

Abstract
Matrix metalloproteinase-8 (MMP-8) is a potent interstitial collagenase thought to be expressed mainly by PMNs. To determine whether Mmp-8 regulates lung inflammatory or fibrotic responses to bleomycin, we delivered bleomycin by the intratracheal (IT) route to wild type (WT) vs. Mmp-8−/− mice and quantified Mmp-8 expression, and inflammation and fibrosis in the lung samples. Mmp-8 steady-state mRNA and protein levels increase in whole lung and bronchoalveolar lavage samples when WT mice are treated with bleomycin. Activated murine lung fibroblasts express Mmp-8 in vitro. MMP-8 expression is increased in leukocytes in the lungs of patients with idiopathic pulmonary fibrosis compared with control lung samples. Compared with bleomycin-treated WT mice, bleomycin-treated Mmp-8−/− mice have greater lung inflammation, but reduced lung fibrosis. While bleomycin-treated Mmp-8−/− and WT mice have similar lung levels of several pro- and anti-fibrotic mediators (Tgf-β, Il-13, JE, and Ifn-γ), Mmp-8−/− mice have higher lung levels of Ip-10 and Mip-1α. Genetically deleting either Ip-10 or Mip-1α in Mmp-8−/− mice abrogates their lung inflammatory response to bleomycin but reconstitutes their lung fibrotic response to bleomycin. Studies of bleomycin-treated Mmp-8 bone marrow-chimeric mice show that both leukocytes and lung parenchymal cells are sources of pro-fibrotic Mmp-8 during bleomycin-mediated lung fibrosis. Thus, during bleomycin-mediated lung injury, Mmp-8 dampens the lung acute inflammatory response but promotes lung fibrosis by reducing lung levels of Ip-10 and Mip-1α. These data indicate therapeutic strategies to reduce lung levels of MMP-8 may limit fibroproliferative responses to injury in the human lung.
Keywords: MMP-8, fibrosis, inflammation, IP-10, interferon-gamma, mice, bleomycin
INTRODUCTION
Fibrosing lung diseases are some of the most devastating diseases of the lung. Among fibrosing lung diseases, idiopathic pulmonary fibrosis (IPF) is the most common and carries the poorest prognosis. Currently, we lack any effective treatments for this disease. The prevailing hypothesis for the pathogenesis of IPF is that an initial injury to the lung epithelium produces an aberrant and exuberant wound healing process. A characteristic feature of IPF is the presence of fibroblastic foci, which are aggregates of proliferating fibroblasts and myofibroblasts, located beneath breaks in the epithelial basement membrane, which deposit interstitial collagens and other extracellular matrix (ECM) proteins in the lung.
ECM proteins (collagen, elastin, fibronectin, and laminin) accumulate in the lungs of patients with IPF. This process depends on the balance between lung levels of: 1) growth factors and pro-fibrotic cytokines that activate fibroblasts and increase their production of ECM; 2) anti-fibrotic cytokines that inhibit ECM production by fibroblasts; and 3) proteinases, especially MMPs, that degrade ECM proteins. Pro-fibrotic growth factors include TGF-β, which stimulates fibroblasts to migrate, proliferate, transform into myofibroblasts, and synthesize interstitial collagens and other ECM proteins (1). Th-2 cytokines (IL-4 and IL-13) and other mediators such as Mip-1α can also promote fibrotic responses to lung injury (2,3). Anti-fibrotic mediators include interferon-γ (IFN-γ) and IP-10 (IFN-γ inducible protein-10, CXCL10) (4). Mice genetically deficient in either Ip-10 or a key Ip-10 receptor (Cxcr3) by gene targeting have worse fibrosis, and Cxcr3−/− mice have higher mortality after bleomycin instillation when compared with WT mice (4,5). Proteinases, especially MMPs, have important activities in regulating lung inflammatory and fibrotic responses to injury. Mmps cleave and thereby regulate the activities of pro-inflammatory mediators (6–10) and activate latent growth factors such as TGF-β (11,12). In addition, MMPs degrade components of the ECM. The interstitial collagenase subfamily of MMPs (MMP-1,-8, -13, and -14 in man; and Mmp-8, -13, and -14 (13) in mouse) are the key proteinases that degrade interstitial collagens (types I-III). As an interstitial collagenase, MMP-8 cleaves collagen at a single locus, and this cleavage step is rate limiting in collagen degradation (14,15). Interstitial collagenases have been thought to limit fibrotic responses to injury based upon their potent collagen-degrading activities in vitro (15,16), but these findings have not been confirmed in vivo.
MMP-8 (collagenase-2, neutrophil collagenase) is transcribed and translated in PMN precursors in bone marrow in both humans and mice, and stored as latent pro-MMP-8 in the specific granules of mature PMNs (17–19). Upon PMN activation, pro-MMP-8 is released into the extracellular space where it is activated by the cysteine switch mechanism (20–22). Mmp-8 has potent anti-inflammatory activities during LPS-mediated acute lung injury and in allergic airway inflammation in mice (10,23). During LPS-mediated acute lung injury, Mmp-8 reduces lung inflammatory responses to LPS by cleaving and inactivating Mip-1α (10). Mmp-8 also reduces mortality in the hyperoxic model of lung injury in mice, which is mediated in part by Mmp-8 cleaving and inactivating Mip-1α (10). In the ovalbumin (OVA) alloimmune murine model of allergic airway inflammation, Mmp-8 deficient mice have increased granulocytic lung inflammation likely due to reduced inflammatory cell apoptosis in the absence of Mmp-8 (23). However, in a murine model of skin inflammation, Mmp-8 has pro-inflammatory activity and this is mediated by Mmp-8 cleaving and activating the PMN chemokine, lipopolysaccharide-induced CXC chemokine (LIX) (6,24). Mmp-8 also has crucial activities in wound healing because Mmp-8−/− mice have delayed neutrophil infiltration in full thickness skin wounds, delayed resolution of inflammation, and delayed wound healing compared with WT mice due to altered Tgf-β signaling (25). MMP-8 contributes to the generation of the neutrophil chemoattractant proline-glycine-proline (PGP) which promotes emphysema pathogenesis in mice (26,27). Recently an association was found between MMP-8 gene variation and the extent of atherosclerosis in patients with coronary artery disease (28).
Although MMP-8 is a potent type I collagen-degrading proteinase which might be expected to reduce lung fibrotic responses to injury, Garcia-Prieto et al. showed recently that Mmp-8 reduces lung inflammation but promotes lung fibrotic responses to bleomycin in mice by cleaving il-10 (29). Our previous work has shown that Mmp-8 regulates the accumulation of PMNs and macrophages in the lung during LPS-mediated lung injury, at least in part, by cleaving and inactivating Mip-1α (10). Herein, we have built upon the prior studies of Garcia Prieto by identifying which leukocyte subsets in the lung are regulated by Mmp-8 during bleomycin-mediated acute lung injury and the mechanisms involved. We also assessed whether Mmp-8 regulates lung inflammatory and fibrotic responses to injury by reducing lung levels of Mip-1α and/or other mediators. Additionally, to identify the crucial cellular sources of Mmp-8 in the lung mediating the activities of this proteinase in this model, we measured lung fibrotic response to bleomycin in Mmp-8 bone marrow-chimeric mice. We found that bleomycin-treated Mmp-8−/− mice have higher lung macrophage and CD4+ T cells than bleomycin-treated WT mice. When compared with bleomycin-treated WT mice, Mmp-8−/− mice are protected from bleomycin-induced lung fibrosis and have reduced accumulation of myofibroblasts in the lung, and this is associated with higher lung levels of Mip-1α and Ip-10 in bleomycin-treated Mmp-8−/− mice. Genetic deletion of either Ip-10 or Mip-1α in Mmp-8−/− mice reduces their lung inflammatory response to bleomycin, and restores their fibroproliferative responses to bleomycin. These data indicate that Ip-10 and Mip-1α are the key molecules in the lung regulated by Mmp-8 during bleomycin-mediated lung injury. We have also shown for the first time that both bone marrow-derived leukocytes and lung parenchymal cells are crucial cellular sources of pro-fibrotic Mmp-8 during bleomycin-mediated lung injury. Our results indicate that strategies to inhibit MMP-8 activity or reduce MMP-8 levels in the lungs may limit lung fibrotic responses to injury. Thus, MMP-8 may be a novel therapeutic target for IPF and other fibrotic lung diseases.
MATERIALS AND METHODS
Materials
Recombinant human MMP-8 and rabbit anti-MMP-8 IgG was purchased from Millipore (Billerica, MA). Murine Mmp-8, human IP-10, and the ELISA kit for TGF-β were purchased from R & D Systems (Minneapolis, MN). The ELISA kit for measuring lung levels of Mmp-8 in mice was purchased from MyBioSource, Inc. (San Diego, CA). Recombinant murine Il-4 and Il-9, and the ELISA kits for measuring Mip-1α, Ip-10, and Ifn-γ were purchased from PeproTech (Rocky Hill, NJ). The ELISA kits for quantifying Il-13, Il-4, Il-9, and JE were purchased from eBioscience (San Diego, CA). The p-aminophenylmercuric acetate (APMA), 1,10 phenanthroline, Sigma-Proteinase Inhibitor Cocktail, phenylmethylsulphonyl fluoride (PMSF), alkaline phosphatase coupled monoclonal mouse anti-smooth muscle actin clone 1A4, Masson’s Trichrome stain kit, Bouin’s solution, Weigert’s iron hematoxylin solutions, and dithiothreitol (DTT) were purchased from Sigma-Aldrich (St. Louis, MO). The Silver Xpress silver staining kit was purchased from Invitrogen (Carlsbad, CA). Bleomycin was purchased from Henry Schein Animal Health (Melville, NY). Ketamine and xylazine were purchased from Webster Veterinary (Sterling, MA). The DNeasy DNA extraction kit was purchased from Qiagen (Valencia, CA). The Vector Red alkaline phosphatase substrate kit was purchased from Vector Laboratories (Burlingame, CA). Anti-CD-4 IgG, CD-8, and Gr-1 antibodies were purchased from BD Biosciences (San Jose, CA). Goat anti-rabbit IgG-horseradish peroxidase conjugate was purchased from Bio-Rad (Hercules, CA).
Animals
All procedures performed on mice were approved by the Harvard Medical School Animal Care1 and Use Committee. All mice were housed in a barrier facility under specific pathogen-free conditions. Mmp-8−/− mice were generated in the mixed SVeV129 X C57BL/6 strain (6) and backcrossed 10 generations into the pure C57BL/6 strain. The Mmp-8−/− mice have normal lifespan, fertility, and lung development, and no abnormality in the unchallenged state (6). We initially studied parental C57BL/6 wild type littermate mice as our experimental controls for C57BL/6 Mmp-8−/− mice. Ip-10−/− mice in the pure C57BL/6 strain were purchased from The Jackson Laboratory (Bar Harbor, ME). Mmp-8−/− X Ip-10−/− mice were generated by crossing Mmp-8−/− and Ip-10−/− mice. Mip-1α−/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mmp-8−/− X Mip-1α−/− mice were generated by crossing Mmp-8−/− and Mip-1α−/− mice.
Genotyping
The genotypes of all mice bred in house were confirmed by PCR-based genotyping protocols on DNA extracted from murine tails using Qiagen DNeasy extraction kits.
