

Recent studies have revealed the impact of animal–bacterial symbioses on physiology and disease. In this review, the authors consider how model system studies have influenced our understanding of host–microbiota interactions and highlight the importance of the gut microbiome to human health.
Keywords: microbiome, microbiota, model organisms, symbiosis
Abstract
The animal and bacterial kingdoms have coevolved and coadapted in response to environmental selective pressures over hundreds of millions of years. The meta'omics revolution in both sequencing and its analytic pipelines is fostering an explosion of interest in how the gut microbiome impacts physiology and propensity to disease. Gut microbiome studies are inherently interdisciplinary, drawing on approaches and technical skill sets from the biomedical sciences, ecology, and computational biology. Central to unraveling the complex biology of environment, genetics, and microbiome interaction in human health and disease is a deeper understanding of the symbiosis between animals and bacteria. Experimental model systems, including mice, fish, insects, and the Hawaiian bobtail squid, continue to provide critical insight into how host–microbiota homeostasis is constructed and maintained. Here we consider how model systems are influencing current understanding of host–microbiota interactions and explore recent human microbiome studies.
The distinctive body plans of animals offer many niches for members of the archaeal and bacterial kingdoms. While the lumen of the human distal gut is one of the most densely populated ecosystems on our planet, humans and other animals harbor several microbiomes on and within their body surfaces such as the respiratory and urogenital tracts and the skin. As the gut has evolved from the relatively simple tube of the ancient cyclostomatida to the more highly compartmentalized gastrointestinal tracts of mammalian species, the diversity and complexity of the microbial inhabitants of those spaces has increased as well (Fig. 1). Use of model systems with differing levels of microbial complexity reveals how host genes impact the microbiome and how the microbiome regulates host genetic programs (Table 1). Here we broadly explore not just the gut microbiota, but the chronic colonization of mucosal epithelia by extracellular microbes.
Figure 1.
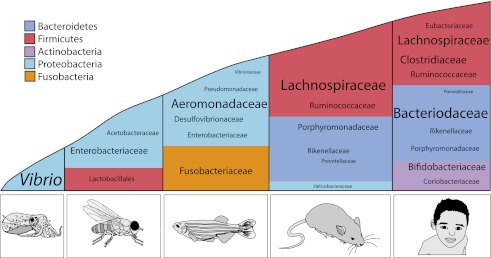
The structure of the microbiota across species. Although there can be significant interindividual variation in the composition of the microbiota, broad trends exist within a given species, particularly at the phylum level. Phyla are represented by color, and the relative abundance of the lower taxonomic levels is indicated by font size. This figure was produced with data adapted from Arumugam et al. (2011), Brinkman et al. (2011), Chandler et al. (2011), and Roeselers et al. (2011).
Table 1.
Considerations in choosing a model organism for studying the microbiome

Model organisms provide opportunities to perturb, engineer, and study host–microbiome interplay with a level of experimental control that is not achievable in human studies. Both vertebrates and invertebrates have been essential for discovering the microbial-associated molecular patterns and host pattern recognition receptors required for sensing of pathogenic and symbiotic microbes. In many cases, tractable genetics in the host and microbes have proven essential for identifying both microbial and host factors that enable symbiosis. Model systems are also revealing roles for the microbiome in host physiology ranging from mate selection (Sharon et al. 2010) to skeletal biology (Cho et al. 2012; Sjögren et al. 2012) and lipid metabolism (Wang et al. 2011; Semova et al. 2012). With an ever-increasing number of human microbiome studies completed and under way, experimental systems employing models organisms will prove essential tools to interrogate and validate the associations identified between the human microbiome and disease.
Model systems
Hawaiian bobtailed squid
The coastal waters of Hawaii teem with microbial life. Less than 0.1% belong to the species Vibrio fischeri (Lee and Ruby 1992; Ruby and Lee 1998). Despite this numerical challenge, the tiny Hawaiian bobtail squid (Euprymna scolopes) selectively acquires V. fischeri from its environment to create one of the most well-understood models of bacterial–animal symbiosis. Unlike the systems discussed later in this review, E. scolopes does not harbor V. fischeri within its gut, and this symbiosis does not contain a consortium of microbes. Instead the squid forms a naturally occurring one-on-one relationship with V. fischeri within a ventrally located cavity called the light organ.
Bobtail squid feed at night in shallow open stretches of water, where the presence of moonlight or starlight can cast shadows revealing the squids' presence to predators lying below. To camouflage themselves, squid use counterillumination from light generated by V. fischeri contained within the light organ. Juvenile squid are born without V. fischeri and must rapidly acquire and selectively populate their light organ with these symbiotic bacteria if they are to survive in the wild (McFall-Ngai and Ruby 1991). However, in the laboratory, both squid and V. fischeri can be grown independently, thereby permitting detailed study of the initiation, developmental consequences, and molecular steps necessary for this highly specific microbial symbiosis.
The juvenile light organ is morphologically very different from the adult organ, since its primary function is to harvest V. fischeri from surrounding seawater and initiate the symbiosis. Newly hatched squid have light organs with two sets of ciliated appendages that beat seawater toward pores leading to the interior of the organ. Within 1–2 h of hatching, peptidoglycan from environmental microbes trigger secretion of mucus outside the light organ (Nyholm et al. 2002). This mucus further aggregates Gram-negative bacteria outside the light organ, which, within a few hours, becomes dominated by V. fischeri (Nyholm et al. 2000; Nyholm and McFall-Ngai 2003). By 4–6 h after the squid hatch, V. fischeri begin to migrate into the pores of the light organ through the epithelial-lined ducts and settle into the deep crypts (McFall-Ngai and Ruby 1998; Nyholm et al. 2000). Once inside the light organ, V. fischeri “signal” their presence to the host by release of lipopolysaccharide (LPS) and peptidoglycan, classic examples of microbial-associated molecular patterns (Foster et al. 2000; Koropatnick et al. 2004). In response to the bacteria, the light organ undergoes a maturation process resulting in rapid attenuation of nitric oxide production, constriction of the ducts, and a dramatic morphogenesis that includes full regression of the surface appendages and cilia (Nyholm and McFall-Ngai 2004). While human–microbial symbiosis is far more complex, such observations in the squid raise questions of how much of human postnatal development is influenced by the microbes.
The development of the light organ only occurs with a specific association with V. fischeri, as squid raised without V. fischeri remain uncolonized by other bacteria, and the light organ fails to mature (McFall-Ngai and Ruby 1991, 1998). The enrichment for squid-specific symbionts is so selective that a fish-associated strain of V. fischeri is unable to initiate colonization. Genomic comparison of V. fischeri strains that colonize either fish or squid identified a gene encoding a two-component sensor kinase as ubiquitously present in the genomes of squid symbionts, while this gene is absent in half of the fish-associated strains (Mandel et al. 2009). Expression of this single gene in a fish symbiont conferred the ability to colonize squid at levels comparable with the natural squid symbiont. This finding illustrates the important and widely applicable concept of how minor genetic changes in a bacterium can influence the evolution of symbiont host range and specificity (Mandel et al. 2009).
Interestingly, the squid and V. fischeri symbiosis follows a diel rhythm, temporal pattern, where each dawn, the squid expel the majority of bacteria from their light organ, allowing the remaining V. fischeri to regrow during the day. Both the squid and the symbiont undergo profound changes throughout this diel cycle (Wier et al. 2010), but perhaps the most obvious difference is the increased microbial luminescence that occurs at night and during the squid's active period. These observations raise curiosity about how day–night cycle changes in light, temperature, and nutrient availability impact animal–microbe symbioses.
Mutants in the quorum-sensing system of V. fischeri fail to produce light but can initially colonize the light organ. Light production may represent a metabolic cost to V. fischeri, since quorum-sensing mutants outcompete wild-type strains in carbon-limited growth medium (Bose et al. 2008). This potential metabolic advantage for nonluminescent bacteria could undermine the function of the light organ if these microbes chronically colonized the squid. However, several studies have demonstrated that the quorum-sensing mutants are unable to initiate the normal light organ developmental program and are significantly less capable of persistent colonization (Visick et al. 2000; Lupp et al. 2003; Bose et al. 2008; Chun et al. 2008). How the squid monitors bacterial luminesce is not as well understood, but the light organ does share some similarities to the eye. Both organs have a lens and reflective tissue, and the light organ expresses many of the same photodetection genes found in the eye (Tong et al. 2009).
The symbiosis of the squid and V. fischeri is a fascinating illustration of the mutualism that exists between microbes and animals. In particular, this model offers several experimental advantages, as it is a naturally occurring one-on-one relationship where each participant can be grown independently. The use of microbial genetics with V. fischeri has provided surprising and powerful insights into the molecular and even photoluminescent dialog that mediates a productive relationship between microbes and their hosts.
Fruit fly
The fruit fly Drosophila melanogaster has contributed greatly to the understanding of basic cellular and developmental biology over the past decades. Owing to their tremendous experimental tractability and wealth of genetic tools, Drosophila are a powerful model to explore innate immunity and microbial pathogenesis (Dionne and Schneider 2008; O'Callaghan and Vergunst 2010). Scientists first discovered Toll receptors while studying Drosophila embryogenesis almost 30 years ago (Anderson et al. 1985), and it is now apparent that toll-like receptors (TLRs) function as critical innate immune sensors in evolutionarily diverse organisms, including humans. However, in contrast to their use in host–pathogen studies, the interaction of Drosophila with their symbiotic microbiota is only beginning to emerge as a model system for symbiosis.
Numerous studies have surveyed the gut microbes of Drosophila and identified bacterial communities far less complex than mammals (Corby-Harris et al. 2007; Cox and Gilmore 2007; Ren et al. 2007; Ryu et al. 2008; Chandler et al. 2011; Wong et al. 2011). An analysis of both laboratory-raised and wild-caught flies found a maximum of 30 operational taxonomic units (OTUs), a proxy for species commonly employed in 16S rRNA gene sequencing analysis, associated with some wild Drosophila, and an average of 6.3 OTUs with laboratory-reared flies (Chandler et al. 2011). The most prevalent microbes identified in the Drosophila gut microbiota belong to the families of Acetobacteraceae, Lactobacillales, and Enterobacteriaceae, with diet being the most powerful determinant of species composition of these gut microbial communities (Fig. 1; Corby-Harris et al. 2007; Chandler et al. 2011). These microbes are either aerotolerant or obligate aerobes, suggesting that, unlike mammals, oxygen is able to penetrate across the entire diameter of the Drosophila gut (Chandler et al. 2011; Charroux and Royet 2012). The aerobic growth of these gut microbes combined with their relative taxonomic simplicity has allowed laboratory culturing of virtually the entire gut microbiota in some laboratory Drosophila stocks (Shin et al. 2011). The ability to culture large proportions of the Drosophila microbiota combined with the vast collections of mutant flies and rapid growth and high reproductive capacity of Drosophila has produced a model of symbiosis with enormous potential to reveal new insights into host–symbiont interactions.
