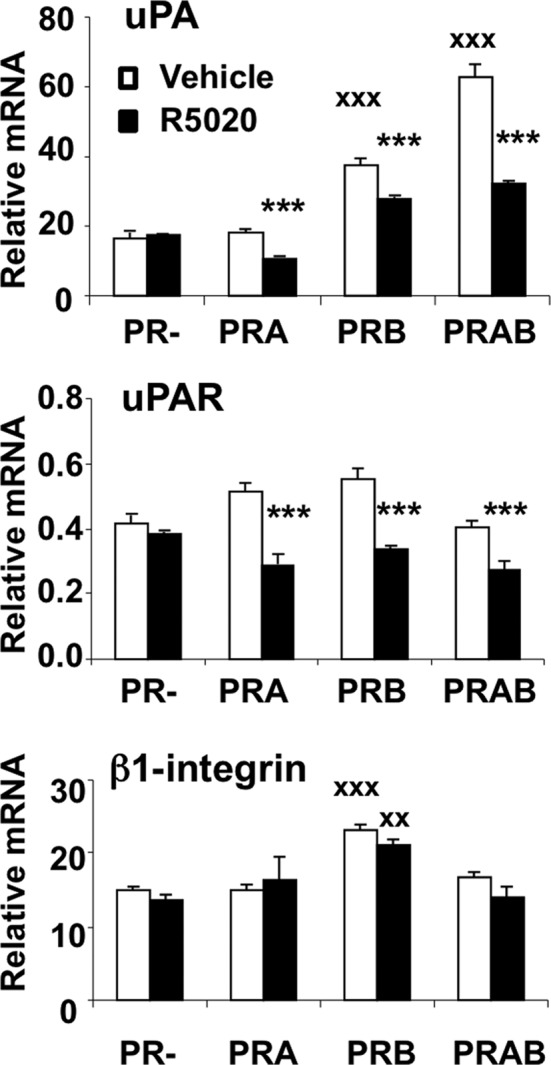
PRB regulates key genes of the PA system. After 24 h induction by Dox and/or RSL1 to induce or PRA and/or PRB, MDA-iPRAB cells were treated (black bars) or not (white bars) by R5020 (10−8 M) for 6 h. uPA, uPAR, and β1-integrin were quantified by real-time qRT-PCR as described in Materials and Methods. The data were normalized to 18S rRNA and expressed as mean ± SEM (n = 6). Statistical analyses using the Student's two-tailed test are shown by either crosses, referring to PR− cells with vehicle, or stars, referring to PR+ (PRA and/or PRB) cells with R5020.
We next determined whether PRA and PRB can regulate transcription of PAI-1, the main inhibitor of uPA proteolytic functions. PAI-1 mRNA was induced by R5020 but not by the unliganded PRs (Figure 3A), suggesting the possible effect of this factor in the relative antimigratory action of hormone observed in PRB-expressing cells. As shown in Figure 3B, RU486 inhibited the R5020-induced expression of the PAI-1 gene, supporting the PRB specificity of the mechanism. We also determined that neither R5020 nor RU486 had any effect on such transcription in PR− cells (data not shown). Furthermore, as measured by enzyme-linked immunosorbent assay (ELISA; Figure 3C), transcriptional induction of PAI-1 transcript by R5020 was translated into secretion of PAI-1 protein in the culture medium, which was inhibited by RU486. To test the effect of PAI-1 on cell migration, we performed wound-healing repair assays on PR− cells treated by increasing amounts of recombinant PAI-1 (Figure 3D, left). Surprisingly, up to 100 ng/ml PAI-1 strongly enhanced migration, whereas higher doses led to decreasing effects, likely through cell surface desensitization. This promigratory effect of PAI-1 on malignant cells is supported by previous data (Waltz et al., 1997; Croucher et al., 2007; Fabre-Guillevin et al., 2008). Moreover, as shown in Figure 3E, high amounts of PAI-1 failed to decrease migration of PRB cells. Therefore this ruled out that R5020-mediated down-regulation of PRB-dependent cell migration could act via PAI-1 stimulation.
FIGURE 3:

PR isoforms regulate expression of PAI-1. (A) After 24 h induction by Dox and/or RSL1 or vehicle, MDA-iPRAB cells were treated by 10−8 M R5020 (black bars) or vehicle (white bars) for 6 h. PAI-1 mRNA was measured by real-time qRT-PCR as described in Materials and Methods. Results and statistical analyses are calculated as in Figure 2. (B) MDA-iPRAB cells were treated with Dox for 24 h to induce PRB expression and as indicated by 10−8 M R5020 and/or 10−6 M RU486 or vehicle for 6 h. PAI-1 transcript was measured as in A. (C) MDA-iPRAB cells were induced as in B and treated with 10−8 M R5020 and/or 10−6 M RU486 or vehicle for 14 h. PAI-1 concentration was measured in conditioned medium by ELISA (see Materials and Methods) (D) MDA-iPRAB cells were grown without inductors for 24 h, and increasing amounts of soluble recombinant PAI-1 were added to the conditioned medium as described in Materials and Methods. Cell migration was quantified after 10-h treatment as in Figure 1, and results are expressed as percentage of basal migration obtained in the absence of PAI-1 (mean ± SEM, n = 3). (E) MDA-iPRAB cells were grown with Dox to express PRB or with vehicle (PR−) for 24 h. After addition of PAI-1 (200 ng/ml) or vehicle in the conditioned medium, cell migration was measured after 10 h as in D.
Taken together, these results show that PRA and PRB regulate the PA system to different extents depending on ligand status. Mainly, PRB up-regulates uPA and β1-integrin in the absence of ligand, thus potentially inducing promigratory effects by facilitating proteolysis of ECM and activating uPAR signaling. In contrast, although ligand-bound PRB switched off uPA signal, in agreement with its effect on migration, at the same time it induced PAI-1 gene transcription and enhanced secretion of PAI-1 protein, having a promigratory effect on MDA-MB-231 cells. Such effects on promigratory gene expression are consistent with a global promigratory mechanism triggered by PRB expression in cancer cells, irrespective of ligand condition.
PRA and PRB differentially affect regulation of FAK activity
Recent studies showed that P4 enhances T47D breast cancer cell migration via extranuclear activation of FAK (Fu et al., 2010) resulting from initial phosphorylation of Y397 residue. Because FAK signaling is involved in the regulation of FA assembly/disassembly, we asked whether conditional induction of PRA and PRB in MDA-iPRAB cells could interfere with it. We analyzed FAK phosphorylation at the Tyr-397 key residue (FAKY397p) and total FAK (FAKtotal) expression. After 24-h induction of expression (Figure 4A), unliganded PRB but not PRA was able to enhance both FAKY397p and total FAKtotal to a similar extent, that is, without inducing any change in their ratio. This suggested that PRB but not PRA might selectively increase FAK expression in the absence of ligand. Furthermore, the 1-h time course of hormone-dependent FAK phosphorylation (Figure 4B) revealed that the liganded PRA was unable to activate FAK, in contrast to PRB inducing FAK phosphorylation as early as 5 min. Although PRB but not PRA expression slightly enhanced FAK mRNA level as compared with PR− cells (Supplemental Figure S2), addition of hormone did not significantly decrease this level, excluding that hormone-dependent down-regulation of cell migration resulted from any drastic transcriptional repression of the FAK gene.