Bleomycin-mediated acute lung injury in mice
Age- and gender-matched adult mice (10–16 weeks old) were anesthetized with ketamine (200 mg/Kg) and xylazine (10 mg/Kg) and then given 60, 75, or 100 mU of bleomycin in 30 μl of endotoxin-free normal saline or 30 μl of normal saline alone by the IT route. Mice were euthanized by CO2 narcosis followed by cervical dislocation at various intervals after IT bleomycin or saline. Preliminary experiments confirmed that the dose of bleomycin tested and the time points studied were optimal for each endpoint being studied. Twenty one days after treating mice with 60 mU of bleomycin, right lungs were inflated with PBS to 25 cm H20 and fixed in 10% (v/v) buffered formalin, and left lungs were snap frozen in liquid nitrogen and stored at −80°C for either hydroxyproline assays or subsequently homogenized to quantify growth factors, cytokines, and chemokines using ELISAs. Hematoxylin and eosin and Masson’s Trichrome staining was performed on fixed and inflated lung sections. Three, 10, and 14 days after treating mice with 75 mU of bleomycin by the IT route, we performed bronchoalveolar lavage (BAL) and prepared lung digests and lung homogenates to measure lung inflammation. At intervals after treating mice with 100 mU of bleomycin by the IT route, we prepared homogenates of lungs in PBS containing 0.5% triton to measure Ip-10 and Mip-1α using ELISAs, or extracted RNA from lung or bronchoalveolar (BAL) leukocyte samples to measure steady state Mmp-8 mRNA levels using real time RT-PCR. To measure lung inflammation in bleomycin-treated mice, BAL was performed using eight 0.5-ml aliquots of sterile PBS. The BAL cell and supernatant fractions were separated by centrifugation (500 g for 3 min). The BAL supernatant fractions were frozen to −80°C for quantitation of chemokines and cytokines using ELISAs. Erythrocytes were removed from the cell fraction by hypotonic lysis. Total and differential WBC counts were performed on the BAL leukocyte samples.
Real time RT-PCR to quantify Mmp-8 and Ip-10 expression in lung samples
Lungs were removed from WT mice 3, 5, and 7 days after delivering either 100 mU of bleomycin or normal saline by the IT route. BAL was performed on WT mice 7–21 days after delivering 100 mU of bleomycin or saline by the IT route to isolate lung leukocytes. Primary lung fibroblasts were isolated from the lungs of unchallenged WT mice, cultured at 37°C until they were 80% confluent. Cells were then incubated at 37°C for up to 24 h with or without varying concentrations (1–10 ng/ml) of recombinant active human TGF-β1. RNA was isolated from lung, BAL leukocytes, and murine lung fibroblasts using a TRIzol® reagent method (30). Reverse transcription of the RNA samples was performed using a RETROscript® kit from Applied Biosystems (Carlsbad, CA) according to kit instructions. Real-time RT-PCR analysis was performed using a Stratagene MxPro instrument (Agilent Technologies, Santa Clara, CA) and a TaqMan® Mmp-8 or Ip-10 gene expression assay (Invitrogen). We used the comparative cycle threshold method, cyclophilin, GAPDH, or 18 S as endogenous reference genes, and FAM as the fluorophore.
Quantification of Mmp-8 protein levels in BAL fluid (BALF) samples from mice
WT mice were treated with 100 mU of bleomycin or saline and 3, 7, 14, and 21 days later, BAL was performed using 1 ml of PBS per mouse. BAL cells were removed by centrifuging the samples (500 g for 5 min). Mmp-8 protein levels were measured in cell-free BALF samples using a commercially available ELISA kit.
Immunohistochemistry
Formalin-fixed lung sections from patients with IPF and from normal lung were deparafinized. Antigen retrieval was performed by incubating slides in boiling 0.01 M Tris containing 1 mM EDTA (pH 9.0) in a pressure cooker in a microwave for 3 min. Slides were then blocked with 1% (w/v) BSA and 10% (v/v) goat serum in Tris buffered saline (TBS; 0.05M Tris containing 0.15 M NaCL and 0.02 M CaCl2) for 2 h at room temperature. Slides were then incubated with either rabbit anti-MMP-8 IgG or non-immune rabbit IgG for 18 h at 4°C. Slides were then rinsed twice with TBS. Slides were incubated in 3% hydrogen peroxide for 20 min, washed, incubated again with hydrogen peroxide, washed, and then incubated for 1 h at room temperature with goat anti-rabbit horseradish peroxidase conjugate (Bio-Rad) in serum block. Slides were then rinsed, incubated in avidin biotin complex (ABC) for 1 h at room temperature, rinsed again, and developed with 3,3′-diaminobenzidine (DAB). Slides were then counterstained with 1% (wt/v) methyl green, dehydrated, and mounted.
Enzymatic lung digestion and cell staining
Lungs were removed from mice after flushing the right ventricle with 30 mL PBS, minced, incubated with 0.1% (w/v) type IV collagenase and 0.05% (w/v) DNAase for 1 h at 37°C. The digestion solution was then run through a 70 μm cell strainer, and red cells were removed using a hypotonic lysis step. Cells were then resuspended in 44% (v/v) Percoll, overlaid on 67% (v/v) Percoll, and centrifuged at 710 g for 20 min. Cells were harvested from the interphase, washed, counted, and resuspended at 10 million cells per mL in PBS containing 2% (v/v) FBS with rat anti-mouse CD16/32 IgG2b κ for 20 min at 4°C to block non-specific binding of antibodies. Cells were then double immunostained at 4°C with either PE-conjugated anti-CD4 IgG 2b κ, anti-CD8 IgG 2b κ, or anti-Gr-1 IgG 2bκ; and FITC-conjugated anti-CD45 IgG 2b κ, or appropriate isotype-matched control antibodies (PE-conjugated anti-rat IgG 2b κ and FITC-conjugated anti-rat IgG 2b κ). The percentage of PE and FITC double positive leukocytes was measured using flow cytometry after subtracting non-specific staining detected on cells incubated with the isotype control matched antibodies.
Hydroxyproline assay for lung collagen content
The hydroxyproline lung content of murine lungs was measured exactly as previously described (31).
Histology
The right lungs of bleomycin- or saline-treated mice were inflated to 25 cm H20 and fixed in 10% (v/v) buffered formalin and embedded in paraffin. Mid-saggital lung sections were stained with hematoxylin and eosin or Masson’s Trichrome stain. Other lung sections were stained for smooth muscle actin using a Vector Red alkaline phosphatase developing kit according to kit instructions and counterstaining with 1% (w/v) methyl green.
Lung levels of pro- and anti-inflammatory mediators and pro- and anti-fibrotic mediators
WT and Mmp-8−/− mice were treated with either bleomycin or saline via the IT route. Lungs were removed at intervals and homogenized in PBS containing 0.5% (v/v) Triton X-100 and Sigma-Mammalian Proteinase Inhibitor Cocktail, 1 mM PMSF, and 1mM 1,10 phenanthroline. Tgf-β, Ifn-γ, Ip-10, JE, Il-9, Il-4, Il-10, and Il-13 were quantified in BAL samples and lung homogenates using commercially-available ELISAs.
In vitro assays to determine whether Mmp-8 cleaves mediators that regulate lung inflammation and fibrosis
Recombinant human pro-MMP-8 (Millipore, 170 nM) or recombinant murine pro-Mmp-8 (R&D Systems, 0.96μM) was incubated with 1mM amino-phenyl mercuric acetate [to activate pro-Mmp-8 (10)] with or without recombinant human IP-10 (R&D Systems, 5μM), recombinant murine Il-4 (PeproTech, 1.85 μM), or recombinant murine Il-9 (PeproTech, 1.75 μM) in TBS [50 mM Tris containing 150 mM NaCl, 20 mM CaCl2; and 0.05% (v/v) Triton; pH 7.5] with for 18 h at 37°C or buffer alone as a control. Reaction products were then separated on 16.5% (v/v) Tris Tricine gels at 60 V for 5 h and developed with a SilverXpress® (Invitrogen) staining kit according to the kit manufacturer’s instructions.
Bone marrow chimeric mice experiments
WT and Mmp-8−/− recipient mice in the pure C57BL/6 background aged 10–16 weeks were irradiated twice with 450 cGy 4 h apart. Bone marrow (BM) was isolated from WT or Mmp-8−/− donor mice, and 2 million BM cells were injected into the tail veins of each mouse in a volume of 20 μl of PBS. Mice were housed for 8–10 weeks to permit engraftment of BM, and Mmp-8 BM chimeric mice were then treated with either saline or 60 mU of bleomycin by the IT route. Mmp-8 BM chimeric mice (WT BM donors into WT recipients, Mmp-8−/− BM donors into Mmp-8−/− recipients, WT BM donors into Mmp-8−/− recipients, and Mmp-8−/− BM donors into WT recipients) were euthanized 21 days after delivering either saline or 60 mU of bleomycin via the IT route. The right lungs were inflated with PBS to 25 cm H20 and fixed in 10% (v/v) buffered formalin. The left lungs were snap frozen in liquid nitrogen and stored at −80°C for hydroxyproline assay.
Statistics
Data are expressed as mean ± SEM or mean ± SD. The results for paired and unpaired data were compared by the Student’s t-test for parametric data and the Mann-Whitney rank sum test for non-parametric data; p values less than 0.05 were considered significant.
RESULTS
Mmp-8 expression in the lung is increased during bleomycin-mediated lung injury
In order to investigate whether Mmp-8 expression is regulated during bleomycin-mediated lung injury, we delivered bleomycin or saline by the IT route to WT mice and 3, 5, and 7 days later, measured Mmp-8 steady state mRNA levels in whole lung samples using real time RT-PCR. Bleomycin-treated WT mice have a mean ~4-fold increase in steady state Mmp-8 mRNA levels in their lungs on day 7 compared with saline-treated WT mice (Fig. 1A). In addition, Mmp-8 protein levels in BALF samples significantly increase 7 days after delivering bleomycin, levels peak at 14 days, and Mmp-8 protein levels remained elevated 21 days after delivering bleomycin to WT mice (Fig. 1B).
Figure 1. Mmp-8 levels increase in the lungs during bleomycin-mediated lung injury in mice.
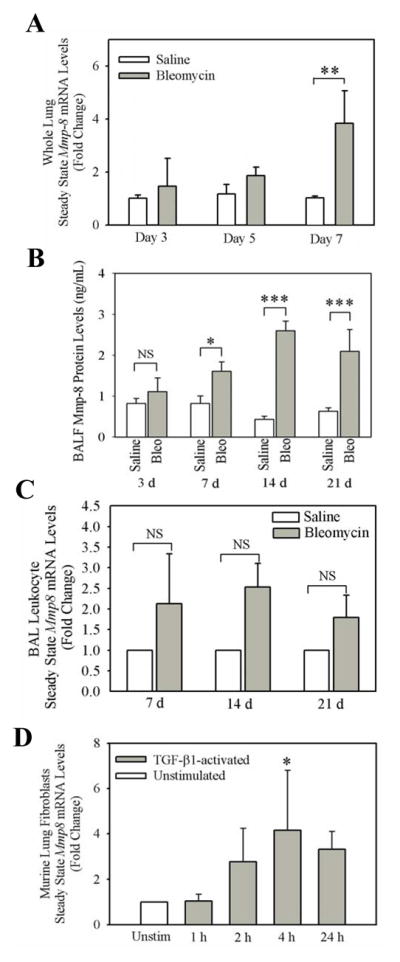
In A, real time RT-PCR was performed to measure Mmp8 steady-state mRNA levels in WT lung 3, 5, and 7 d after delivering 100 mU of bleomycin in normal saline or normal saline by the IT route using cyclophilin as the housekeeping gene and the ΔΔCT method. Data are expressed as mean fold change in expression ± SEM; n = 3–13 mice/group. Double asterisk indicates p = 0.008. In B, WT mice were treated with 100 mU of bleomycin in normal saline or normal saline by the IT route. After 3–21 days, BAL was performed on the mice and Mmp-8 protein levels were quantified in cell-free BALF samples using an ELISA. Data are mean ± SEM; n = 3–10 mice/group for saline-treated mice, n = 4–9 mice/group for bleomycin-treated mice. In A and B, asterisk indicates p < 0.05; **, p <0.01; and ***, p <0.005. In C, real time RT-PCR was performed to measure Mmp-8 steady-state mRNA levels in leukocytes in BAL samples isolated from WT mice 7, 14, or 21 days after 100 mU of bleomycin (n = 4–6 mice/group) or saline (n = 2–5 mice/group) was delivered by the IT route. Fold change in Mmp-8 gene expression was measured using 18S as the housekeeping gene and the ΔΔCT method. In D, Mmp8 steady-state mRNA levels were measured in primary murine lung fibroblasts (isolated from 4–5 mice/group) that were incubated at 37°C with or without 10 ng/mL recombinant active TGF-β1 for 1–24 h using real time RT-PCR, cyclophilin as the housekeeping gene, and the ΔΔCT method. Data in C and D are expressed as mean fold change in expression ± SEM. In D, asterisk indicates p = 0.016 compared with unstimulated fibroblasts.