An example of one such unexpected interaction between flies and bacterial symbionts concerns mate selection: Drosophila prefer to mate with flies harboring similar gut microbes (Sharon et al. 2010). Initially, the investigators found that flies maintained on separate diets of either starch or molasses preferred mating with flies fed the same diet, yet this preference could be abolished with antibiotics. To precisely define the contribution of microbes to mating preference, flies were treated with antibiotics and then recolonized with a defined microbiota. Remarkably, colonization with a single symbiotic organism, Lactobacillus plantarum, was sufficient to influence Drosophila mating preference (Sharon et al. 2010). While this study used antibiotics to reduce the indigenous microbiota, other studies have used axenic or germ-free (GF) flies grown from the time of birth without any bacteria.
To generate GF flies, eggs are washed with sodium hypochlorite solution (bleach) and hatched under sterile conditions. The resulting GF flies are viable and can be easily maintained in laboratory conditions (Ridley et al. 2012). However, several studies show that GF larva grow significantly more slowly than conventional larva, leading to delayed time to pupariation (Shin et al. 2011; Storelli et al. 2011; Ridley et al. 2012). This delay of growth is further exaggerated when GF larva are fed a nutritionally limited diet (Storelli et al. 2011) and can even result in death at first instar when larva are raised on a more minimal diet (<0.1% yeast or casamino acids substituted for yeast) (Shin et al. 2011). By culturing the five principal bacteria found in their laboratory Drosophila stock, Shin et al. (2011) demonstrated that a single bacterial species, Acetobacter pomorum, was able to restore normal growth by stimulating insulin/insulin-like growth factor. To discover the bacterial factors that aid larval growth, the investigators screened a transposon mutant library of A. pomorum in developing flies. Bacterial mutants in the pyrroloquinoline quinone-dependent alcohol dehydrogenase (PQQ-ADH)-dependent oxidative respiratory chain were able to colonize Drosophila at levels commensurate with wild-type bacteria but produced larva with developmental defects and adults with smaller body size. In Acetobacter, PQQ-ADH is the primary dehydrogenase in the ethanol oxidative respiratory chain that generates acetic acid (Yakushi and Matsushita 2010), and accordingly, the developmental defect in flies colonized by PQQ-ADH mutants could be complemented by the addition of acetic acid (Shin et al. 2011). Interestingly, feeding acetic acid alone to GF flies does not restore growth to conventional levels, indicating that Drosophila developmental physiology requires both acetic acid-dependent and -independent microbial metabolism.
Much like mammals, the microbiota of Drosophila contributes to gut homeostasis by promoting the integrity of the epithelial barrier, which separates gut luminal contents from the rest of an animal's body. Under conventional conditions, the gut epithelium of adult flies is renewed by intestinal stem cells (ISCs) every 1–2 wk (Micchelli and Perrimon 2006). In GF Drosophila, both the number of mitotic ISCs and the rate of epithelial renewal are reduced, since gut bacteria activate JAK–STAT signaling to induce ISC proliferation (Buchon et al. 2009). However the rate of cell turnover in the Drosophila gut is dynamic and responds to pathogenic infection or damage by dramatically increasing ISC proliferation to renew the entire mid-gut in 2–3 d (Buchon et al. 2009; Jiang et al. 2009). Balancing a capacity to respond appropriately to pathogens while not mounting an inappropriate, inflammatory response to symbiotic microbes is critical for both Drosophila and mammals. Understanding the cellular and molecular mechanisms regulating the differential responses to pathogens and symbionts remains a central goal of immunology and microbiology.
A particular challenge arises in trying to distinguish pathogenic microbes from microbes that transiently and chronically colonize a host, especially since they share so many molecular features. Drosophila are proving a useful model system for understanding this aspect of host–microbe interaction. Peptidoglycan, a component of bacterial cell walls that is common to both pathogenic and commensal microbes, can activate the innate immune response in both Drosophila and mammals (Royet and Dziarski 2007). In flies, the recognition of peptidoglycan occurs through peptidoglycan recognition proteins (PGRPs), which can induce either the Toll or immune deficiency (IMD) pathways, leading to release of antimicrobial peptides (AMPs) and reactive oxygen species (ROS) (Tzou et al. 2000; Ha et al. 2005), However it is vital that flies have the capacity to negatively regulate this immune response because of the endogenous microbes they harbor (Zaidman-Rémy et al. 2006; Ryu et al. 2008; Paredes et al. 2011). While some members of the PGRP family can initiate an immune response, other members of this family can act as negative regulators of the IMD pathway (Zaidman-Rémy et al. 2006; Lhocine et al. 2008; Paredes et al. 2011; Bosco-Drayon et al. 2012). Mutation of PGRPs such as PGRP-LB, the PGRP-SC family, or the interacting partner PIMS (PGRP-LC-interacting inhibitor of Imd signaling) results in aberrantly high levels of AMPs in the presence of their resident microbiota and higher fly mortality when colonized by relatively innocuous Gram-negative bacteria (Lhocine et al. 2008; Paredes et al. 2011).
Even with the negative regulation of IMD pathways, the presence of indigenous Drosophila gut microbes can still induce activation of inflammatory pathways—specifically, the nuclear translocation of the NF-κB homolog relish. However, while pathogenic infection drives translocation of relish to the nucleus and triggers expression of immune factors, indigenous Drosophila gut microbes do not trigger production of AMPs, as would be expected (Ryu et al. 2008). To understand how some microbes avoid triggering production of AMPs, Ryu et al. (2008) analyzed expression of the homeobox transcription factor caudal, which regulates AMP production in some epithelial cell types. They found that caudal was specifically expressed in the gut epithelium of adult flies and that knockdown of caudal with RNAi led to AMP production in the presence of resident Drosophila gut microbes. In turn, the overexpression of AMPs due to disruption of caudal dramatically restructured the gut microbiota, thereby resulting in high levels of epithelial cell apoptosis and death of the flies.
The Drosophila model leverages substantial genetic tools with a relatively simple microbiota to create an experimentally tractable system to discover the molecular underpinnings of host–commensal interactions. In some instances, Drosophila can reveal relationships between hosts and microbiota that are applicable to other insects, including those that act as vectors of infectious diseases or are of importance to agriculture (Douglas et al. 2011). In other cases, the implications of Drosophila–microbiota interaction may uncover broader concepts of mutualism that are conserved among higher-order organisms.
Zebrafish
A distinguishing feature that marks the evolution of vertebrates, in contrast to the squid and the fruit fly, is the appearance of adaptive immunity. The cells of the adaptive immune system, principally B and T lymphocytes, express antigen-specific receptors that permit vertebrates to recognize and remember microbes. Vertebrates also exhibit a complex, resident microbiota, whereas the invertebrate gut microbiota is composed of only a small number of species and tends to be shaped by the microbial composition of the environment (McFall-Ngai 2007). It has been proposed that the ability of the adaptive immune system to recognize and remember specific organisms has facilitated the expansion of the resident microbiota in vertebrates (McFall-Ngai 2007; Maynard et al. 2012). Therefore, the zebrafish Danio rerio, with a diverse microbiota but still among the simplest vertebrate models, is emerging as a powerful model system for studying the complexities of host–microbiota interactions.
The zebrafish has a number of features that make it an attractive experimental system. These include external fertilization and optical transparency of zebrafish embryos and larvae that allow for the visualization of developing cells as well as colonizing microbes by time-lapse microscopy. The rapid development of organ primordia (within 5 d post-fertilization [dpf]) and onset of adult metamorphosis (within 14 dpf), together with small size, make zebrafish an invaluable resource for forward genetic and chemical screening. Methods for genetic screening include ENU mutagenesis, retrovirus-based insertional mutagenesis, zinc finger nucleases (Amacher 2008), morpholino-based gene knockdown (Nasevicius and Ekker 2000), conditional loss of function by RNAi (De Rienzo et al. 2012), and, most recently, genome editing with the use of the TALEN system (Bedell et al. 2012). There is a high degree of homology between zebrafish and mammals not only in the adaptive immune system, but also in the digestive system. Zebrafish have a pancreas, gall bladder, liver, and intestine. Furthermore, the cells of the intestinal epithelium include absorptive enterocytes, goblet cells, and enteroendocrine cells similar to mammals. Relatively simple methods have been developed for rearing GF and gnotobiotic zebrafish during early post-embryonic development (Pham et al. 2008) and up to 30 dpf (Milligan-Myhre et al. 2011). To date, most GF zebrafish studies have centered on the early post-embryonic period and therefore focused on the impact of the microbiota on the innate immune system, as the adaptive immune system has yet to fully develop at that time.
To investigate geographic variation in the zebrafish gut microbiota and determine whether there is a shared or core microbiota in zebrafish, 16S rRNA gene sequencing of gut microbial contents was carried out on zebrafish collected from their natural habitat and from several research laboratories in different geographic locations (Roeselers et al. 2011). Members of the γ-Proteobacteria and Fusobacteria classes were the most dominant constituents of the microbiota and were shared by all fish, despite the relatively large geographical and generational distances that separated them. The selective pressures of the zebrafish intestinal environment appear to favor a highly specific collection of microbes influenced by host anatomy, physiology, nutrient availability, and immunology (referred to as gut habitat effects) much more strongly than the effects of dietary differences or environment (for example, an animal's parents [referred to as legacy effects]), which may be expected to be important factors in mammals.
In an elegant test of gut habitat versus legacy effects, a reciprocal gut microbiota transplant was performed between GF zebrafish and mice (Rawls et al. 2006). The gut microbes of the zebrafish microbe-transplanted mice resembled the gut microbes of conventional mice, rather than that of the mouse gut microbe-transplanted zebrafish. Similarly, the gut microbes of conventional zebrafish resembled the gut microbes of mouse gut microbe-transplanted zebrafish. These experiments demonstrate that the gut sculpts the community it has to work with into a predefined shape heavily influenced by the host. The observations of Rawls et al. (2006) raise the question of what host factors perform this sculpting and suggest that zebrafish will be a very useful model system to identify such factors.
Studies that compare conventionally raised versus GF zebrafish larvae have revealed insights into many mechanisms of host–microbiota relationships. The gut microbiota stimulates intestinal epithelial cell proliferation (Rawls et al. 2004; Bates et al. 2006) through MyD88 signaling and not inflammation (Cheesman et al. 2011), promotes a shift in epithelial glycan expression (Bates et al. 2006), and stimulates the infiltration of gut-associated immune cells (Rawls et al. 2004; Bates et al. 2007).
Zebrafish sense microbes by detecting microbe-associated molecular patterns (MAMPs), principally by the use of TLRs and nucleotide oligomerization domain (NOD)-like receptors (NLRs) operating at the cell surface or the cytoplasm. Therefore, one possible mechanism to promote regulated and appropriate responses to symbiotic bacteria is to limit the exposure to and detection of MAMPs. Intestinal alkaline phosphatase (IAP) is an enzyme that is expressed at the apical surface of the brush border of intestinal epithelial cells that serves to detoxify the lipid A moiety of bacterial LPS by dephosphorylation. IAP activity is induced in response to microbial colonization in conventionally reared zebrafish but is not induced in GF zebrafish (Bates et al. 2006). In a seminal set of experiments, Bates et al. (2007) demonstrate that the detoxifying activity of IAP prevents an inappropriate inflammatory response to the resident gut microbiota in zebrafish. Bates et al. (2007) also demonstrate that IAP forms a negative feedback loop that prevents excessive TLR activity and inflammation. Interestingly, subsequent studies by other groups support that IAP is a highly conserved symbiotic factor promoting host–microbe homeostatic interactions in squid and mice (Goldberg et al. 2008; Rader et al. 2012).
