

Abstract
IL-6 plays a central role in supporting pathological TH2 and TH17 cell development and inhibiting the protective T regulatory cells in allergic asthma. TH17 cells have been demonstrated to regulate allergic asthma in general and T-bet-deficiency-induced asthma in particular. Here we found an inverse correlation between T-bet and Il-6 mRNA expression in asthmatic children. Moreover, experimental subcutaneous immunotherapy (SIT) in T-bet(−/−) mice inhibited IL-6, IL-21R and lung TH17 cells in a setting of asthma. Finally, local delivery of an anti-IL-6R antibody in T-bet(−/−) mice resulted in the resolution of this allergic trait. Noteworthy, BATF, crucial for the immunoglobulin-class-switch and TH2,TH17 development, was found down-regulated in the lungs of T-bet(−/−) mice after SIT and after treatment with anti-IL-6R antibody, indicating a critical role of IL-6 in controlling BATF/IRF4 integrated functions in TH2, TH17 cells and B cells also in a T-bet independent fashion in allergic asthma.
Allergic asthma is a world-wide increasing disease characterized by chronic airway inflammation associated with recurrent episodes of wheezing, breathlessness and coughing in response to otherwise innocuous environmental stimuli1. Subcutaneous Immunotherapy (SIT) has been used successfully in the last two decades as therapy for this disease2,3,4,5. Interleukin-6 (IL-6) is a pro-inflammatory cytokine influencing T and B cell functions relevant also to asthma exacerbation in children6. IL-6 is produced by dendritic cells upon allergen challenge that induces both, TH2 and TH17 differentiation in allergic asthma7. In fact, IL-6 in conjunction with IL-21 induces TH17 cells8. It has been demonstrated that TH17 cells are involved in the pathogenesis of allergic asthma, especially in the absence of T-bet9,10,11,12,13. Targeted deletion of T-bet, a T-box transcription factor that trans-activates the Interferon-gamma (IFN-γ) gene in TH1 cells, is associated with an aggravated asthmatic trait14. We previously demonstrated that patients with asthma have increased soluble IL-6R in their airways. Local treatment with α-IL-6R antibodies led to a 50% reduction of STAT-3 but not STAT-1 phosphorylation in the lung of treated mice as compared to control treated mice. Moreover, we showed that blockade of IL-6R signaling leads to cell death of lung effector T cells by activating regulatory T cells in experimental asthma15,16. Here we found that in asthmatic children, an increase of IL-6 mRNA values coexists with low values of T-bet mRNA expression in their PBMCs. Furthermore, experimental SIT decreased IL-6, IL-21R, as well as Interferon regulatory factor 4 (IRF4) encoded by the Irf4 gene and lung TH17 cells in T-bet(−/−) mice in a setting of asthma. Finally, local treatment of T-bet(−/−) mice with an antibody against the IL-6R resulted in the resolution of the allergic trait. Notably, Basic leucine zipper transcription factor ATF-like, also known as BATF, a transcription factor essential for the development of TH2 and TH17 cells and immunoglobulin-class-switch of B cells17,18,19,20 was found down-regulated in the lungs of T-bet(−/−) mice after SIT and after in vitro stimulation with α-IL-6R antibody. These results indicate an important role of IL-6 in controlling integrated functions of BATF in TH2, TH17 and B cells also in a T-bet independent manner in allergic asthma21,22,23.
Results
Here, we found an inverse correlation between Il-6 and T-bet mRNA expression in the peripheral blood mononuclear cells (PBMC) of small children with asthma (Figure 1a and Supplementary Table 1). T-bet has been previously reported to be down-regulated in CD4+ T cells in asthmatic children24 and IL-6 was found to be up-regulated in asthmatic patients25,26,27.
Figure 1. Increased IL-6 in asthma in the absence of T-bet.

(a) Correlation between mRNA values of healthy pre-school control children (left panel) and asthmatic (right panel) children.(b) Experimental design of a murine model of allergic asthma in wild-type and T-bet(−/−) mice. Mice received 100 μg OVA/Alum intraperitoneally (i.p.) and 2 mg/ml OVA intranasally (i.n.). (c) Increased expression of Il-6 mRNA in murine lung tissue by qPCR in T-bet(−/−) naïve (PBS) and asthmatic mice (OVA). (d) Increased IL-6 in murine lung CD4+ T cells in T-bet(−/−) asthmatic mice after intracellular flow cytometric analysis.