Mmp-8 expression increases murine lung fibroblasts activated with TGF-β1 in vitro
Next, we investigated which cells in the lung produce Mmp-8 during bleomycin-mediated lung injury. We first attempted to immunostain lung sections from bleomycin-treated WT mice for Mmp-8. However, we were not able to detect specific staining for Mmp-8 in lung sections despite testing several commercial antibodies on lungs fixed with different fixatives. Next, we isolated and studied leukocytes from the lungs of bleomycin- vs. saline-treated WT mice as PMNs are known to be an important source of Mmp-8, and macrophages can be induced to express Mmp-8 transcripts when activated in vitro (32,33). Although BAL leukocytes express Mmp-8 transcripts, steady state Mmp-8 mRNA levels did not significantly increase in BAL leukocytes isolated from the mice 7–21 days after bleomycin was delivered (Fig. 1C).
Human fibroblasts isolated from the synovium of patients with rheumatoid arthritis and gingival fibroblasts express MMP-8 (34,35), but it is not known whether lung fibroblasts express MMP-8 in health or during pathologic processes. Accordingly, we measured Mmp-8 expression in primary murine lung fibroblasts which were either not stimulated or activated in vitro with an agonist that is relevant to the pathogenesis of IPF (active TGF-β1). Unstimulated murine primary lung fibroblasts express low levels of Mmp-8 mRNA transcripts and levels do not increase over time when cells are incubated in the absence of agonists (as assessed by calculating ΔCT; data not shown). However, incubating fibroblasts with 10 ng/ml recombinant active TGF-β1 induces ~4-fold increases in Mmp-8 steady state mRNA levels after 4 h (Fig. 1D). Time course and dose response experiments confirmed that our experimental conditions are optimal for inducing Mmp-8 expression in lung fibroblasts using TGF-β1 (Supplement Fig. 1). Thus, Mmp-8 gene expression is up-regulated in leukocytes recruited to the lung and likely activated lung fibroblasts in bleomycin-treated mice.
Mmp-8 expression increases in leukocytes in the lungs of human IPF patients
We immunostained lung sections from human IPF patients (n = 4) with an antibody to human MMP-8 or an isotype control matched primary antibody. As a control, we immunostained sections of normal human lung tissue around surgically excised benign lung tumors (n = 2). We observed prominent staining for MMP-8 in leukocytes in the airways and the lung parenchyma in IPF lung samples. Representative immunostaining results are shown in Fig. 2. In contrast, there is minimal or no staining of leukocytes in the lungs of the IPF lung sections stained with the non-immune control primary antibody, and no positive staining in normal lung sections stained with the anti-MMP-8 antibody (Fig. 2). Although bronchial epithelial cells are stained when we incubate IPF lung sections with the anti-MMP-8 antibody, similar staining is detected in bronchial epithelial cells stained with the non-immune control antibody (as assessed by a senior pathologist; LK) indicating that this airway epithelial staining is not specific for MMP-8.
Figure 2. MMP-8 expression is increased in leukocytes in the lungs of patients with IPF.

The photomicrographs show lung sections from an IPF patient (top panels) and normal lung (bottom panels) stained with either rabbit anti-MMP-8 IgG (left panels) or non-immune rabbit IgG (right panels). Magnification is X 400. Note the positive (brown) staining for MMP-8 in leukocytes in lumen of the airway shown. The photomicrographs shown are representative of immunostained lung sections from 4 IPF patients and 2 control cases.
Mmp-8 promotes fibrosis during bleomycin-mediated lung injury
To determine whether Mmp-8 regulates lung fibrotic responses to lung injury, we compared lung fibroproliferative responses of WT vs. Mmp-8−/− mice harvested 21 days after the mice were treated with either bleomycin or saline by the IT route. Saline-treated WT and Mmp-8−/− mice have no evidence of pathology in hematoxylin and eosin-stained lung sections, as expected (Fig. 3A). Bleomycin-treated WT mice have robust cellular infiltrates in their lungs after 21 days. However, the lungs of bleomycin-treated Mmp-8−/− mice are markedly less cellular than those of bleomycin-treated WT mice after 21 days. In addition, bleomycin-treated Mmp-8−/− mice have reduced lung collagen deposition after 21 days as assessed by Masson’s Trichrome-stained lung sections (Fig. 3B) and quantitative hydroxyproline assays (Fig. 3D). Immunostaining of lung sections for alpha smooth muscle actin (a marker of myofibroblasts) shows minimal staining in saline-treated WT and Mmp-8−/− mice, as expected. There is robust staining for alpha smooth muscle actin in the lungs of bleomycin-treated WT mice, but this is markedly diminished in bleomycin-treated Mmp-8−/− mice (Fig. 3C). These results confirm those of an earlier study by Garcia-Prieto et al. that Mmp-8 promotes lung fibrotic responses to bleomycin and increases lung myofibroblast numbers (29).
Figure 3. Mmp-8 has pro-fibrotic activities in the lungs of bleomycin-treated mice.
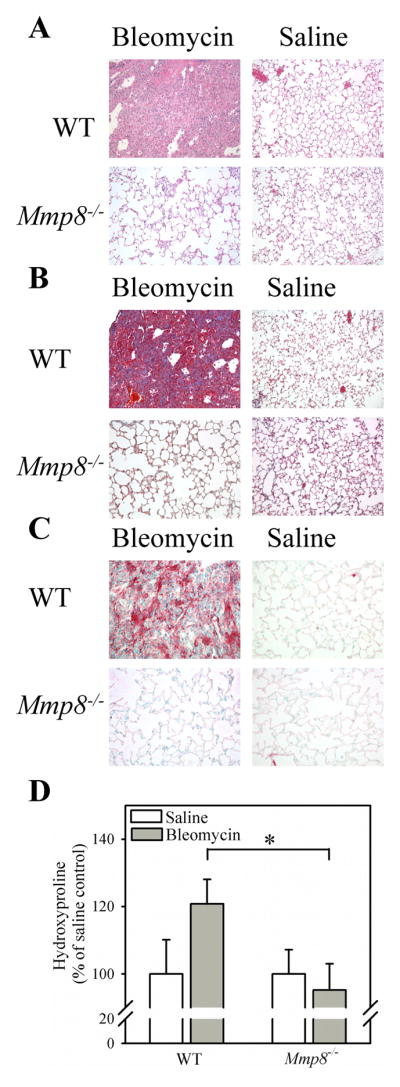
In A–C, WT and Mmp-8−/− mice in the pure C57BL/6 strain were given 75 mU of bleomycin in normal saline or normal saline by the IT route. After 14 days, right lungs were inflated and fixed in formalin and lung sections were stained with hematoxylin and eosin (in A), with Masson’s Trichrome stain (in B), or with anti-α-smooth muscle actin (in C). In A–C, lung sections are representative of 6 mice in each group. In D, collagen was quantified in left lungs using hydroxyproline assays. Data are mean ± SEM; n = 16–19 mice per group. Asterisk indicates p = 0.028.
Mmp-8 reduces acute lung inflammation after bleomycin-mediated lung injury
Mmp-8 regulates the accumulation of PMNs and macrophages in the lung during LPS-mediated acute lung injury (10). Mmp-8 also increases total leukocyte counts 7 days after delivering IT bleomycin, but the leukocyte subsets regulated by Mmp-8 in this model were not determined in our earlier study (10). Lung inflammation regulates lung fibrosis in the murine bleomycin model (36). Therefore, we quantified leukocyte subsets in the lungs of WT and Mmp-8−/− mice 3–14 days after delivering bleomycin or saline by the IT route. Bleomycin induces a robust and complex lung inflammatory response in both WT and Mmp-8−/− mice which peaks at 7 days and consists predominantly of macrophages (mean = 67% and 65% for bleomycin-treated WT mice and Mmp-8−/− mice, respectively) and lymphocytes (mean = 24% and 26% for bleomycin-treated WT mice and Mmp-8−/− mice, respectively). PMNs represent only ~9% of BAL leukocytes on average in both genotypes 7 days after II instillation of bleomycin. However, lung inflammatory responses are significantly higher in bleomycin-treated Mmp-8−/− mice compared with bleomycin-treated WT mice on day 7 (Fig. 4A). The increased lung inflammatory response in bleomycin-treated Mmp-8−/− mice is predominantly due to higher BAL macrophage counts on day 7 in Mmp-8−/− mice (Fig. 4B). We also found a strong trend (p = 0.074) towards higher BAL lymphocyte counts on day 7 (Fig. 4C) in bleomycin-treated Mmp-8−/− mice versus WT mice, but BAL PMN counts are similar in bleomycin-treated WT and Mmp-8−/− mice at all time points examined (Fig. 4D). Lymphocytes (37–41) and PMNs (42–46) have crucial activities in regulating lung fibrotic responses to injury. However, PMN and lymphocyte counts are relatively low in BAL samples from the mice. Thus, we also quantified these leukocytes and lymphocyte subsets in lung digests from bleomycin-treated WT vs. Mmp-8−/− mice.
Figure 4. Mmp-8 reduces acute lung inflammation during bleomycin-mediated lung injury.

WT and Mmp-8−/− mice both in the pure C57BL/6 strain were given 75 mU of bleomycin in normal saline or normal saline by the IT route and BAL was performed 3–10 days after bleomycin was delivered. In A, all leukocytes were counted in BAL samples. Data are expressed as mean ± SEM; n = 3–7 mice/group for saline-treated mice and n = 8–18 mice/group for bleomycin-treated mice. B shows absolute macrophage counts, C shows absolute lymphocyte counts, and D shows absolute PMN counts in BAL samples 7 days after mice were treated with either bleomycin or saline. In B–D, data are expressed as mean ± SEM; n = 4–7 mice/group for saline-treated animals and n = 11–18 mice/group for bleomycin-treated animals. Asterisk indicates p < 0.05; **, p <0.01; and ***, p <0.005.
Mmp-8 increases CD4+ T cells in lungs after bleomycin-mediated lung injury
On day 7 after IT instillation of bleomycin, lungs from WT and Mmp-8−/− mice were enzymatically digested and stained with FITC for CD45, and with PE for CD4, CD8, or the PMN markers, Ly6C and Ly6G. Double immunostained cells were then analyzed by flow cytometry. We found significantly higher numbers of CD4+ T cells in the lungs of bleomycin-treated Mmp-8−/− mice compared with bleomycin-treated WT mice (Fig. 5). However, bleomycin-treated WT and Mmp-8−/− mice do not differ significantly in the numbers of CD8+ T cells (data not shown) or PMNs in enzymatic lung digest samples.
Figure 5. Mmp-8 reduces the number of CD4+ T cells in bleomycin-injured lungs.