The composition of the zebrafish gut microbiota can have direct impacts on disease pathogenesis. Inflammatory bowel disease can be modeled in zebrafish using a chemical called oxazolone, which induces intestinal inflammation (Brugman et al. 2009). By treating zebrafish with the antibiotic vancomycin, Fusobacteria became the dominant phyla in the gut microbiota, and the inflammatory response observed in response to oxazolone was markedly decreased. Treatment with colistin sulfate treatment results in a γ-Proteobacteria-dominated gut microbiota, and these zebrafish developed intestinal inflammation in response to oxazolone treatment. These results demonstrate that certain members of the microbiota, such as the γ-Proteobacteria, may help drive intestinal inflammation in an experimental model of colitis. In human diseases, it is often difficult to discern whether a change in the gut microbiota is a cause or effect of the underlying disease, and expansions of gut microbiota Proteobacteria have been observed in human chronic inflammatory diseases. This study in zebrafish suggests that members of the Proteobacteria may increase propensity for inflammatory responses in the gastrointestinal tract.
In a recent study of the contributions of the microbiota to dietary lipid absorption, Semova et al. (2012) effectively used the transparent zebrafish embryo to monitor how fatty acids are absorbed in the presence and absence of microbes. Conventionalized zebrafish show increased fatty acid absorption compared with GF fish under both fed and starved dietary conditions. Conventionalized fish also exhibit higher fatty acid export to the liver and increased lipid droplet size regardless of diet, suggesting that the microbiota may directly affect fatty acid storage. Previous studies have shown that caloric intake can influence the relative abundance of Firmicutes and Bacteroidetes (Ley et al. 2006, 2008; Turnbaugh et al. 2008b); generally, Firmicutes are expanded in high-calorie diets. GF zebrafish were monocolonized with a Firmicutes representative, a Bacteroidetes representative, or a Proteobacteria representative. The Firmicutes representative was below the level of detection under starved conditions but robustly colonized under fed conditions, and only the Firmicutes-colonized zebrafish had an increase in lipid droplet number relative to GF fish. This study demonstrates that the intestinal microbiota is integral to fatty acid metabolism and absorption and that specific components of the microbiota carry out prescribed roles in this process. It also provides insight into observations about the altered gut microbiota in obesity—specifically, how the gut microbe of obese humans may contribute to a maladaptive fatty acid metabolism in these individuals.
Mice
The laboratory mouse has been instrumental for establishing roles for the gut microbiota in many aspects of mammalian physiology: angiogenesis (Stappenbeck et al. 2002; Reinhardt et al. 2012), bone mineral density (Cho et al. 2012; Sjögren et al. 2012), brain development and behavior (Sudo et al. 2004; Bravo et al. 2011; Diaz Heijtz et al. 2011; Ezenwa et al. 2012), obesity and malnutrition (Turnbaugh et al. 2006; Smith et al. 2013), hepatic function (Dapito et al. 2012; Henao-Mejia et al. 2012), intestinal response to injury and repair (Rakoff-Nahoum et al. 2004; Swanson et al. 2011), and innate and adaptive immune function (for reviews, see Garrett et al. 2010b; Littman and Pamer 2011; Hooper et al. 2012). Ninety-nine percent of mouse genes are shared with humans at the host genetic level, and they share key similarities with the human gut microbiome at the phylum through family levels (Fig. 1), making them a powerful model system for evaluating host–microbiota interactions applicable to human biology (Spor et al. 2011). Mouse genetics; the availability of both inbred and outbred strains and collections of knockout, knock-in, and transgenic mutants (International Knockout Mouse Consortium [IKMC, http://www.knockoutmouse.org], Knockout Mouse Project [KOMP, https://www.komp.org], and Mutant Mouse Regions Resource Center [MMRRC, http://www.mmrrc.org]); and forward genetic approaches (e.g., in vivo RNAi, insertional mutagenesis, and N-ethyl-N-nitrosurea [ENU]-based mutagenesis-based screens) make mice a useful model system for studying the role of host genetics in host–microbiota interactions. An ability to select breeding strategies, alter housing conditions and littermates, manipulate diet, regulate light cycles, and subject mice to defined exposure facilitates study of how environmental exposures impact host–microbiota interactions. However, all of these experimental opportunities come with some caveats. Mouse skin, fur, orapharyneal structures, compartmentalization of the gastrointestinal tract, and behavior (e.g., coprophagia) clearly differ significantly from humans and may influence microbial communities. Experimental and animal husbandry practices, including bedding materials and exposure of immunodeficient lines to prophylactic antibiotics, can all impact the microbiome, too. When aware of the environmental and legacy effects in mouse models and their implications for microbiome experimental design, mice can still be a stalwart tool for unraveling mechanisms of host–microbiota interactions relevant to humans.
Using mice to parse the contribution of genetics to configurations of the gut microbiota
A major question in microbiome studies is what host genetic features impact the assembly, temporal stability, and resilience of microbial communities. Mouse models would appear to provide a straightforward approach to understanding the impact of host genotype of the microbiome. However, in some cases, confounding factors have made drawing conclusions from the impact of mouse genetics on the microbiome far from clear. Acknowledging this complexity and the potential pitfalls is not meant to suggest that using mice for host–microbiota studies is a flawed approach; rather, the point is to highlight that studying host–microbiota interactions in mice requires careful experimental design. Single and double gene knockout studies, often but not always in the context of disease phenotype, have provided insight into how genetics regulating host immune function and energy balance impact the gut microbiome. Of the many studies examining the impact of single and compound gene knockouts in mice on the microbiome, we highlight a few that focus on genes involved in sensing conserved features of the microbiota. Generally, these one to two genes or “fine-scale” host genotype and microbiota studies use mice with hypomorphic gene function, global loss of gene function, or cell type-specific deletion using Cre-lox technology.
Many mouse genotype perturbation–microbiome studies have examined the gut microbiota of mice deficient in genes involved in host response to microbe-associated molecular patterns. Several studies have examined the effects of TLR and NOD family members on the gut microbiota. Effects of Nod2 mutations have been a focus because Nod2 mutations are a risk factor for human Crohn's disease, a disease that exemplifies dysregulated host–microbiota interactions. Nod2 expression in the intestine is dependent on the presence of a gut microbiota, and Nod2 knockout mice are more susceptible to colonization by intestinal mouse pathogens (Petnicki-Ocwieja et al. 2009). Mice deficient in Nod1 and Nod2 have altered gut microbiota composition as compared with their heterozygous littermates (Natividad et al. 2012). NLRP6 is a NLR family member that functions in inflammasome, type 1 interferon, and NF-κB signaling. Inflammasomes are multicomponent protein complexes that detect infection and cellular injury. Inflammasome activation results in caspase-1 activation and subsequent caspase-1 proteolytic activation of two proinflammatory cytokines: IL-1β and IL-18 (Franchi et al. 2012). NLRP6 inflammasome-deficient mice have an altered gut microbiota notable for an expansion of Prevotellaceae. Although why this bacterial family expands remains unclear, this microbiota is functionally significant, as it increased colitis severity induced by a chemical that disrupts the epithelial barrier and causes colitis (Elinav et al. 2011). NLRP6 is a negative regulator of inflammatory signaling, and its loss activates MAP kinase and NF-κB signaling pathways downstream from TLRs and increases circulating monocyte numbers (Anand et al. 2012). These cellular and signaling alterations may explain why NLRP6-deficient mice have an increased resistance to a number of pathogenic bacteria and an altered endogenous microbiota (Anand et al. 2012). Caspases are cysteine proteases that play wide-ranging roles functioning in apoptosis to inflammation. Caspase-1 cleavage of IL-1β and IL-18 contributes to regulating inflammatory tone in the gut and thus may modulate the microbiome. The gut microbiota of Capsase-1-, Capsase-3-, and Capsase-7-deficient mice were recently evaluated in comparison with wild-type mice using 16S rRNA gene fecal profiling, and significant differences were observed across several families, including Lachnospiraceae, Porphyromonodaceae, and Prevotellaceae (Brinkman et al. 2011).
MyD88 is an adaptor protein common to IL-1 and IL-18 signaling and to several TLRs (TLR1, TLR2, TLR4, TLR5, TLR6, TLR9, and TLR10). Thus, loss of MyD88 may be expected to impact a wide range of innate immune sensing of the microbiota. The absence of MyD88 in the nonobese diabetic mouse strain led to an altered microbiota composition with enriched abundance of Lactobacillaceae, Rikenellaceae, and Porphyromonadaceae (Shen et al. 2010). In another recent study, loss of MyD88 was examined from both a microbial ecology and host transcriptome perspective along the length of the small intestine and colon (Larsson et al. 2011). The small intestinal microbiota of MyD88-deficient mice was notable for an enrichment of segmented filamentous bacteria (SFB) and greater interindividual variation. Recently, SFB have garnered increased attention, as they promote a population of T cells, called T helper 17 (Th17) cells, which function in immunity against extracellular bacteria and fungi. The host transcriptome data set of Larsson et al. (2011), which uses both GF (discussed below) and MyD88-sufficient and -deficient conventional mice, is available as a community-wide resource at Encyclopedia of Tut Microbiota Regulated Genes (http://microbiota.wall.gu.se). Targeted intestinal epithelial deletion of MyD88 using Villin-Cre × MyD88-Flox mice has revealed that such mice have reduced levels of the polymeric immunoglobulin receptor mucin-2 and antibacterial peptides (Frantz et al. 2012). MyD88 regulation of the expression of RegIIIγ, an antibacterial lectin that lyses Gram-positive bacteria, is of special importance, as RegIIIγ restricts the localization of bacteria and ensures its proper segregation from the inner mucus layer of the intestinal mucosa (Vaishnava et al. 2011).