In this study, in a murine model of asthma (Figure 1b), we found a spontaneous significant up-regulation of IL-6 in lung tissue as well as in lung CD4+ T cells from asthmatic T-bet(−/−) mice as compared to those isolated from wild type littermates (Figure 1c and d, respectively).
IL-6 up-regulates BATF, a transcription factor involved in both TH17 development and immunoglobulin class switch18,20. We thus next looked at the serum level of IgE of wild type and T-bet(−/−) mice in a murine model of allergic asthma. We found a statistically significant up-regulation of IgE in the serum of asthmatic T-bet(−/−) mice (Figure 2a). We next investigated whether BATF was induced in lung CD4+ T cells isolated from T-bet(−/−) mice. As shown in Figure 2b, BATF was spontaneously up-regulated in lung CD4+ T cells isolated from T-bet(−/−) mice and both wild-type and T-bet(−/−) asthmatic mice had a significant up-regulation of BATF in lung CD4+ T cells (Figure 2b).
Figure 2. IL-6 induces BATF in the absence of T-bet in lung CD4+ T cells from naïve and asthmatic mice.

(a) Increased levels of IgE in murine blood serum in T-bet(−/−) mice (n = 9–17 mice per group). (b) Increased Batf mRNA expression measured by qPCR in isolated murine lung CD4+ T cells from T-bet(−/−) mice (n = 3–5 mice per group). (c) Increased expression of Rorγt mRNA in murine lung tissue of T-bet(−/−) mice. (d, e) Batf and Rorγt mRNA expression after quantitative real time PCR in naïve lung murine CD4+ T cells cultured with α-CD3 and α-CD28 alone (Med) or with IL-6 (20 ng/ml) (Med+IL-6) (n = 3–5 mice per group). BATF and not RORγt were induced by IL-6 in lung CD4+ T cells isolated from T-bet(−/−) mice. Students t test was used to calculate statistical significances. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Results are expressed as mean ± s.e.m.
We then looked for the TH17 cell signature transcription factor, RAR-related orphan receptor (ROR)γt8,28. RORγt was found to be up-regulated but only in the lungs of naïve T-bet(−/−) mice and in the lungs of asthmatic wild type mice (Figure 2c).
Because BATF was found to be induced in conjunction with IL-6 in T-bet(−/−) mice, we isolated lung CD4+ T cells from naïve wild-type and T-bet(−/−) mice and stimulated them with IL-6. We found that BATF was induced by IL-6 in T-bet(−/−) CD4+ T cells but not in wild-type CD4+ T cells (Figure 2d). However, we observed a significant increase of RORγt in the T-bet(−/−) CD4+ T cells compared to the wild-type, but no further up-regulation in either wild-type or T-bet(−/−) CD4+ T cells after IL-6 stimulation (Figure 2e), indicating that RORγt is controlled by other factors and by T-bet29,30. TH17 cells can be induced by IL-6 in conjunction with IL-2129,30and BATF is known to play an important role in this context21,23. We then analyzed IL-21 and found that this cytokine was up-regulated in asthma in wild-type and T-bet(−/−) mice (Figure 3a). Similarly, the IL-21R was found up-regulated in the lungs of T-bet(−/−) mice both spontaneously and in a setting of asthma (Figure 3b). Interferon regulatory factor 4 is another transcription factor involved in TH17 cell differentiation31,32. Moreover, IL-21 has been shown to induce IRF4 and BATF integrated functions in TH17 cells leading to IL-10 production in these cells23,32. IRF4 has been recently found elevated in asthmatic patients33. Consistently, we found an up-regulation of Irf4 mRNA expression in the lung of both wild-type and T-bet(−/−) OVA sensitized and challenged mice (Figure 3c). Consistent with our findings at the transcriptional level, we found significantly higher IL-17A protein levels in the supernatants of cultured CD4+ T cells