FIGURE 4:
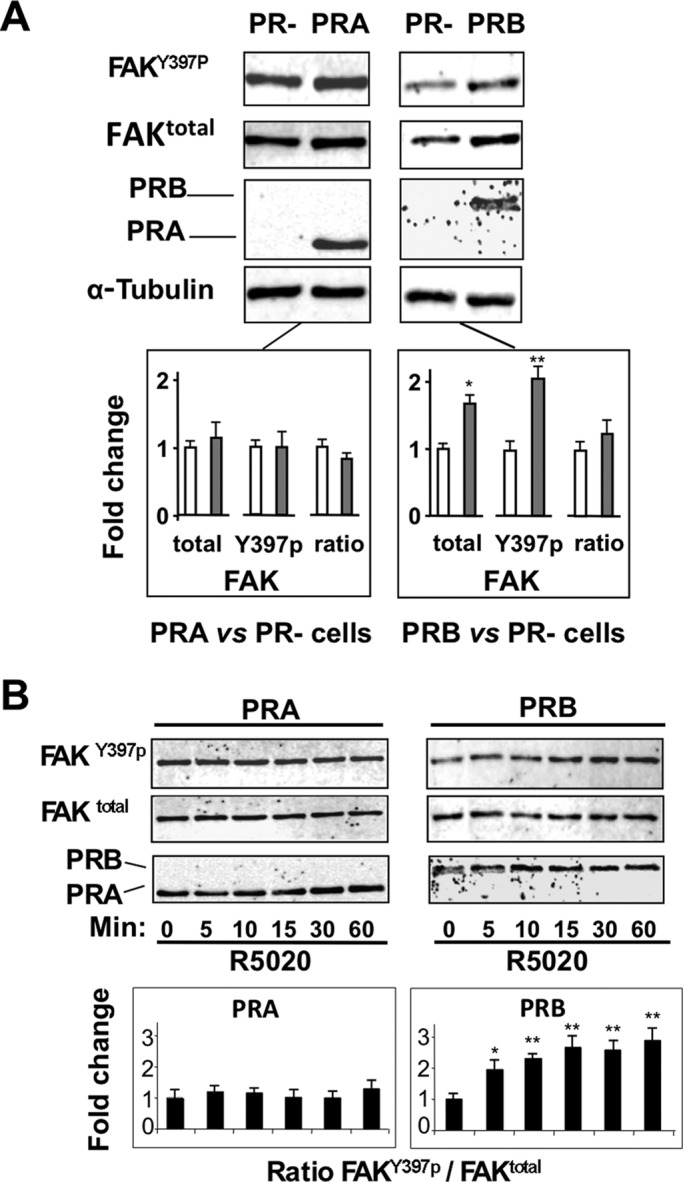
PRA and PRB differentially affect regulation of FAK activity. (A) MDA-iPRAB cells were grown in the presence of Dox (PRB), RSL1 (PRA), or vehicle (PR−) for 24 h and analyzed by Western blot using alternately anti-FAKY397p, anti-FAKtotal, and anti-tubulin antibodies as described in Materials and Methods. PR isoforms were analyzed on a separated gel using anti-PR antibody. A representative immunoblot is shown, and FAKY397p, FAKtotal, and FAKY397p/FAKtotal ratio were quantified and normalized to tubulin from three independent experiments. The corresponding graphs show fold change of values obtained in PR− cells (means ± SEM, Mann–Whitney statistical test). (B) Cells were induced as in A and then treated or not by 10−8 M R5020 for the indicated times (minutes). Cell lysates were analyzed as in A, and data are presented as fold change of values obtained for the corresponding PR isoform at zero time point.
These results show that PRA and PRB inductions differentially affect FAK activity, depending on ligand status. In the absence of ligand, PRB but not PRA stabilizes FAKY397p, in agreement with unliganded PRB-dependent cell migration observed in wound-healing assays. In contrast, R5020 leads to rapid increase of PRB-dependent FAK phosphorylation, excluding that its down-regulating effect on migration results from direct PRB-dependent inhibition of FAK activity.
PRB and FAKY397p are colocalized at focal adhesion points
Because PRB-dependent alteration of cell migration was detected as early as 3 h after exposure to R5020, the early activation of FAK by liganded PRB should result in enhanced cell migration, contrasting with the data obtained in wound-healing assays. We hypothesized that PRB-specific, FAK-dependent migration could be affected by variation in cytoplasmic PRB expression. To clarify this point, using immunofluorescence experiments at various time intervals after hormone treatment, we compared PR isoform and FAKY397p distributions in iPRAB cells. High motility of MDA-MB-231 cells has already been correlated with their ability to produce cellular protrusions such as filipodia and lamellipodia. These structures lead to sustained cell orientation with growing FAs at the leading edge contributing to cell expansion, as well as cell movement. These elements were clearly visible in PR− cells (Figure 5, left). Whereas ligand-free PRB was found to be equally distributed within cytoplasmic and nuclear compartments, the hormone-bound PRB was, as expected, fully translocated in the nucleus in 30 min. In the absence of PR, FAK was expressed in the cytoplasm and the nucleus and was especially condensed in submembrane speckles corresponding to FAs in the pseudopodia involved in migration. Of interest, after 24-h induction of PRB expression, the ligand-free PRB was repeatedly found in such structures containing FAK, especially at the leading edge of migration and also in filamentous radiant elements such as lamellipodia (Figure 5). In addition, the number of cell protrusions with FAK-containing FAs was increased, supporting the notion that the unliganded PRB could somehow potentiate FAK-dependent migration. However, although FAKY397p, as well as the apo PRB, was also present in the nuclei, no colocalized speckles were detected in merged images of this compartment. Of interest, upon hormone exposure, PRB-FAKY397p colocalization was impaired concomitant with nuclear translocation of PRB. Time-course experiments showed that the subcellular localization of PRB was modified as early as 5 min after R5020 exposure (Supplemental Figure S3). PRB-FAKY397p colocalization in FAs was completely abolished after 30 min for 90% of the cells owing to the complete nuclear translocation of PRB. Of note, some nuclear speckles were visible in the perinuclear region of a few cells, suggesting that PRB-FAKY397p complexes might be transiently present in the nuclear compartment. As opposed to R5020, RU486 antagonist resulted in slower PRB nuclear translocation, with RU486-bound PRB remaining in the cytoplasmic compartment at 30 min (Figure 5, right). Of interest, PRB-RU486 complexes were also clearly present in FAKY397p-containing FAs, similar to unliganded PRB. These characteristics were thus well correlated with the effects on cell migration observed in both ligand conditions. In contrast, the hormone-dependent decrease in PRB-mediated migration was likely related to PRB nuclear translocation, limiting its effect on FAK phosphorylation.
FIGURE 5:

PRB and FAKY397p are colocalized in focal adhesion sites. MDA-iPRAB cells were induced (PRB) or not (PR−) by Dox for 24 h and then treated with either 10−8 M R5020 or 10−6 M RU486 or vehicle for 30 min. Immunofluorescence microscopy was performed as described in Materials and Methods by analyzing FAKY397P (red) and PRB (green). The nuclei were counterstained with DAPI (blue). Photographs were taken by using a confocal microscope at 400× magnification. (a–c) Magnifications to focus on representative structures.