There have been several studies of the impact of the TLRs upstream of MyD88. Investigations of TLR5-deficient mice have revealed phenotypes resulting in metabolic syndrome (Vijay-Kumar et al. 2010) and colitis (Vijay-Kumar et al. 2007), with coincident alterations in the microbiota and transient elevations in the proteobacteria and, in particular, enterobacteria species in colitic TLR5 knockout mice (Vijay-Kumar et al. 2010; Carvalho et al. 2012). While there have been other observations supporting that alterations in TLR signaling impact the microbiota (e.g., that there is an altered colonic mucosal microbiota in TLR2-deficient mice) (Kellermayer et al. 2011), other studies have challenged the magnitude of the impact of TLR signaling perturbations on the gut microbiome. A denaturing gradient gel electrophoresis and fluorescence in situ hybridization (FISH)-based investigation (Loh et al. 2008) and a more recent study using deep 16S rRNA gene surveys both call into question whether TLRs or MyD88 alter the gut microbiota in a genotype-dependent fashion (Ubeda et al. 2012). Ubeda et al. (2012) generated MyD88-, TLR-2-, TLR-4-, TLR-6-, and TLR-9-deficient mouse lines from heterozygote × heterozygote breeding strategies. Interestingly, they did not detect statistically significant differences in community composition or diversity in cecal luminal, ileal luminal, or ileal 16S rRNA gene surveys in these mice compared with their littermate controls. They also did not detect statistically significant differences in microbial community response after an antibiotic (vancomycin) perturbation. However, differences were observed between wild-type and TLR mutant colonies that had been maintained as separate lines from homozygous × homozygous crosses for many years. These results raise awareness about the importance of considering lineage or legacy effects in microbiome studies, and it should be noted that these effects were considered in several of the cited studies (Vaishnava et al. 2011).
TLRs and NLRs are not the only molecules involved in host response to the microbiota. Defensins are cationic proteins found in both the animal and plant kingdom that have broad antimicrobial activity against bacteria, fungi, and viruses, and the microbiota of mice with altered enteric defensin activity have been investigated. In mice and humans, defensins are principally produced by small intestinal Paneth cells and also by intestinal absorptive enterocytes. Intestinal tissue and luminal samples from transgenic mice expressing one or two copies of the human α defensin (DEFA5) and knockout mice deficient in the metalloprotease MMP7, which proteolytically activates α-defensins, were analyzed using 16S rRNA gene surveys, quantitative PCR (qPCR), and FISH and carried out with appropriate experimental genotype, housing, and lineage controls (Salzman et al. 2010). Several statistically significant differences were observed at the phylum to species level, and notably, SFB were markedly reduced in the DEFA5 transgenic mice. While it may seem intuitive that antimicrobial molecules, TLRs, and NLRs would impact the microbiome, the microbiome field is relatively young. Thus, there is a need to define the impact of such genes on the microbiota and clarify whether changes are stochastic or host genotype driven.
Beyond TLRs, NLRs, and defensins, several genetic mouse models of colitis have been interrogated regarding their microbiome patterns before, after, and during active inflammation, which can perturb the microbiota as oxidative stress and antimicrobial molecules increase during inflammation (Morgan et al. 2012). Mouse models of colitis, including FVB.mdr1a−/− (Nones et al. 2009), BALB/c.T-bet−/− × RAG2−/− (Garrett et al. 2010a) and IL-10R2−/− × TGFBRII−/− (Bloom et al. 2011) mice, have been profiled using multiple methodologies inclusive of culture-dependent and -independent techniques and revealed distinctive patterns with the gut microbiota. The Enterobacteriaceae Klebsiella pneumoniae and Proteus mirabilis were associated with colitis in T-bet−/− × RAG2−/− mice (Garrett et al. 2010a), while Bacteroides thetaiotaomicron was associated with disease in IL-10R2−/− × TGFBRII−/− mice (Bloom et al. 2011), suggesting that a variety of genes may impact colonization and fitness of specific gut microbes in the setting of an inflamed epithelium and genetic immune perturbations. Mice provide significant opportunities in GWAS (genome-wide association studies) and MWAS (microbiome-wide association studies) to interrogate the contribution of genetic risk alleles to disease phenotype and the presence of genetic risk alleles to microbiome patterns. While inflammatory bowel disease has been a focus of such studies in mice (Parkes 2012), hopefully, mice can be used to assess causality of genetic risk alleles and their associated microbiota in a variety of disease states. While the studies highlighted here have focused on single or double gene knockouts or transgenics, mice also provide opportunities for investigating the impact of host genetics on a larger scale.
Using mice to parse the contribution of genetics to configurations of the microbiota
The first inbred mouse strain was generated >90 years ago, and there are now >450 inbred strains described. Inbred strains are a valuable tool for genetics and immunology research not only because of isogenicity within a given strain, but also because of the differences in genotype and phenotype between different inbred strains (Beck et al. 2000). Mouse genetic background has been observed to impact the composition, diversity, and richness of the gut microbiota in both wild-type and knockout mice (Esworthy et al. 2010; Büchler et al. 2012). More than 10 years ago, a collaborative effort was initiated to establish a resource for mouse experimental populations for high-resolution quantitative trait loci (QTL) analysis. This consortium, called the Collaborative Cross (CC) project (http://compgen.unc.edu), provides a powerful platform not only for mouse systems biology, but also for unparalleled opportunities for examining host genotype effects on the microbiome (Threadgill and Churchill 2012; Welsh et al. 2012). Although not completed, the CC is already providing important insight into complex trait analysis of the microbiome. Campbell et al. (2012) analyzed the gut microbiota of the eight core inbred CC using 16S rRNA gene-based surveys. While litter and cohousing effects on the microbiota were discernible, there were significant correlations and distinctive community clustering observed between the mouse strains. Strain-specific microbiota patterns were detected. Some strains have more interindividual variability than others, and other strains have more sex-dependent clustering in their microbiomes. A microbiome QTL investigation examining >600 mice from an intercross between C57Bl/6J (inbred) and an ICR-derived (outbred) line has provided a rich data set on host genetic factors influencing the microbiome (Benson et al. 2010). In Benson et al. (2010) as in Campbell et al. (2012), the litter and cohort had a significant effect on gut microbiota composition; however, there were discernible host genotype effects on microbiome variation as well. Numerous QTL were observed at the family and genus levels across Bacteroidetes, Clostridia, and Bacilli. There were also QTL signals for Cortiobacteriaceae Turicibacter and Helicobacter. Notably, no QTL were detected at the genus level for Lactobacillus, and, as lactobacilli are a genera remarkable for many species host-adapted populations, this prompted further trait mapping for the Lactobacillus reuteri, johnsonii/gasseri, and animalis/murinus groups that did reveal two significant QTLs. Collectively, these two studies by Campbell et al. (2012) and Benson et al. (2010) provide a strong foundation for integrating genetic, phenotypic and microbiome data and identifying QTL and genes that shape microbiome function from large limbs of the phylogenetic tree out to its strain-level branchlets.
Approaches to perturb and design the microbiome in mice
Conventional mice, those in traditional mouse colonies and with wide availability from commercial sources, have provided insight into the gut microbiota. For several decades, immunologists have appreciated that some inflammatory-associated phenotypes have varied across vivaria and vendors, and considering these environmental influences and microbiome differences is useful for understanding functional consequence of the microbiota. Vendor differences between CD4+ IL-17- and IL-22-producing cells (Th17) in the intestine of the same mouse strain led to the recognition that SFB play a critical role in driving Th17 development in mice (Ivanov et al. 2009). A whole-genome shotgun sequencing analysis of mouse microbiota across international vivaria will be available in the near future and will provide a very useful resource for mouse microbiome studies and understanding phenotypic differences across isogenic strains at different locations (SD Ehrlich, pers. comm.).
Antibiotics are an effective means to perturb the gut microbiota. By following communities before, during, and after treatment with antibiotics, significant insight can be gained into not only basic questions of microbial ecology and microbiome function (Maurice et al. 2013), but also how antibiotics—among the most widely used pharmaceuticals—impact susceptibility to infectious microbes and disease susceptibility. The temporal changes in luminal and mucosal microbial populations and their coincident impact on intestinal lymphoid populations, epithelial gene expression, and peripheral lymphoid tissues have been examined in response to antibiotics (vancomycin, metronidazole, neomycin, and ampicillin ± amphotericin-B) provided in the drinking water or by gavage (Hill et al. 2010; Reikvam et al. 2011). Antibiotic perturbations of the mouse microbiota have shed insight into how vancomycin-resistant enterococcus (VRE), an organism that causes significant morbidity and mortality, can come to dominate the gut microbiome and set the stage for VRE bacteremias, which are increasingly seen in hospitalized patients (Ubeda et al. 2010). Recently, mice have been used to understand the mechanisms underpinning how low-dose antibiotics promote growth in livestock, and this study raises concern for the judicious use of antibiotics in pediatric populations as juvenile obesity is becoming a global epidemic (Cho et al. 2012). Cho et al. (2012) examined the impact of several different classes and combinations of antibiotics (at low doses similar to those given to livestock) as an early-life exposure in mice. They examined several host metrics—weight, body fat, bone density, and metabolic parameters—by microarray and serum measurements. Concurrently, Cho et al. (2012) examined microbiota responses using 16S rRNA gene and internal transcribed spacer (ITS) sequencing and qPCR to assess composition and gas chromatography to examine microbiota metabolic capacity, focusing on short chain fatty production. Many human infants are exposed during parturition, post-partum, in their first year of life, and throughout childhood to antibiotics directly and indirectly through the food supply. The findings of Cho et al. (2012) fuel current interest in how antibiotics may impact obesity, skeletal development, and insulin sensitivity in children.
The impact of many early-life exposures on the microbiota can be evaluated in mice. Breast milk represents an early-life exposure common to all mammals, and the impact of breast milk on the microbiome has been investigated in mice by culture-independent profiling of microbiome composition in mouse neonates (Schloss et al. 2012). Milk has been manipulated in vivo by using lactating female mice deficient in sialyltransferase genes, which influence milk silalylactose levels. Mouse pups exposed to these altered milks showed changes in their microbiota composition and a heightened colitis susceptibility in later life (Fuhrer et al. 2010). Interestingly, maternal diet as well as maternal inflammatory state lead to milk with proinflammatory properties that can influence the health of exposed litters (Madsen et al. 2002; Garrett et al. 2010a; Du et al. 2012). Dietary interventions in mouse models have been the topic of several outstanding recent reviews and papers, and mice have been an excellent model to examine intersections of the microbiome, diet, genetics, and obesity and metabolic syndrome as well as intestinal inflammation (Ley et al. 2005; Turnbaugh et al. 2006, 2008a, 2009; Hildebrandt et al. 2009; Faith et al. 2011; Tilg and Kaser 2011; Devkota et al. 2012; Tremaroli and Bäckhed 2012). Housing, lineage, and legacy effects all impact the microbiome. While these variables may appear to just confound experimental interpretation, they can be used to study the assembly and stability of the microbiome (for review, see Spor et al. 2011).
Engineered microbiomes in mice
Gnotobiotics is the science of well-controlled microbial environments within and for biological specimens (Ward and Trexler 1958), encompassing the generation and maintenance of both GF and defined microbial community animals (Ward and Trexler 1958). While gnotobiotic mice are discussed here, there are gnotobiotic fish, flies, rats, pigs, and foals. Gnotobiotic techniques have been essential to mechanistically interrogate host–microbiota interactions in mice (Yi and Li 2012). Rederivation of any combination of genetic mutant mice is possible via embryo transfer into GF pseudopregnant mice or aseptic harvesting of a gestational uterine package and transfer of the fetuses to a GF foster female. It is important to realize that although GF mice do not harbor live bacteria or archaea, they are not naive to microbial-associated molecular patterns, as they encounter them in their sterilized (by autoclave or irradiation) food, water, and bedding. Microarray-based comparisons of host tissues from GF mice and their conventional counterparts have been a successful approach to understand the broad impact of the microbiome on physiology (Cash et al. 2006; Larsson et al. 2011; Yamamoto et al. 2012). Microarrays of GF and conventionalized mice even have shown that the gut microbiota modulate host gene expression post-transcriptionally by altering expression of host microRNAs within the small and large intestine (Dalmasso et al. 2011).