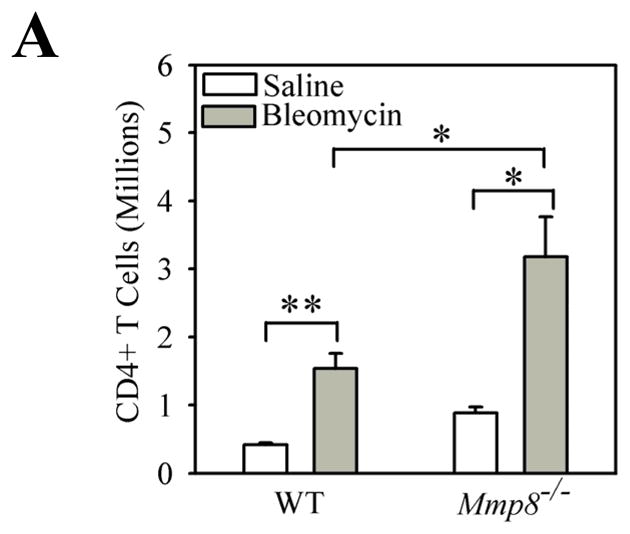
WT and Mmp-8−/− mice were treated with bleomycin or saline by the IT route as outlined above, and after 7 days lungs were removed, and digested with collagenase and DNAse. Cells were double immunostained with FITC for CD45 to identify leukocytes and with PE for CD4 as described in methods. The percentage of lung leukocytes that stained positively for CD4 was determined by flow cytometry and absolute number of cells calculated. Data are mean ± SEM; n = 4 mice/group. Asterisk indicates p < 0.05 and **, p <0.01.
Mmp-8 does not regulate lung levels of growth factors that promote lung fibrosis in bleomycin-treated mice
Next, we investigated the mechanism by which Mmp-8 promotes lung fibrotic responses to injury. We considered the possibility that Mmp-8 cleaves and thereby regulates the levels or biologic activities of growth factors or anti-fibrotic mediators in the lung. Growth factors such as Tgf-β drive lung fibrotic responses to injury and are produced in latent forms which require activation to attain full activity (47–49). Mmps other than Mmp-8 activate latent growth factors in vitro (11). Thus, we measured lung levels of latent and active growth factors and anti-fibrotic mediators in lungs removed from bleomycin-treated WT and Mmp-8−/− mice. Levels of several pro-fibrotic mediators (total Tgf-β and active Tgf-β, Il-13, and JE) are similar in bleomycin-treated WT and Mmp-8−/− mice (Figs. 6A and B, and Supplement Table I). Lung levels of Il-4 and Il-9 are higher in bleomycin-treated Mmp-8−/− mice compared with bleomycin-treated WT mice (Supplement Table I). There is a strong trend towards lower lung levels of Il-10 in Mmp-8−/− mice 48 h after instilling bleomycin, but WT and Mmp-8−/− mice do not differ in lung Il-10 levels 21 days after delivering bleomycin (Supplement table 1). When we incubate purified active murine Mmp-8 with recombinant murine Il-4 or Il-9, Mmp-8 does not cleave these mediators as assessed by analysis of the reaction products separated on silver-stained Tris Tricine gels (supplemental Figs. 2A and 2B). Thus, it is unlikely that Mmp-8 reduces lung levels of Il-4 or Il-9 by proteolytically cleaving them. The increased lung levels of Il-4 and Il-9 in bleomycin-treated Mmp-8−/− versus WT mice is quite modest in magnitude and could be secondary to the increased lymphocyte counts in bleomycin-treated Mmp-8−/− mice because T cells produce these mediators (50,51).
Figure 6. Compared with bleomycin-treated WT mice, bleomycin-treated Mmp-8−/− mice have similar lung levels of total and active Tgf-β protein but higher lung levels of anti-fibrotic Ip-10 protein.

In A and B, WT and Mmp-8−/− mice were treated with 75 mU of bleomycin in normal saline or normal saline by the IT route. After 2–10 days, lungs were removed and active and total Tgf-β (A and B, respectively), and Ip-10 (C) levels quantified in homogenates of lungs using ELISAs. Data are mean ± SEM; n = 2 mice/group for saline-treated mice, n = 4–20 mice/group for bleomycin-treated mice. In D, real time RT-PCR was performed to measure Ip-10 steady-state mRNA levels in BAL leukocytes isolated from WT and Mmp8−/− mice 7 days after either saline (n=2–3 different preparations per genotype) or 100 mU of bleomycin (n = 4 WT mice and n = 4 Mmp-8−/− mice) was delivered by the IT route. We used 18 S as the housekeeping gene and the ΔΔCT method to calculate fold change in Ip-10 mRNA levels. Data are expressed as mean fold change in expression ± SEM. In A–D, ** indicates p <0.01; and ***; p <0.005.
Mmp-8−/− mice have higher lung levels of the anti-fibrotic mediator, Ip-10, during bleomycin-mediated lung injury
To test whether Mmp-8 promotes lung fibrosis by reducing lung levels of anti-fibrotic mediators, we measured lung levels of Ip-10 and Ifn-γ in lung homogenates from bleomycin-treated WT and Mmp-8−/− mice. Bleomycin-treated WT and Mmp-8−/− mice have similar lung levels of Ifn-γ in lung homogenates (Supplement Table I). Mmp-8−/− mice have significantly higher Ip-10 protein levels in lung homogenates than WT mice after they receive bleomycin (Fig. 6C) although the difference between the genotypes is modest in magnitude (less than 2-fold). When we measured Ip-10 levels in BALF samples from the mice, we found a strong trend towards higher Ip-10 protein levels in BALF from bleomycin-treated Mmp-8−/− mice compared with BALF from WT mice (Supplement Fig. 3). However, BALF Ip-10 protein levels are low in both genotypes relative to levels detected in lung homogenates from bleomycin-treated WT and Mmp8−/− mice. Measuring Ip-10 protein levels in whole lung or BALF samples potentially could dilute out substantial signals derived from specific groups of cells in the lungs. Thus, we also measured Ip-10 steady state mRNA levels in BAL leukocytes isolated from bleomycin-treated WT and Mmp8−/− mice at the time point corresponding to peak lung inflammation (day 7). BAL leukocytes from bleomycin-treated Mmp-8−/− mice have substantially (~9.6-fold) higher Ip-10 steady state mRNA levels than BAL leukocytes from bleomycin-treated WT mice (Fig. 6D). Thus, Mmp-8 potently reduces the expression of Ip-10 in leukocytes recruited to the lung during bleomycin-mediated lung injury.
Genetic deletion of Ip-10 in Mmp-8−/− mice reconstitutes their fibrotic lung responses to bleomycin-mediated lung injury
Lung levels of anti-fibrotic Ip-10 are higher in bleomycin-treated Mmp-8−/− mice. This could potentially explain the resistance of Mmp-8−/− mice to bleomycin-induced lung fibrosis. We tested this possibility by genetically deleting Ip-10 in Mmp-8−/− mice and comparing lung fibrotic responses to bleomycin in these Mmp-8−/− X Ip-10−/− compound mutant mice vs. single mutant mice. Genetic deletion of Ip-10 in Mmp-8−/− mice reconstitutes the lung fibroproliferative response of Mmp-8−/− mice to bleomycin. Lung collagen deposition in Mmp-8−/− X Ip-10−/− mice is similar to that in bleomycin-treated WT mice as assessed by histology in hematoxylin and eosin- (Fig. 7A), Masson’s Trichrome- (Fig. 7B) stained lung sections, and quantitative hydroxyproline assays performed on lung hydrolysates (Fig. 7D). Genetic deletion of Ip-10 in Mmp-8−/− mice also restores myofibroblasts accumulation in their lungs (Fig. 7C). Thus, Mmp-8’s pro-fibrotic activities are due, at least in part, to Mmp-8 decreasing Ip-10 levels in vivo. When we tested the activity of active MMP-8 against IP-10 in vitro, we found that neither human MMP-8 nor murine Mmp-8 cleaves IP-10 (a representative gel is shown in Supplement Fig. 1C).
Figure 7. Genetic deletion of Ip-10 in Mmp-8−/− mice reconstitutes their fibrotic lung responses to bleomycin-mediated lung injury.

In A–D, WT, Mmp-8−/−, Mmp-8−/− X Ip-10−/−, and Ip-10−/− mice were treated with either 60 mU of bleomycin or saline by the IT route and 21 days later, right lungs were inflated, fixed, and stained with hematoxylin and eosin in A, Masson’s Trichrome stain in B, and with anti-α-smooth muscle actin antibody in C. Original magnification X100. Hematoxylin and eosin-stained lung sections (A) are representative of 2–4 mice/group for saline-treated mice and 6–16 mice/group for bleomycin-treated mice. Masson’s Trichrome- and anti-α-smooth muscle actin antibody-stained sections are representative of 4 mice in each group. In D, collagen was quantified in left lungs using hydroxyproline assays. Data are mean ± SEM; n = 2–4 mice/group for saline-treated mice, n = 6–16 mice/group for bleomycin-treated mice. Asterisk indicates p < 0.05 and **, p <0.01.
Genetic deletion of Ip-10 in Mmp-8−/− mice abrogates the increased BAL leukocyte counts observed in bleomycin-treated Mmp-8−/− mice
We also investigated whether Ip-10 regulates inflammation in bleomycin-mediated lung injury because: 1) Ip-10 is a chemokine for monocytes/macrophages, T cells, and NK cells (52–54); and 2) the intensity of the lung inflammatory response to bleomycin can regulate subsequent lung fibrosis in this model (3,37,55,56). Compared with bleomycin-treated WT mice, bleomycin-treated Ip-10−/− mice have similar total leukocyte, macrophage, PMN, and lymphocyte counts in BAL samples (Fig. 8A – 8D). However, the increased total leukocyte count and the elevated macrophage, PMN, and lymphocyte counts in BAL samples in bleomycin-treated Mmp-8−/− mice are all attenuated by genetically deleting Ip-10 in Mmp-8−/− mice (Fig. 8A – 8D).
Figure 8. Genetic deletion of Ip-10 in Mmp-8−/− mice abrogates their increased lung inflammatory response to bleomycin.
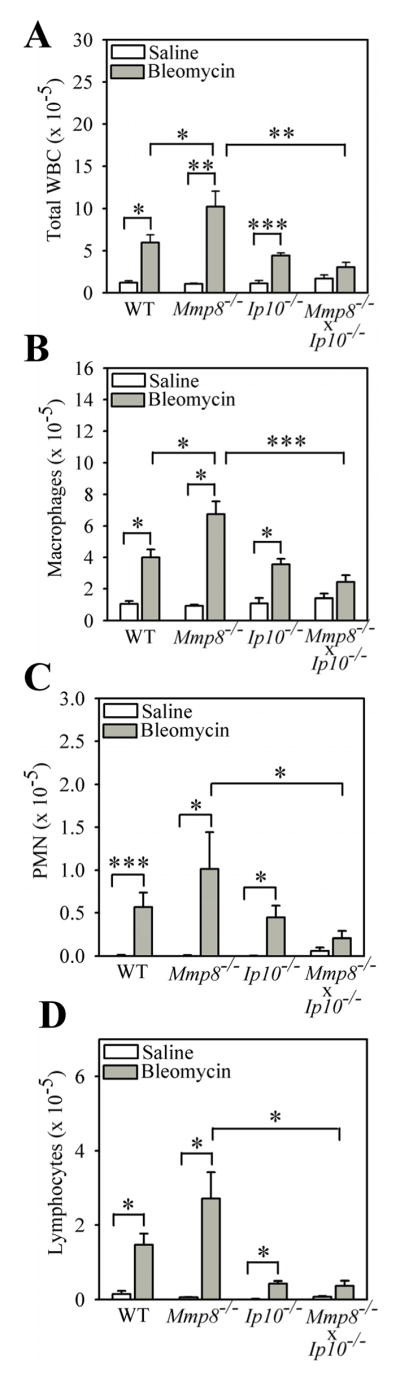
In A–D, WT, Mmp-8−/−, Mmp-8−/− X Ip-10−/−, and Ip-10−/− mice were treated with either 75 mU of bleomycin or saline by the IT route and 7 days later, BAL was performed and absolute numbers of all leukocytes (A), macrophages (B), PMNs (C), and lymphocytes (D) in BAL samples were enumerated. Data are expressed as mean ± SEM; n = 3–7 mice/group for saline-treated mice, n = 4–18 mice/group for bleomycin-treated mice. Asterisk indicates p < 0.05; **, p <0.01; and ***, p <0.005.