Similar experiments were performed in cells treated by RSL1 to induce PRA expression (Supplemental Figure S4). In contrast to PRB, unbound PRA was essentially localized in the nuclei with a perinuclear distribution. Although low expression of PRA was also slightly visible in the cytoplasm, we failed to identify any condensation points containing unliganded PRA with FAKY397p in pseudopodia. However, several colocalized speckles were found into the nucleus, supporting that PRA–FAK complexes could be assembled there. R5020 treatment did not alter cellular distribution of PRA. Confocal analysis profiles corresponding to overlain images of Figure 5 and Supplemental Figure S4 are shown in Supplemental Figure S5, clearly indicating that PRB but not PRA could be detected in the submembrane FAs present in the leading edges of migrating cells grown in the absence of ligand or in the presence of RU486.
Collectively these experiments showed that unliganded PRB but not PRA is colocalized with autophosphorylated FAKY397p in FAs at the leading edge of migration, strongly suggesting that PRB can constitutively enhance cell motility via a FAK-dependent mechanism independent of transcriptional regulations. In contrast, nuclear translocation of liganded PRB well correlated with the reduction of PRB-dependent cell motility by R5020.
PRA and PRB interactions with FAKY397p differentially regulate FAK turnover
To determine whether ligand-dependent cellular colocalization of PRB and FAKY397p might underlie a protein–protein interaction within the FAs, we immunoprecipitated FAKtotal complexes from lysates of either PRB- or PRA-expressing cells using anti-FAKtotal or anti-PR antibodies. Of interest, as shown in Figure 6A, PRA and PRB were refolded in FAK-specific immunoprecipitates in the presence, as well as the absence, of hormone. Reverse coimmunoprecipitation (coIP) experiments were performed (Supplemental Figure S6) confirming that FAKY397p was present only when PR isoforms were induced. Therefore these coIPs provide evidence for the abilities of both isoforms to interact with complexes containing FAK, independent of ligand status. This raised the question of whether PRA and PRB might alternately interact with a shared component of the FAK complexes. As shown in Figure 6 (right inset), induction of PRA expression in PRB-expressing cells resulted in a drastic decrease in PRB–FAK complexes, suggesting that PRA could compete with PRB for the same binding sites.
FIGURE 6:

PRA and PRB interact with FAK complexes and regulate their turnover. (A) MDA-iPRAB cells were induced by RSL1 and/or Dox for 24 h and then exposed to 10–8 M R5020 or vehicle for 1 h. Cell lysates were incubated with either total FAK antibody or nonrelated antibody (immunoglobulin G). Lysates (input) and immunoprecipitates (IPs) were analyzed by Western blot for FAKY397P, FAKtotal, PRA, PRB, and tubulin. Framed inset: the coIP experiments were repeated using iPRAB cells induced by either Dox or RSL1 + Dox to induce PRB or PRA + PRB for 24 h. (B) iPRAB cells were induced by either RSL1 or Dox or both of them and then treated or not by 10−8 M R5020 for 10 h. Western blots were performed and quantified as described in Figure 4A for FAKY397P, FAKtotal, PRA, PRB, and tubulin (mean ± SEM, Mann–Whitney statistical test). (C) Ishikawa cells stably transfected by either PRA or PRB and T47D cells endogenously expressing PRA and PRB were treated either by 10−8 M R5020 or vehicle for 24 h. Western blot analyses were performed as in B.
Given that proteasome-mediated degradation of agonist-bound PRB is required for its transcriptional activity (Dennis et al., 2005), we next asked whether PRB-dependent FAK activity could be also controlled by such dynamic processes. FAK expression was analyzed in iPRAB cells after 10 h of hormonal treatment (Figure 6B). In PRB-expressing cells and in agreement with previous reports (Lange et al., 2000; Khan et al., 2011), R5020 induced proteasome-dependent degradation of PRB, with a 50% decrease after 10 h in the MDA-iPRAB cell line. In contrast, liganded PRA remained stable after 10 h of hormone treatment in these cells. Of interest, PRB but not PRA expression clearly led to hormone-dependent decrease of total FAK as well as FAKY397p expression, whereas coexpression of PRA strikingly prevented this effect. Therefore FAK turnover was at least partially coupled to hormone-induced PRB degradation long after PRB nuclear translocation was achieved. Because turnover dynamics might be cell specific, experiments were repeated in two different cell lines expressing PRA or PRB (Figure 6C). As previously reported (Khan et al., 2011), in endometrial cancer Ishikawa cells (ERα−) stably transfected by PRB or PRA, both isoforms are rapidly down-regulated after hormone exposure. In mammary cancer T47D cells, PRA and PRB are endogenously expressed independent of estrogen regulation, although ERα is present. In both cell lines, FAK down-regulation was induced by hormone treatment.
These results, taken together, provide evidence for ligand-independent interactions of both PRA and PRB with FAK and establish a biological link between FAK and PR isoform turnovers depending on PRA and PRB degradation kinetics.
DISCUSSION
P4 was reported to induce cell spreading and adhesion in MDA-MB-231 stably expressing both PRA and PRB (Lin et al., 2000, 2001). In T47D cells endogenously expressing PRA and PRB, P4 increased cell migration (Fu et al., 2008b, 2010), whereas PRA but not PRB enhanced migration in PR-inducible T47D YiA and YiB cells (Jacobsen et al., 2005). In sharp contrast, our unique bi-inducible cell line allowed us to unambiguously evaluate the relative contributions of both isoform expressions in the absence, as well as in the presence, of hormone. In our model, PRB and PRA clearly cooperate through multiple mechanisms accelerating migration. PRB but not PRA enhances uPA transcript level, consistent with a positive effect on cell migration in the absence of progestins. Moreover, because addition of hormone decreased PRB-dependent up-regulation of uPA, this gene might be only sensitive to unliganded PRB but switched off by hormone. Therefore unliganded PRB-dependent cell migration would result from synergetic activations of uPA signaling and FAK cascade via both transcriptional and nongenomic processes. Moreover, upon hormone addition, PAI-1 synthesis and secretion were enhanced in PRA- and PRB-expressing cells. Although PAI-1 was initially considered as an inhibitor of both uPA-mediated ECM proteolysis and uPA-inducible uPAR signaling, several reports argued in favor of other PAI-1 functions linked to endocytic receptor LRP and integrin signaling, leading to stimulate adhesion/deadhesion dynamics (Waltz et al., 1997; Croucher et al., 2007; Fabre-Guillevin et al., 2008). Moreover, PAI-1 is considered a metastasis prognostic marker (Leissner et al., 2006). In this context, because PAI-1 strongly increased migration of MDA-MB-231 cells at low concentration, this factor might potentially mediate PRB-dependent migration through a paracrine mechanism affecting FAK activity.