Colonization of GF mice with one or two bacteria has been useful both to understand the features of microbes needed to colonize the intestinal ecosystem and to dissect how specific microbes contribute to immune system development and disease. Monoassociations of mice with Bacteroides fragilis have shown that B. fragilis contributes to immune homeostasis by balancing CD4+ Th1 and Th2 lymphocyte populations and guiding FoxP3+ T regulatory cell populations in the intestine. In addition, gnotobiotics have been a helpful tool for establishing the specific role of B. fragilis polysaccharide A (PsA) in these processes (Mazmanian et al. 2005; Round and Mazmanian 2010; Round et al. 2011). PsA is an example par excellence of using mouse models to identify symbiotic factors with important roles in host immune regulation. Similarly, monoassociations of mice with SFB have been important in establishing how SFB can drive expansion of intestinal IgA-producing plasma cell populations and Th17 cells (Klaasen et al. 1993; Gaboriau-Routhiau et al. 2009; Ivanov et al. 2009). The host pathways engaged by SFB and identification of specific features of SFB that influence host immune responses remain an area of active interest. Monoassociations with the model human gut model symbiont B. thetaiotaomicron were an integral part of groundbreaking experiments performed more than a decade ago by Hooper et al. (2001), wherein they examined microarrays from B. thetaiotaomicron-associated mice versus GF mice. B. thetaiotaomicron was found to influence host responses spanning xenobiotic metabolism, angiogenesis, intestinal maturation, mucosal barrier function, and nutrient absorption, and, using laser capture microdissection, the responses observed were linked to specific intestinal cell subsets (Hooper et al. 2001). While a précis of the discoveries stemming from monoassociation experiments of mice is beyond the scope of this review, a few technical innovations that are adding to the insight that can be gained from monoassociations are worth noting. Recently, Goodman et al. (2009) employed insertion sequencing along with genome-wide transposon mutant libraries of B. thetaiotaomicron and used wild-type, mutant GF, and defined community mice to identify the genetic determinants of B. thetaiotaomicron fitness in the gut. Such approaches hold tremendous promise for unraveling the contributions of both host and microbiota genetics for coadaptation (Goodman et al. 2011). Hapfelmeier et al. (2010) developed a system of “reversible colonization” by developing a triple mutant strain of Escherichia coli K12 with three auxotrophic deletions. When fed to gnotobiotic mice, these live but nonreplicating bacteria resulted in mice that were transiently colonized and, as such, provided a system to probe questions of immunological memory regarding antibacterial IgA production in the gut (Hapfelmeier et al. 2010). As a result of the facile nature of bacterial genetics, rapidly evolving meta'omics technology, and the surge of interest in gnotobiotics, continued technical ingenuity in highly simplified microbial community mouse model systems should be expected.
Gnotobiotic mice can be colonized with several bacterial strains and maintained with these communities over many generations. One model microbial community used since the 1960s and introduced to standardize the microbiota of laboratory mouse strains is referred to as the Altered Schaedler Flora (ASF). The ASF consists of eight species: Five are from the genera Clostridium, Eubacterium, and Bacteroides; one is a spirochete from the Flexistipes group (Mucispirillum schaederli); and two are Lactobacillus species. The laboratory of Jeffrey Gordon (Goodman et al. 2009; Faith et al. 2011; McNulty et al. 2011) has employed consortia of 10–15 human gut bacteria in several studies and used these communities to predict and test the response of the microbiota to diets and to unravel factors involved in model bacterial symbiont fitness in vivo (Goodman et al. 2009; Faith et al. 2011; McNulty et al. 2011). The laboratory of Kenya Honda (Atarashi et al. 2011) has used chloroform treatment of intestinal contents, which enriches for spore formers, as inocula into GF recipients as well as a cocktail of Clostridia strains. These approaches were useful for establishing that Clostridium species play an important role in colonic T regulatory cell development (Atarashi et al. 2011). Communities can be constructed in several ways, and with the sequencing and annotation of an increasing number of gut bacteria, the possibilities for more complex and rationally constructed communities becomes more achievable.
Inoculating mice with consortia of human gut bacteria or human feces is a way to construct humanized microbiome mice (HMM) (Faith et al. 2010). HMM have been useful for examining questions about the functionality of the microbiome and establish that human microbiomes can alter the metabolic capacity of mice. Specifically, when microbiota have been transplanted from obese human donors or, in another recent study, from women in the third trimester of pregnancy, these microbiota conferred some of their host's phenotypes to the mouse recipients: weight gain, increased adiposity, and insulin resistance (Turnbaugh et al. 2006; Koren et al. 2012). In contrast to prior studies of obesity and Western dietary patterns, a study of the microbiota of malnutrition was recently published. Smith et al. (2013) performed fecal surveys of the microbiota in a Malawian twin cohort study of severe malnutrition and generated HMM from afflicted children. These HMM mice lost a substantial amount of weight and also provided important data on how the microbiota responds to dietary interventions used to treat malnutrition. Thus, HMM can be an extremely useful tool to understand the role of human gut microbiomes in disease, how diet reconfigures gut microbiota composition and function, and how nutritional interventions can be optimized to restore human health.
Under certain circumstances, it appears that there is host specificity for which members of the human or rat microbiota can colonize the mouse small intestine; when these microbiome differences are linked to easily measurable immune phenotypes, as was observed by Chung et al. (2012), it provides the chance to explore the time scales, environmental factors, and genetics (both host and microbe) required for coadaptation. GF mice, simplified microbial community mice, and humanized microbiota mice are an “artificial” system. However, they have provided and will continue to provide insight into the wide-ranging effects of the gut microbiota on mammalian physiology and the mechanisms that permit and regulate relationships between animals and their microbiota.
Humans
Microbes and human development across the stages of life
An integral aspect of human development is the acquisition of our microbiota. The human infant is born with an essentially sterile gut, and although there can be dramatic variation in microbial diversity in early childhood, the adult gut microbiota eventually takes on a configuration that is “conserved” across humans, featuring the dominance of the Bacteroidetes and Firmicutes phyla (Fig. 2). Therefore, there are strong evolutionary forces acting on both humans and microbes, which operate in the development of a very specific constellation of the gut microbiota. Although many of the mechanisms by which these forces operate remain to be elucidated, recent metagenomics studies have shed light on the major features of the developing gut microbiota.
Figure 2.
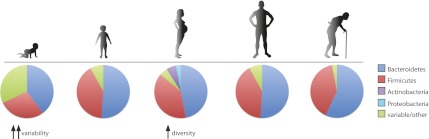
The structure of the human intestinal microbiota across the life cycle. The composition of the gut microbiome changes throughout the course of life. The infant microbiome shows great interindividual variability and relatively low diversity but becomes more diverse and converges into an “adult-like” structure by 3 yr after birth. Pregnancy is associated with an increase in Actinobacteria and Proteobacteria and increased diversity, but the gut microbiota returns to its original structure sometime after delivery. Old age (>65 yr) is associated with a number of changes in the microbiota, including an increase in the abundance of Bacteroidetes.
Prior to birth, a major remodeling of the mother's gut microbiota occurs throughout the course of pregnancy (Koren et al. 2012). The interindividual diversity in the gut microbiota is dramatically increased in the third trimester of pregnancy relative to the first trimester, with a particular increase in Proteobacteria and Actinobacteria. However, this new bacterial community acquired late in pregnancy is not transferred to the newborn; the child's gut microbiota is more similar to its mother's microbiota in the first trimester than in the third. This is consistent with other studies that have reported that the infant's gut microbiota is shaped by the mother's vaginal, rather than gut, microbiota (Dominguez-Bello et al. 2010) and that similarities between the child's and mother's microbiota increase with the age of the child (Yatsunenko et al. 2012).
Although historically the infant has been considered sterile prior to birth, recent evidence shows that the amniotic fluid is host to a diversity of microorganisms (DiGiulio et al. 2008). Nevertheless, the meconium, the first stool sample produced by the infant, has both a very low bacterial density (Palmer et al. 2007) and a very low phylogenetic diversity (Koenig et al. 2011). In a study that measured bacterial load using qRT–PCR, most babies had an overall fecal bacterial density of <106 rDNA gene copies per gram of stool in the first 2 d after birth, and the only two babies delivered by planned cesarean section in the study had low bacterial counts that continued until the seventh day after birth, as might be expected from newborns that were not exposed to a burst amniotic sac or to the vaginal canal (Palmer et al. 2007). After ∼1 wk, the bacterial density rose and then stabilized to 108–109 rDNA copies per gram of stool for most infants.
Mode of delivery appears to play a substantial role in determining the structure of the newborn's microbiota not only in the gut, but also in other body habitats. Dominguez-Bello et al. (2010) collected samples from skin, oral mucosa, nasopharyngeal aspirate, and stool from a set of 10 newborn infants as well as skin, oral, and vaginal samples from their mothers immediately prior to delivery and analyzed them by 16S rDNA pyrosequencing. The composition of the microbiota was relatively homogenous across all sampled body sites of the newborns. Notably, the newborn's bacterial community was determined by mode of delivery, closely resembling the bacterial composition of the mother's skin for a baby delivered by caesarian section (in which Staphylococcus and Propionibacterium spp. dominated) or the mother's vagina for those babies delivered vaginally (which in turn were principally composed of Lactobacillus, Prevotella, Atopobium, or Sneathia spp.). A mother's vaginal community was more similar to her own baby's microbiota than to that of other babies; however, this was not true of the mothers' skin microbiota in those newborns that were delivered by caesarian section, suggesting that nonmaternal sources contributed significantly to shaping the microbiota of these babies.
The composition of the gut microbiota is unstable in the first 3 yr of life. Following initial colonization, the maturation of microbiota involves an increase in diversity and stability; communities undergo several consecutive changes until an “optimal” community is acquired (Lozupone et al. 2012). Palmer et al. (2007) followed the gut microbiota in 14 infants through their first year of life using a 16S rRNA gene-based microarray. Although the phylum-level diversity is still relatively limited, there was an enormous amount of variation at the genus level between newborns (Palmer et al. 2007). The early colonizers tend to be strict aerobes (for example, Streptococcus and Staphylococcus), whereas strict anaerobes generally colonized later (most notably Clostridia spp.). Large fluctuations in the composition of the microbiota were common in the first few weeks, as were “uneven” populations (communities dominated by a single taxonomic group), but the populations became more stable over time. The infants' gut microbiota converged to a profile that were more similar to one another and to the microbiota of adult stool samples, suggesting that the gut microbial communities converge to a generic “adult-like” microbiota after 1 yr of age. This “adult-like” community was characterized by dominance of the Bacteroidetes and Firmicutes and a very low abundance of Proteobacteria and Gram-negative bacteria (see Fig. 2).