isolated from T-bet(−/−) mice after asthma induction as compared to the levels measured in the CD4+ T cell supernatants from wild-type littermates (Figure 3d). TH17 cells were shown to produce IL-10 which is regulated by BATF-JUN-IRF4 and c-maf21,22,23,34,35, a possible mechanism by which TH17 cells auto-regulate themselves34. When we analyzed IL-10 in this context, we found increased levels of this cytokine in CD4+ T cells from T-bet(−/−) mice compared to wild-type mice (Figure 3e and f). In conclusion, we found an up-regulation of TH17 cells expressing both pro-inflammatory and anti-inflammatory cytokines in the lung of asthmatic T-bet(−/−) mice in a setting of allergic asthma. These cells probably developed via IL-6 and IL-21. T-bet(−/−) mice have increased CD4+CD25+ effector T cells in their lungs after allergen sensitization but in the absence of allergen challenge36. Moreover, IL-2 inhibits T regulatory cell development when given intranasally in experimental asthma37. Here, we found that IL-2 was up-regulated in the lungs of T-bet deficient asthmatic mice under tolerogenic conditions (without costimulation) as compared to their wild-type littermates (Figure 4a and Supplementary Figure 1). Since TH17 cells and Treg cells are controlled reciprocally and high levels of IL-6 induce TH17 cells while inhibiting Treg cells, we next analyzed CD4+CD25+Foxp3+ T cells in the lungs of wild-type and T-bet(−/−) mice. We observed a reduced number of Foxp3+ Treg cells in both wild-type and especially in T-bet(−/−) mice after sensitization and challenge with OVA (Figure 4b). This decrease represents a significant proportion of the T regulatory cells that in the lung represent only 5% of the total CD4+ T cells. Moreover, T-bet(−/−) mice showed significantly lower numbers of Treg cells in the asthma model compared to wild-type mice (Figure 4b). These data indicate that TH17 skewing and T regulatory inhibiting conditions are sustained environmental conditions in the lungs of T-bet(−/−) mice in an experimental asthma model.
Figure 3. Increased IL-21, IL-17A and IL-10 expression in lung CD4+ T cells of T-bet(−/−) asthmatic mice.

(a) Increased Il-21 mRNA expression in lung CD4+ T cells isolated from asthmatic T-bet(−/−) mice. (b) Increased Il-21R mRNA expression in murine lung tissue from asthmatic T-bet(−/−) mice. (c) Increased Expression of Irf4 mRNA in asthmatic murine lung tissue. (d) IL-17A level measured in the supernatants of murine lung CD4+ T cells isolated from T-bet(−/−) asthmatic mice and cultured for 24 h with α-CD3 and α-CD28 (n = 4–6 mice per group). (e) Increased number of lung CD4+IL-10+ T cells in T-bet(−/−) asthmatic mice after intracellular flow cytometric analysis. (f) Increased IL-10 release by lung CD4+ T cells from T-bet(−/−) mice measured by ELISA in the supernatants of these cells after 24 hours of cell culture with α-CD3, α-CD28 antibody challenge. Students t test was used to calculate statistical significances. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Results are expressed as mean ± s.e.m.
Figure 4. IL-2 expression is increased in the lung of T-bet(−/−) asthmatic mice.
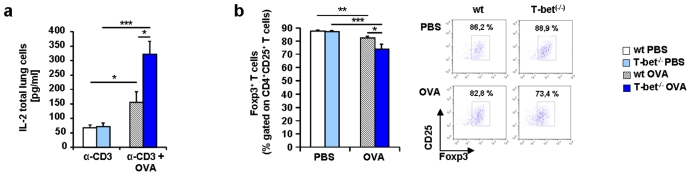
(a) Increased IL-2 protein expression in the supernatants of lung cells isolated from T-bet(−/−) asthmatic mice and cultured with α-CD3 and OVA (500 μg/ml) for 24 hours as indicated. (b) Decreased CD4+CD25+Foxp3+ Treg cells in T-bet(−/−) mice sensitized and challenged with OVA as described in Figure 1 and after flow cytometric analysis. Students t test was used to calculate statistical significances. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Results are expressed as mean ± s.e.m.
