

Abstract
The genes contributing to childhood obesity are categorized into three different types based on distinct genetic and phenotypic characteristics. These types of childhood obesity are represented by rare monogenic forms of syndromic or non-syndromic childhood obesity, and common polygenic childhood obesity. In some cases, genetic susceptibility to these forms of childhood obesity may result from different variations of the same gene. Although the prevalence for rare monogenic forms of childhood obesity has not increased in recent times, the prevalence of common childhood obesity has increased in the United States and developing countries throughout the world during the past few decades. A number of recent genome-wide association studies and mouse model studies have established the identification of susceptibility genes contributing to common childhood obesity. Accumulating evidence suggests that this type of childhood obesity represents a complex metabolic disease resulting from an interaction with environmental factors, including dietary macronutrients. The objective of this article is to provide a review on the origins, mechanisms, and health consequences of obesity susceptibility genes and interaction with dietary macronutrients that predispose to childhood obesity. It is proposed that increased knowledge of these obesity susceptibility genes and interaction with dietary macronutrients will provide valuable insight for individual, family, and community preventative lifestyle intervention, and eventually targeted nutritional and medicinal therapies.
Keywords: Childhood obesity, Energy balance, Mouse model, Refined carbohydrates, Saturated fat
Introduction
Childhood obesity is a major health problem in the United States and developing countries throughout the world. The most recent National Health and Nutrition Examination Surveys (NHANES) indicate that childhood obesity in the United States has approximately doubled during the past three decades and adolescent obesity has more than tripled during the same period (Ogden et al. 2010, 2012). These studies also report that although the prevalence of childhood and adolescent obesity has not changed since 2007–2008, the prevalence of obesity among 2–5-year-olds (12.1 %), 6–11-year-olds (18.0 %), and 12–19-year-olds (18.4 %) remains at the highest recorded levels since establishment of the NHANES (Ogden et al. 2002). However, it should be noted that some epidemiological studies indicate that the prevalence of childhood obesity is continuing to increase in certain sex, age, ethnic, and socioeconomic status groups within the United States and that the current childhood obesity epidemic will contribute to an increase in the number of obese adults (Serdula et al. 1993; Wang et al. 2012). It was reported that ~80 % of overweight children between 10 and 15 years of age become obese adults by 25 years of age and that overweight children before 8 years of age usually result in more severe adult obesity (Whitaker et al. 1997). With respect to the global prevalence of childhood obesity, it was estimated that 43 million preschool children (6.7 %) were overweight or obese in 2010 and projected to reach ~60 million preschool children (9.1 %) in 2020 (de Onis et al. 2010). A graph showing the increasing percentage of overweight children in different countries with the highest known prevalence of childhood obesity is provided (Fig. 1).
Fig. 1.

The increasing percentage of overweight children in eight countries with the highest prevalence of childhood obesity
The childhood obesity epidemic is also anticipated to culminate in a large number of weight-associated complications affecting the neurological, cardiovascular, endocrine, musculoskeletal, renal, gastrointestinal, and pulmonary systems in addition to psychosocial problems (Fig. 2). These medical complications will further burden an already fragile national healthcare system as children progress through adulthood (Yach et al. 2006). It is projected that these complications will ultimately decrease the life expectancy of people in the United States by 2–5 years for the first time in modern history (Olshansky et al. 2005). To address and hopefully lessen the impact of this major health problem, experts from diverse fields of study are attempting to understand the etiology of childhood obesity. It is well accepted that the timing of this epidemic parallels an increased availability of calorie-dense foods and a more sedentary lifestyle in what is referred to as an “obesogenic environment” (Chaput et al. 2011). However, the cause of this epidemic is obscured because not all individuals become overweight or obese while living in the same environment. Therefore, variability among individuals is suspected to result from heritability of obesity susceptibility genes that interact with known and unknown components in the obesogenic environment to promote positive energy balance responsible for weight gain (Wardle et al. 2008b; Hofker and Wijmenga 2010).
Fig. 2.

The major complications associated with childhood obesity
The objective of this article is to provide a review on the origins, mechanisms, and health consequences of obesity susceptibility genes and the interaction with dietary macronutrients that predispose to childhood obesity. It is proposed that increased knowledge of these obesity susceptibility genes and interaction with dietary macronutrients will provide valuable insight for individual, family, and community preventative lifestyle intervention and eventually targeted nutritional and medicinal therapies.
Definition of energy balance
The ultimate cause of weight gain is usually considered in terms of altered energy balance, the basic components of which include energy consumption, energy expenditure, and the storage of excess energy in the form of triacylglycerol within tissues, primarily adipose (Hill et al. 2012). For instance, when energy in the form of calories derived from food and drink exceeds energy expenditure involving the combination of resting metabolic rate, energy necessary for absorption and metabolism of dietary macronutrients, and energy expended during physical activity, a state of positive energy balance occurs characterized by the storage of this energy (~60 to 80 %) within adipose tissue. In contrast, when energy consumption is less than the energy expenditure, a state of negative energy balance occurs characterized by the mobilization of energy (~60 to 80 %) from adipose tissue. Therefore, any genetic or environmental factor that alters body weight must involve the basic components of energy balance over a period of time (Hill et al. 2003).
Heritability of body weight and interaction with environmental factors
It has been recognized for several decades that obesity is a heritable disorder. The Hereditary Abilities Study initiated in 1952 was a comprehensive study performed in the United States to investigate heritability of physical traits, including measures of adiposity (birth weight, body weight, and waist circumference) among monozygous and dizygous twins, which demonstrated that the greater part of variance for these traits was genetically determined (Clark 1956). These findings were consistent with other studies reported over 20 years later indicating a high heritability of body weight among monozygous and dizygous twins (Brook et al. 1975; Borjeson 1976; Feinleib et al. 1977). However, it became apparent that genetic susceptibility interacts with undefined environmental factors to increase adiposity and body weight, in what has formally become known as a “gene–environment interaction” and defined as “a response or adaptation to an environmental agent, a behavior, or a change in behavior conditional on the genotype of the individual” (Bouchard 2009). An early study clearly demonstrating a gene–environment interaction in relation to weight gain was performed using 12 monozygotic twins who consumed a 1,000 kcal/day surplus of calories for a period of 100 days while maintaining a sedentary lifestyle (Bouchard et al. 1990). The results from this study showed a significant within twin-pair resemblance in adaptation to the excess calories (3 times more variance in response between twin-pairs than within twin-pairs in relation to increased body weight) suggesting that genetic susceptibility influenced the amount of stored fat. A study designed to assess genetic and environmental influences using 114 monozygotic twins, 81 dizygotic twins, and 98 virtual twins (same age but unrelated siblings) indicated that genetic variation contributed ~65 % to heritability of body mass index (BMI) while undefined environmental factors contributed to the remaining balance (Segal and Allison 2002). The heritability of childhood obesity was more closely examined and confirmed in a study using 8,234 children, which demonstrated a fourfold increased risk of childhood obesity if one parent was obese and a 10-fold increased risk of childhood obesity if both parents were obese (Reilly et al. 2005). Another study using 672 twin pairs indicated that genetic variation contributed 84–88 % to heritability of body weight when measured at 5 months and 5 years of age (Dubois et al. 2007). Therefore, it has been estimated that obesity susceptibility genes contribute an estimated 40–70 % to variation in BMI within populations (Day and Loos 2011). It should also be emphasized that heritability estimates have been shown to increase from early childhood through adolescence due to genetic susceptibility genes interacting more strongly with environmental factors (Lajunen et al. 2009; Dubois et al. 2012).
Missing or hidden heritability of common diseases
The “common disease, common variant” hypothesis states that genetic risk for common diseases is due to alleles of high frequency (Pritchard 2001; Reich and Lander 2001). It was once believed that common variants of high frequency would explain common disease heritability, defined as the proportion of phenotypic variance in a population due to additive genetic factors. However, after identifying hundreds of different variants associated with common diseases, the variants in combination accounted for only a small proportion of the estimated disease heritability (Hindorff et al. 2009). For example, the estimated heritability for BMI within populations has been reported to be 40–70 %, yet only a few percent of the estimated heritability has been accounted for by the combined phenotypic effect size or penetrance of gene reference variants (Li et al. 2010; Speliotes et al. 2010). The identification of “missing heritability” is now an essential step for determining how allelic variants contribute to common diseases such as obesity and translating this genetic information into clinical practice (Maher 2008; McCarthy et al. 2008). The missing heritability of common diseases may be the result of differences in allelic architecture not detected using genome-wide association studies (GWAS), in which case it may be more appropriately referred to as “hidden heritability” (Gibson 2010). Accumulating evidence now suggests that low-frequency alleles not detected by GWAS and also alleles with insufficient phenotypic effect size not detected by gene-linkage analysis represent “causal variants” responsible for hidden heritability of common diseases (Manolio et al. 2009). In other words, there is now evidence to suggest that partial linkage disequilibrium between reference variants and low-frequency causal variants with a minor allele frequency between 0.5 and 5.0 % and intermediate phenotypic effect sizes can fully account for hidden heritability of common diseases, where linkage disequilibrium is defined as a measure of the non-random association of alleles at two or more loci (Slatkin 2008). To date, several causal variants of intermediate effect size for common diseases have been found to exist in partial linkage disequilibrium (r2 < 0.5) with reference variants (Zhu et al. 2012). This same study also provides evidence that the minor allele frequency for causal variants may range from ~0.5 % (rare) to ~5.0 % (low frequency), more than one causal variant may be in partial linkage disequilibrium with the reference variant, and that causal variants likely represent deleterious mutations that adversely affect encoded protein function and metabolism (Zhu et al. 2012).
Epigenetics and interaction with environmental factors
It should be noted that heritability of body weight and interaction with environmental factors may occur through a different mechanism. This mechanism is referred to as “epigenetics” and generally defined as the study of heritable changes which effect gene expression or function without modifying the DNA sequence (Bird 2007). Recent studies suggest that epigenetic adaptation through increased or decreased methylation of nucleotide bases (primarily cytosine) in addition to more generalized histone modification can alter the transcription of genes involved in regulating energy balance (Russo et al. 2010; Herrera et al. 2011). To date, the methylation status for a number of genes involved in metabolic or endocrine function has been identified that results from dietary changes during prenatal or early postnatal life and associated with childhood or adulthood adiposity (Tobi et al. 2009; Godfrey et al. 2011). However, it should be noted that the definitive role of epigenetic–environment interactions in relation to obesity remains controversial due to difficulties in discriminating the cause and effect for these DNA modifications (does epigenetic modification cause obesity or does obesity cause epigenetic modification) and the central importance of DNA sequence at particular loci (Martin et al. 2011; Youngson and Morris 2012). As a result, it is believed that meaningful clinical application of epigenetics in the prevention or treatment of obesity will remain a vision until further research is performed (Franks and Ling 2010).