A similar study focused on a single infant, monitoring the gut microbiota by 16S rDNA sequencing and shotgun metagenomics of stool specimens for a period of 3 yr (Koenig et al. 2011). The phylogenetic diversity of the infant's microbiota had a steady increasing trend over time, with the lowest diversity on the first day of life. By clustering OTUs identified in the infant's stool across all of the time points tested, a clear, discrete set of community structures emerges that correspond to specific life events: the breast feeding period, the start of rice cereal and table foods and the cessation of breast milk, and the incidence of fever and antibiotic treatments. Metagenomic analysis identified changes in functional gene dynamics that are associated with the developing microbiota, such as the enrichment of lactose, galactose, and sucrose utilization genes during breast feeding; the appearance of genes involved in plant-derived polysaccharide metabolism prior to beginning a diet of solid food; and the presence of genes involved in xenobiotic compound breakdown and vitamin biosynthesis after weaning. This work reveals the remarkable modularity with which the microbiome changes, adding and replacing gene functionalities to fit the metabolism of the ever-fluctuating infant diet.
Other major contributors to the developing microbiota include diet and geography. In a study that looked at variations in the microbiota across geography and age, stool samples were collected from both children and adults from the United States, Malawi, and Amerindians from Venezuela (Yatsunenko et al. 2012). Consistent with other studies (Palmer et al. 2007; Dominguez-Bello et al. 2010), they observed an adult-like configuration of the gut microbiota by 3 yr after birth. The bacterial diversity increased with age at an exponential scale within the first 3 yr and continued to increase, although at a lower rate, thereafter. Interpersonal variation was significantly greater between children than between adults. Monozygotic twins were no more similar in their microbial composition than dizygotic twins, whether from Malawi or from the United States, underscoring the importance of environmental rather than genetic determinants in shaping the microbiota. In a related study, the gut microbiota of young children living in a remote village in Burkina Faso were compared with children living in urban Florence, Italy (De Filippo et al. 2010). The African children (BF children) had a diet high in fiber and low in animal protein, whereas the European children (EU children) had a typical western diet high in animal protein, starch, and sugar and low in fiber. The BF microbiota was dominated by the Bacteroidetes; however, the EU microbiota had a high abundance of Firmicutes and much lower level of Bacteroidetes and also a higher level of Proteobacteria. The BF microbiota also had a higher richness and biodiversity. A high Firmicutes-to-Bacteroidetes ratio and low microbial diversity is indicative of a high-calorie diet and obesity (Ley et al. 2006; Turnbaugh and Gordon 2009), suggesting that the association of the Western diet with poorer health and greater disease risk may be directly linked to the gut microbiota.
Comprehensive characterizations of the “healthy” human adult microbiota have been carried out by the European and Chinese-led Metagenomics of the Human Intestinal Tract (MetaHIT) Consortium and the National Institutes of Health (NIH)-sponsored Human Microbiome Project (HMP). MetaHIT focused on shotgun metagenomic sequencing of fecal samples from 146 European individuals, uncovering the presence of a minimal gut genome and metagenome based on taxonomy and gene functionality (Qin et al. 2010) and the clustering of individuals into one of three enterotypes, identified by variation in the level of the Bacteroides, Prevotella, and Ruminococcus genera (Arumugam et al. 2011). The HMP carried out both 16S rRNA gene sequencing and metagenomic sequencing on 15 (for males) or 18 (for females) body sites on each of 242 healthy adults in the United States samples on three separate visits (The Human Microbiome Project Consortium 2012a). The gut and tooth habitats were found to harbor the greatest between-subject microbial diversity but also the lowest between-visit variability, whereas the skin had lower between-subject diversity but much higher between-visit variability (The Human Microbiome Project Consortium 2012b). Although there was strong variability in bacterial abundance at the taxonomic level in almost every body site, the gene-level metabolic pathways were highly stable, in agreement with previous results that were limited to fecal microbiota (Turnbaugh et al. 2008b).
The gut microbial composition of the elderly shows a decrease in the Firmicutes-to-Bacteroidetes ratio and greater variability than that of younger adults (Claesson et al. 2011), and this trend may be more pronounced in centenarians (Biagi et al. 2010). Recently, the fecal microbiota composition in 178 elderly individuals was assessed by 16S rRNA gene sequencing (Claesson et al. 2012). The investigators observed a clear clustering in gut microbiota patterns according to whether the subjects live in a community residence or a long-term care facility, with a distinctly higher level of Bacteroidetes among the long-term care patients. Diet was a major determinant of microbial diversity, which was highest in those with a “low-fat/high-fiber” diet and lowest with a “moderate- or high-fat/low-fiber” diet. Microbial diversity showed clear health associations using multiple clinical parameters of geriatric health. This work demonstrates that the microbiota continues to change in old age and is closely linked to diet and health status, suggesting the possibility that microbial profiling may serve as a clinically useful biomarker in geriatric care.
Conclusions
Model systems and the microbiota
The microbiome field has undergone a renaissance in recent years and is uncovering the many dependencies of the host on the microbiota in the maintenance of health and also a substantial role for symbionts in the initiation and propagation of disease. The similarities of host–microbiota interactions across model systems are striking. Each model plays host to a specific community of microorganisms that is conserved across members of the species and employs a collection of host factors that maintains the structure of this microbiota. The Hawaiian bobtail squid is the ultimate “reduction of complexity” system for understanding host–microbiota interactions on a one-to-one basis and harbors a bacterium with tractable genetics. The fruit fly offers the advantage of a greatly reduced microbiota, <30 species, that is aerobic and culturable, therefore affording microbial manipulability in the context of a highly genetically manipulable host. The zebrafish bears the physiological and immunological features common to all vertebrates as well as a complex microbiota but greater throughput potential and a higher degree of genetic tractability than that of higher vertebrates. The mouse is highly similar to humans at both the host genetic level and the taxonomic levels of the microbiota and has been by far the most productive workhorse for dissecting host–microbiota functional relationships to date. Going forward, the microbiome field can benefit in a synergistic manner by combining these model systems and others to understand the underlying principles that govern host–microbiota interactions.
Perspective and future directions
We have begun to understand the dynamics of the human microbiome through human life stages, largely driven by sequencing technology developments in recent years that have fueled metagenomics. Large systematic efforts such as the MetaHIT and HMP consortia have contributed vastly to our understanding of the composition of the “normal” microbiome and so pave the way for future efforts to study its perturbations in disease. Much work remains to be done. We are moving from a phylogenetic definition of our microbiota and are beginning to appreciate its gene content and functionality and, by extension, are beginning to acquire a dynamic metatranscriptomic and metametabolomic view of our gut. In order to facilitate expansion into these new fields, more effort must be contributed to the sequencing and annotation of new bacterial reference whole genomes, as is currently under way. Recent progress has been made to develop new culturing technologies so that we can grow previously “unculturable” organisms in the laboratory environment (Vartoukian et al. 2010). We have expanded our view of the microbiome beyond bacteria and into the virome (viruses) (Minot et al. 2011) and mycome (fungi) (Ghannoum et al. 2010) and have begun to investigate the reciprocal interactions between these constituents (Kane et al. 2011; Kuss et al. 2011). The microbiome holds great potential for breakthroughs in translational medicine, including the promise of precision tools that will allow us to “sculpt” the microbiome with diet, prebiotics, probiotics, and targeted antibiotics to prevent and treat disease. Going forward, model systems will be essential for conducting the experimental perturbations and interventions required to understand the complexities of host interactions with the microbiota.
Acknowledgments
We thank members of the Garrett laboratory for discussion. Work from our laboratory is supported by grants from the NIH (AI078942, R01CA154426, R01DK092405, and GM099531 to W.S.G., and F32DK098826 to M.H.), the Burroughs Wellcome Fund, the Searle Scholars Program, the Lavietes Fund, and the Cancer Research Institute to W.S.G.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.212522.112.