Mmp-8−/− mice have increased lung levels of Mip-1α during bleomycin-mediated lung injury, but Mmp-8 does not significantly regulate lung levels of other mediators that regulate lung inflammation
We showed previously that Mmp-8 limits lung inflammatory responses to LPS by cleaving and inactivating Mip-1α (10) which is a potent inducer of mononuclear cell migration (57) and PMN recruitment into the lung (58). Therefore, we tested whether Mip-1α protein levels in the lung are increased in the absence of Mmp-8 during bleomycin-mediated lung injury. We found higher levels of Mip-1α protein in homogenates of lungs harvested from bleomycin-treated Mmp-8−/− mice compared with bleomycin-treated WT mice (Fig. 9A). However, levels of Mip-1α protein are below the lower limit of detection of the ELISA when measured in BALF samples from both WT and Mmp-8−/− mice. We also measured lung levels of other pro- and anti-inflammatory mediators (Il-13, Ifn-γ, JE) and found no differences in lung levels of any of these mediators in bleomycin-treated WT vs. Mmp-8−/− mice. There is a strong trend toward lower lung levels of Il-10 in Mmp-8−/− mice compared with WT mice 48 h after IT bleomycin, but lung Il-10 levels are similar in WT and Mmp-8−/− mice 24 h and 21 days after IT bleomycin (Supplement Table I).
Figure 9. Bleomycin-treated Mmp-8−/− mice have higher lung levels of Mip-1α protein and genetic deletion of Mip-1α in Mmp-8−/− mice abrogates their increased lung inflammatory response to bleomycin.

In A, lungs were removed from WT and Mmp-8−/− mice 3–10 days after saline or bleomycin was delivered by the IT route. Mip-1α was measured in lung homogenates using an ELISA. Data are mean ± SEM; n = 4–5 mice/group for saline-treated mice and n = 10–16 mice/group for bleomycin-treated mice. In B–D, WT, Mmp-8−/−, Mip-1α−/−, and Mmp-8−/− X Mip-1α−/− mice were given saline or bleomycin by the IT route and 7 days later, BAL was performed and absolute numbers of macrophages (B), PMNs (C), and lymphocytes (D) were enumerated in BAL samples. Data in B–D are mean ± SEM; n = 4–7 mice/group for saline-treated mice, n = 11–18 mice/group for bleomycin-treated mice. Asterisk indicates p < 0.05; **, p <0.01; and ***, p <0.005.
Genetic deletion of Mip-1α in Mmp-8−/− mice abrogates the increased inflammation observed in bleomycin-treated Mmp-8−/− mice
To assess whether the higher lung levels of Mip-1α protein in bleomycin-treated Mmp-8−/− mice compared to bleomycin-treated WT mice contribute to the increased lung inflammatory response of Mmp-8−/− mice to IT bleomycin, we compared lung inflammatory responses of WT, Mmp-8−/−, Mip-1α−/−, and Mmp-8−/− X Mip-1α−/− mice to IT bleomycin vs. IT saline. Mip-1α−/− mice have a markedly attenuated lung inflammatory response to IT bleomycin, with reduced BAL macrophage, PMN, and lymphocyte counts (Figs. 9B–9D). Both the increased total leukocyte and the increased absolute macrophage, PMN, and lymphocyte counts in BAL samples in bleomycin-treated Mmp-8−/− mice are significantly abrogated by genetic deletion of Mip-1α in Mmp-8−/− mice (Figs. 9B–9D). These data indicate that Mmp-8 restrains the lung inflammatory response to bleomycin, in part, by reducing lung levels of Mip-1α.
Genetic deletion of Mip-1α in Mmp-8−/− mice reconstitutes their fibrotic lung responses to bleomycin-mediated lung injury
Next, we investigated whether the protection of Mmp-8−/− mice from bleomycin-mediated lung fibrosis is linked to their higher lung levels of Mip-1α. Genetic deletion of Mip-1α in Mmp-8−/− mice reconstitutes their lung fibroproliferative responses to bleomycin as assessed by analysis of lung sections stained with hematoxylin and eosin and Masson’s trichrome stain or for α smooth muscle actin, and hydroxyproline assays performed on lung hydrolysates (Fig. 10A-D).
Figure 10. Genetic deletion of Mip-1α in Mmp-8−/− mice reconstitutes their fibrotic lung responses to bleomycin.

In A–D, WT, Mmp-8−/−, Mmp-8−/− X Ip-10−/−, Mmp-8−/− X Mip-1α−/−, Ip-10−/−, and Mip-1α−/− mice were treated with either 60 mU of bleomycin or saline by the IT route and 21 days later, right lungs were inflated and fixed, and lung sections were stained with hematoxylin and eosin in A, Masson’s Trichrome stain in B, and with anti-α-smooth muscle actin antibody in C. Original magnification X100. Trichrome-stained and anti-α-smooth muscle actin antibody-stained lung sections are representative of 4 mice in each group. Hematoxylin and eosin stained lung sections are representative of 2–4 mice/group for saline-treated mice and 6–16 mice/group for bleomycin-treated mice. In D, collagen was quantified in left lungs using hydroxyproline assays. Data are mean ± SEM; n = 2–4 mice/group for saline-treated mice, n = 6–16 mice/group for bleomycin-treated mice. Asterisk indicates p < 0.05
Both bone marrow-derived cells and lung parenchymal cells are a source of pro-fibrotic Mmp-8 during bleomycin-mediated lung injury
To investigate whether bone marrow-derived cells or lung parenchymal cells are a source of pro-fibrotic Mmp-8, we generated Mmp-8 bone marrow chimeric mice. When the bone marrow in WT mice is replaced with Mmp-8−/− bone marrow there is a significant (but incomplete) reduction in the lung fibrotic response to bleomycin compared with WT recipients transplanted with WT bone marrow (Figs. 11A–D). When the bone marrow in Mmp-8−/− mice is replaced with WT bone marrow, the defective lung fibrotic response to bleomycin in Mmp-8−/− recipients is partially restored as assessed by analysis of lung sections from the mice stained with hematoxylin and eosin (Fig. 11A) and Masson’s Trichrome-stained (Fig. 11B), or for smooth muscle actin (Fig. 11C). There is also a strong trend towards a significant increase in lung collagen content in bleomycin-treated Mmp-8−/− recipients transplanted with WT bone marrow compared with bleomycin-treated Mmp-8−/− recipients transplanted with Mmp-8−/− bone marrow as assessed by quantitative hydroxyproline assays performed on lung hydrolysates (Fig. 11D). These results indicate that both bone marrow-derived cells and lung parenchymal cells are sources of pro-fibrotic Mmp-8 during bleomycin-mediated lung injury.
Figure 11. Bone marrow-derived cells and lung parenchymal cells are crucial sources of pro-fibrotic Mmp-8 during bleomycin-mediated lung fibrosis.

Mmp-8 bone marrow chimeric mice were generated as described in Methods. Mice were allowed to reconstitute their bone marrow with donor cells for 8 weeks, and then given 60 mU of bleomycin or saline by the IT route. After 21 days, right lungs were inflated, fixed, and stained with hematoxylin and eosin (in A), Masson’s Trichrome stain (in B), and with anti-α-smooth muscle actin antibody (in C). Original magnification X100. Lung sections are representative of 4–6 mice in Masson’s Trichrome stained and anti-α-smooth muscle actin antibody stained groups. Hematoxylin and eosin stained lung sections are representative of 4–8 mice/group for saline-treated mice and 9–20 mice/group for bleomycin-treated mice. In D, collagen was quantified in left lungs using hydroxyproline assays. Data are mean ± SEM; n = 4–8 mice/group for saline- treated mice, n = 9–20 mice/group for bleomycin-treated mice. Asterisk indicates p < 0.05, and ***, p <0.005.
DISCUSSION
Herein, we have confirmed the recent finding of Garcia-Prieto et al that Mmp-8 promotes bleomycin-mediated lung fibrosis in mice. We show for the first time that Mmp-8 expression is upregulated in lung samples during bleomycin-mediated lung injury and in murine lung fibroblasts activated with Tgf-β ex vivo. Additionally, our novel data show that Mmp-8 decreases the accumulation of macrophages and CD4+ lymphocytes in the lung during the acute phase of the bleomycin model by reducing lung levels of Mip-1α and Ip-10. Our studies of Mmp-8 bone marrow-chimeric mice show that both bone marrow-derived cells and lung parenchymal cells are key sources of pro-fibrotic Mmp-8 during bleomycin-mediated lung fibrosis.
Prior studies have shown that levels of other members of the MMP family are increased in blood or lung samples from patients with IPF and that these other MMPs contribute to lung fibrotic responses to injury in mice. For example, MMP-1, -7, and -8 protein levels are increased in plasma samples (59), and MMP-2 and MMP-9 protein levels are elevated in BAL fluid from patients with IPF vs. control subjects without lung disease (60). MMP-3 mRNA and protein levels are increased in lung tissue from patients with IPF compared with controls (61). Furthermore, Mmp-3, -7, and -13 promote lung fibrotic responses to injury in mice (61–63).
Mechanisms by which Mmp-8 promotes lung fibrosis in mice
We investigated whether Mmp-8 activates growth factors during bleomycin-mediated lung injury because other MMPs activate latent TGF-β in vitro (11), and Mmp-8 promotes wound healing in murine skin by altering Tgf-β signaling (25). However, we found no significant differences in lung levels of active or total Tgf-β in bleomycin-treated WT and Mmp-8−/− mice. We report for the first time that Mmp-8’s pro-fibrotic activity during bleomycin-mediated ALI is linked to Mmp-8 reducing lung levels of Mip-1α and Ip-10 because: 1) bleomycin-treated Mmp-8−/− mice have higher lung levels of Mip-1α and Ip-10 compared with bleomycin-treated WT mice; and 2) genetic deletion of either Mip-1α or Ip-10 in Mmp-8−/− mice restores the lung fibroproliferative responses of these mice to bleomycin to levels similar to those observed in bleomycin-treated WT mice.