Thrifty gene hypothesis
A presentation of gene–environment interactions should include the concept of “thrifty genes” first introduced and later modified by the American geneticist Neel (1962, 1999). In brief, the thrifty gene hypothesis states that certain groups of people with hunter-gatherer evolutionary lifestyles may have experienced repeated periods of feast and famine, which through adaptation, resulted in the natural selection of thrifty genes, thereby eventually predisposing these groups to chronic diseases of civilization such as obesity and diabetes. The thrifty genes encode proteins directly or indirectly involved in maintaining energy balance, such as the conversion of food calories into fat when food supplies are plentiful (feasting period). These genes were proposed to contain certain variants that were believed to enhance this conversion. The increased storage of fat was proposed to be used as an energy source when food calories become limited (fasting period). A key feature of the thrifty gene hypothesis is that selective advantage of these gene variants become a disadvantage (susceptibility to obesity and diabetes) for individuals living in an obesogenic environment. It must be emphasized that this particular feature suggests that thrifty genes represent gene variants present at a frequency greater than 1 % in a population due to natural selection, in contrast to gene variants present at lower frequency and responsible for rare monogenic forms of syndromic and non-syndromic obesity (Kagawa et al. 2002).
The thrifty gene hypothesis is consistent with higher frequency gene variants interacting with environmental factors to promote weight gain among certain populations. The most studied and documented population referenced in relation to thrifty genes includes the Pima Indians of Arizona. This population at one time had a subsistence lifestyle but is now predisposed to weight gain and diabetes as a result of a modern obesogenic lifestyle (Knowler et al. 1983, 1991). Moreover, a number of well-designed studies using both monozygotic and dizygotic twins have provided strong evidence demonstrating that children with obesity susceptibility genes living in an obesogenic environment (adopted family) are at increased risk of developing childhood obesity (Borjeson 1976; Silventoinen and Kaprio 2009; Silventoinen et al. 2010). These results are consistent with recent anthropological studies verifying that gene–environment interactions are responsible for marked differences among populations genetically susceptible to weight gain (Casazza et al. 2011).
It should be noted, however, that the thrifty gene hypothesis remains controversial due to suggestions that famines were neither long nor severe enough to select for thrifty genes and that no evidence exists for excessive weight gain between famines or during the feast periods (Prentice et al. 2008; Speakman 2008). In addition, it has been shown that the average daily energy expenditure is similar for modern hunter-gatherers and Westerners after controlling for body size, in contrast to previous assumptions that early hunter-gatherers had an increased daily energy expenditure (Pontzer et al. 2012). Other studies have also found no evidence for the natural selection of obesity and diabetes susceptibility genes identified using GWAS and that such genes are unlikely to exist in a stable polymorphic state (Southam et al. 2009; Baig et al. 2011). An alternative explanation that is gaining recognition for the current obesity epidemic involves a nonadaptive approach referred to as the “predation release” or “drifty hypothesis”, which suggests that adiposity may result from random genetic mutations and drift that interact with environmental factors (Speakman 2008; Speakman and O’Rahilly 2012). Regardless of the evolutionary mechanism, it is clear that obesity susceptibility genes interact with known and unknown environmental factors that result in positive energy balance responsible for weight gain (Fig. 3).
Fig. 3.

The interaction of obesity susceptibility genes with single-nucleotide polymorphisms or variants with known and unknown environmental factors that predispose to weight gain
The high-fat diet as a major component in the obesogenic environment
The obesogenic environment consists of a complex interplay of contributing factors that influence behavior thereby effecting dietary choice, physical activity, or metabolism responsible for maintaining energy balance (Patrick et al. 2004). A number of recent studies suggest that both sedentary behavior (viewing television, playing video games, doing cognitive work, and listening to music) and reduced overall physical activity along with shorter sleep duration promote the overconsumption of dietary macronutrients, particularly fats and refined carbohydrates (Stroebele and de Castro 2006; Graves et al. 2007; Temple et al. 2007; Chaput et al. 2008; Westerlund et al. 2009). The increased consumption of a high-fat diet, particularly a high-fat diet enriched with saturated fatty acids, has been found to be strongly associated with increased adiposity in overweight and obese children (Aeberli et al. 2006, 2008). Moreover, another recent study performed with 810 participants indicated a highly significant association of saturated fatty acid consumption (but not plant protein, carbohydrates, or other types of fat) at 6 months with body weight at 18 months of age (Lin et al. 2012). Consistent with these results, studies indicate that obesity susceptibility genes tend to preferentially interact with saturated fatty acids, but not monounsaturated fatty acids or polyunsaturated fatty acids, to promote weight gain (Razquin et al. 2010; Corella et al. 2011). For these reasons, it is widely accepted that high-fat diets, characterized by enhanced palatability and high energy density, may be primarily responsible for the current obesity epidemic. However, it should be noted that studies suggest that increased consumption of carbohydrates, particularly refined carbohydrates and sugar-sweetened beverages, during the past 30 years better parallels the increased prevalence of obesity (Gross et al. 2004; Malik et al. 2006, 2010).
The importance of gene–diet interactions believed responsible for chronic nutrition-related diseases, such as obesity and type 2 diabetes, has prompted the development of a relatively new scientific discipline referred to as “nutritional genetics” and “nutritional genomics” (Ordovas and Corella 2004). Although current childhood obesity intervention programs have traditionally focused only on generalized population guidelines, further investigation and insight into gene–diet interactions may serve an important role in both the prevention and treatment of childhood obesity (Papoutsakis and Dedoussis 2007; Hetherington and Cecil 2010). This will be possible by enhancing our knowledge surrounding the etiology and pathophysiology of childhood obesity and developing methods to prevent the onset and improve treatment of childhood obesity using targeted nutritional and medicinal therapies.
Childhood obesity susceptibility genes
During the course of a decade (1996–2005), an extensive amount of work was performed to identify candidate obesity susceptibility genes responsible for heritability of obesity phenotypes. The culmination of these studies resulted in the identification of 127 candidate obesity susceptibility genes (Rankinen et al. 2006). However, only a limited number (~25 %) of these candidate genes have been validated using independent studies. This small percentage of candidate obesity susceptibility genes in addition to many other genes recently identified using GWAS now comprise a comprehensive list of approximately 69 obesity susceptibility genes (34 genes from candidate studies and 35 genes from GWAS) that predispose to increased body weight, BMI, or body fat percentage (Hofker and Wijmenga 2010; Day and Loos 2011; Fernandez et al. 2012). There are three different types of childhood obesity (syndromic, non-syndromic, and common) based on distinct genetic and phenotypic characteristics that will be described in the following sections (Fig. 4).
Fig. 4.
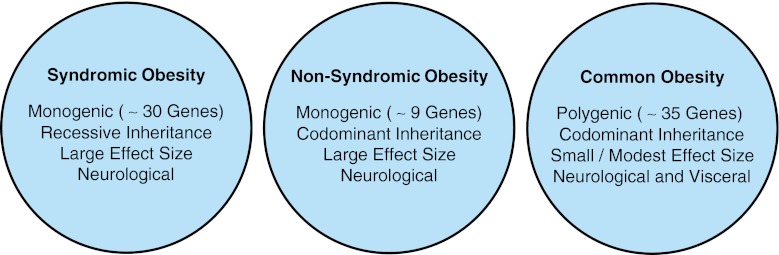
The three different types of childhood obesity based on distinct genetic and phenotypic characteristics
Syndromic childhood obesity
The first type of childhood obesity is represented by approximately 30 unidentified susceptibility genes responsible for rare monogenic forms of syndromic obesity. The best-known examples of syndromic obesity are represented by Prader–Willi, Bardet–Biedl, Alstrom, Carpenter, Rubinstein–Taybi, and Cohen syndromes. In general, children with syndromic obesity have extreme adiposity, physical dysmorphology, and intellectual disabilities, some with undefined neuroendocrine abnormalities. It is the latter abnormality believed responsible for adversely affecting function of the hypothalamus which serves as the brain appetite center regulating energy balance through food consumption and energy expenditure (Farooqi and O’Rahilly 2005; Mutch and Clement 2006; Goldstone and Beales 2008; Schaefer et al. 2010). As a result, children with syndromic obesity are usually characterized with severe hyperphagia and diminished satiety which promotes weight gain (Bray 1992; Sahoo et al. 2008; Marshall et al. 2011). These particular disorders are genetically complex and involve several overlapping and undefined loci believed responsible for the altered regulation of energy balance. With respect to a possible gene–diet interaction responsible for the best characterized form of syndromic obesity, in this case Prader–Willi syndrome, a recent study has indicated that a strict low-fat and modified carbohydrate diet (25 % protein, 20 % fat, and 55 % modified carbohydrate) can successfully prevent or at least reduce weight gain among patients with this disease (Schmidt et al. 2008). The reason being is that children with Prader–Willi syndrome have delayed gastric emptying due to ineffective stomach contractions. The therapeutic diet allows for increased and timely absorption of carbohydrates, otherwise somewhat inhibited by a high-fat diet, to prevent hypoglycemia and resultant food-craving behavior marked by hyperphagia. However, it should be noted that children with Prader–Willi syndrome receiving this therapeutic diet were of significantly decreased stature beginning at 2 years of age, suggesting that growth hormone may be a useful additional treatment for these patients. It is therefore suspected that this concern prompted authors from including children without Prader–Willi syndrome into the study.
Non-syndromic childhood obesity
The second type of childhood obesity is represented by approximately 8 susceptibility genes responsible for rare monogenic forms of non-syndromic obesity, defined by weight gain in the absence of other clinical symptoms (Choquet and Meyre 2010). The eight genes responsible for non-syndromic obesity include brain-derived neurotrophic factor (BDNF), leptin (LEP), leptin receptor (LEPR), melanocortin-4 receptor (MC4R), neurotrophic tyrosine kinase receptor type 2 (NTRK2), prohormone convertase 1 (PCSK1), proopiomelanocortin (POMC), and single-minded homolog 1 (SIM1). These 8 obesity susceptibility genes code for proteins which have a central role in the integration of peripheral and neuronal signals through the leptin/melanocortin pathway present in the hypothalamus and therefore also responsible for maintaining energy balance through food consumption and energy expenditure (Farooqi and O’Rahilly 2008). In the case of LEP, genetic mutations result in leptin deficiency, and administering leptin has been shown to have beneficial effects by restoring satiety and promoting weight loss (Farooqi et al. 2002). Mutations in these genes also cause severe hyperphagia and a lack of satiety that ultimately manifests in extreme forms of childhood obesity. To date, studies performed with patients possessing the best characterized form of non-syndromic obesity, in this case leptin deficiency, have shown that although restriction of a high-fat diet may partially or temporarily be successful, long-term weight management is difficult and usually unsuccessful (Erez et al. 2011).