References
- Amacher SL 2008. Emerging gene knockout technology in zebrafish: Zinc-finger nucleases. Brief Funct Genomics Proteomics 7: 460–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, Kanneganti T-D 2012. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KV, Jürgens G, Nüsslein-Volhard C 1985. Establishment of dorsal–ventral polarity in the Drosophila embryo: Genetic studies on the role of the Toll gene product. Cell 42: 779–789 [DOI] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al. 2011. Enterotypes of the human gut microbiome. Nature 473: 174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. 2011. Induction of colonic regulatory T cells by indigenous clostridium species. Science 331: 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates JM, Mittge E, Kuhlman J, Baden KN, Cheesman SE, Guillemin K 2006. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol 297: 374–386 [DOI] [PubMed] [Google Scholar]
- Bates JM, Akerlund J, Mittge E, Guillemin K 2007. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2: 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF, Fisher EM 2000. Genealogies of mouse inbred strains. Nat Genet 24: 23–25 [DOI] [PubMed] [Google Scholar]
- Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG II, Tan W, Penheiter SG, Ma AC, Leung AYH, et al. 2012. In vivo genome editing using a high-efficiency TALEN system. Nature 491: 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, et al. 2010. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci 107: 18933–18938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagi E, Nylund L, Candela M, Ostan R, Bucci L, Pini E, Nikkïla J, Monti D, Satokari R, Franceschi C, et al. 2010. Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 5: e10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, Dunne WM, Allen PM, Stappenbeck TS 2011. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 9: 390–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco-Drayon V, Poidevin M, Boneca IG, Narbonne-Reveau K, Royet J, Charroux B 2012. Peptidoglycan sensing by the receptor PGRP-LE in the Drosophila gut induces immune responses to infectious bacteria and tolerance to microbiota. Cell Host Microbe 12: 153–165 [DOI] [PubMed] [Google Scholar]
- Bose JL, Rosenberg CS, Stabb EV 2008. Effects of luxCDABEG induction in Vibrio fischeri: Enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch Microbiol 190: 169–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, Bienenstock J, Cryan JF 2011. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci 108: 16050–16055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman BM, Hildebrand F, Kubica M, Goosens D, Del Favero J, Declercq W, Raes J, Vandenabeele P 2011. Caspase deficiency alters the murine gut microbiome. ISME J 2: e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugman S, Liu KY, Lindenbergh Kortleve D, Samsom JN, Furuta GT, Renshaw SA, Willemsen R, Nieuwenhuis EES 2009. Oxazolone-induced enterocolitis in zebrafish depends on the composition of the intestinal microbiota. Gastroenterology 137: 1757–1767.e1 [DOI] [PubMed] [Google Scholar]
- Büchler G, Wos-Oxley ML, Smoczek A, Zschemisch N-H, Neumann D, Pieper DH, Hedrich HJ, Bleich A 2012. Strain-specific colitis susceptibility in IL10-deficient mice depends on complex gut microbiota-host interactions. Inflamm Bowel Dis 18: 943–954 [DOI] [PubMed] [Google Scholar]
- Buchon N, Broderick NA, Chakrabarti S, Lemaitre B 2009. Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev 23: 2333–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JH, Foster CM, Vishnivetskaya T, Campbell AG, Yang ZK, Wymore A, Palumbo AV, Chesler EJ, Podar M 2012. Host genetic and environmental effects on mouse intestinal microbiota. ISME J 6: 2033–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Koren O, Goodrich JK, Johansson MEV, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, Gonzalez A, et al. 2012. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 12: 139–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash HL, Whitham CV, Behrendt CL, Hooper LV 2006. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313: 1126–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A 2011. Bacterial communities of diverse Drosophila species: Ecological context of a host-microbe model system. PLoS Genet 7: e1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charroux B, Royet J 2012. Gut-microbiota interactions in non-mammals: What can we learn from Drosophila? Semin Immunol 24: 17–24 [DOI] [PubMed] [Google Scholar]
- Cheesman SE, Neal JT, Mittge E, Seredick BM, Guillemin K 2011. Epithelial cell proliferation in the developing zebrafish intestine is regulated by the Wnt pathway and microbial signaling via Myd88. Proc Natl Acad Sci 108: 4570–4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I, Yamanishi S, Cox L, Methé BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, et al. 2012. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488: 621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun CK, Troll JV, Koroleva I, Brown B, Manzella L, Snir E, Almabrazi H, Scheetz TE, Bonaldo M de F, Casavant TL, et al. 2008. Effects of colonization, luminescence, and autoinducer on host transcription during development of the squid-vibrio association. Proc Natl Acad Sci 105: 11323–11328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al. 2012. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149: 1578–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, et al. 2011. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci 108: 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, Harris HMB, Coakley M, Lakshminarayanan B, O'Sullivan O, et al. 2012. Gut microbiota composition correlates with diet and health in the elderly. Nature 488: 178–184 [DOI] [PubMed] [Google Scholar]
- Corby-Harris V, Pontaroli AC, Shimkets LJ, Bennetzen JL, Habel KE, Promislow DEL 2007. Geographical distribution and diversity of bacteria associated with natural populations of Drosophila melanogaster. Appl Environ Microbiol 73: 3470–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CR, Gilmore MS 2007. Native microbial colonization of Drosophila melanogaster and its use as a model of Enterococcus faecalis pathogenesis. Infect Immun 75: 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmasso G, Nguyen HTT, Yan Y, Laroui H, Charania MA, Ayyadurai S, Sitaraman SV, Merlin D 2011. Microbiota modulate host gene expression via microRNAs. PLoS ONE 6: e19293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapito DH, Mencin A, Gwak G-Y, Pradere J-P, Jang M-K, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, et al. 2012. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21: 504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P 2010. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci 107: 14691–14696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rienzo G, Gutzman JH, Sive H 2012. Efficient shRNA-mediated inhibition of gene expression in zebrafish. Zebrafish 9: 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB 2012. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487: 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Heijtz R, Wang S, Anuar F, Qian Y, Björkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S 2011. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci 108: 3047–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, Gotsch F, Kim CJ, Erez O, Edwin S, Relman DA 2008. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: A molecular and culture-based investigation. PLoS ONE 3: e3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionne MS, Schneider DS 2008. Models of infectious diseases in the fruit fly Drosophila melanogaster. Dis Mod Mech 1: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R 2010. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci 107: 11971–11975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas AE, Douglas AE, Douglas AE 2011. Lessons from studying insect symbioses. Cell Host Microbe 10: 359–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Yang M, Lee S, Behrendt CL, Hooper LV, Saghatelian A, Wan Y 2012. Maternal western diet causes inflammatory milk and TLR2/4-dependent neonatal toxicity. Genes Dev 26: 1306–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. 2011. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145: 745–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esworthy RS, Smith DD, Chu F-F 2010. A strong impact of genetic background on gut microflora in mice. Int J Inflamm 2010: 986046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezenwa VO, Gerardo NM, Inouye DW, Medina M, Xavier JB 2012. Animal behavior and the microbiome. Science 338: 198–199 [DOI] [PubMed] [Google Scholar]
- Faith JJ, Rey FE, O'Donnell D, Karlsson M, McNulty NP, Kallstrom G, Goodman AL, Gordon JI 2010. Creating and characterizing communities of human gut microbes in gnotobiotic mice. ISME J 4: 1094–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FE, Gordon JI 2011. Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science 333: 101–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JS, Apicella MA, McFall-Ngai MJ 2000. Vibrio fischeri lipopolysaccharide induces developmental apoptosis, but not complete morphogenesis, of the Euprymna scolopes symbiotic light organ. Dev Biol 226: 242–254 [DOI] [PubMed] [Google Scholar]
- Franchi L, Muñoz-Planillo R, Núñez G 2012. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13: 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz AL, Rogier EW, Weber CR, Shen L, Cohen DA, Fenton LA, Bruno MEC, Kaetzel CS 2012. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol 5: 501–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer A, Sprenger N, Kurakevich E, Borsig L, Chassard C, Hennet T 2010. Milk sialyllactose influences colitis in mice through selective intestinal bacterial colonization. J Exp Med 207: 2843–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaboriau-Routhiau V, Rakotobe S, Lécuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, et al. 2009. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31: 677–689 [DOI] [PubMed] [Google Scholar]
- Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN 2010a. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8: 292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Gordon JI, Glimcher LH 2010b. Homeostasis and inflammation in the intestine. Cell 140: 859–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, Gillevet PM 2010. Characterization of the oral fungal microbiome (Mycobiome) in healthy individuals. PLoS Pathog 6: e1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg RF, Austen WG, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, et al. 2008. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci 105: 3551–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI 2009. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6: 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, Wu M, Gordon JI 2011. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nat Protoc 6: 1969–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha E-M, Oh C-T, Bae YS, Lee W-J 2005. A direct role for dual oxidase in Drosophila gut immunity. Science 310: 847–850 [DOI] [PubMed] [Google Scholar]
- Hapfelmeier S, Lawson MAE, Slack E, Kirundi JK, Stoel M, Heikenwalder M, Cahenzli J, Velykoredko Y, Balmer ML, Endt K, et al. 2010. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328: 1705–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. 2012. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482: 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen Y-Y, Knight R, Ahima RS, Bushman F, Wu GD 2009. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137: 1716–1724.e1–e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, Artis D 2010. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol 3: 148–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI 2001. Molecular analysis of commensal host-microbial relationships in the intestine. Science 291: 881–884 [DOI] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ 2012. Interactions between the microbiota and the immune system. Science 336: 1268–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium 2012a. A framework for human microbiome research. Nature 486: 215–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium 2012b. Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA 2009. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137: 1343–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, Golovkina TV 2011. Successful transmission of a retrovirus depends on the commensal microbiota. Science 334: 245–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellermayer R, Dowd SE, Harris RA, Balasa A, Schaible TD, Wolcott RD, Tatevian N, Szigeti R, Li Z, Versalovic J, et al. 2011. Colonic mucosal DNA methylation, immune response, and microbiome patterns in Toll-like receptor 2-knockout mice. FASEB J 25: 1449–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaasen HL, Van der Heijden PJ, Stok W, Poelma FG, Koopman JP, Van den Brink ME, Bakker MH, Eling WM, Beynen AC 1993. Apathogenic, intestinal, segmented, filamentous bacteria stimulate the mucosal immune system of mice. Infect Immun 61: 303–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE 2011. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci 108: 4578–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Kling Bäckhed H, Gonzalez A, Werner JJ, Angenent LT, Knight R, et al. 2012. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150: 470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropatnick TA, Engle JT, Apicella MA, Stabb EV, Goldman WE, McFall-Ngai MJ 2004. Microbial factor-mediated development in a host-bacterial mutualism. Science 306: 1186–1188 [DOI] [PubMed] [Google Scholar]
- Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK 2011. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334: 249–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson E, Tremaroli V, Lee YS, Koren O, Nookaew I, Fricker A, Nielsen J, Ley RE, Backhed F 2011. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 61: 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Ruby EG 1992. Detection of the light organ symbiont, Vibrio fischeri, in Hawaiian seawater by using lux gene probes. Appl Environ Microbiol 58: 942–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI 2005. Obesity alters gut microbial ecology. Proc Natl Acad Sci 102: 11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI 2006. Microbial ecology: Human gut microbes associated with obesity. Nature 444: 1022–1023 [DOI] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone CA, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, et al. 2008. Evolution of mammals and their gut microbes. Science 320: 1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhocine N, Ribeiro PS, Buchon N, Wepf A, Wilson R, Tenev T, Lemaitre B, Gstaiger M, Meier P, Leulier F 2008. PIMS modulates immune tolerance by negatively regulating Drosophila innate immune signaling. Cell Host Microbe 4: 147–158 [DOI] [PubMed] [Google Scholar]