IP-10 (CXCL10) is expressed by IFN-γ-activated monocytes, fibroblasts, and endothelial cells (64) and has anti-fibrotic activities following bleomycin-induced lung injury by inhibiting chemotaxis of lung fibroblasts (4). Interestingly, IP-10 production is impaired in fibroblasts in the lungs of IPF patients due to defective histone deacetylation and hypermethylation and this may contribute to lung fibrosis in patients with IPF (65). We detected modestly (but significantly) higher levels of Ip-10 protein in homogenates of lungs and BALF samples from Mmp-8−/− mice compared with WT mice at early time points after delivering bleomycin. However, lung leukocytes from bleomycin-treated Mmp-8−/− mice have substantially (9.6-fold) higher Ip-10 steady state mRNA levels compared with lung leukocytes from bleomycin-treated WT mice at the time point corresponding to peak lung inflammation. Likely measuring Ip-10 levels in whole lung samples dilutes out more impressive signals generated by subsets of cells such as leukocytes in the lungs. The high lung levels of anti-fibotic Ip-10 in bleomycin-treated Mmp-8−/− mice likely contributes to the protection of these mice from lung fibrosis by inhibiting myofibroblast accumulation in the lung. Consistent with this hypothesis, our study shows that myofibroblasts numbers are lower in the lungs of bleomycin-treated Mmp-8−/− mice when compared with those in the lungs of WT mice, and the accumulation of myofibroblasts in the lungs of bleomycin-treated Mmp-8−/− mice is restored by genetic deletion of Ip-10 in Mmp-8−/− mice. Although Mmp-8 cleaves other CXC motif chemokines such as LIX and Mip-1α (10,24), we found no evidence that either human or murine MMP-8 cleaves human or murine IP-10 in vitro.
The increased lung levels of Mip-1α in bleomycin-treated Mmp-8−/− mice also contribute to the protection of Mmp-8−/− mice from bleomycin-mediated lung fibrosis compared with WT mice, because genetic deletion of Mip-1α in Mmp-8−/− mice reconstitutes lung fibroproliferative responses of Mmp-8−/− mice to bleomycin. A previous study reported that Mip-1α has pro-fibrotic activities in the lung (3), but our study did not confirm this finding. It is possible that the exuberant acute inflammatory response in the lungs of bleomycin-treated Mmp-8−/− mice (due to increases in lung levels of both Mip-1α and Ip-10) limits subsequent progression to fibroproliferation in Mmp-8−/− mice. Consistent with this hypothesis, genetic deletion of Ip-10 or Mip-1α in Mmp-8−/− mice reduces lung inflammation in Mmp-8−/− mice during the acute phase of bleomycin-mediated ALI and subsequently restores their lung fibroproliferative response to bleomycin in the sub-acute phase of the model. Lung inflammation is linked to lung fibrosis during bleomycin-mediated ALI because antibody-mediated PMN depletion in bleomycin-treated hamsters worsens the animals’ lung fibrotic responses to bleomycin (55). Thus, in bleomycin-treated Mmp-8−/− mice, the increased lung levels of Ip-10 and/or Mip-1α may inhibit lung fibrosis, in part, by increasing lung inflammatory responses to bleomycin leading to increased clearance of provisional matrix by urokinase-type plasminogen activator which is highly expressed by leukocytes (66,67). Alternatively, the anti-fibrotic activities of Ip-10 may exceed the pro-fibrotic activity of Mip-1α in the lung, thereby leading to overall protection of Mmp-8−/− mice from bleomycin-induced lung fibrosis. These possibilities will be investigated in our future studies.
Mechanisms underlying Mmp-8’s anti-inflammatory activities during bleomycin-mediated lung injury
Our novel studies of Mip-1a−/− X Mmp-8−/− and Ip-10−/− X Mmp-8−/− mice showed that the anti-inflammatory activities of Mmp-8 in bleomycin-treated mice are due to Mmp-8 reducing lung levels of Mip-1α and Ip-10. Mip-1α is known to be a potent chemokine for macrophages (68) and lymphocytes (69), and Ip-10 increases the accumulation of macrophages and lymphocytes in other models of lung inflammation (70). Our study provides evidence that the increased lung levels of Mip-1α and Ip-10 in bleomycin-treated Mmp-8−/− mice contribute to the increased lung macrophage and CD4+ T lymphocyte counts in these mice. Likely, Mmp-8 reduces lung levels of Mip-1α in bleomycin-treated mice by directly cleaving and inactivating this chemokine in the lung because our prior studies showed that Mmp-8 cleaves Mip-1α and reduces its chemotactic activity in vitro (10). Macrophages express receptors for both Mip-1α (Ccr1 and Ccr5) and Ip-10 (Cxcr3) and both mediators are produced by macrophages and induce recruitment and activation of monocytes and macrophages (70–72). Although our study shows that Mmp-8 does not cleave Ip-10, a possible explanation for Mmp-8 reducing Ip-10 levels in the bleomycin-treated lung is that Mip-1α is upstream of Ip-10 in this model because Mip-1α activates macrophages, and activated macrophages are a significant source of Ip-10 (73). In support of this concept, Mip-1a−/− mice have a severely blunted systemic inflammatory response in a trauma-hemorrhage model of shock and this is associated with reduced serum levels of various pro-inflammatory mediators that are produced by macrophages (74). Although Ip-10 levels were not measured in Mip-1α−/− mice with shock, it is likely that macrophage production of Ip-10 is reduced in mice lacking Mip-1α (74)]. In bleomycin-treated Mmp-8−/− mice it is likely that loss of Mmp-8-mediated proteolytic activation of Mip-1α leads to enhanced Mip-1α-induced Ip-10 generation in the lung associated with increased lung inflammation but reduced progression to lung fibrosis in the sub-acute phase of the bleomycin model. This possibility will be the focus of our future studies.
Macrophage counts are increased in the lungs of bleomycin-treated Mmp-8−/− mice compared with bleomycin-treated WT mice. Although alveolar macrophages are activated in the lungs of IPF patients, and have the potential to promote lung fibrosis because they produce increased amounts of fibronectin and platelet-derived growth factor (75–77), more recent studies show that alveolar macrophages have multiple phenotypes with respect to fibrosis, with alternatively activated macrophages (M2 macrophages) promoting fibrosis and classically activated macrophages (M1 macrophages) promoting resolution of fibrosis (78,79). M2 macrophages in the lungs of patients with obstructive lung disease have increased expression of MMP-2 and -7, but MMP-8 expression by these cells was not assessed (80). We also found increased accumulation of CD4+ T cells in bleomycin-treated Mmp-8−/− mice. Although CD4+ T cells have pro-fibrotic activities during some tissue responses to injury (81), CD4+ T cells inhibit or promote renal fibrosis in mice depending on the context of CD4+ T cell activation (82). Future studies will investigate whether the increased lung CD4+ T cell or macrophage counts in bleomycin-treated Mmp-8−/− mice contribute to their protection from fibrosis and/or whether Mmp-8 regulates the macrophage skewing from an M1 to a pro-fibrotic M2 phenotype.
Cellular sources of pro-fibrotic Mmp-8 in the lung
We detected robust staining for MMP-8 protein in leukocytes in the lungs of patients with IPF. Although we did not detect significant increases in Mmp-8 steady state mRNA levels in leukocytes isolated from the lungs of bleomycin-treated WT mice, PMNs store Mmp-8 protein within their specific granules but do not synthesize Mmp-8 de novo. It is likely that PMNs recruited to the bleomycin-injured murine lung contribute to the increases in Mmp-8 protein levels that we detected in BALF samples. Macrophages represent most of the leukocytes present in BAL samples during bleomycin-mediated lung injury, and express Mmp-8 mRNA transcript when activated ex vivo (33). However, our results indicate that they do not significantly contribute to the increase in Mmp-8 steady state mRNA levels in the lung during bleomycin-mediated lung injury because steady state mRNA levels do not increase significantly in BAL leukocytes isolated from bleomycin-treated WT mice. Although other studies have shown that MMP-8 is expressed by fibroblasts isolated from organs other than the lung (34,35), we report for the first time that lung fibroblasts can be induced to express Mmp-8 when stimulated ex vivo with Tgf-β. Although the concentration of TGF-β1 that we used to induce murine lung fibroblasts to express Mmp-8 is relatively high (10 ng/ml), other studies have used this concentration to activate fibroblasts (83–85). Additionally, 10 ng/ml of TGF-β1 is optimal for inducing human fibroblasts to express MMP-13 (86). This suggests that optimal TGF-β1-mediated induction of collagenase gene expression (vs. other genes regulated by TGF-β1) in lung fibroblasts may require relatively high concentrations of TGF-β1. Our study is the first to show that both bone marrow-derived cells and lung parenchymal cells are crucial sources of pro-fibrotic Mmp-8 during bleomycin-mediated lung injury based upon the lung fibroproliferative responses of Mmp-8 bone marrow-chimeric mice to IT bleomycin. Potential bone marrow-derived sources of Mmp-8 include macrophages (33), PMNs (10), and fibrocytes (87)]. Our results also indicate that fibroblasts are potential sources of pro-fibrotic Mmp-8 among lung parenchymal cells.
Limitations of our study include the shortcomings of the bleomycin murine model as a model of IPF. For example, lung fibrosis induced by bleomycin eventually resolves in marked contrast to human IPF in which lung fibrosis is progressive. Bleomycin induces robust lung inflammation whereas minimal inflammation is present in the lungs of most patients with IPF. Also, therapeutics that have efficacy in the bleomycin model in mice have not been effective in treating human IPF (88). Our future studies will evaluate Mmp-8−/− mice in newer models of lung fibrosis, including the repeat dosing bleomycin model which leads to persistent lung fibrosis (89), lung-specific over-expression of TGF-β models, and radiation-induced lung fibrosis models.
Other studies of Mmp-8 in the context of fibrosis
In a murine model of obliterative bronchiolitis, Mmp-8 has pro-fibrotic activities associated with increased migration of PMNs into the airway lumen (90). However, in a model of liver fibrosis in rats, PMN-derived Mmp-8 activity was increased during the repair phase of this model and this was associated with resolution of fibrotic scarring (91). Recent studies from Garcia-Prieto et al. (29) also report that Mmp-8 has anti-inflammatory and pro-fibrotic in the lungs of bleomycin-treated mice. Our study agrees with these findings, and adds to this literature by providing insights into the mechanisms underlying the anti-inflammatory activities of Mmp-8 during bleomycin-mediated lung injury and the leukocyte subsets that are regulated in the lung by Mmp-8. Garcia-Prieto et al attributed the protection from lung fibrosis of bleomycin-treated Mmp-8−/− mice to lower lung levels of Il-10 in Mmp-8−/− mice than WT mice which they detected three and six week after instilling bleomycin. While we found slightly lower lung levels of Il-10 in Mmp-8−/− mice 48 h after delivering bleomycin by the IT route, we found similar levels lung levels of Il-10 in WT vs. Mmp-8−/− mice three weeks after instilling bleomycin. It is noteworthy in this respect that there are conflicting prior reports in the literature on the effects of Il-10 in regulating lung fibrotic responses to injury as bleomycin-treated Il-10−/− and WT mice have similar lung fibrotic responses (92) but transgenic mice over-expressing Il-10 in the lung in an inducible manner develop lung fibrosis (93). Additional studies are needed to address the reasons for these conflicting reports underlying the activities of Il-10 in regulating lung fibrosis.
We now report that Mmp-8 has anti-inflammatory and pro-fibrotic activities during bleomycin-mediated lung injury in mice by decreasing lung levels of Mip-1α and Ip-10. We have shown for the first time that both bone marrow-derived cells and lung parenchymal cells are crucial sources of pro-fibrotic Mmp-8. Our data suggest that treatment strategies aimed at reducing lung levels of MMP-8 or inhibiting MMP-8’s pro-fibrotic activity in the lung may have therapeutic potential in IPF patients.
Supplementary Material
Acknowledgments
We thank Drs. Ivan O. Rosas and William G. Richards for providing the IPF and normal lung tissue samples. We also thank Dr. Augustine M. K. Choi for critically reading the manuscript. We thank Anja Hergrueter, B.A. for proof reading the manuscript.