Independent of regulation of the PA system, PRB directly targets the FAs through interaction with FAK complexes. PRB clearly colocalized with FAKY397p, which is required for focal adhesion disassembly (Hamadi et al., 2005; Deramaudt et al., 2011). P4 has been found to rapidly enhance phosphorylation of Tyr-397-FAK in T47D cells (Fu et al., 2010), and we confirmed these results here using R5020 progestin in our bi-inducible cell model. However, in contrast to this report, our studies argue for a direct interaction of PRB with FAK complexes, which likely enhances FAK-mediated motility. Other converging data indicate that steroid receptors are able to cross-talk with FAK signaling. In this regard, the ligand-free ER has been identified in the same complex as FAK via interaction with Src tyrosine kinase, which was disrupted by estrogens (Le Romancer et al., 2008). Moreover, evidence was reported for the interaction of FAK complexes with steroid receptor coactivator 3 (SRC3) in MDA-MB-231 cells (Long et al., 2010). Tumor suppressor BRCA1 colocalizes in FAs, leading to decreased cell motility (Coene et al., 2011). Of interest, PRB interacts with ER (Ballare et al., 2003), c-Src (Boonyaratanakornkit et al., 2001), SRC3 (Long et al., 2010), and BRCA1 (Ma et al., 1999; Poole et al., 2006). It is possible that, even in the absence of ER, cytoplasmic PRB might be directly recruited by FAK concomitant to Src and other partners, leading to modulation of cell migration in a coordinated manner.
Under physiological conditions, PRB trafficking and shuttling are finely tuned through hormone-sensitive regulation (Guiochon-Mantel et al., 1991; Tyagi et al., 1998), which might strongly influence cell migration dynamics. In our cell model, hormone-induced PRB nuclear translocation led to rapid depletion of PRB in sub–plasma membrane structures, as well as in cytoplasm, which was correlated with decreased migration rate after a few hours of hormone treatment. It was also surprising that hormones induced FAK degradation in PRB cells. PRB transcriptional hyperactivity is tightly coupled to its proteasome-dependent turnover (Dennis et al., 2005). In a similar manner, the increase in hormone-dependent degradation of PRB–FAK complexes might be the signature of PRB-dependent FA disassembly. FAKY397p translocates to the nucleus, where it can interact with p53 (Golubovskaya et al., 2005) and down-regulate its turnover by enhancing p53 ubiquitination (Lim et al., 2008). Because PRA strongly interacted with FAK in cell lysates but neither colocalized with FAK at FAs nor activated FAK at Tyr-397, PRA interaction with soluble FAK could mainly take place in the nuclear compartment. The fact that PRA interfered with PRB-dependent FAK turnover and could compete with PRB for binding FAK suggests that such cross-talk might transiently occur into the nucleus, with important consequences for FAK stability in this compartment.
We previously reported that RU486 stabilizes PRB through a MAPK-dependent mechanism (Khan et al., 2011) and concomitantly stabilizes SRC-1, a major coactivator of PRB (Amazit et al., 2011). RU486 also inhibited the hormone-dependent spread of MDA-MB-231 cells expressing PRB (Lin et al., 2001). Accordingly, we found here that this antagonist induced delayed nuclear translocation compared with R5020-treated cells and abolished the relative down-regulating effect of hormones on cell migration. The promigratory action of RU486 likely resulted from both increased uPA expression and PRB stabilization, which might favor its interaction with FAK in the cytoplasmic compartment. This highlights the need for selective PR antagonists that neither induce MAPK-dependent stabilization of PRB nor inhibit proteasome-dependent turnover, in contrast to RU486 and most of its derivatives.
Our results are schematically summarized in Figure 7. Induction of PRB expression enhances cell migration through transcriptional, as well as nongenomic, processes involving FAK-dependent signaling. Based on the mechanistic model previously proposed for FAK assembly/disassembly kinetics (Hamadi et al., 2005; Deramaudt et al., 2011), cell migration might be correlated with FAK time residence within FAs. In PR-expressing cells, association of stable unliganded PRB with FAK would enhance FAK-dependent cell migration in a sustained manner by favoring FAK recruitment within FAs and subsequent activation of migration. Hormone-bound PRB transiently leads to rapid stimulation of FAKY397p and enhancement in cell motility as compared with PR− cells. However, hormone-bound PRB cells migrate at a lower speed than unliganded PRB cells due to enhanced nuclear localization. PRB-mediated cell migration is thus dependent on cytoplasmic PRB abundance, which is an intricate function of PRB neosynthesis, nucleocytoplasmic shuttling, and proteasome-dependent degradation, as well as hormone status. Of importance, antagonist ligands such as RU486 that stabilize PR lead to sustained stimulation of migration compared with hormone-treated cells. In addition, PRA would extend hormone-dependent migration of PRB-expressing cells by stabilizing FAK, leading to constitutively activate migration at high level. Our model predicts that the more PRB is stable and shuttles in cytoplasm, the more it interacts with FAK and enhances migration. PRB overexpression would thus favor mammary cancer cell expansion, especially in the context of low P4 status or disturbed PRB trafficking or any associated treatment with RU486-like antagonists. Of interest, the more PRA is expressed, the more it stabilizes FAK and amplifies PRB-dependent cell migration. Therefore high expression of PRA might play a critical role in setting output signals for PRB-dependent cell migration independent of progestins, in agreement with the deleterious effects of high PRA expression level found in a majority of PR+ ER+ breast cancers.
FIGURE 7:

Model for PRB-dependent regulation of cell migration. Impaired FAK phosphorylation at the Y397 site decreases FAK time residence at FAs, leading to reduce cell retractile activity and migration (migration OFF). Increased stabilization of FAK at FAs leads to sustained stimulation of phosphorylation cascades initiated by FAKY397 phosphorylation, interactions with protein partners, and disassembly of FAs (migration ON). Interaction of FAK with cytoplasmic (cyto) PRB at FAs leads to enhance FAK time residence within FAs (PRB-dependent migration HIGH). The hormone transiently enhances Y397 phosphorylation, enhancing migration, and PRB-FAK is released from the FAs and is thus translocated into the nucleus and/or degraded (PRB-dependent migration LOW). The exchange of PRB with PRA within soluble FAK complexes inhibits FAK/PRB codegradation, potentiating the effect of hormone on PRB-dependent migration (PRB-dependent migration HIGH). The indicated PR-dependent transcriptional regulations affecting migration may further strengthen these effects at delayed time. RU486 antagonist counteracts all the previous hormone-dependent effects and potentiates PRB-dependent cell migration. The triangles indicate R5020 (black), RU486 (gray), or PRA expression (white) variations leading to extended migration rate at the indicated high or low level.
In sum, PRB and PRA differentially affect cell migration through multiple mechanisms activating FAK as a function of their relative expression level in cell compartments. Our findings argue for the important roles that PRA/PRB ratio might play in regulation of cellular movements of PR-expressing cells and suggest a mechanistic scheme for cell motility and metastatic dissemination of ER+ PR+ breast cancers.