Common childhood obesity
The third type of childhood obesity is represented by many undefined susceptibility genes believed responsible for contributing to common polygenic forms of childhood obesity (Zhao et al. 2011; Zhao and Grant 2011). These obesity susceptibility genes are associated with both common childhood and adult obesity, as shown in large population-based or case–control GWAS performed during the past several years (Meyre et al. 2009; den Hoed et al. 2010; Sandholt et al. 2010; Wu et al. 2010; Zhao et al. 2011) (Table 1). It is interesting to note that among the obesity susceptibility genes that have been identified using GWAS, different variants of the same gene (FTO, MC4R, and BNDF) may also be responsible for rare non-syndromic forms of childhood obesity. However, unlike the genes contributing to monogenic forms of childhood obesity characterized by large phenotypic effect sizes, genes contributing to common obesity have small to modest phenotypic effect sizes (~0.17 to 1.13 kg or 0.14–0.33 % body fat per risk allele) (Speliotes et al. 2010; Kilpelainen et al. 2011). That being the case, common obesity susceptibility genes function in an additive fashion and interact with environmental factors to promote positive energy balance resulting in substantial weight gain, consistent with common obesity being a complex genetic and metabolic disorder (Levin 2009; Sandholt et al. 2010). As indicated earlier, the prevalence of common childhood obesity has more than tripled during the past few decades and represents a major health problem as a result of numerous complications.
Table 1.
A list of 35 obesity susceptibility genes, reference SNPs, susceptible subjects, and phenotypes identified using GWAS adapted from Meyre et al. (2009), den Hoed et al. (2010), Sandholt et al. (2010), Wu et al. (2010), and Zhao et al. (2011)
| Chromosome | Nearest gene | Reference SNP | Subjects | Phenotypes |
|---|---|---|---|---|
| 1 | NEGR1 | rs2815752 | Adults | Weight, obesity, BMI |
| 1 | TNN13K | rs1514175 | Adults, children | BMI |
| 1 | PTBP2 | rs1555543 | Adults | BMI |
| 1 | SEC16B | rs543874 | Adults, children | BMI |
| 2 | TMEM18 | rs2867125 | Adults, children | Weight, obesity, BMI |
| 2 | RBJ | rs713586 | Adults, children | BMI |
| 2 | FANCL | rs887912 | Adults | BMI |
| 2 | LRP1B | rs2890652 | Adults | BMI |
| 3 | CADM2 | rs13078807 | Adults | BMI |
| 3 | ETV5 | rs9816226 | Adults | Weight, obesity, BMI |
| 4 | GNPDA2 | rs10938397 | Adults, children | Weight, obesity, BMI |
| 4 | SLC39A8 | rs13107325 | Adults | BMI |
| 5 | FLJ35779 | rs2112347 | Adults, children | BMI |
| 5 | ZNF608 | rs4836133 | Adults | BMI |
| 6 | NUDT3 | rs206936 | Adults | BMI |
| 6 | TFAP2B | rs987237 | Adults | BMI |
| 9 | LRRN6C | rs10968576 | Adults, children | BMI |
| 10 | PTER | rs10508503 | Adults, children | BMI |
| 11 | RPL27A | rs4929949 | Adults | BMI |
| 11 | BDNF | rs10767664 | Adults, children | Weight, obesity, BMI |
| 11 | MTCH2 | rs3817334 | Adults | BMI |
| 12 | FAIM2 | rs7138803 | Adults, children | Weight, obesity, BMI |
| 13 | MTIF3 | rs4771122 | Adults | BMI |
| 14 | PRKD1 | rs11847697 | Adults | BMI |
| 14 | NRXN3 | rs10150332 | Adults, children | BMI |
| 15 | MAP2K5 | rs2241423 | Adults | BMI |
| 16 | GPRC5B | rs12444979 | Adults | BMI |
| 16 | SH2B1 | rs7359397 | Adults | Weight, obesity, BMI |
| 16 | MAF | rs1424233 | Adults, children | BMI |
| 16 | FTO | rs1558902 | Adults, children | Weight, obesity, BMI |
| 18 | MC4R | rs571312 | Adults, children | Weight, obesity, BMI |
| 18 | NPC1 | rs1805081 | Adults, children | BMI |
| 19 | KCTD15 | rs299941 | Adults | BMI |
| 19 | QPCTL | rs2287019 | Adults, children | BMI |
| 19 | TMEM160 | rs3810291 | Adults | BMI |
Gene–diet interactions predisposing to common childhood obesity
A list of 16 well-established obesity susceptibility genes associated with common childhood obesity within American, Chinese, and European populations identified using GWAS include brain-derived neurotrophic factor (BNDF), cardiac troponin I-interacting kinase (TNNI3K), Fas apoptotic inhibitory molecule 2 (FAIM2), homo sapiens hypothetical protein FLJ35779 (FLJ35779), fat-mass and obesity associated (FTO), glucosamine-6-phosphate deaminase 2 (GNPDA2), leucine-rich repeat neuronal 6C (LRRN6C), melanocortin-4 receptor (MC4R), musculoaponeurotic fibrosarcoma oncogene homolog (MAF), Niemann-Pick C1 (NPC1), neurexin-3-alpha (NRXN3), phosphotriesterase-related (PTER), glutaminyl-peptide cyclotransferase-like (QPCTL), Rab and DnaJ domain containing (RBJ), Saccharomyces cerevisiae 16 homolog B (SEC16B), and transmembrane protein 18 (TMEM18). Five of these obesity susceptibility genes (FTO, MC4R, MAF, NPC1, and PTER) were identified in the first or second case–control GWAS for early-onset (less than 6 years of age) and morbid-adult obesity (BMI ≥ 40 kg/m2) (Hinney et al. 2007; Meyre et al. 2009). More recent meta-analysis of several case–control GWAS using ~40,000 individuals has determined that three of these obesity susceptibility genes (FTO, MC4R, and NPC1) are also associated with body fat percentage which serves as an accurate measure for whole body adiposity (Kilpelainen et al. 2011; den Hoed et al. 2012). It has been hypothesized that individuals possessing these obesity susceptibility genes associated with extreme (early-onset and morbid-adult) obesity and body fat percentage may either possess more than the average number and/or enhance phenotypic expression of other common obesity susceptibility genes (Meyre et al. 2009). The following sections will provide information describing what is now understood about obesity susceptibility genes interacting with either a high-fat diet or sugar-sweetened beverages to promote weight gain.
Obesity susceptibility genes that interact with dietary fats
The FTO gene is localized on chromosome 16q12.2 and encodes a Fe II- and 2-oxoglutarate-dependent dioxygenase that functions to catalyze demethylation of nucleotide bases (Gerken et al. 2007). The same study also indicated that the Fto gene in mice is expressed at high levels in the appetite center (arcurate nucleus) of the hypothalamus and regulated by periods of fasting/feeding. A direct Fto gene–diet interaction in relation to weight gain was first established in rats when fasting was shown to reduce the amounts of Fto mRNA and promote food consumption, while in contrast, feeding had the opposite result to increase amounts of Fto mRNA and decrease food consumption (Tung et al. 2010). These results provided the first indication that expression and functional amounts of the encoded FTO protein in the hypothalamus influenced appetite. The common FTO gene variant (rs9939609) was subsequently used in a population-based GWAS to identify this gene as the first to be associated with common childhood obesity (Frayling et al. 2007). Consistent with this result, a later case–control GWAS performed with lean and extremely obese German children indicated that this particular FTO gene variant was associated with common childhood obesity (Hinney et al. 2007). A number of other studies performed with both children and adults indicated that this FTO gene variant was associated with increased and preferential consumption of energy-dense macronutrients, particularly foods enriched with saturated fatty acids (Cecil et al. 2008; Timpson et al. 2008; Bauer et al. 2009). Studies have also provided evidence that weight gain among children and adults with this FTO gene variant results from an unusual eating behavior characterized by loss-of-control eating episodes for high-fat foods (Wardle et al. 2008a; den Hoed et al. 2009; Tanofsky-Kraff et al. 2009). Interestingly, the weight gain was found to be independent of alterations in energy expenditure typically associated with hypothalamic abnormalities and was unlike any other eating behavior previously reported in the general population (Speakman et al. 2008; Haupt et al. 2009; Jonassaint et al. 2011). At the same time, studies performed using two different Fto mouse models characterized by complete inactivation and a dominant point mutation of the Fto gene revealed a complex and contradictory phenotype compared to humans possessing the common FTO gene variant (Church et al. 2009; Fischer et al. 2009). In brief, these Fto mouse models were shown to be lean and protected from obesity despite being fed a basal or high-fat diet and having consumed increased amounts of these diets after adjustment for body weight. Consistent with the decreased relative amounts or function of the encoded FTO protein being associated with a lean phenotype for these mice, a subsequent study performed using a mouse model characterized by overexpression of the Fto gene marked by increased amounts of encoded FTO protein indicated a dose-dependent increase in adiposity, weight gain, and glucose intolerance resulting from increased food consumption when adjusted for body weight (Church et al. 2010). Therefore, the common FTO gene variant in the human population is a gain-of-function mutation that interacts with a high-fat diet to promote common childhood obesity through an unusual behavioral response characterized by an increased consumption of high-fat diets.
The MC4R gene is localized on chromosome 18q21.32 and encodes a complex plasma membrane protein that belongs to the seven transmembrane G protein-coupled receptor family that activates adenylate cyclase to produce cyclic adenosine monophosphate (cAMP) during signal transduction (Magenis et al. 1994; Yang et al. 2000). The first evidence indicating that the MC4R gene was associated with regulating energy balance resulted from mice possessing a targeted disruption of the Mc4r gene (Bultman et al. 1992; Huszar et al. 1997). These studies demonstrated that Mc4r heterozygous (Mc4r+/−) mice were susceptible to intermediate weight gain compared to Mc4r normal (Mc4r+/+) and Mc4r homozygous (Mc4r−/−) mice, suggesting a codominant mode of inheritance for the phenotype. Moreover, in comparison with Mc4r+/+ mice fed a high-fat diet, the Mc4r−/− mice were found to have hyperphagia and altered energy expenditure characterized by decreased diet-induced activity and thermogenesis (Butler et al. 2001; Weide et al. 2003). Consistent with these results, the Mc4r−/− mice developed hyperphagia when fed a high-fat diet, but not when fed a low-fat diet, providing evidence for a gene–diet interaction in relation to weight gain (Butler and Cone 2003; Sutton et al. 2006). An additional study performed using adenovirus to preferentially knockdown expression of the Mc4r gene in hypothalamus of rats revealed that a high-fat diet induces hyperphagia and weight gain (Garza et al. 2008). With respect to studies that have been performed investigating the MC4R gene in relation to weight gain in humans, a frameshift mutation in the MC4R gene provided the first compelling evidence that this gene was associated with extreme childhood obesity (Vaisse et al. 1998; Yeo et al. 1998). In support of these studies, additional studies found that individuals possessing MC4R gene variants were at increased risk of obesity and that binge eating was a major phenotype responsible for weight gain (Branson et al. 2003; Farooqi et al. 2003). These results were confirmed in a GWAS performed to identify chromosomal regions contributing to increased consumption of dietary components among 1,030 Hispanic children. This study found that MC4R gene variants have a key role in regulating body weight through both increased energy consumption and decreased energy expenditure (Cai et al. 2006; Cole et al. 2010). A more recent study suggests that individuals possessing MC4R gene variants consume increased amounts of food enriched with total and saturated fatty acids (Bauer et al. 2009). Therefore, similar to children with FTO gene variants, children possessing MC4R gene variants have an increased preference for calorie-dense foods enriched with fat, in addition to a decreased propensity for energy expenditure, both of which promote weight gain.