Abbreviations used
- ECM
extracellular matrix
- IPF
idiopathic pulmonary fibrosis
- PMN
polymorphonuclear cell
- IP-10
interferon gamma inducible protein-10
- IT
intratracheal
- WT
wild type
- MMP-8
matrix metalloproteinase-8
- MIP-1α
macrophage inflammatory protein-1α
- BAL
bronchoalveolar lavage
- Lix
lipopolysaccharide-induced CXC chemokine
- PGP
Proline-Glycine-Proline
- APMA
p-aminophenylmercuric acetate
- ABC
avidin biotin complex
- DAB
3,3′-diaminobenzidine
- Mig
monokine induced by gamma interferon
- Itac
interferon inducible T-cell alpha chemoattractant
- ALI
acute lung injury
Footnotes
This work was supported by the Public Health Service, National Heart, Lung, and Blood Institute Grants HL063137, HL086814, HL 835111 and NIH T32 HL007633, and the BWH-Lovelace Respiratory Research Institute Consortium.
References
- 1.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 2.Huaux F, Liu T, McGarry B, Ullenbruch M, Phan SH. Dual roles of IL-4 in lung injury and fibrosis. J Immunol. 2003;170:2083–2092. doi: 10.4049/jimmunol.170.4.2083. [DOI] [PubMed] [Google Scholar]
- 3.Ishida Y, Kimura A, Kondo T, Hayashi T, Ueno M, Takakura N, Matsushima K, Mukaida N. Essential roles of the CC chemokine ligand 3-CC chemokine receptor 5 axis in bleomycin-induced pulmonary fibrosis through regulation of macrophage and fibrocyte infiltration. Am J Pathol. 2007;170:843–854. doi: 10.2353/ajpath.2007.051213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tager AM, Kradin RL, LaCamera P, Bercury SD, Campanella GS, Leary CP, Polosukhin V, Zhao LH, Sakamoto H, Blackwell TS, Luster AD. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. 2004;31:395–404. doi: 10.1165/rcmb.2004-0175OC. [DOI] [PubMed] [Google Scholar]
- 5.Jiang D, Liang J, Hodge J, Lu B, Zhu Z, Yu S, Fan J, Gao Y, Yin Z, Homer R, Gerard C, Noble PW. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J Clin Invest. 2004;114:291–299. doi: 10.1172/JCI16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 7.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167:1083–1089. doi: 10.1164/rccm.200212-1396OC. [DOI] [PubMed] [Google Scholar]
- 8.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 9.McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
- 10.Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA. Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol. 2010;184:1575–1588. doi: 10.4049/jimmunol.0900290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson TJ, Singh RK. Proteases as modulators of tumor-stromal interaction: primary tumors to bone metastases. Biochim Biophys Acta. 2008;1785:85–95. doi: 10.1016/j.bbcan.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Owen CA, Campbell EJ. The cell biology of leukocyte-mediated proteolysis. J Leukoc Biol. 1999;65:137–150. doi: 10.1002/jlb.65.2.137. [DOI] [PubMed] [Google Scholar]
- 14.Gross J, Nagai Y. Specific degradation of the collagen molecule by tadpole collagenolytic enzyme. Proc Natl Acad Sci U S A. 1965;54:1197–1204. doi: 10.1073/pnas.54.4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welgus HG, Jeffrey JJ, Eisen AZ. The collagen substrate specificity of human skin fibroblast collagenase. J Biol Chem. 1981;256:9511–9515. [PubMed] [Google Scholar]
- 16.Hasty KA, Jeffrey JJ, Hibbs MS, Welgus HG. The collagen substrate specificity of human neutrophil collagenase. J Biol Chem. 1987;262:10048–10052. [PubMed] [Google Scholar]
- 17.Hasty KA, Hibbs MS, Kang AH, Mainardi CL. Secreted forms of human neutrophil collagenase. J Biol Chem. 1986;261:5645–5650. [PubMed] [Google Scholar]
- 18.Wright DG, Gallin JI. Secretory responses of human neutrophils: exocytosis of specific (secondary) granules by human neutrophils during adherence in vitro and during exudation in vivo. J Immunol. 1979;123:285–294. [PubMed] [Google Scholar]
- 19.Horwitz AL, Hance AJ, Crystal RG. Granulocyte collagenase: selective digestion of type I relative to type III collagen. Proc Natl Acad Sci U S A. 1977;74:897–901. doi: 10.1073/pnas.74.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knauper V, Kramer S, Reinke H, Tschesche H. Characterization and activation of procollagenase from human polymorphonuclear leucocytes. N-terminal sequence determination of the proenzyme and various proteolytically activated forms. Eur J Biochem. 1990;189:295–300. doi: 10.1111/j.1432-1033.1990.tb15489.x. [DOI] [PubMed] [Google Scholar]
- 21.Gruber BL, Schwartz LB, Ramamurthy NS, Irani AM, Marchese MJ. Activation of latent rheumatoid synovial collagenase by human mast cell tryptase. J Immunol. 1988;140:3936–3942. [PubMed] [Google Scholar]
- 22.Knauper V, Wilhelm SM, Seperack PK, DeClerck YA, Langley KE, Osthues A, Tschesche H. Direct activation of human neutrophil procollagenase by recombinant stromelysin. Biochem J. 1993;295(Pt 2):581–586. doi: 10.1042/bj2950581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gueders MM, Balbin M, Rocks N, Foidart JM, Gosset P, Louis R, Shapiro S, Lopez-Otin C, Noel A, Cataldo DD. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol. 2005;175:2589–2597. doi: 10.4049/jimmunol.175.4.2589. [DOI] [PubMed] [Google Scholar]
- 24.Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, Lopez-Otin C, Overall CM. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS One. 2007;2:e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutierrez-Fernandez A, Inada M, Balbin M, Fueyo A, Pitiot AS, Astudillo A, Hirose K, Hirata M, Shapiro SD, Noel A, Werb Z, Krane SM, Lopez-Otin C, Puente XS. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8) FASEB J. 2007;21:2580–2591. doi: 10.1096/fj.06-7860com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, Clancy JP, Blalock JE. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180:5662–5669. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braber S, Koelink PJ, Henricks PA, Jackson PL, Nijkamp FP, Garssen J, Kraneveld AD, Blalock JE, Folkerts G. Cigarette smoke-induced lung emphysema in mice is associated with prolyl endopeptidase, an enzyme involved in collagen breakdown. Am J Physiol Lung Cell Mol Physiol. 2011;300:L255–L265. doi: 10.1152/ajplung.00304.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laxton RC, Hu Y, Duchene J, Zhang F, Zhang Z, Leung KY, Xiao Q, Scotland RS, Hodgkinson CP, Smith K, Willeit J, Lopez-Otin C, Simpson IA, Kiechl S, Ahluwalia A, Xu Q, Ye S. A role of matrix metalloproteinase-8 in atherosclerosis. Circ Res. 2009;105:921–929. doi: 10.1161/CIRCRESAHA.109.200279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Prieto E, Gonzalez-Lopez A, Cabrera S, Astudillo A, Gutierrez-Fernandez A, Fanjul-Fernandez M, Batalla-Solis E, Puente XS, Fueyo A, Lopez-Otin C, Albaiceta GM. Resistance to bleomycin-induced lung fibrosis in MMP-8 deficient mice is mediated by interleukin-10. PLoS One. 2010;5:e13242. doi: 10.1371/journal.pone.0013242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 31.WOESSNER JF., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 32.Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172:7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- 33.Herman MP, Sukhova GK, Libby P, Gerdes N, Tang N, Horton DB, Kilbride M, Breitbart RE, Chun M, Schonbeck U. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation. 2001;104:1899–1904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- 34.Hanemaaijer R, Sorsa T, Konttinen YT, Ding Y, Sutinen M, Visser H, van Hinsbergh VW, Helaakoski T, Kainulainen T, Ronka H, Tschesche H, Salo T. Matrix metalloproteinase-8 is expressed in rheumatoid synovial fibroblasts and endothelial cells. Regulation by tumor necrosis factor-alpha and doxycycline. J Biol Chem. 1997;272:31504–31509. doi: 10.1074/jbc.272.50.31504. [DOI] [PubMed] [Google Scholar]
- 35.Xu L, Yu Z, Lee HM, Wolff MS, Golub LM, Sorsa T, Kuula H. Characteristics of collagenase-2 from gingival crevicular fluid and peri-implant sulcular fluid in periodontitis and peri-implantitis patients: pilot study. Acta Odontol Scand. 2008;66:219–224. doi: 10.1080/00016350802183393. [DOI] [PubMed] [Google Scholar]
- 36.Baran CP, Opalek JM, McMaken S, Newland CA, O’Brien JM, Jr, Hunter MG, Bringardner BD, Monick MM, Brigstock DR, Stromberg PC, Hunninghake GW, Marsh CB. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:78–89. doi: 10.1164/rccm.200609-1279OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pociask DA, Chen K, Choi SM, Oury TD, Steele C, Kolls JK. gammadelta T cells attenuate bleomycin-induced fibrosis through the production of CXCL10. Am J Pathol. 2011;178:1167–1176. doi: 10.1016/j.ajpath.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collins SL, Chan-Li Y, Hallowell RW, Powell JD, Horton MR. Pulmonary vaccination as a novel treatment for lung fibrosis. PLoS One. 2012;7:e31299. doi: 10.1371/journal.pone.0031299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Groot KT, Pardo A, Knipping K, Buendia-Roldan I, Garcia-de-Alba C, Blokhuis BR, Selman M, Redegeld FA. Immunoglobulin free light chains are increased in hypersensitivity pneumonitis and idiopathic pulmonary fibrosis. PLoS One. 2011;6:e25392. doi: 10.1371/journal.pone.0025392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasse P, Riteau N, Vacher R, Michel ML, Fautrel A, di PF, Fick L, Charron S, Lagente V, Eberl G, Le BM, Quesniaux VF, Huaux F, Leite-de-Moraes M, Ryffel B, Couillin I. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS One. 2011;6:e23185. doi: 10.1371/journal.pone.0023185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol. 2008;83:237–244. doi: 10.1189/jlb.0707504. [DOI] [PubMed] [Google Scholar]
- 42.Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE., Jr Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest. 2008;133:226–232. doi: 10.1378/chest.07-1948. [DOI] [PubMed] [Google Scholar]
- 43.Lawson WE, V, Polosukhin V, Stathopoulos GT, Zoia O, Han W, Lane KB, Li B, Donnelly EF, Holburn GE, Lewis KG, Collins RD, Hull WM, Glasser SW, Whitsett JA, Blackwell TS. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol. 2005;167:1267–1277. doi: 10.1016/S0002-9440(10)61214-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dona M, Dell’Aica I, Calabrese F, Benelli R, Morini M, Albini A, Garbisa S. Neutrophil restraint by green tea: inhibition of inflammation, associated angiogenesis, and pulmonary fibrosis. J Immunol. 2003;170:4335–4341. doi: 10.4049/jimmunol.170.8.4335. [DOI] [PubMed] [Google Scholar]
- 45.Pardo A, Ruiz V, Arreola JL, Ramirez R, Cisneros-Lira J, Gaxiola M, Barrios R, Kala SV, Lieberman MW, Selman M. Bleomycin-induced pulmonary fibrosis is attenuated in gamma-glutamyl transpeptidase-deficient mice. Am J Respir Crit Care Med. 2003;167:925–932. doi: 10.1164/rccm.200209-1007OC. [DOI] [PubMed] [Google Scholar]
- 46.Yamada H. ANCA: associated lung fibrosis. Semin Respir Crit Care Med. 2011;32:322–327. doi: 10.1055/s-0031-1279828. [DOI] [PubMed] [Google Scholar]
- 47.Annes JP, Rifkin DB, Munger JS. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett. 2002;511:65–68. doi: 10.1016/s0014-5793(01)03280-x. [DOI] [PubMed] [Google Scholar]