MATERIALS AND METHODS
Cell cultures and treatments
MDA-iPRAB cell line deriving from MDA-MB-231 human breast cancer cell line (American Type Culture Collection, Manassas, VA) was previously described (Khan et al., 2012). Briefly, these cells stably express all components of Rheoswitch (New England BioLabs, Ipswich, MA) and T-Rex (Invitrogen, Carlsbad, CA) systems, allowing controlled expression using RSL1 and Dox nonsteroidal inducers. They were also stably transfected by two vectors conditionally expressing either PRA in the presence of RSL1 or PRB in the presence of Dox in a dose-dependent manner. Cells were grown in DMEM supplemented with 5% fetal bovine serum (FBS) as previously described (Khan et al., 2012) in the presence of Geneticin (500 μg/ml), blasticidin (2 μg/ml), and Zeocin (100 μg/ml) to maintain selective pressure on plasmid expression (Khan et al., 2012). At 24 h before each experiment, cells were starved in DMEM without phenol red with 5% FBS stripped using the dextran-coated charcoal method with 100 U/ml penicillin and 100 μg/ml streptomycin. Then RSL1 (0.25 μM) and/or Dox (2 μM) was added in the medium to induce PRA and/or PRB expression, respectively. After induction for 24 h, steroids or vehicle (0.01% ethanol) were added in the medium as indicated. Ishikawa cells expressing PRA or PRB and T47D cells were grown as previously described (Khan et al., 2011).
Real-time qRT-PCR
Total RNA was extracted from iPRAB cells treated or not by the indicated ligands for a given time using TRIZOL reagent (Invitrogen), and equal amounts (1 μg) were reverse transcribed for real-time qRT-PCR analysis as previously described (Khan et al., 2011). Primers (300 nM) used are listed in Supplemental Table S1. Quantification of gene expression was normalized to 18S rRNA and expressed as means ± SEM from six experiments.
In vitro wound-healing repair assays
MDA-iPRAB cell migration was assessed according to a previously reported method (Chen et al., 2011). Briefly, 105 cells were grown to confluence into inserts onto graduated plastic microdishes (Ibidi, Munich, Germany). After induction of PRA and/or PRB expression by RSL1 and/or Dox for 24 h, cell proliferation was arrested by adding 10 μg/ml mitomycin C (Sigma-Aldrich, St. Louis, MO) for 1 h. The insert was pulled out, and cell debris was removed by washing with phosphate-buffered saline (PBS). Width of each wounded area was measured using grids at three marked positions. The cells were then treated by 10−8 M R5020, 10−6 M RU486, or vehicle. The cultures were kept at 37°C in a humidified incubator and photographed (40× magnification) at the indicated times to monitor migration of cells into the wounded area. Cell migration was quantified as the distance covered by cells in wound-healed surface from the marked positions. Results are expressed as mean ± SEM of three independent experiments.
Immunoblots and immunoprecipitations
MDA-iPRAB cells were harvested in cold PBS and the pellet resuspended in lysis buffer (0.1% Triton X-100, 50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 150 mM, NaCl, 0.2% NaF, 1.3% sodium pyrophosphate) containing a mixture of phosphatases and proteases inhibitors (Sigma-Aldrich). PAGE electrophoresis and immunoblotting were performed as previously described (Khan et al., 2011). The membranes were incubated with the indicated primary antibodies at 4°C overnight and then incubated with secondary antibody conjugated to horseradish peroxidase for fluorescence before being developing using enhanced chemiluminescence plus detection reagents (Amersham Biosciences, Piscataway, NJ) or scanning using the Odyssey system (LI-COR, Lincoln, NE). For coimmunoprecipitation experiments, the cells were lysed in lysis buffer containing 0.5% Nonidet-P40. Supernatants (1 mg of total proteins) were incubated with 2 μg of antibodies for 5 h at 4°C. The samples were then mixed with protein G magnetic beads (Millipore, Billerica, MA) according to manufacturer's instructions. Bound immunocomplexes were boiled in Laemmli loading buffer for 10 min and analyzed by Western blot. The antibodies used were monoclonal anti-PR (NCL-LPGR-312/2; Novocastra, Newcastle upon Tyne, United Kingdom), rabbit polyclonal anti–FAK C-terminal domain (C20; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti–phospho Tyr397-FAK (ab4803, Abcam), and anti–α-tubulin (Sigma-Aldrich).
Plasminogen activator inhibitor-1 antigen assays
Cells were grown at 80% confluence in medium containing either RSL1 or Dox for 24 h. The cells were then treated by 10−8 M R5020, or/and 10−6 M RU486, or vehicle for 16 h. Conditioned media were immediately transferred to −20°C until the next step. Total PAI-1 was measured using ELISA-Kit (Gentaur, Paris) as described by the manufacturer. Results are expressed as mean ± SEM from three independent experiments.
Immunocytochemistry and confocal imaging
MDA-iPRAB cells were plated onto chambered slides (Lab-Tek, Rochester, NY), fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton, and saturated with 5% BSA and 0.1% casein in PBS. The slides were incubated with primary anti-FAKY397P rabbit polyclonal antibody (ab4803; Abcam, Cambridge, MA), anti-PRB monoclonal antibody let126 (Lorenzo et al., 1988), anti–PRB and PRA monoclonal antibody (Novocastra, NCL-LPGR-312/2), Cy5-conjugated secondary anti-rabbit antibody, and Alexa Fluor green–conjugated secondary anti-mouse (Invitrogen). Nuclei were stained using To-PRO3 (Invitrogen) or 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen), and the slides were mounted using Fluorescence Mounting Medium (Dako, Trappes, France). The cells were analyzed by confocal fluorescence microscopy as previously described (Amazit et al., 2011).
Statistical analysis
All data are representative of at least three independent experiments and are presented as mean ± SEM. Nonparametric Mann–Whitney or Student's two-tailed tests were used to determine statistical significance of difference between groups using the software InVivoStat (www.invivostat.co.uk). Statistical significance: ***p < 0.001,**p < 0.01, *p < 0.05.
Supplementary Material
Acknowledgments
We are grateful to Claude Labrie (Laval University Medical Research Center, Quebec, Canada) for his support with regard to the inducible expression system. We thank Philippe Leclerc for confocal photographs and technical assistance. This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale, Université Paris-Sud 11, and the Association pour la Recherche sur le Cancer. C.B. was the recipient of a fellowship from the Conseil Régional de la Martinique, France. J.A.K. was on study leave from the Department of Physiology and Pharmacology, University of Agriculture, Faisalabad, Pakistan, and was the recipient of a doctoral scholarship from the Higher Education Commission, Islamabad, Pakistan, and a fellowship from La Ligue Contre le Cancer, France.
Abbreviations used:
- ER
estrogen receptor
- FAK
focal adhesion kinase
- FAKY397p
FAK phosphorylated at Tyr-397 residue
- P4
progesterone
- PAI-1
plasminogen activator inhibitor type 1
- PR
progesterone receptor
- PRA
progesterone receptor isoform A
- PRB
progesterone receptor isoform B
- R5020
17,21-dimethyl-19-norpregna-4,9-dien-3,20-dione
- RU486
11β-(4-dimethyl-amino)-phenyl-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one
- uPA
urokinase plasminogen activator
- uPAR
urokinase plasminogen activator receptor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-11-0807) on March 13, 2013.
C.B. performed and designed experiments and participated in the drafting of the article. J.A.K. designed the experiments and characterized the cell lines. H.L. and M.L. conceived the studies and wrote the article. A.G.M. participated in discussions and data interpretation.