Finally, the NPC1 gene is localized on chromosome 18q11.2 and encodes a complex multi-spanning transmembrane protein that possesses structural homology with members of the resistance-nodulation-division family of prokaryotic permeases (Carstea et al. 1997; Davies et al. 2000). Studies have demonstrated that the NPC1 protein has a central role in regulating the transport of lipoprotein-derived lipids, such as cholesterol and fatty acids, from late endosomes/lysosomes to other cellular compartments (Garver et al. 2002; Chen et al. 2005). Although the NPC1 gene has been primarily investigated in relation to an autosomal-recessive lipid-storage disorder characterized by hepatosplenomegaly and neurological degeneration, four independent GWAS have now reported that a variant of the NPC1 gene (rs1805081 encoding H215R) is associated with measures of common childhood obesity (BMI and body fat percentage) among European and Chinese populations (Meyre et al. 2009; Wu et al. 2010; Kilpelainen et al. 2011; den Hoed et al. 2012). In addition, a study performed using an Npc1 mouse model indicated that Npc1 heterozygous (Npc1+/−) mice are predisposed to weight gain when fed a high-fat diet, but not when fed a low-fat diet, consistent with a gene–diet interaction responsible for promoting weight gain (Jelinek et al. 2009). These studies were confirmed using a different strain of mice that became obese and developed metabolic features associated with insulin resistance (Jelinek et al. 2010). Together, reanalysis of the combined data derived from both studies revealed that the Npc1 gene interacts with both modifying genes and a high-fat diet to promote weight gain, features that are consistent with common and complex diseases such as obesity (Jelinek et al. 2011). However, unlike the Fto and Mc4r genes, these mouse studies clearly indicate that the Npc1 gene does not promote weight gain through increased consumption of food or decreased energy expenditure, thereby excluding potential involvement of the hypothalamus. It is interesting to note that a number of earlier studies performed using both NPC1 human fibroblasts and the Npc1 mouse model provided information suggesting potential involvement of the NPC1 protein in regulating energy balance. For instance, NPC1 heterozygous (NPC1+/−) human fibroblasts have increased expression of caveolin-1, which serves as a protein marker for obesity and diabetes (Garver et al. 1997b; Catalán et al. 2008). These results were confirmed and extended using Npc1 heterozygous (Npc1+/−) mice, which compared to Npc1 normal (Npc1+/+) and Npc1 homozyous (Npc1−/−) mice, had livers with an increased expression of caveolin-1 and concentration of triacylglycerol, both of which serve as markers for obesity and diabetes (Garver et al. 1997a, 1999, 2007). More recent studies indicate that the Npc1 gene is downregulated by dietary fatty acids, but not dietary cholesterol, through feedback inhibition of the sterol regulatory element-binding protein (SREBP) pathway (Jelinek et al. 2012). Therefore, additional studies must be performed to further characterize the NPC1 gene in relation to common childhood obesity.
Obesity susceptibility genes that interact with dietary carbohydrates
It has been reported that three candidate obesity susceptibility genes, beta-2 adrenergic receptor (ADRB2), perilipin 1 (PLIN1), and peroxisome proliferator-activated receptor gamma (PPARG), which have not been found to be associated with either childhood or adult obesity using GWAS, interact with dietary carbohydrates to promote measures of obesity (Marti et al. 2002; Martinez et al. 2003; Smith et al. 2008). Moreover, a recent study performed using three different adult cohorts (Nurses’ Health Study, Health Professionals Follow-up Study, and Women’s Genome Health Study) indicated that six of 32 obesity susceptibility genes identified using GWAS interact with dietary carbohydrates (sugar-sweetened beverages including colas, fruit drinks/punches, and lemonades) to increase BMI when one or more servings are consumed per day (Qi et al. 2012). In this study, the obesity susceptibility genes found to interact with dietary carbohydrates to increase BMI included the FAIM2, FLJ35779, FTO, LRRN6C, RBJ, and SEC16B genes.
Preventative lifestyle intervention for common childhood obesity
The American Academy of Pediatrics proposed that prevention should be the first step in addressing the childhood obesity epidemic (Krebs and Jacobson 2003). However, systematic review of the literature provides conflicting results as to effectiveness of preventative lifestyle interventions for childhood obesity. For instance, a recent systemic review with meta-analysis for 33 studies indicated significant immediate and post-treatment improvements in weight and cardio-metabolic outcomes for childhood obesity, but suggested that further research was needed to determine the optimal length, intensity, and long-term effectiveness of these interventions (Ho et al. 2012). Moreover, another recent systematic review based on 37 independent studies with 27,946 children (mostly 6–12 years of age) provides evidence that intensive school-based programs serve as the most promising type of preventative lifestyle intervention (Waters et al. 2011). However, this same systematic review and others provide evidence indicating that obesity prevention programs for older preschool children and adolescents (13–19 years of age) are less effective (Bond et al. 2009; Hesketh and Campbell 2010; Waters et al. 2011). Consistent with these later systematic reviews, a number of studies have reported that the cornerstone for preventative lifestyle intervention, namely modification of dietary and exercise habits, has been largely ineffective (Miller 1999; Birch and Ventura 2009). A recent meta-analysis of randomized controlled trials determined that school-based physical activity interventions did not improve BMI compared to pre-existing physical education activity, thereby suggesting that mandated increased physical activity does not have a significant effect on preventing childhood obesity (Harris et al. 2009). This result is consistent with a more recent non-intervention prospective study examining children between 7 and 10 years of age suggesting that physical inactivity is the result rather than the cause of childhood obesity (Metcalf et al. 2011). While many obesity interventions have focused only on behavioral change in relation to more appropriate eating and exercise habits, few interventions have taken into consideration the emerging role of obesity susceptibility genes and interaction with environmental factors. There is now a crucial need to understand the molecular basis responsible for childhood obesity to provide more effective preventative lifestyle intervention and clinical care. A decade ago John R. Speakman, a leading obesity expert, stated that “addressing the genetic side of this (gene–environment) interaction is potentially a far more tractable problem than addressing the environmental component by reengineering society, because the level at which interventions might ultimately be made is the individual rather than the society as a whole. There is a clear need, therefore, to understand the genetic basis of food intake, energy expenditure and hence, energy balance variations” (Speakman 2004). These recommendations have only recently gained momentum due to the limited success of conventional preventative lifestyle intervention methods in the face of our growing knowledge concerning gene–environment interactions and targeted intervention for genetically predisposed individuals, families, and communities (Khoury et al. 2005; Gluckman et al. 2011).
One way in which individual preventative lifestyle interventions could utilize genetic information is by providing individuals with personalized information to influence behavior. Many behavioral change strategies aim to increase the perception of risk in individuals to improve motivation. The genetic information would expand on the risk perception model to provide individualized risk information, as well as personalize the harms of the health condition. If individuals are aware that they have the possibility of becoming obese and developing chronic complications, they may be more likely to adhere to behavioral change intervention (McBride et al. 2002). It has been determined that the availability of genetic information results in better compliance and longer-term weight reduction when using personalized diet plans to treat patients with a history of failed weight reduction (Arkadianos et al. 2007). A more recent study demonstrated that genetic testing for the FTO gene variant serves as a useful preventative and clinical tool when combined with other information by providing an explanation for increased body weight (Meisel et al. 2011). On the other hand, there may be a problem in using this model, because if tested individuals are found not to have an obesity susceptibility gene variant the effect is a perception for decreased risk of obesity. This may actually defeat the original purpose for providing a technologically advanced intervention in that individuals may be less likely to change behaviors if they have a perceived decreased risk of obesity. In addition to personalizing medical information for patients, genetic information could also be used to stratify individuals based on increased risk. By doing this, individuals who have obesity susceptibility gene variants would be categorized as “high risk” and therefore provided more intensive preventative lifestyle intervention strategies than individuals categorized as “low risk”. In this way, children who are genetically predisposed could be more closely monitored and potential preventative lifestyle interventions would become more cost-effective by targeting individuals at high risk (Johnson et al. 2005).
Unfortunately, although current individual preventative lifestyle interventions have only limited effectiveness, the benefits can be even less within a pediatric population. The reason is because most children have little control over their food choices and behaviors, and these choices impact weight status (Faith et al. 2004). A more logical place to intervene is at the family level. Family preventative lifestyle intervention strategies could have multiple positive effects as families not only control feeding practices of children, but often share cultural, environmental, and genetic predisposition for diseases such as obesity (Johnson et al. 2005). Interventions that target susceptible families could benefit the children at high risk for obesity while also benefiting close family members. This benefit could occur through targeting group-focused health promotion strategies that aim to affect social norms, increase social support, and strengthen familial bonds (Teufel-Shone 2006). Finally, as more information is obtained concerning the unique distribution of specific obesity susceptibility gene variants among diverse populations, community preventative lifestyle intervention strategies can be applied to lessen the impact of childhood obesity. It must be noted that targeted individuals in the community who do not possess suspected obesity susceptibility gene variants will likewise benefit from such interventions, as environmental factors still have a role in the etiology of childhood obesity.
Nutritional and medicinal intervention for genetically predisposed children
The goal for successful nutritional and medicinal intervention of common childhood obesity depends on understanding the molecular basis or mechanism for how gene variants predispose to weight gain. For instance, the FTO and MC4R gene variants tend to increase preference for calorie-dense foods enriched with fat and decrease satiety. A nutritional or medicinal therapy may soon be identified that stimulate regions of the brain (arcurate nucleus of the hypothalamus) to promote satiety. Although only recently approved by the Food and Drug Administration (FDA), the medicinal therapies lorcaserin hydrochloride (Belviq) and the combination of phenteramine/topiramate (Qsymia) may prove useful in this regard. Similar therapies may also be developed that regulate tissue and whole body lipid metabolism (decrease lipogenesis or increase lipolysis within select lipid-storage tissues) as suspected for children with NPC1 gene variations.
Conclusion and perspectives
The current epidemic of common childhood obesity represents a complex metabolic disease characterized by the interaction of obesity susceptibility gene variants with certain dietary macronutrients (saturated fatty acids or refined carbohydrates) and a sedentary lifestyle. The continued investigation of gene–diet interactions responsible for this health problem will be important for several reasons. First, diseases such as obesity and associated complications result from an undefined and complex interaction between susceptibility gene variants and various environmental components (Perusse and Bouchard 2000). The obesity susceptibility gene variants described in this article interact with the diet to either increase consumption of saturated fat and refined carbohydrates, decrease energy expenditure, or alter regulation of lipid metabolism to increase weight gain and adiposity (Bauer et al. 2009; Garver 2011). Second, the identification of gene–diet interactions should be at the forefront in attempts to understand the etiology and pathophysiology of nutrition-related diseases, particularly common childhood obesity (Levin 2009). Third, the interaction of specific gene variants with known dietary macronutrients will allow for more effective individual, family, and community preventative lifestyle intervention and eventually the development of targeted nutritional or medicinal therapies (Yang and Khoury 1997; Khoury et al. 2005). The overarching goal for investigating gene–diet interactions is to provide a plausible mechanism-based approach to personalized nutritional or medicinal therapy that will more effectively address the current epidemic of common childhood obesity.