A long-term cure for asthma might be the SIT2. To investigate a potential role of IL-6 in SIT and in T-bet(−/−) mice, we applied the protocol of subcutaneous immunotherapy (SIT) in a second murine model of asthma in T-bet(−/−) mice as shown in Figure 5a. We found that Il-6 mRNA was also significantly induced in this model of allergic asthma in the lung of T-bet(−/−) mice as compared to non asthmatic mice (Figure 5b). Moreover, immunotherapy led to a down-regulation of Il-6 mRNA in the lungs of treated mice (Figure 5b). To analyze the role of TH17 cells in SIT, we next analyzed the expression of Rorγt mRNA in the lung, which we found increased in asthma and decreased after SIT (Figure 5c). IL-6 induces BATF downstream of the IL-6R pathway38. Consistent with a reduction of IL-6, we found that BATF was decreased (Figure 5d) after SIT. IRF4, which acts together with BATF at the IL-10 promoter23, was also found down-regulated after immunotherapy (Figure 5e) along with IL-21 and IL-21R (Figure 5f and g). Because the factors inducing TH17 cells were down-regulated in immunotherapy in T-bet(−/−) mice, we looked for IL-17A and IL-10. Both cytokines were found decreased after immunotherapy (Figure 5h und i). Finally, IL-2, a marker of TH cell differentiation was also found down-regulated (Figure 5j). Considering that we could not find an up-regulation of T regulatory cells in this model, these results indicate a possible role for IL-6 in controlling TH17 cells producing IL-17A and IL-10 in asthma that could be inhibited by SIT in the absence of T-bet.
Figure 5. Decreased mRNA expression of different transcription factors and cytokines involved in TH17 differentiation after immunotherapy (SIT) in T-bet(−/−) mice.

(a) Experimental design of the murine asthma model with immunotherapy.Mice received 100 μg OVA/Alum intraperitoneally (i.p.), 1000 μg OVA subcutaneous (s.c.) and 2 mg/ml OVA intranasally (i.n.). (b–j) mRNA expression of Il-6 (b), Rorγt (c), Batf (d), Irf4 (e), Il-21 (f), Il-21R (g), Il-17A (h), Il-10 (i) and Il-2 (j) in lung tissue of T-bet(−/−) mice before and after SIT (n = 3–6 mice per group). Results in this figure are expressed as mean ± s.e.m. Students t test was used to calculate statistical significances in this figure. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
We have previously demonstrated that local blockade of IL-6R in the lung of wild-type mice led to amelioration of the allergic asthmatic reaction15. In this study we found that T-bet(−/−) mice treated with α-IL-6R antibody, as indicated in Figure 6a, had reduced airway inflammation (Figure 6b) and decreased IgE levels in the serum (Figure 6c). IL-6 is known to play an important role in the differentiation of activated B cells into antibody-producing cells39. We found here that B cells (CD19+) were decreased after α-IL-6R antibody treatment (Figure 6d), that they express the IL-6R and are induced in the asthma model (Supplementary Figure 2). We also observed a down-regulation of CD4+ T cells (Figure 6e) as well as of the TH2 cytokines IL-4, IL-5 and IL-13 after α-IL-6R antibody treatment in T-bet(−/−) mice (Figure 6f, g, h, respectively).
Figure 6. Reduction of IgE, inflammation and TH2 cytokines in α-IL-6R antibody treated asthmatic T-bet(−/−) mice.

(a) Experimental design of a murine asthma model and the α-IL-6R antibody therapy.Mice received 100 μg OVA/Alum intraperitoneally (i.p.) and 2 mg/ml OVA intranasally (i.n.). Some of the mice also received 75 μg of α-IL-6R antibody or IgG. (b) Decreased pathological Score of the inflammation and corresponding histological sections of the lungs stained with H&E in anti-IL-6R antibody treated mice. (c) Decreased levels of IgE in blood serum of T-bet(−/−) mice treated with α-IL-6R antibody in vivo. One of three similar experiments are shown with three to five mice per group. (d,e) Decreased number of CD19+ B cells (d) and CD4+ T cells (e) isolated from the lungs of T-bet(−/−) sensitized and challenged mice (n = 4–21 mice per group) and in vivo treated with anti-IL6R antibody. (f–h) ELISA of IL-4 (f), IL-5 (g) and IL-13 (h) in the supernatants of total lung cell cultures from OVA-challenged and α-IL-6R antibody treated mice. Cells were cultured for 24 h with α-CD3 and α-CD28 (n = 7–8 mice per group). Statistical significances in this figure were evaluated with a Students t test. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Data are mean ± s.e.m.