- Littman DR, Pamer EG 2011. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 10: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh G, Brodziak F, Blaut M 2008. The Toll-like receptors TLR2 and TLR4 do not affect the intestinal microbiota composition in mice. Environ Microbiol 10: 709–715 [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R 2012. Diversity, stability and resilience of the human gut microbiota. Nature 489: 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupp C, Urbanowski M, Greenberg EP, Ruby EG 2003. The Vibrio fischeri quorum-sensing systems ain and lux sequentially induce luminescence gene expression and are important for persistence in the squid host. Mol Microbiol 50: 319–331 [DOI] [PubMed] [Google Scholar]
- Madsen KL, Fedorak RN, Tavernini MM, Doyle JS 2002. Normal breast milk limits the development of colitis in IL-10-deficient mice. Inflamm Bowel Dis 8: 390–398 [DOI] [PubMed] [Google Scholar]
- Mandel MJ, Wollenberg MS, Stabb EV, Visick KL, Ruby EG 2009. A single regulatory gene is sufficient to alter bacterial host range. Nature 458: 215–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice CF, Haiser HJ, Turnbaugh PJ 2013. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152: 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard CL, Elson CO, Hatton RD, Weaver CT 2012. Reciprocal interactions of the intestinal microbiota and immune system. Nature 489: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122: 107–118 [DOI] [PubMed] [Google Scholar]
- McFall-Ngai M 2007. Adaptive immunity: Care for the community. Nature 445: 153. [DOI] [PubMed] [Google Scholar]
- McFall-Ngai M, Ruby E 1991. Symbiont recognition and subsequent morphogenesis as early events in an animal-bacterial mutualism. Science 254: 1491–1494 [DOI] [PubMed] [Google Scholar]
- McFall-Ngai MJ, Ruby EG 1998. Sepiolids and vibrios: When first they meet. Bioscience 48: 257–265 [Google Scholar]
- McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, Henrissat B, Oozeer R, Cools-Portier S, Gobert G, et al. 2011. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci Transl Med 3: 106ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micchelli CA, Perrimon N 2006. Evidence that stem cells reside in the adult Drosophila midgut epithelium. ISME J 439: 475–479 [DOI] [PubMed] [Google Scholar]
- Milligan-Myhre K, Charette JR, Phennicie RT, Stephens WZ, Rawls JF, Guillemin K, Kim CH 2011. Study of host–microbe interactions in zebrafish. In The zebrafish: Disease models and chemical screens (ed. WH Detrich et al.), pp. 87–116, Academic Press, Burlington, MA [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, Bushman FD 2011. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res 21: 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, Leleiko N, Snapper SB, et al. 2012. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC 2000. Effective targeted gene ‘knockdown' in zebrafish. Nat Genet 26: 216–220 [DOI] [PubMed] [Google Scholar]
- Natividad JMM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, Philpott D, Garcia Rodenas CL, McCoy KD, Verdu EF 2012. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1−/−; Nod2−/− mice. Inflamm Bowel Dis 18: 1434–1446 [DOI] [PubMed] [Google Scholar]
- Nones K, Knoch B, Dommels YEM, Paturi G, Butts C, McNabb WC, Roy NC 2009. Multidrug resistance gene deficient (mdr1a−/−) mice have an altered caecal microbiota that precedes the onset of intestinal inflammation. J Appl Microbiol 107: 557–566 [DOI] [PubMed] [Google Scholar]
- Nyholm SV, Tzou P, Stabb EV, Ohresser S, Ruby EG, Ferrandon D, McFall-Ngai MJ, Capovilla M, Reichhart JM, Lemaitre B, et al. 2000. Establishment of an animal-bacterial association: Recruiting symbiotic vibrios from the environment. Proc Natl Acad Sci 97: 10231–10235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholm SV, Deplancke B, Gaskins HR, Apicella MA, McFall-Ngai MJ 2002. Roles of Vibrio fischeri and nonsymbiotic bacteria in the dynamics of mucus secretion during symbiont colonization of the Euprymna scolopes light organ. Appl Environ Microbiol 68: 5113–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholm SV, McFall-Ngai MJ 2003. Dominance of Vibrio fischeri in secreted mucus outside the light organ of Euprymna scolopes: The first site of symbiont specificity. Appl Environ Microbiol 69: 3932–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholm SV, McFall-Ngai MJ 2004. The winnowing: Establishing the squid-vibrio symbiosis. Nat Rev Microbiol 2: 632–642 [DOI] [PubMed] [Google Scholar]
- O'Callaghan D, Vergunst A 2010. Non-mammalian animal models to study infectious disease: Worms or fly fishing? Curr Opin Microbiol 13: 79–85 [DOI] [PubMed] [Google Scholar]
- Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO 2007. Development of the human infant intestinal microbiota. PLoS Biol 5: e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes JC, Welchman DP, Poidevin M, Lemaitre B 2011. Negative regulation by amidase PGRPs shapes the Drosophila antibacterial response and protects the fly from innocuous infection. Immunity 35: 770–779 [DOI] [PubMed] [Google Scholar]
- Parkes M 2012. Evidence from genetics for a role of autophagy and innate immunity in IBD pathogenesis. Dig Dis 30: 330–333 [DOI] [PubMed] [Google Scholar]
- Petnicki-Ocwieja T, Hrncir T, Liu Y-J, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS 2009. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci 106: 15813–15818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham LN, Kanther M, Semova I, Rawls JF 2008. Methods for generating and colonizing gnotobiotic zebrafish. Nat Protoc 3: 1862–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader BA, Kremer N, Apicella MA, Goldman WE, McFall-Ngai MJ 2012. Modulation of symbiont lipid A signaling by host alkaline phosphatases in the squid-vibrio symbiosis. MBio 3: e00093-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R 2004. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118: 229–241 [DOI] [PubMed] [Google Scholar]
- Rawls JF, Samuel BS, Gordon JI 2004. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc Natl Acad Sci 101: 4596–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Mahowald MA, Ley RE, Gordon JI 2006. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127: 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reikvam DH, Erofeev A, Sandvik A, Grcic V, Jahnsen FL, Gaustad P, McCoy KD, Macpherson AJ, Meza-Zepeda LA, Johansen F-E 2011. Depletion of murine intestinal microbiota: Effects on gut mucosa and epithelial gene expression. PLoS ONE 6: e17996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt C, Bergentall M, Greiner TU, Schaffner F, Östergren-Lundén G, Petersen LC, Ruf W, Bäckhed F 2012. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 483: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Webster P, Finkel SE, Tower J 2007. Increased internal and external bacterial load during Drosophila aging without life-span trade-off. Cell Metab 6: 144–152 [DOI] [PubMed] [Google Scholar]
- Ridley EV, Wong AC-N, Westmiller S, Douglas AE 2012. Impact of the resident microbiota on the nutritional phenotype of Drosophila melanogaster. PLoS ONE 7: e36765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF 2011. Evidence for a core gut microbiota in the zebrafish. ISME J 5: 1595–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK 2010. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci 107: 12204–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK 2011. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332: 974–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royet J, Dziarski R 2007. Peptidoglycan recognition proteins: Pleiotropic sensors and effectors of antimicrobial defences. Nat Rev Microbiol 5: 264–277 [DOI] [PubMed] [Google Scholar]
- Ruby EG, Lee KH 1998. The Vibrio fischeri–Euprymna scolopes light organ association: Current ecological paradigms. Appl Environ Microbiol 64: 805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J-H, Kim S-H, Lee H-Y, Bai JY, Nam Y-D, Bae J-W, Lee DG, Shin SC, Ha E-M, Lee W-J 2008. Innate immune homeostasis by the homeobox gene caudal and commensal-gut mutualism in Drosophila. Science 319: 777–782 [DOI] [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjöberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, et al. 2010. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 11: 76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Schubert AM, Zackular JP, Iverson KD, Young VB, Petrosino JF 2012. Stabilization of the murine gut microbiome following weaning. Gut Microbes 3: 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semova I, Carten JD, Stombaugh J, Mackey LC, Knight R, Farber SA, Rawls JF 2012. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12: 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon G, Segal D, Ringo JM, Hefetz A, Zilber-Rosenberg I, Rosenberg E 2010. Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proc Natl Acad Sci 107: 20051–20056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, Jovov B, Abdo Z, Sandler RS, Keku TO 2010. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes 1: 138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SC, Kim SH, You H, Kim B, Kim AC, Lee KA, Yoon JH, Ryu JH, Lee WJ 2011. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334: 670–674 [DOI] [PubMed] [Google Scholar]
- Sjögren K, Engdahl C, Henning P, Lerner UH, Tremaroli V, Lagerquist MK, Bäckhed F, Ohlsson C 2012. The gut microbiota regulates bone mass in mice. J Bone Miner Res 27: 1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al. 2013. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 339: 548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spor A, Koren O, Ley R 2011. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9: 279–290 [DOI] [PubMed] [Google Scholar]
- Stappenbeck TS, Hooper LV, Gordon JI 2002. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci 99: 15451–15455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storelli G, Defaye A, Erkosar B, Hols P, Royet J, Leulier F 2011. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through tor-dependent nutrient sensing. Cell Metab 14: 403–414 [DOI] [PubMed] [Google Scholar]
- Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu X-N, Kubo C, Koga Y 2004. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 558: 263–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson PA, Kumar A, Samarin S, Vijay-Kumar M, Kundu K, Murthy N, Hansen J, Nusrat A, Neish AS 2011. Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc Natl Acad Sci 108: 8803–8808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill DW, Churchill GA 2012. Ten years of the collaborative cross. G3 2: 153–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H, Kaser A 2011. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest 121: 2126–2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong D, Rozas NS, Oakley TH, Mitchell J, Colley NJ, McFall-Ngai MJ 2009. Evidence for light perception in a bioluminescent organ. Proc Natl Acad Sci 106: 9836–9841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V, Bäckhed F 2012. Functional interactions between the gut microbiota and host metabolism. Nature 489: 242–249 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Gordon JI 2009. The core gut microbiome, energy balance and obesity. J Physiol 587: 4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI 2008a. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. 2008b. A core gut microbiome in obese and lean twins. Nature 457: 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI 2009. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1: 6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzou P, Ohresser S, Ferrandon D, Capovilla M, Reichhart JM, Lemaitre B, Hoffmann JA, Imler JL 2000. Tissue-specific inducible expression of antimicrobial peptide genes in Drosophila surface epithelia. Immunity 13: 737–748 [DOI] [PubMed] [Google Scholar]
- Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MRM, Kamboj M, et al. 2010. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 120: 4332–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, Khanin R, Pamer EG 2012. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med 209: 1445–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV 2011. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science 334: 255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartoukian SR, Palmer RM, Wade WG 2010. Strategies for culture of ‘unculturable' bacteria. FEMS Microbiol Lett 309: 1–7 [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, et al. 2007. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest 117: 3909–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT 2010. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328: 228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visick KL, Foster J, Doino J, McFall-Ngai M, Ruby EG 2000. Vibrio fischeri lux genes play an important role in colonization and development of the host light organ. J Bacteriol 182: 4578–4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung Y-M, et al. 2011. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward TG, Trexler PC 1958. Gnotobiotics: A new discipline in biological and medical research. Perspect Biol Med 1: 447–456 [DOI] [PubMed] [Google Scholar]
- Welsh CE, Miller DR, Manly KF, Wang J, McMillan L, Morahan G, Mott R, Iraqi FA, Threadgill DW, de Villena FP-M 2012. Status and access to the Collaborative Cross population. Mamm Genome 23: 706–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier AM, Micchelli CA, Nyholm SV, Perrimon N, Mandel MJ, Massengo-Tiasse RP, Schaefer AL, Koroleva I, Splinter-BonDurant S, Brown B, et al. 2010. Transcriptional patterns in both host and bacterium underlie a daily rhythm of anatomical and metabolic change in a beneficial symbiosis. Proc Natl Acad Sci 107: 2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CNA, Ng P, Douglas AE 2011. Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environ Microbiol 13: 1889–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakushi T, Matsushita K 2010. Alcohol dehydrogenase of acetic acid bacteria: Structure, mode of action, and applications in biotechnology. Appl Microbiol Biotechnol 86: 1257–1265 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Yamaguchi R, Munakata K, Takashima K, Nishiyama M, Hioki K, Ohnishi Y, Nagasaki M, Imoto S, Miyano S, et al. 2012. A microarray analysis of gnotobiotic mice indicating that microbial exposure during the neonatal period plays an essential role in immune system development. BMC Genomics 13: 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. 2012. Human gut microbiome viewed across age and geography. Nature 486: 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi P, Li L 2012. The germfree murine animal: An important animal model for research on the relationship between gut microbiota and the host. Vet Microbiol 157: 1–7 [DOI] [PubMed] [Google Scholar]
- Zaidman-Rémy A, Hervé M, Poidevin M, Pili-Floury S, Kim M-S, Blanot D, Oh B-H, Ueda R, Mengin-Lecreulx D, Lemaitre B 2006. The Drosophila amidase PGRP-LB modulates the immune response to bacterial infection. Immunity 24: 463–473 [DOI] [PubMed] [Google Scholar]