- 48.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 49.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, OHynes R, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 50.Torbett BE, Laxer JA, Glasebrook AL. Frequencies of T cells secreting IL-2 and/or IL-4 among unprimed CD4+ populations. Evidence that clones secreting IL-2 and IL-4 give rise to clones which secrete only IL-4. Immunol Lett. 1990;23:227–233. doi: 10.1016/0165-2478(90)90197-x. [DOI] [PubMed] [Google Scholar]
- 51.Uyttenhove C, Brombacher F, Van SJ. TGF-beta interactions with IL-1 family members trigger IL-4-independent IL-9 production by mouse CD4(+) T cells. Eur J Immunol. 2010;40:2230–2235. doi: 10.1002/eji.200940281. [DOI] [PubMed] [Google Scholar]
- 52.Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engelhardt E, Toksoy A, Goebeler M, Debus S, Brocker EB, Gillitzer R. Chemokines IL-8, GROalpha, MCP-1, IP-10, and Mig are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. Am J Pathol. 1998;153:1849–1860. doi: 10.1016/s0002-9440(10)65699-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taub DD, Sayers TJ, Carter CR, Ortaldo JR. Alpha and beta chemokines induce NK cell migration and enhance NK-mediated cytolysis. J Immunol. 1995;155:3877–3888. [PubMed] [Google Scholar]
- 55.Clark JG, Kuhn C., III Bleomycin-induced pulmonary fibrosis in hamsters: effect of neutrophil depletion on lung collagen synthesis. Am Rev Respir Dis. 1982;126:737–739. doi: 10.1164/arrd.1982.126.4.737. [DOI] [PubMed] [Google Scholar]
- 56.Thrall RS, Phan SH, McCormick JR, Ward PA. The development of bleomycin-induced pulmonary fibrosis in neutrophil-depleted and complement-depleted rats. Am J Pathol. 1981;105:76–81. [PMC free article] [PubMed] [Google Scholar]
- 57.Fillion I, Ouellet N, Simard M, Bergeron Y, Sato S, Bergeron MG. Role of chemokines and formyl peptides in pneumococcal pneumonia-induced monocyte/macrophage recruitment. J Immunol. 2001;166:7353–7361. doi: 10.4049/jimmunol.166.12.7353. [DOI] [PubMed] [Google Scholar]
- 58.Ramos CD, Canetti C, Souto JT, Silva JS, Hogaboam CM, Ferreira SH, Cunha FQ. MIP-1alpha[CCL3] acting on the CCR1 receptor mediates neutrophil migration in immune inflammation via sequential release of TNF-alpha and LTB4. J Leukoc Biol. 2005;78:167–177. doi: 10.1189/jlb.0404237. [DOI] [PubMed] [Google Scholar]
- 59.Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, Lindell KO, Cisneros J, Macdonald SD, Pardo A, Sciurba F, Dauber J, Selman M, Gochuico BR, Kaminski N. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93. doi: 10.1371/journal.pmed.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suga M, Iyonaga K, Okamoto T, Gushima Y, Miyakawa H, Akaike T, Ando M. Characteristic elevation of matrix metalloproteinase activity in idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2000;162:1949–1956. doi: 10.1164/ajrccm.162.5.9906096. [DOI] [PubMed] [Google Scholar]
- 61.Yamashita CM, Dolgonos L, Zemans RL, Young SK, Robertson J, Briones N, Suzuki T, Campbell MN, Gauldie J, Radisky DC, Riches DW, Yu G, Kaminski N, McCulloch CA, Downey GP. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol. 2011;179:1733–1745. doi: 10.1016/j.ajpath.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flechsig P, Hartenstein B, Teurich S, Dadrich M, Hauser K, Abdollahi A, Grone HJ, Angel P, Huber PE. Loss of matrix metalloproteinase-13 attenuates murine radiation-induced pulmonary fibrosis. Int J Radiat Oncol Biol Phys. 2010;77:582–590. doi: 10.1016/j.ijrobp.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 63.Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, Lollini L, Morris D, Kim Y, DeLustro B, Sheppard D, Pardo A, Selman M, Heller RA. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A. 2002;99:6292–6297. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luster AD, Unkeless JC, Ravetch JV. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- 65.Coward WR, Watts K, Feghali-Bostwick CA, Jenkins G, Pang L. Repression of IP-10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol Cell Biol. 2010;30:2874–2886. doi: 10.1128/MCB.01527-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sisson TH, Hattori N, Xu Y, Simon RH. Treatment of bleomycin-induced pulmonary fibrosis by transfer of urokinase-type plasminogen activator genes. Hum Gene Ther. 1999;10:2315–2323. doi: 10.1089/10430349950016960. [DOI] [PubMed] [Google Scholar]
- 67.Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest. 1996;97:232–237. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uguccioni M, D’Apuzzo M, Loetscher M, Dewald B, Baggiolini M. Actions of the chemotactic cytokines MCP-1, MCP-2, MCP-3, RANTES, MIP-1 alpha and MIP-1 beta on human monocytes. Eur J Immunol. 1995;25:64–68. doi: 10.1002/eji.1830250113. [DOI] [PubMed] [Google Scholar]
- 69.Roth SJ, Carr MW, Springer TA. C-C chemokines, but not the C-X-C chemokines interleukin-8 and interferon-gamma inducible protein-10, stimulate transendothelial chemotaxis of T lymphocytes. Eur J Immunol. 1995;25:3482–3488. doi: 10.1002/eji.1830251241. [DOI] [PubMed] [Google Scholar]
- 70.Zeng X, Moore TA, Newstead MW, Deng JC, Lukacs NW, Standiford TJ. IP-10 mediates selective mononuclear cell accumulation and activation in response to intrapulmonary transgenic expression and during adenovirus-induced pulmonary inflammation. J Interferon Cytokine Res. 2005;25:103–112. doi: 10.1089/jir.2005.25.103. [DOI] [PubMed] [Google Scholar]
- 71.Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis DE, Kheradmand F. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1:e8. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Standiford TJ, Kunkel SL, Lukacs NW, Greenberger MJ, Danforth JM, Kunkel RG, Strieter RM. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol. 1995;155:1515–1524. [PubMed] [Google Scholar]
- 73.Korpi-Steiner NL, Bates ME, Lee WM, Hall DJ, Bertics PJ. Human rhinovirus induces robust IP-10 release by monocytic cells, which is independent of viral replication but linked to type I interferon receptor ligation and STAT1 activation. J Leukoc Biol. 2006;80:1364–1374. doi: 10.1189/jlb.0606412. [DOI] [PubMed] [Google Scholar]
- 74.Hsieh CH, Frink M, Hsieh YC, Kan WH, Hsu JT, Schwacha MG, Choudhry MA, Chaudry IH. The role of MIP-1 alpha in the development of systemic inflammatory response and organ injury following trauma hemorrhage. J Immunol. 2008;181:2806–2812. doi: 10.4049/jimmunol.181.4.2806. [DOI] [PubMed] [Google Scholar]
- 75.Rennard SI, Hunninghake GW, Bitterman PB, Crystal RG. Production of fibronectin by the human alveolar macrophage: mechanism for the recruitment of fibroblasts to sites of tissue injury in interstitial lung diseases. Proc Natl Acad Sci U S A. 1981;78:7147–7151. doi: 10.1073/pnas.78.11.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lacronique JG, Rennard SI, Bitterman PB, Ozaki T, Crystal RG. Alveolar macrophages in idiopathic pulmonary fibrosis have glucocorticoid receptors, but glucocorticoid therapy does not suppress alveolar macrophage release of fibronectin and alveolar macrophage derived growth factor. Am Rev Respir Dis. 1984;130:450–456. doi: 10.1164/arrd.1984.130.3.450. [DOI] [PubMed] [Google Scholar]
- 77.Nagaoka I, Trapnell BC, Crystal RG. Upregulation of platelet-derived growth factor-A and -B gene expression in alveolar macrophages of individuals with idiopathic pulmonary fibrosis. J Clin Invest. 1990;85:2023–2027. doi: 10.1172/JCI114669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Varin A, Gordon S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology. 2009;214:630–641. doi: 10.1016/j.imbio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 79.Gibbons MA, Mackinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van RN, Haslett C, Howie SE, Simpson AJ, Hirani N, Gauldie J, Iredale JP, Sethi T, Forbes SJ. Ly6Chi Monocytes Direct Alternatively Activated Pro-fibrotic Macrophage Regulation of Lung Fibrosis. Am J Respir Crit Care Med. 2011 doi: 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 80.Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O’Connor TP, Crystal RG. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183:2867–2883. doi: 10.4049/jimmunol.0900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hu H, Stein-Streilein J. Hapten-immune pulmonary interstitial fibrosis (HIPIF) in mice requires both CD4+ and CD8+ T lymphocytes. J Leukoc Biol. 1993;54:414–422. doi: 10.1002/jlb.54.5.414. [DOI] [PubMed] [Google Scholar]
- 82.Niedermeier M, Reich B, Rodriguez GM, Denzel A, Schmidbauer K, Gobel N, Talke Y, Schweda F, Mack M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A. 2009;106:17892–17897. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med. 2009;180:122–133. doi: 10.1164/rccm.200811-1730OC. [DOI] [PubMed] [Google Scholar]
- 84.Cunnington RH, Wang B, Ghavami S, Bathe KL, Rattan SG, Dixon IM. Antifibrotic properties of c-Ski and its regulation of cardiac myofibroblast phenotype and contractility. Am J Physiol Cell Physiol. 2011;300:C176–C186. doi: 10.1152/ajpcell.00050.2010. [DOI] [PubMed] [Google Scholar]
- 85.Gonzalez AV, Le BF, Ludwig MS. Imbalance of receptor-regulated and inhibitory Smads in lung fibroblasts from bleomycin-exposed rats. Am J Respir Cell Mol Biol. 2007;36:206–212. doi: 10.1165/rcmb.2006-0132OC. [DOI] [PubMed] [Google Scholar]
- 86.Uria JA, Jimenez MG, Balbin M, Freije JM, Lopez-Otin C. Differential effects of transforming growth factor-beta on the expression of collagenase-1 and collagenase-3 in human fibroblasts. J Biol Chem. 1998;273:9769–9777. doi: 10.1074/jbc.273.16.9769. [DOI] [PubMed] [Google Scholar]
- 87.Garcia-de-Alba C, Becerril C, Ruiz V, Gonzalez Y, Reyes S, Garcia-Alvarez J, Selman M, Pardo A. Expression of matrix metalloproteases by fibrocytes: possible role in migration and homing. Am J Respir Crit Care Med. 2010;182:1144–1152. doi: 10.1164/rccm.201001-0028OC. [DOI] [PubMed] [Google Scholar]
- 88.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;299:L442–L452. doi: 10.1152/ajplung.00026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Khatwa UA, Kleibrink BE, Shapiro SD, Subramaniam M. MMP-8 promotes polymorphonuclear cell migration through collagen barriers in obliterative bronchiolitis. J Leukoc Biol. 2010;87:69–77. doi: 10.1189/jlb.0509361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harty MW, Huddleston HM, Papa EF, Puthawala T, Tracy AP, Ramm GA, Gehring S, Gregory SH, Tracy TF., Jr Repair after cholestatic liver injury correlates with neutrophil infiltration and matrix metalloproteinase 8 activity. Surgery. 2005;138:313–320. doi: 10.1016/j.surg.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 92.Kradin RL, Sakamoto H, Jain F, Zhao LH, Hymowitz G, Preffer F. IL-10 inhibits inflammation but does not affect fibrosis in the pulmonary response to bleomycin. Exp Mol Pathol. 2004;76:205–211. doi: 10.1016/j.yexmp.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 93.Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, Shanley TP. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L341–L353. doi: 10.1152/ajplung.00122.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.