Acknowledgments
This work was supported in part by the National Center for Research Resources and the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) through grant number UL1 TR000041, the Diabetes Research Center at the University of Washington through grant number P30 DK017047 as an institutional affiliate, a grant from Dedicated Health Research Funds of the University of New Mexico School of Medicine, a grant from the Tohono O’odham of Southwest Arizona Nation, and private donations for the investigation of childhood genetic and metabolic diseases.
Conflict of interest
No competing financial interests related to the content of this article exist.
Contributor Information
William S. Garver, Phone: 1-505-272-4790, FAX: 1-505-272-6587, Email: wgarver@salud.unm.edu
Robert A. Orlando, Phone: 1-505-272-4790, FAX: 1-505-272-6587, Email: rorlando@salud.unm.edu
References
- Aeberli I, Molinari L, Spinas G, Lehmann R, I’Allemand D, Zimmerman MB. Dietary intakes of fat and antioxidant vitamins are predictors of subclinical inflammation in overweight Swiss children. Am J Clin Nutr. 2006;84:748–755. doi: 10.1093/ajcn/84.4.748. [DOI] [PubMed] [Google Scholar]
- Aeberli I, Beljean N, Lehmann D, Spinas GA, Zimmerman MB. The increase of fatty acid-binding protein aP2 in overweight and obese children: interactions with dietary fat and impact on measures of subclinical inflammation. Int J Obes. 2008;32:1513–1520. doi: 10.1038/ijo.2008.128. [DOI] [PubMed] [Google Scholar]
- Arkadianos I, Valdes AM, Marinos E, Florou A, Gill RD, Grimaldi KA. Improved weight management using genetic information to personalize a calorie controlled diet. Nutr J. 2007;6:29–36. doi: 10.1186/1475-2891-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig U, Belsare P, Watve M, Jog M. Can thrifty gene(s) or predictive fetal programming for thriftiness lead to obesity? J Obes. 2011;2011:861049. doi: 10.1155/2011/861049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer F, Elbers CC, Adan RAH, Loos RJF, Onland-Moret NC, Grobbee DE, van Vliet-Ostaptchouk JV, Wijmenga C, van der Schouw YT. Obesity genes identified in genome-wide association studies are associated with adiposity measures and potentially with nutrient-specific food preference. Am J Clin Nutr. 2009;90:951–959. doi: 10.3945/ajcn.2009.27781. [DOI] [PubMed] [Google Scholar]
- Birch LL, Ventura AK. Preventing childhood obesity: what works? Int J Obes. 2009;33:S74–S81. doi: 10.1038/ijo.2009.22. [DOI] [PubMed] [Google Scholar]
- Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- Bond M, Wyatt K, Lloyd J, Welch K, Taylor R. Systematic review of the effectiveness and cost-effectiveness of weight management schemes for the under fives: a short report. Health Technol Assess. 2009;13:1–75. doi: 10.3310/hta13610. [DOI] [PubMed] [Google Scholar]
- Borjeson M. The aetiology of obesity in children: a study of 101 twin pairs. Acta Paediatr Scand. 1976;65:279–287. doi: 10.1111/j.1651-2227.1976.tb04887.x. [DOI] [PubMed] [Google Scholar]
- Bouchard C. Childhood obesity: are genetic differences involved? Am J Clin Nutr. 2009;89:1494–1501. doi: 10.3945/ajcn.2009.27113C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Tremblay A, Despres JP, Nadeau A, Lupien PJ, Theriault G, Dussault J, Moorjani S, Pinault S, Fournier G. The response to long-term overfeeding in identical twins. N Engl J Med. 1990;322:1477–1482. doi: 10.1056/NEJM199005243222101. [DOI] [PubMed] [Google Scholar]
- Branson R, Potoczna N, Kral JG, Lentes KU, Hoehe MR, Horber FF. Binge eating as a major phenotype of melanocortin-4 receptor gene mutations. N Engl J Med. 2003;348:1096–1103. doi: 10.1056/NEJMoa021971. [DOI] [PubMed] [Google Scholar]
- Bray GA. Genetic, hypothalamic, and endocrine features of clinical and experimental obesity. Prog Brain Res. 1992;93:333–340. doi: 10.1016/S0079-6123(08)64583-5. [DOI] [PubMed] [Google Scholar]
- Brook CGD, Huntley RMC, Slack J. Influence of heredity and environment in determination of skinfold thickness in children. Br Med J. 1975;2:719–721. doi: 10.1136/bmj.2.5973.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman SJ, Michaud EJ, Woychik RP. Molecular characterization of the mouse agouti locus. Cell. 1992;71:1195–1204. doi: 10.1016/S0092-8674(05)80067-4. [DOI] [PubMed] [Google Scholar]
- Butler AA, Cone RD. Knockout studies defining different roles for melanocortin receptors in energy homeostasis. Ann NY Acad Sci. 2003;994:240–245. doi: 10.1111/j.1749-6632.2003.tb03186.x. [DOI] [PubMed] [Google Scholar]
- Butler AA, Marks DL, Fan W, Kuhn CM, Bartolome M, Cone RD. Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat Neurosci. 2001;4:605–611. doi: 10.1038/88423. [DOI] [PubMed] [Google Scholar]
- Cai G, Cole SA, Butte N, Bacino C, Diego V, Tan K, Goring HH, O’Rahilly S, Farooqi IS, Comuzzie AG. A quantitative trait locus on chromosome 18q for physical activity and dietary intake in Hispanic children. Obesity. 2006;14:1596–1604. doi: 10.1038/oby.2006.184. [DOI] [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF, III, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O’Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Casazza K, Hanks LJ, Beasley M, Fernandez JR. Beyond thriftiness: independent and interactive effects of genetic and dietary factors on variations in fat disposition and distribution across populations. Am J Phys Anthro. 2011;145:181–191. doi: 10.1002/ajpa.21483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán V, Gómez-Ambrosi J, Rodríguez A, Silva C, Rotellar F, Gil MJ, Cienfuegos JA, Salvador J, Frühbeck G. Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin Endocrinol. 2008;68:213–219. doi: 10.1111/j.1365-2265.2007.03021.x. [DOI] [PubMed] [Google Scholar]
- Cecil JE, Tavendale R, Watt P, Hetherington MM, Palmer CNA. An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med. 2008;359:2558–2566. doi: 10.1056/NEJMoa0803839. [DOI] [PubMed] [Google Scholar]
- Chaput JP, Drapeau V, Poirier P, Teasdale N, Tremblay A. Glycemic instability and spontaneous energy intake: association with knowledge-based work. Psychosom Med. 2008;70:797–804. doi: 10.1097/PSY.0b013e31818426fa. [DOI] [PubMed] [Google Scholar]
- Chaput JP, Klingenberg L, Astrup A, Sjodin AM. Modern sedentary activities promote overconsumption of food in our current obesogenic environment. Obes Rev. 2011;12:e12–e20. doi: 10.1111/j.1467-789X.2010.00772.x. [DOI] [PubMed] [Google Scholar]
- Chen FW, Gordon RE, Ioannou YA. NPC1 late endosomes contain elevated levels of non-esterified (‘free’) fatty acids and an abnormally glycosylated form of the NPC2 protein. Biochem J. 2005;390:549–561. doi: 10.1042/BJ20050236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet H, Meyre D. Genomic insights into early-onset obesity. Genome Med. 2010;2:36. doi: 10.1186/gm157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church C, Lee S, Bagg EAL, McTaggart JS, Deacon R, Gerken T, Lee A, Moir L, Mecinovic J, Quwailid MM, Schofield CJ, Ashcroft FM, Cox RD. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS ONE. 2009;5:e1000599. doi: 10.1371/journal.pgen.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, Wells S, Bruning JC, Nolan PM, Ashcroft FM, Cox RD. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42:1086–1092. doi: 10.1038/ng.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PJ. The heritability of certain anthropometric characters as ascertained from measurements of twins. Am J Hum Genet. 1956;8:49–54. [PMC free article] [PubMed] [Google Scholar]
- Cole SA, Butte NF, Voruganti VS, Cai G, Haack K, Kent JW, Blangero J, Coumuzzie AG, McPherson JD, Gibbs RA. Evidence that multiple genetic variations of MC4R play a functional role in the regulation of energy expenditure and appetite in Hispanic children. Am J Clin Nutr. 2010;91:191–199. doi: 10.3945/ajcn.2009.28514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corella D, Arnett DK, Tucker KL, Kabagambe EK, Tsai M, Parnell LD, Lai CC, Lee YC, Warodomwichit D, Hopkins PN, Ordovas JM. A high intake of saturated fatty acids strengthens the association between the fat mass and obesity-associated gene and BMI. J Nutr. 2011;141:2219–2225. doi: 10.3945/jn.111.143826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JP, Chen FW, Ioannou YA. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science. 2000;290:2295–2298. doi: 10.1126/science.290.5500.2295. [DOI] [PubMed] [Google Scholar]
- Day FR, Loos RJR. Developments in obesity genetics in the era of genome-wide association studies. J Nutrigenet Nutrigenomics. 2011;4:222–238. doi: 10.1159/000332158. [DOI] [PubMed] [Google Scholar]
- de Onis M, Blossner M, Borghi E. Global prevalence and trends of overweight and obesity among preschool children. Am J Clin Nutr. 2010;92:1257–1264. doi: 10.3945/ajcn.2010.29786. [DOI] [PubMed] [Google Scholar]
- den Hoed M, Westerterp-Plantenga MS, Bouwman FG, Mariman ECM, Westerterp KR. Postprandial responses in hunger and satiety are associated with the rs9939609 single nucleotide polymorphism in FTO. Am J Clin Nutr. 2009;90:1426–1432. doi: 10.3945/ajcn.2009.28053. [DOI] [PubMed] [Google Scholar]
- den Hoed M, Ekelund U, Brage S, Grontved A, Zhao JH, Sharp SJ, Ong KK, Wareham NJ, Loos RJ. Genetic susceptibility to obesity and related traits in childhood and adolescence. Influence of loci identified by genome-wide association studies. Diabetes. 2010;59:2980–2988. doi: 10.2337/db10-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hoed M, Luan J, Langenberg C, Cooper C, Sayer AA, Jameson K, Kumari M, Kivimaki M, Hingorani AD, Grontved A, Khaw KT, Ekelund U, Wareham NJ, Loos RJF. Evaluation of common genetic variants identified by GWAS for early onset and morbid obesity in population-based samples. Int J Obes. 2012;34:1–6. doi: 10.1038/ijo.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois L, Girard M, Girard A, Tremblay R, Boivin M, Perusse D. Genetic and environmental influences on body size in early childhood: a twin birth-cohort study. Twin Res Hum Genet. 2007;10:479–485. doi: 10.1375/twin.10.3.479. [DOI] [PubMed] [Google Scholar]