REFERENCES
- Amazit L, Roseau A, Khan JA, Chauchereau A, Tyagi RK, Loosfelt H, Leclerc P, Lombes M, Guiochon-Mantel A. Ligand-dependent degradation of SRC-1 is pivotal for progesterone receptor transcriptional activity. Mol Endocrinol. 2011;25:394–408. doi: 10.1210/me.2010-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Talukder AH, Wang RA, Vadlamudi RK, Kumar R. Metastasis-associated protein 1 deregulation causes inappropriate mammary gland development and tumorigenesis. Development. 2004;131:3469–3479. doi: 10.1242/dev.01213. [DOI] [PubMed] [Google Scholar]
- Ballare C, Uhrig M, Bechtold T, Sancho E, Di Domenico M, Migliaccio A, Auricchio F, Beato M. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol Cell Biol. 2003;23:1994–2008. doi: 10.1128/MCB.23.6.1994-2008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard-Trifilo JA, Lim ST, Hou S, Schlaepfer DD, Ilic D. Analyzing FAK and Pyk2 in early integrin signaling events. Curr Protoc Cell Biol. 2006 doi: 10.1002/0471143030.cb1407s30. Chapter 14, Unit 14.17. [DOI] [PubMed] [Google Scholar]
- Biermann JC, Holzscheiter L, Kotzsch M, Luther T, Kiechle-Bahat M, Sweep FC, Span PN, Schmitt M, Magdolen V. Quantitative RT-PCR assays for the determination of urokinase-type plasminogen activator and plasminogen activator inhibitor type 1 mRNA in primary tumor tissue of breast cancer patients: comparison to antigen quantification by ELISA. Int J Mol Med. 2008;21:251–259. [PubMed] [Google Scholar]
- Bogina G, Bortesi L, Marconi M, Venturini M, Lunardi G, Coati F, Massocco A, Manfrin E, Pegoraro C, Zamboni G. Comparison of hormonal receptor and HER-2 status between breast primary tumours and relapsing tumours: clinical implications of progesterone receptor loss. Virchows Arch. 2011;459:1–10. doi: 10.1007/s00428-011-1097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonyaratanakornkit V, Bi Y, Rudd M, Edwards DP. The role and mechanism of progesterone receptor activation of extra-nuclear signaling pathways in regulating gene transcription and cell cycle progression. Steroids. 2008;73:922–928. doi: 10.1016/j.steroids.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Boonyaratanakornkit V, McGowan E, Sherman L, Mancini MA, Cheskis BJ, Edwards DP. The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol Endocrinol. 2007;21:359–375. doi: 10.1210/me.2006-0337. [DOI] [PubMed] [Google Scholar]
- Boonyaratanakornkit V, Scott MP, Ribon V, Sherman L, Anderson SM, Maller JL, Miller WT, Edwards DP. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol Cell. 2001;8:269–280. doi: 10.1016/s1097-2765(01)00304-5. [DOI] [PubMed] [Google Scholar]
- Busso N, Masur SK, Lazega D, Waxman S, Ossowski L. Induction of cell migration by pro-urokinase binding to its receptor: possible mechanism for signal transduction in human epithelial cells. J Cell Biol. 1994;126:259–270. doi: 10.1083/jcb.126.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevale RP, et al. Progestin effects on breast cancer cell proliferation, proteases activation, and in vivo development of metastatic phenotype all depend on progesterone receptor capacity to activate cytoplasmic signaling pathways. Mol Endocrinol. 2007;21:1335–1358. doi: 10.1210/me.2006-0304. [DOI] [PubMed] [Google Scholar]
- Chapman HA. Plasminogen activators, integrins, and the coordinated regulation of cell adhesion and migration. Curr Opin Cell Biol. 1997;9:714–724. doi: 10.1016/s0955-0674(97)80126-3. [DOI] [PubMed] [Google Scholar]
- Chazaud B, Ricoux R, Christov C, Plonquet A, Gherardi RK, Barlovatz-Meimon G. Promigratory effect of plasminogen activator inhibitor-1 on invasive breast cancer cell populations. Am J Pathol. 2002;160:237–246. doi: 10.1016/S0002-9440(10)64367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, et al. A novel sialyltransferase inhibitor suppresses FAK/paxillin signaling and cancer angiogenesis and metastasis pathways. Cancer Res. 2011;71:473–483. doi: 10.1158/0008-5472.CAN-10-1303. [DOI] [PubMed] [Google Scholar]
- Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med. 2008;359:2814–2823. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebowski RT, et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. J Am Med Assoc. 2010;304:1684–1692. doi: 10.1001/jama.2010.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebowski RT, et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women's Health Initiative Randomized Trial. J Am Med Assoc. 2003;289:3243–3253. doi: 10.1001/jama.289.24.3243. [DOI] [PubMed] [Google Scholar]
- Coene ED, Gadelha C, White N, Malhas A, Thomas B, Shaw M, Vaux DJ. A novel role for BRCA1 in regulating breast cancer cell spreading and motility. J Cell Biol. 2011;192:497–512. doi: 10.1083/jcb.201004136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher DR, Saunders DN, Stillfried GE, Ranson M. A structural basis for differential cell signalling by PAI-1 and PAI-2 in breast cancer cells. Biochem J. 2007;408:203–210. doi: 10.1042/BJ20070767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czekay RP, Loskutoff DJ. Unexpected role of plasminogen activator inhibitor 1 in cell adhesion and detachment. Exp Biol Med (Maywood) 2004;229:1090–1096. doi: 10.1177/153537020422901102. [DOI] [PubMed] [Google Scholar]
- Dennis AP, Lonard DM, Nawaz Z, O'Malley BW. Inhibition of the 26S proteasome blocks progesterone receptor-dependent transcription through failed recruitment of RNA polymerase II. J Steroid Biochem Mol Biol. 2005;94:337–346. doi: 10.1016/j.jsbmb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Deramaudt TB, Dujardin D, Hamadi A, Noulet F, Kolli K, De Mey J, Takeda K, Ronde P. FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol Biol Cell. 2011;22:964–975. doi: 10.1091/mbc.E10-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre-Guillevin E, et al. PAI-1 and functional blockade of SNAI1 in breast cancer cell migration. Breast Cancer Res. 2008;10:R100. doi: 10.1186/bcr2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Flamini M, Sanchez AM, Goglia L, Giretti MS, Genazzani AR, Simoncini T. Progestogens regulate endothelial actin cytoskeleton and cell movement via the actin-binding protein moesin. Mol Hum Reprod. 2008a;14:225–234. doi: 10.1093/molehr/gan010. [DOI] [PubMed] [Google Scholar]
- Fu XD, Giretti MS, Baldacci C, Garibaldi S, Flamini M, Sanchez AM, Gadducci A, Genazzani AR, Simoncini T. Extra-nuclear signaling of progesterone receptor to breast cancer cell movement and invasion through the actin cytoskeleton. PLoS One. 2008b;3:e2790. doi: 10.1371/journal.pone.0002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Goglia L, Sanchez AM, Flamini M, Giretti MS, Tosi V, Genazzani AR, Simoncini T. Progesterone receptor enhances breast cancer cell motility and invasion via extranuclear activation of focal adhesion kinase. Endocr Relat Cancer. 2010;17:431–443. doi: 10.1677/ERC-09-0258. [DOI] [PubMed] [Google Scholar]
- Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–25021. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- Graham JD, Yager ML, Hill HD, Byth K, O'Neill GM, Clarke CL. Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol. 2005;19:2713–2735. doi: 10.1210/me.2005-0126. [DOI] [PubMed] [Google Scholar]
- Graham JD, Yeates C, Balleine RL, Harvey SS, Milliken JS, Bilous AM, Clarke CL. Characterization of progesterone receptor A and B expression in human breast cancer. Cancer Res. 1995;55:5063–5068. [PubMed] [Google Scholar]
- Guiochon-Mantel A, Delabre K, Lescop P, Milgrom E. Nuclear localization signals also mediate the outward movement of proteins from the nucleus. Proc Natl Acad Sci USA. 1994;91:7179–7183. doi: 10.1073/pnas.91.15.7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiochon-Mantel A, Lescop P, Christin-Maitre S, Loosfelt H, Perrot-Applanat M, Milgrom E. Nucleocytoplasmic shuttling of the progesterone receptor. EMBO J. 1991;10:3851–3859. doi: 10.1002/j.1460-2075.1991.tb04954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamadi A, Bouali M, Dontenwill M, Stoeckel H, Takeda K, Ronde P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J Cell Sci. 2005;118:4415–4425. doi: 10.1242/jcs.02565. [DOI] [PubMed] [Google Scholar]
- Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev. 2007;28:726–741. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- Harbeck N, Kates RE, Schmitt M, Gauger K, Kiechle M, Janicke F, Thomassen C, Look MP, Foekens JA. Urokinase-type plasminogen activator and its inhibitor type 1 predict disease outcome and therapy response in primary breast cancer. Clin Breast Cancer. 2004;5:348–352. doi: 10.3816/cbc.2004.n.040. [DOI] [PubMed] [Google Scholar]
- Hiscox S, Barnfather P, Hayes E, Bramble P, Christensen J, Nicholson RI, Barrett-Lee P. Inhibition of focal adhesion kinase suppresses the adverse phenotype of endocrine-resistant breast cancer cells and improves endocrine response in endocrine-sensitive cells. Breast Cancer Res Treat. 2010;125:659–669. doi: 10.1007/s10549-010-0857-4. [DOI] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Jacobsen BM, Schittone SA, Richer JK, Horwitz KB. Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer: PR isoform-specific gene regulation and tumor biology. Mol Endocrinol. 2005;19:574–587. doi: 10.1210/me.2004-0287. [DOI] [PubMed] [Google Scholar]
- Janicke F, Prechtl A, Thomssen C, Harbeck N, Meisner C, Untch M, Sweep CG, Selbmann HK, Graeff H, Schmitt M. Randomized adjuvant chemotherapy trial in high-risk, lymph node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. J Natl Cancer Inst. 2001;93:913–920. doi: 10.1093/jnci/93.12.913. [DOI] [PubMed] [Google Scholar]
- Jo M, Thomas KS, Marozkina N, Amin TJ, Silva CM, Parsons SJ, Gonias SL. Dynamic assembly of the urokinase-type plasminogen activator signaling receptor complex determines the mitogenic activity of urokinase-type plasminogen activator. J Biol Chem. 2005;280:17449–17457. doi: 10.1074/jbc.M413141200. [DOI] [PubMed] [Google Scholar]
- Kato S, et al. Progesterone increases tissue factor gene expression, procoagulant activity, and invasion in the breast cancer cell line ZR-75-1. J Clin Endocrinol Metab. 2005;90:1181–1188. doi: 10.1210/jc.2004-0857. [DOI] [PubMed] [Google Scholar]
- Khan JA, Amazit L, Bellance C, Guiochon-Mantel A, Lombes M, Loosfelt H. p38 and p42/44 MAPKs differentially regulate progesterone receptor A and B isoform stabilization. Mol Endocrinol. 2011;25:1710–1724. doi: 10.1210/me.2011-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan JA, Bellance C, Guiochon-Mantel A, Lombes M, Loosfelt H. Differential regulation of breast cancer-associated genes by progesterone receptor isoforms PRA and PRB in a new bi-inducible breast cancer cell line. PLoS One. 2012;7:e45993. doi: 10.1371/journal.pone.0045993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labriola L, Salatino M, Proietti CJ, Pecci A, Coso OA, Kornblihtt AR, Charreau EH, Elizalde PV. Heregulin induces transcriptional activation of the progesterone receptor by a mechanism that requires functional ErbB-2 and mitogen-activated protein kinase activation in breast cancer cells. Mol Cell Biol. 2003;23:1095–1111. doi: 10.1128/MCB.23.3.1095-1111.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, Goddard S, Gobert-Gosse S, Corbo L. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008;31:212–221. doi: 10.1016/j.molcel.2008.05.025. [DOI] [PubMed] [Google Scholar]
- Leissner P, Verjat T, Bachelot T, Paye M, Krause A, Puisieux A, Mougin B. Prognostic significance of urokinase plasminogen activator and plasminogen activator inhibitor-1 mRNA expression in lymph node- and hormone receptor-positive breast cancer. BMC Cancer. 2006;6:216. doi: 10.1186/1471-2407-6-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo JC, Wang SM, Guo CH, Aw SE, Zhao Y, Li JM, Hui KM, Lin VC. Gene regulation profile reveals consistent anticancer properties of progesterone in hormone-independent breast cancer cells transfected with progesterone receptor. Int J Cancer. 2005;117:561–568. doi: 10.1002/ijc.21186. [DOI] [PubMed] [Google Scholar]
- Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin VC, Eng AS, Hen NE, Ng EH, Chowdhury SH. Effect of progesterone on the invasive properties and tumor growth of progesterone receptor-transfected breast cancer cells MDA-MB-231. Clin Cancer Res. 2001;7:2880–2886. [PubMed] [Google Scholar]
- Lin VC, Ng EH, Aw SE, Tan MG, Bay BH. Progesterone induces focal adhesion in breast cancer cells MDA-MB-231 transfected with progesterone receptor complementary DNA. Mol Endocrinol. 2000;14:348–358. doi: 10.1210/mend.14.3.0426. [DOI] [PubMed] [Google Scholar]
- Long W, Yi P, Amazit L, LaMarca HL, Ashcroft F, Kumar R, Mancini MA, Tsai SY, Tsai MJ, O'Malley BW. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol Cell. 2010;37:321–332. doi: 10.1016/j.molcel.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo F, Jolivet A, Loosfelt H, Thu vu Hai M, Brailly S, Perrot-Applanat M, Milgrom E. A rapid method of epitope mapping. Application to the study of immunogenic domains and to the characterization of various forms of rabbit progesterone receptor. Eur J Biochem. 1988;176:53–60. doi: 10.1111/j.1432-1033.1988.tb14250.x. [DOI] [PubMed] [Google Scholar]