- Dubois L, Kyvik KO, Girard M, Tatone-Tokuda F, Perusse D, Hjelmborg J, Skytthe A, Rasmussen F, Wright MJ, Lichtenstein P, Martin NG. Genetic and environmental contributions to weight, height, and BMI from birth to 19 years of age: an international study of over 12,000 twin pairs. PLoS ONE. 2012;7:e30153. doi: 10.1371/journal.pone.0030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez G, Tirosh A, Rudich A, Meiner V, Schwarzfuchs D, Sharon N, Shpitzen S, Bluher M, Stumvoll M, Thiery J, Fiedler GM, Friedlander Y, Leitersdorf E, Shai I. Phenotypic and genetic variation in leptin as determinants of weight regain. Int J Obes. 2011;35:785–792. doi: 10.1038/ijo.2010.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith MS, Scanlon KS, Birch LL, Francis LA, Sherry B. Parent-child feeding strategies and their relationships to child eating and weight status. Obes Res. 2004;12:1711–1722. doi: 10.1038/oby.2004.212. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, O’Rahilly S. Monogenic obesity in humans. Ann Rev Med. 2005;56:443–458. doi: 10.1146/annurev.med.56.062904.144924. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, O’Rahilly S. Mutations in ligands and receptors of the leptin-melanocortin pathway that leads to obesity. Nat Clin Pract Endocrinol Metab. 2008;4:569–577. doi: 10.1038/ncpendmet0966. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, Depaoli AM, O’Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, Keogh JM, Yeo GSH, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- Feinleib M, Garrison RJ, Fabsitz R, Christian JC, Hrubec Z, Borhani NO, Kannel WB, Rosenman R, Schwarz JT, Wagner JO. The NHLBI twin study of cardiovascular disease risk factors: methodology and summary of results. Am J Epidemiol. 1977;106:284–295. doi: 10.1093/oxfordjournals.aje.a112464. [DOI] [PubMed] [Google Scholar]
- Fernandez JR, Klimentidis YC, Dulin-Keita A, Casazza K. Genetic influences in childhood obesity: recent progress and recommendations for experimental designs. Int J Obes. 2012;36:479–484. doi: 10.1038/ijo.2011.236. [DOI] [PubMed] [Google Scholar]
- Fischer J, Kock L, Emmerling C, Vierkotten J, Peters T, Bruning JC, Ruther U. Inactivation of the Fto gene protects from obesity. Nature. 2009;458:894–899. doi: 10.1038/nature07848. [DOI] [PubMed] [Google Scholar]
- Franks PW, Ling C. Epigenetics and obesity: the devil is in the details. BMC Med. 2010;8:88. doi: 10.1186/1741-7015-8-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJF, Barroso I, Wareham NJ, Karpe F, Owens KR, Cardon LR, Walker M, Hitman GA, Palmer CNA, Doney ASF, Morris AD, Smith GD, Hattersley AT, McCarthy MI. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver WS. Gene-diet interactions in childhood obesity. Curr Genom. 2011;12:180–189. doi: 10.2174/138920211795677903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver WS, Erickson RP, Wilson JM, Colton TL, Hossain GS, Kozloski MA, Heidenreich RA. Altered expression of caveolin-1 and increased cholesterol in detergent insoluble membrane fractions from liver in mice with Niemann-Pick disease type C. Biochim Biophys Acta. 1997;1361:272–280. doi: 10.1016/S0925-4439(97)00047-1. [DOI] [PubMed] [Google Scholar]
- Garver WS, Hsu SC, Erickson RP, Greer WL, Byers DM, Heidenreich RA. Increased expression of caveolin-1 in heterozygous Niemann-Pick type II human fibroblasts. Biochem Biophys Res Comm. 1997;236:189–193. doi: 10.1006/bbrc.1997.6929. [DOI] [PubMed] [Google Scholar]
- Garver WS, Hossain GS, Winscott MM, Heidenreich RA. The Npc1 mutation causes an altered expression of caveolin-1, annexin II and protein kinases and phosphorylation of caveolin-1 and annexin II in murine livers. Biochim Biophys Acta. 1999;1453:193–206. doi: 10.1016/S0925-4439(98)00101-X. [DOI] [PubMed] [Google Scholar]
- Garver WS, Krishnan K, Gallagos JR, Michikawa M, Francis GA, Heidenreich RA. Niemann-Pick C1 protein regulates cholesterol transport to the trans-Golgi network and plasma membrane caveolae. J Lipid Res. 2002;43:579–589. [PubMed] [Google Scholar]
- Garver WS, Jelinek D, Oyarzo JN, Flynn J, Zuckerman M, Krishnan K, Chung BH, Heidenreich RA. Characterization of liver disease and lipid metabolism in the Niemann-Pick C1 mouse. J Cell Biochem. 2007;101:498–516. doi: 10.1002/jcb.21200. [DOI] [PubMed] [Google Scholar]
- Garza JC, Kim CS, Liu J, Zhang W, Lu XY. Adeno-associated virus-mediated knockdown of melanocortin-4 receptor in the paraventricular nucleus of the hypothalamus promotes high-fat diet induced hyperphagia and obesity. J Endocrinol. 2008;197:471–482. doi: 10.1677/JOE-08-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerken T, Girard CA, Tung YCL, Webby CJ, Saudek V, Hewitson KSH, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O’Rahilly S, Schofield CJ. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. Hints of hidden heritablity in GWAS. Nat Genet. 2010;42:558–560. doi: 10.1038/ng0710-558. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson M, Zimmet P, Forrester T. Losing the war against obesity: the need for a developmental perspective. Sci Trans Med. 2011;3:1–3. doi: 10.1126/scitranslmed.3002554. [DOI] [PubMed] [Google Scholar]
- Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR, Emerald BS, Gale CR, Inskip HM, Cooper C, Hanson MA. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60:1534. doi: 10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstone AP, Beales PL. Genetic obesity syndromes. Front Horm Res. 2008;2008:37–60. doi: 10.1159/000115336. [DOI] [PubMed] [Google Scholar]
- Graves L, Stratton G, Ridgers ND, Cable NT. Comparison of energy expenditure in adolescents when playing new generation and sedentary computer games: cross sectional study. BMJ. 2007;335:1282–1284. doi: 10.1136/bmj.39415.632951.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross LS, Li L, Ford ES, Liu S. Increased consumption of refined carbohydrates and the epidemic of type 2 diabetes in the United States: an ecologic assessment. Am J Clin Nutr. 2004;79:774–779. doi: 10.1093/ajcn/79.5.774. [DOI] [PubMed] [Google Scholar]
- Harris KC, Kuramoto LK, Schulzer M, Retallack JE. Effect of school-based physical activity interventions on body mass index in children: a meta-analysis. Can Med Asso J. 2009;180:719–726. doi: 10.1503/cmaj.080966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt A, Thamer C, Staiger H, Tschritter O, Kirchhoff K, Machicao F, Haring HU, Stefan N, Fritsche A. Variation in the FTO gene influences food intake but not energy expenditure. Exp Clin Endocrinol Diabetes. 2009;117:194–197. doi: 10.1055/s-0028-1087176. [DOI] [PubMed] [Google Scholar]
- Herrera BM, Keildson S, Lindgren CM. Genetics and epigenetics of obesity. Maturitas. 2011;69:41–49. doi: 10.1016/j.maturitas.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh KD, Campbell KJ. Interventions to prevent obesity in 0–5 year olds: an updated systematic review of the literature. Obesity. 2010;18:S27–S35. doi: 10.1038/oby.2009.429. [DOI] [PubMed] [Google Scholar]
- Hetherington MM, Cecil JE. Gene-environment interactions in obesity. Forum Nutr. 2010;63:195–203. doi: 10.1159/000264407. [DOI] [PubMed] [Google Scholar]
- Hill JO, Levine JS, Saris WHM. Energy expenditure and physical activity. In: Bray G, Bouchard C, editors. Handbook of obesity. 2. New York City, NY: Marcel Dekker, Inc.; 2003. pp. 631–654. [Google Scholar]
- Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation. 2012;126:126–132. doi: 10.1161/CIRCULATIONAHA.111.087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinney A, Nguyen TT, Scherag A, Friedel S, Bronner G, Muller TD, Grallert H, Illig T, Wichmann HE, Rief W, Schafer H, Hebebrand J. Genome wide association (GWA) study for early onset extreme obesity supports the role of fat mass and obesity associated gene (FTO) variants. PLoS ONE. 2007;2:e1361. doi: 10.1371/journal.pone.0001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M, Garnett SP, Baur L, Burrows T, Stewart L, Neve M, Collins C. Effectiveness of lifestyle interventions in child obesity: systematic review with meta-analysis. Pediatrics. 2012;130:e1647. doi: 10.1542/peds.2012-1176. [DOI] [PubMed] [Google Scholar]
- Hofker M, Wijmenga C. A supersized list of obesity genes. Nat Genet. 2010;41:139–140. doi: 10.1038/ng0209-139. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/S0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Jelinek D, Heidenreich RA, Erickson RP, Garver WS. Decreased Npc1 gene dosage in mice is associated with weight gain. Obesity. 2009;18:1457–1459. doi: 10.1038/oby.2009.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek D, Millward V, Birdi A, Trouard TP, Heidenreich RA, Garver WS. Npc1 haploinsufficiency promotes weight gain and metabolic features associated with insulin resistance. Hum Mol Genet. 2010;20:312–321. doi: 10.1093/hmg/ddq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek D, Heidenreich RA, Garver WS. The Niemann-Pick C1 gene interacts with a high-fat diet and modifying genes to promote weight gain. Am J Med Genet. 2011;155:2317–2319. doi: 10.1002/ajmg.a.34135. [DOI] [PubMed] [Google Scholar]
- Jelinek D, Castillo JJ, Richardson LM, Luo L, Heidenreich RA, Garver WS. The Niemann-Pick C1 gene is downregulated in livers of C57BL/6J mice by dietary fatty acids, but not dietary cholesterol, through feedback inhibition of the SREBP pathway. J Nutr. 2012;142:1935–1942. doi: 10.3945/jn.112.162818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J, Giles RT, Larsen L, Ware J, Adams T, Hunt SC. Utah’s family high risk program: bridging the gap between genomics and public health. Prev Chronic Dis. 2005;2:1–7. doi: 10.1191/1479972305cd059ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassaint CR, Szatkiewicz JP, Bulik CM, Thornton LM, Bloss C, Berrettini W, Kaye WH, Bergen AW, Magistretti P, Strober M, Keel PK, Brandt H, Crawford S, Crow S, Fichter MM, Goldman D, Halmi KA, Johnson G, Kaplan AS, Klump KL, La Via M, Mitchell J, Rotondo A, Treasure J, Woodside DB. Specific common variants of the obesity-associated FTO gene are not associated with psychological and behavioral eating disorder phenotypes. Am J Med Genet. 2011;156B:454–461. doi: 10.1002/ajmg.b.31182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa Y, Yanagisawa Y, Hasegawa K, Suzuki H, Tasuda K, Kudo H, Abe M, Matsuda S, Ishikawa Y, Tsuchiya N, Sato A, Umetsu K, Kagawa Y. Single nucleotide polymorphisms of thrifty genes for energy metabolism: evolutionary origins and prospects for intervention to prevent obesity-related diseases. Biochem Biophys Res Comm. 2002;295:207–222. doi: 10.1016/S0006-291X(02)00680-0. [DOI] [PubMed] [Google Scholar]