- Luo M, Guan JL. Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010;289:127–139. doi: 10.1016/j.canlet.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Hong H, Huang SM, Irvine RA, Webb P, Kushner PJ, Coetzee GA, Stallcup MR. Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol Cell Biol. 1999;19:6164–6173. doi: 10.1128/mcb.19.9.6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbaix E, Donnez J, Courtoy PJ, Eeckhout Y. Progesterone regulates the activity of collagenase and related gelatinases A and B in human endometrial explants. Proc Natl Acad Sci USA. 1992;89:11789–11793. doi: 10.1073/pnas.89.24.11789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011;13:202. doi: 10.1186/bcr2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan EM, Clarke CL. Effect of overexpression of progesterone receptor A on endogenous progestin-sensitive endpoints in breast cancer cells. Mol Endocrinol. 1999;13:1657–1671. doi: 10.1210/mend.13.10.0356. [DOI] [PubMed] [Google Scholar]
- McMahon GA, Petitclerc E, Stefansson S, Smith E, Wong MK, Westrick RJ, Ginsburg D, Brooks PC, Lawrence DA. Plasminogen activator inhibitor-1 regulates tumor growth and angiogenesis. J Biol Chem. 2001;276:33964–33968. doi: 10.1074/jbc.M105980200. [DOI] [PubMed] [Google Scholar]
- Michael KE, Dumbauld DW, Burns KL, Hanks SK, Garcia AJ. Focal adhesion kinase modulates cell adhesion strengthening via integrin activation. Mol Biol Cell. 2009;20:2508–2519. doi: 10.1091/mbc.E08-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio A, Piccolo D, Castoria G, Di Domenico M, Bilancio A, Lombardi M, Gong W, Beato M, Auricchio F. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 1998;17:2008–2018. doi: 10.1093/emboj/17.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Lim ST, Chi A, Schlaepfer DD. Intrinsic focal adhesion kinase activity controls orthotopic breast carcinoma metastasis via the regulation of urokinase plasminogen activator expression in a syngeneic tumor model. Oncogene. 2006;25:4429–4440. doi: 10.1038/sj.onc.1209482. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat. 2002;72:163–172. doi: 10.1023/a:1014820500738. [DOI] [PubMed] [Google Scholar]
- Olson D, Pollanen J, Hoyer-Hansen G, Ronne E, Sakaguchi K, Wun TC, Appella E, Dano K, Blasi F. Internalization of the urokinase-plasminogen activator inhibitor type-1 complex is mediated by the urokinase receptor. J Biol Chem. 1992;267:9129–9133. [PubMed] [Google Scholar]
- Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314:1467–1470. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- Proietti C, et al. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 (Stat3) via a Jak- and Src-dependent mechanism in breast cancer cells. Mol Cell Biol. 2005;25:4826–4840. doi: 10.1128/MCB.25.12.4826-4840.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Providence KM, Higgins PJ. PAI-1 expression is required for epithelial cell migration in two distinct phases of in vitro wound repair. J Cell Physiol. 2004;200:297–308. doi: 10.1002/jcp.20016. [DOI] [PubMed] [Google Scholar]
- Rosenthal DT, Iyer H, Escudero S, Bao L, Wu Z, Ventura AC, Kleer CG, Arruda EM, Garikipati K, Merajver SD. p38gamma promotes breast cancer cell motility and metastasis through regulation of RhoC GTPase, cytoskeletal architecture, and a novel leading edge behavior. Cancer Res. 2011;71:6338–6349. doi: 10.1158/0008-5472.CAN-11-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossouw JE, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. J Am Med Assoc. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Sakakibara T, Hibi K, Kodera Y, Ito K, Akiyama S, Nakao A. Plasminogen activator inhibitor-1 as a potential marker for the malignancy of esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10:1375–1378. doi: 10.1158/1078-0432.ccr-03-0196. [DOI] [PubMed] [Google Scholar]
- Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123:1007–1013. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- Schneider J, Lucas R, Sanchez J, Tejerina A, Ruibal A. Cellular accumulation of uPA-PAI-1 [correction of UPA-PAI-1] and uPA-PAI-2 [correction of UPA-PAI-2] complexes in early (pT1) breast cancer: a new link in the uPA-UPAr-PAI chain. In Vivo. 2000;14:507–511. [PubMed] [Google Scholar]
- Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- Sood AK, Coffin JE, Schneider GB, Fletcher MS, DeYoung BR, Gruman LM, Gershenson DM, Schaller MD, Hendrix MJ. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am J Pathol. 2004;165:1087–1095. doi: 10.1016/S0002-9440(10)63370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl A, Mueller BM. Binding of urokinase to its receptor promotes migration and invasion of human melanoma cells in vitro. Cancer Res. 1994;54:3066–3071. [PubMed] [Google Scholar]
- Tang H, Kerins DM, Hao Q, Inagami T, Vaughan DE. The urokinase-type plasminogen activator receptor mediates tyrosine phosphorylation of focal adhesion proteins and activation of mitogen-activated protein kinase in cultured endothelial cells. J Biol Chem. 1998;273:18268–18272. doi: 10.1074/jbc.273.29.18268. [DOI] [PubMed] [Google Scholar]
- Tyagi RK, Amazit L, Lescop P, Milgrom E, Guiochon-Mantel A. Mechanisms of progesterone receptor export from nuclei: role of nuclear localization signal, nuclear export signal, and ran guanosine triphosphate. Mol Endocrinol. 1998;12:1684–1695. doi: 10.1210/mend.12.11.0197. [DOI] [PubMed] [Google Scholar]
- Vincent AJ, Zhang J, Ostor A, Rogers PA, Affandi B, Kovacs G, Salamonsen LA. Decreased tissue inhibitor of metalloproteinase in the endometrium of women using depot medroxyprogesterone acetate: a role for altered endometrial matrix metalloproteinase/tissue inhibitor of metalloproteinase balance in the pathogenesis of abnormal uterine bleeding. Hum Reprod. 2002;17:1189–1198. doi: 10.1093/humrep/17.5.1189. [DOI] [PubMed] [Google Scholar]
- Waltz DA, Natkin LR, Fujita RM, Wei Y, Chapman HA. Plasmin and plasminogen activator inhibitor type 1 promote cellular motility by regulating the interaction between the urokinase receptor and vitronectin. J Clin Invest. 1997;100:58–67. doi: 10.1172/JCI119521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA. Regulation of integrin function by the urokinase receptor. Science. 1996;273:1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- Wei Y, Tang CH, Kim Y, Robillard L, Zhang F, Kugler MC, Chapman HA. Urokinase receptors are required for alpha 5 beta 1 integrin-mediated signaling in tumor cells. J Biol Chem. 2007;282:3929–3939. doi: 10.1074/jbc.M607989200. [DOI] [PubMed] [Google Scholar]
- Weigelt B, Peterse JL, van‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- Wilkins-Port CE, Higgins CE, Freytag J, Higgins SP, Carlson JA, Higgins PJ. PAI-1 is a critical upstream regulator of the TGF-beta1/EGF-induced invasive phenotype in mutant p53 human cutaneous squamous cell carcinoma. J Biomed Biotechnol. 2007:85208. doi: 10.1155/2007/85208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Huang J, Zhou K, Xiang Q, Zhang Y, Tan Z, Simoncini T, Fu X, Wang T. Progesterone enhances vascular endothelial cell migration via activation of focal adhesion kinase. J Cell Mol Med. 2012;16:296–305. doi: 10.1111/j.1582-4934.2011.01305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.