- Khoury MJ, Davis R, Gwinn M, Lindegren ML, Yoon P. Do we need genomic research for the prevention of common diseases with environmental causes? Am J Epidemiol. 2005;161:799–805. doi: 10.1093/aje/kwi113. [DOI] [PubMed] [Google Scholar]
- Kilpelainen TO, Zillikens MC, Stancakova A, Finucane FM, Ried JS, Langenberg C, Zhang W, Beckmann JS, Luan J, Vandenput L, Styrkarsdottir U, Zhou Y, Vernon Smith A, Zhao JH, Amin N, Vedantam S, Shin SY. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43:753–760. doi: 10.1038/ng.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowler WC, Pettitt DJ, Bennett PH, Williams RC. Diabetes mellitus in the Pima Indians: genetic and evolutionary considerations. Am J Phys Anthropol. 1983;62:107–114. doi: 10.1002/ajpa.1330620114. [DOI] [PubMed] [Google Scholar]
- Knowler WC, Pettitt DJ, Saad MF, Charles MA, Nelson RG, Howard BV, Bogardus C, Bennett PH. Obesity in the Pima Indians: its magnitude and relationship with diabetes. Am J Clin Nutr. 1991;53:1543S–1551S. doi: 10.1093/ajcn/53.6.1543S. [DOI] [PubMed] [Google Scholar]
- Krebs NF, Jacobson MS. Prevention of pediatric overweight and obesity. Pediatrics. 2003;112:424–430. doi: 10.1542/peds.112.2.424. [DOI] [PubMed] [Google Scholar]
- Lajunen HR, Kaprio J, Keski-Rahkonen A, Rose RJ, Pulkkinen L, Rissanen A, Silventoinen K. Genetic and environmental effects on body mass index during adolescence: a prospective study among Finnish twins. Int J Obes. 2009;33:559–567. doi: 10.1038/ijo.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin BE. Synergy of nature and nurture in the development of childhood obesity. Int J Obes. 2009;33:S53–S56. doi: 10.1038/ijo.2009.18. [DOI] [PubMed] [Google Scholar]
- Li S, Zhao JH, Luan J, Luben RN, Rodwell SA, Khaw KT, Ong KK, Wareham NJ, Loos RJF. Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association studies. Am J Clin Nutr. 2010;91:184–190. doi: 10.3945/ajcn.2009.28403. [DOI] [PubMed] [Google Scholar]
- Lin PH, Wang Y, Grambow SC, Goggins W, Almirall D. Dietary saturated fat intake is negatively associated with weight maintenance among the PREMIER participants. Obesity. 2012;20:571–575. doi: 10.1038/oby.2011.17. [DOI] [PubMed] [Google Scholar]
- McBride CM, Bepler G, Lipkus IM, Lyna P, Samsa G, Albright J, Datta S, Rimer BK (2002) Incorporating genetic susceptibility feedback into a smoking cessation program for African-American smokers with low income. Cancer Epidemiol Biomarkers Prev 11:521–528 [PubMed]
- Magenis RE, Smith L, Nadeau JH, Johnson KR, Mountjoy KG, Cone RD. Mapping of the ACTH, MSH, and neural (MC3 and MC4) melanocortin receptors in the mouse and human. Mamm Genom. 1994;5:503–508. doi: 10.1007/BF00369320. [DOI] [PubMed] [Google Scholar]
- Maher B. The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- Malik VS, Schulze MB, Hu FB. Intake of sugar-sweetened beverages and weight gain: a systematic review. Am J Clin Nutr. 2006;84:274–288. doi: 10.1093/ajcn/84.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik VS, Popkin BM, Bray GA, Despres JP, Hu FB. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation. 2010;121:1356–1364. doi: 10.1161/CIRCULATIONAHA.109.876185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakraborty A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TFC, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom syndrome: genetics and clinical overview. Curr Genom. 2011;12:225–235. doi: 10.2174/138920211795677912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti A, Corbalan MS, Martinez-Gonzalez MA, Forga L, Martinez JA. CHO intake alters obesity risk associated with Pro12Ala polymorphism of PPAR-gamma gene. J Physiol Biochem. 2002;58:219–220. doi: 10.1007/BF03179859. [DOI] [PubMed] [Google Scholar]
- Martin DI, Cropley JE, Suter CM. Epigenetics in disease: leader or follower? Epigenetics. 2011;6:843–848. doi: 10.4161/epi.6.7.16498. [DOI] [PubMed] [Google Scholar]
- Martinez JA, Corbalan MS, Sanchez-Villegas A, Forga L, Marti A, Martinez-Gonzalez MA. Obesity risk is associated with carbohydrate intake in women carrying the Gln27Glu beta-2 adrenoceptor polymorphism. J Nutr. 2003;133:2549–2554. doi: 10.1093/jn/133.8.2549. [DOI] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Meisel SF, Walker C, Wardle J. Psychological responses to genetic testing for weight gain: a vignette study. Obesity. 2011;20:540–546. doi: 10.1038/oby.2011.324. [DOI] [PubMed] [Google Scholar]
- Metcalf BS, Hosking J, Jeffery AN, Voss LD, Henley W, Wilkin TJ. Fatness leads to inactivity, but inactivity does not lead to fatness: a longitudinal study in children (EarlyBird 45) Arch Dis Child. 2011;96:942–947. doi: 10.1136/adc.2009.175927. [DOI] [PubMed] [Google Scholar]
- Meyre D, Delplanque J, Chevre JC, Lecoeur C, Lobbens S, Gallina S, Durand E, Vatin V, Degraeve F, Proenca C, Gaget S, Korner A, Kovacs P, Kiess W, Tichet J, Marre M, Hartikainen AL, Horber F, Potoczna N, Hercberg S, Levy-Marchal C, Pattou F, Heude B, Tauber M, McCarthy MI, Blakemore AIF, Montpetit A, Polychronakos C, Weill J, Coin LJM, Asher J, Elliott P, Jarvelin MR, Visvikis-Siest S, Balkau B, Sladek R, Balding D, Walley A, Dina C, Froguel P. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41:157–159. doi: 10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
- Miller WC. How effective are traditional dietary and exercise interventions for weight loss? Med Sci Sports Exerc. 1999;31:1129–1134. doi: 10.1097/00005768-199908000-00008. [DOI] [PubMed] [Google Scholar]
- Mutch DM, Clement K. Unraveling the genetics of human obesity. PLoS Genet. 2006;2:e88. doi: 10.1371/journal.pgen.0020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. 1962;14:353–362. [PMC free article] [PubMed] [Google Scholar]
- Neel JV. The “thrifty genotype” in 1998. Nutr Rev. 1999;57:S2–S9. doi: 10.1111/j.1753-4887.1999.tb01782.x. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Flegal KM, Carroll MD, Johnson GL. Prevalence and trends in overweight among US children and adolescents, 1999–2000. JAMA. 2002;288:1728–1732. doi: 10.1001/jama.288.14.1728. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Curtin LR, Lamb MM, Flegal KM. Prevalence of high body mass index in US children and adolescents, 2007–2008. JAMA. 2010;303:242–249. doi: 10.1001/jama.2009.2012. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity and trends in body mass index among US children and adolescents, 1999–2010. JAMA. 2012;307:483–490. doi: 10.1001/jama.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshansky SJ, Passaro DJ, Hershow RC, Layden J, Carnes BA, Brody J, Hayflick L, Butler RN, Allison DB, Ludwig DS. A potential decline in life expectancy in the United States in the 21st century. N Engl J Med. 2005;352:1138–1145. doi: 10.1056/NEJMsr043743. [DOI] [PubMed] [Google Scholar]
- Ordovas JM, Corella D. Nutritional genomics. Annu Rev Genomics Hum Genet. 2004;5:71–118. doi: 10.1146/annurev.genom.5.061903.180008. [DOI] [PubMed] [Google Scholar]
- Papoutsakis C, Dedoussis GV. Gene-diet interactions in childhood obesity: paucity of evidence as the epidemic of childhood obesity continues to rise. Pers Med. 2007;4:133–146. doi: 10.2217/17410541.4.2.133. [DOI] [PubMed] [Google Scholar]
- Patrick K, Norman GJ, Calfas KJ, Sallis JF, Zabinski MF, Rupp J, Cella J. Diet, physical activity, and sedentary behaviors as risk factors for overweight in adolescence. Arch Pediatr Adolesc Med. 2004;158:385–390. doi: 10.1001/archpedi.158.4.385. [DOI] [PubMed] [Google Scholar]
- Perusse L, Bouchard C. Gene-diet interactions in obesity. Am J Clin Nutr. 2000;72:1285S–1290S. doi: 10.1093/ajcn/72.5.1285s. [DOI] [PubMed] [Google Scholar]
- Pontzer A, Raichlen DA, Wood BM, Mabulla AZ, Racette SB, Marlowe FW. Hunter-gatherer energetics and human obesity. PLoS ONE. 2012;7:e40503. doi: 10.1371/journal.pone.0040503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice AM, Hennig BJ, Fulford AJ. Evolutionary origins of the obesity epidemic: natural selection of thrifty genes or genetic drift following predation release? Int J Obes. 2008;32:1607–1610. doi: 10.1038/ijo.2008.147. [DOI] [PubMed] [Google Scholar]
- Pritchard JK. Are rare variants responsible for susceptibility for complex diseases? Am J Hum Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Chu AY, Kang JH, Jensen MK, Curhan GC, Pasquale LR, Ridker PM, Hunter DJ, Willett WC, Rimm EB, Chasman DI, Hu FB, Qi L. Sugar-sweetened beverages and genetic risk of obesity. N Eng J Med. 2012;367:1387–1396. doi: 10.1056/NEJMoa1203039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, Walts B, Perusse L, Bouchard C. The human obesity gene map: the 2005 update. Obesity. 2006;14:529–644. doi: 10.1038/oby.2006.71. [DOI] [PubMed] [Google Scholar]
- Razquin C, Martinez JA, Martinez-Gonzalez MA, Bes-Rastrollo M. A 3-year intervention with a Mediterranean diet modified the association between the rs9939609 gene variant in FTO and body weight changes. Int J Obes. 2010;34:266–272. doi: 10.1038/ijo.2009.233. [DOI] [PubMed] [Google Scholar]
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/S0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- Reilly JJ, Armstrong J, Dorosty AR, Emmett PM, Ness A, Rogers I, Steer C, Sherriff A. Early life risk factors for obesity in childhood: Cohort study. BMJ. 2005;330:1357–1363. doi: 10.1136/bmj.38470.670903.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo P, Lauria F, Siani A. Heritability of body weight: moving beyond genetics. Nutr Metabol Cardio Dis. 2010;20:691–697. doi: 10.1016/j.numecd.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719–721. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandholt CH, Sparso T, Grarup N, Albrechtsen A, Almind K, Hansen L, Toft U, Jorgensen T, Hansen T, Pedersen O. Combined analyses of 20 common obesity susceptibility variants. Diabetes. 2010;59:1667–1673. doi: 10.2337/db09-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer E, Zaloszyc A, Lauer J, Durand M, Stutzmann F, Perdomo-Trujillo Y, Redin C, Bennouna Green V, Toutain A, Perrin L, Gerard M, Caillard S, Bei X, Lewis RA, Christmann D, Letsch J, Kribs M, Mutter C, Muller J, Stoetzel C, Fischbach M, Marion V, Katsanis N, Dollfus H. Mutations in SDCCAG8/NPHP10 cause Bardet-Biedl syndrome and are associated with penetrant renal disease and absent polydactyly. Mol Sydromol. 2010;1:273–281. doi: 10.1159/000331268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Pozza SB, Bonfig W, Schwarz HP, Dokoupil K. Successful early dietary intervention avoids obesity in patients with Prader-Willi syndrome: a ten-year follow-up. J Pediatr Endocrinol Metab. 2008;21:651–655. doi: 10.1515/JPEM.2008.21.7.651. [DOI] [PubMed] [Google Scholar]
- Segal NL, Allison DB. Twins and virtual twins: bases of relative body weight revisited. Int J Obes. 2002;26:437–441. doi: 10.1038/sj.ijo.0801941. [DOI] [PubMed] [Google Scholar]
- Serdula MK, Ivery D, Coates RJ, Freedman DS, Williamson DF, Byers T. Do obese children become obese adults? A review of the literature. Prev Med. 1993;22:167–177. doi: 10.1006/pmed.1993.1014. [DOI] [PubMed] [Google Scholar]
- Silventoinen K, Kaprio J. Genetics of tracking of body mass index from birth to late middle age: evidence from twin and family studies. Obes Facts. 2009;3:196–202. doi: 10.1159/000219675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silventoinen K, Rokholm B, Kaprio J, Sorensen TIA. The genetic and environmental influences on childhood obesity: a systematic review of twin and adoption studies. Int J Obes. 2010;34:29–40. doi: 10.1038/ijo.2009.177. [DOI] [PubMed] [Google Scholar]
- Slatkin M. Linkage disequilibrium - understanding the evolutionary past and mapping the medical future. Nature. 2008;9:477–485. doi: 10.1038/nrg2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Tucker KL, Yiannakouris N, Garcia-Bailo B, Mattei J, Lai CQ, Parnell LD, Ordovas JM. Perilipin polymorphism interacts with dietary carbohydrates to modulate anthropometric traits in Hispanics of Caribbean origin. J Nutr. 2008;138:1852–1858. doi: 10.1093/jn/138.10.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southam L, Soranzo N, Montgomery SB, Frayling TM, McCarthy MI, Barroso I, Zeggini E. Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes and obesity susceptibility variants? Diabetologia. 2009;52:1846–1851. doi: 10.1007/s00125-009-1419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman JR. Obesity: the integrated roles of environment and genetics. J Nutr. 2004;134:2090S–2105S. doi: 10.1093/jn/134.8.2090S. [DOI] [PubMed] [Google Scholar]
- Speakman JR. Thrifty genes for obesity, an attractive but flawed idea, and an alternative perspective: the ‘drifty gene’ hypothesis. Int J Obes. 2008;64:1611–1617. doi: 10.1038/ijo.2008.161. [DOI] [PubMed] [Google Scholar]
- Speakman JR, O’Rahilly S. Fat: an evolving issue. Dis Model Mech. 2012;5:569–573. doi: 10.1242/dmm.010553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman JR, Rance KA, Johnstone AM. Polymorphisms of the FTO gene are associated with variation in energy intake, but not energy expenditure. Obesity. 2008;16:1961–1965. doi: 10.1038/oby.2008.318. [DOI] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Magi R, Randall JC, Vedantern S, Winkler TW, Workalernahu T, Heid IM, Steinthorsdottir V, Stringham HM, Weedon MN, Wheeler E, Wood AR, Ferreira T, Weyant AV, Segre AV, Estrada K, Liang L, Nemesh J, Park JH, Gustafsson S, Kilpelainen TO, Yang J, Bouatia-Naji N, Esko T, Feitosa MF, Kutalik Z, Mangino M, Raychaudhuri S, Scherag A, Smith AV, Welch R, Zhao JH, Aben KK, Absher DM, Amin N, Dixon AI, Fisher E, Glazer NI, Goddard ME, Heard-Costa NI, Hoesel V, Hottenga JJ, Johansson A, Johnson T, Ketkar S, Lamina C, Li S, Moffatt MF, Myers RH, Narisu N, Perry JRB, Peters MJ, Preuss M, Ripatti S, Rivadeneira F, Sandholt C, Scott LJ, Timpson NJ, Tyrer JP, van Wingerden S, Watanabe RM, White CC, Wiklund F, Barlassina C, Chasman DI, Cooper MN, Jansson JO, Lawrence RW, Pellikka N, Prokopenko I, Shi J, Thiering E, Alavere H, Alibrandi MTS, Almgren P, Arnold AM, Aspelund T, Atwood LD, Balkau B, Balmforth AJ, Bennett AJ, Ben-Shlomo Y, Bergman RN, Bergmann S, Biebermann H, Blakemore AIF, Boes T, Bonnycastle LL, Bornstein SR, Brown MJ, Buchanan TA, Busonero F, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroebele N, de Castro JM. Listening to music while eating is related to increases in people’s food intake and meal duration. Appetite. 2006;47:285–289. doi: 10.1016/j.appet.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Sutton GM, Trevaskis JL, Hulver MW, McMillian RP, Markward NJ, Babin MJ, Meyer EA, Butler AA. Diet-genotype interactions in the development of the obese, insulin-resistant phenotype of C57BL/6J mice lacking melanocortin-3 or -4 receptors. Endocrinology. 2006;147:2183–2196. doi: 10.1210/en.2005-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanofsky-Kraff M, Han JC, Anandalingam K, Shomaker LB, Columbo KM, Wolkoff LE, Kozlosky M, Elliot C, Ranzenhofer LM, Roza CA, Yanovski SZ, Yanovski JA. The FTO gene rs9939609 obesity-risk allele and loss of control over eating. Am J Clin Nutr. 2009;90:1483–1488. doi: 10.3945/ajcn.2009.28439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple JL, Giacomelli AM, Kent KM, Roemmich JN, Epstein LH. Television watching increases motivated responding for food and energy intake in children. Am J Clin Nutr. 2007;85:355–361. doi: 10.1093/ajcn/85.2.355. [DOI] [PubMed] [Google Scholar]
- Teufel-Shone NI. Promising strategies for obesity prevention and treatment within American Indian communities. J Transcult Nurs. 2006;17:224–229. doi: 10.1177/1043659606288378. [DOI] [PubMed] [Google Scholar]
- Timpson NJ, Emmett PM, Frayling TM, Rogers I, Hattersley AT, McCarthy MI, Smith GD. The fat mass- and obesity-associated locus and dietary intake in children. Am J Clin Nutr. 2008;88:971–978. doi: 10.1093/ajcn/88.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–4053. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung YL, Ayuso E, Shan X, Bosch F, O’Rahilly S, Coll AP, Yeo GSH. Hypothalmus-specific manipulation of Fto, the ortholog of the human obesity gene FTO, affects food intake in rats. PLoS Genet. 2010;5:e8771. doi: 10.1371/journal.pone.0008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- Wang Y, Baker JL, Hill JO, Dietz WH. Controversies regarding reported trends: has the obesity epidemic leveled off in the United States. Adv Nutr. 2012;3:751–752. doi: 10.3945/an.112.002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle J, Carnell S, Haworth CMA, Farooqi IS, O’Rahilly S, Plomin R. Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab. 2008;93:3640–3643. doi: 10.1210/jc.2008-0472. [DOI] [PubMed] [Google Scholar]
- Wardle J, Carnell S, Haworth CMA, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87:398–404. doi: 10.1093/ajcn/87.2.398. [DOI] [PubMed] [Google Scholar]
- Waters E, de Silva-Sanigorski A, Hall BJ, Brown T, Campbell KJ, Gao Y, Armstrong R, Prosser L, Summerbell CD (2011) Interventions for preventing obesity in children. Cochrane Database Syst Rev CD001871 [DOI] [PubMed]
- Weide K, Christ N, Moar KM, Arens J, Hinney A, Mercer JG, Eiden S, Schmidt I. Hyperphagia, not hypometabolism, causes early onset obesity in melanocortin-4 receptor knockout mice. Physiol Genomics. 2003;13:47–56. doi: 10.1152/physiolgenomics.00129.2002. [DOI] [PubMed] [Google Scholar]
- Westerlund L, Ray C, Roos E. Associations between sleeping habits and food consumption patterns among 10–11-year-old children in Finland. Br J Nutr. 2009;102:1531–1537. doi: 10.1017/S0007114509990730. [DOI] [PubMed] [Google Scholar]
- Whitaker RC, Wright JA, Pepe MS, Seidel KD, Dietz WH. Predicting obesity in young adulthood from childhood and parental obesity. N Engl J Med. 1997;337:869–873. doi: 10.1056/NEJM199709253371301. [DOI] [PubMed] [Google Scholar]
- Wu L, Xi B, Zhang M, Shen Y, Zhao X, Cheng H, Hou D, Sun D, Ott J, Wang X, Mi J. Associations of six single nucleotide polymorphisms in obesity-related genes with BMI and risk of obesity in Chinese children. Diabetes. 2010;59:3085–3089. doi: 10.2337/db10-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yach D, Stuckler D, Brownell KD. Epidemiological and economic consequences of the global epidemics of obesity and diabetes. Nat Med. 2006;12:62–66. doi: 10.1038/nm0106-62. [DOI] [PubMed] [Google Scholar]
- Yang Q, Khoury MJ. Evolving methods in genetic epidemiology. III. Gene-environment interaction in epidemiological research. Epidemiol Rev. 1997;19:33–43. doi: 10.1093/oxfordjournals.epirev.a017944. [DOI] [PubMed] [Google Scholar]
- Yang YK, Fong TM, Dickinson CJ, Mao C, Li JY, Tota MR, Mosley R, Van der Ploeg LHT, Gantz I. Molecular determinants of ligand binding to the human melanocortin-4 receptor. Biochemistry. 2000;39:14900–14911. doi: 10.1021/bi001684q. [DOI] [PubMed] [Google Scholar]
- Yeo GSH, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- Youngson NA, Morris MJ. What obesity research tells us about epigenetic mechanisms. Phil Trans R Soc B. 2012;368:20110337. doi: 10.1098/rstb.2011.0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Grant SFA. Genetics of childhood obesity. J Obes. 2011;2011:1–9. doi: 10.1155/2011/845148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bradfield JP, Zhang H, Sleiman PM, Kim CE, Glessner JT, Deliard S, Thomas KA, Frackelton EC, Li M, Chiavacci RM, Berkowitz RI, Hakonarson H, Grant SFA. Role of BMI-associated loci identified in GWAS meta-analyses in the context of common childhood obesity in European Americans. Obesity. 2011;19:2436–2439. doi: 10.1038/oby.2011.237. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Ge D, Heinzen EL, Dickson SP, Urban TJ, Zhu M, Maia JM, He M, Zhao Q, Shianna KV, Goldstein DB. Prioritizing genetic variants for causality on the basis of preferential linkage disequilibrium. Am J Hum Genet. 2012;91:422–434. doi: 10.1016/j.ajhg.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]