BATF deficient mice have been demonstrated to lack TFH cells which are important to prime B cell mediated immune responses17. Moreover, BATF has been recently shown to play a central role in inducing TH2 cells reminiscent of the role of IL-617,18,19. We thus hypothesized that IL-6 controls BATF in allergic asthma. To prove this, we analyzed BATF gene expression in total lung cell cultures isolated from OVA-sensitized T-bet(−/−) mice that were treated with the anti-IL-6R antibody in vitro (Figure 7a) . We found that BATF which was induced in T-bet deficient mice (Figure 7b) was significantly reduced after in vitro stimulation with anti-IL-6R antibody treatment in lung cells of T-bet(−/−) mice. Rorγt mRNA expression was not changed after anti-IL-6R treatment neither in wild-type, nor in T-bet(−/−) mice (Figure 7c). C-maf (Figure 7d) was found down-regulated only in wild-type mice after administration of the anti-IL-6R antibody. IL-6 induces IL-21 via the transcription factors BATF38 and c-maf40,41. Consistently, we found significantly reduced IL-21 in the lung cells that were in vitro treated with anti-IL-6R antibody. We then asked whether the down-regulation of BATF and IL-21 would result in decreased IL-17A expression. IRF4 has been shown to interact with BATF at the IL-17A promoter. However, we could not observe a downregulation of IRF4 after anti-IL6R antibody treatment in vivo (Figure 8a and b). Similarly, IL-17A was not specifically down-regulated by anti-IL-6R antibody treatment in vivo in T-bet deficient mice (Figure 8c).
Figure 7. Reduced BATF, c-maf and IL-21 expression after in vitro treatment with α-IL-6R antibody.

(a) In vitro treatment with OVA (500 μg/ml) and additionally anti-IL-6 R antibody or IgG (15 μg/ml). (b,c) Reduced expression of Batf (b), but not Rorγt (c) after treatment with α-IL-6R antibody.(d) c-maf mRNA expression in total lung cells from untreated, OVA-treated and/or α-IL-6R antibody treated wild-type and T-bet(−/−) mice (n = 3–5 mice per goup). (e) Decreased Il-21 mRNA expression in lung CD4+ T cells isolated from wild-type and T-bet(−/−) mice after α-IL-6R antibody treatment. Cells were cultured for 24 h with α-CD3 and α-CD28 (n = 10–16 mice per group). Data are mean ± s.e.m. Students t test was used to calculate statistical significances. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Figure 8. Decreased IL-21R and IL-10 in T-bet deficient mice treated in vivo with anti-IL-6R antibody.

(a) Experimental design of anti-IL-6R antibody treatment in vivo.100 μg OVA/Alum was administered to the mice intraperitoneally (i.p.) and 2 mg/ml OVA intranasally (i.n.). Some of the mice also received 75 μg of α-IL-6R antibody or IgG. (b) Irf4 mRNA expression in lung tissue of T-bet(−/−) mice (n = 8–9 mice per group). (c) IL-17A measured by ELISA in the supernatants of total lung cell cultures of T-bet(−/−) mice sensitized and challenged in vivo with OVA and treated in vivo with α-IL-6R antibody. (d) Decreased expression of Il-21R mRNA in lung tissue from T-bet deficient mice treated with anti-IL-6R antibody (n = 3–6 mice per group). (d) Decreased IL-10 in the supernatants of total lung cell cultures of T-bet(−/−) mice sensitized with OVA treated in vivo with α-IL-6R antibody . Students t test was used to calculate statistical significances. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Results are expressed as mean ± s.e.m.
It has been recently reported that BATF cooperates with IRF4 and STAT3 in cells stimulated with IL-21 to produce IL-1021–23.We then looked at IL-21R and IL-10 after anti-IL-6R antibody treatment. We found that T-bet(−/−) asthmatic mice had decreased Il-21R mRNA and IL-10 mRNA in their lungs after anti-IL-6R antibody treatment (Figure 8d and e). It is thus possible that IL-6 controls the expression of BATF, IL-21 and IL-21R on TH2 and TH17 cells in the lung in a setting of allergic asthma in T-bet(−/−) mice.
Discussion
IL-6 is a pleiotropic cytokine that plays an important role in inflammation and cancer42. It is released by cells of the innate and adaptive immune-response. As it induces the proliferation of T cells as well as B cells its blockade has an impact on immune responses which lead to IgE hyper-production and to the expansion of effector TH17 and TH2 cells seen in the lungs of asthmatic patients. These findings show the relevant contribution of IL-6 to the pathogenesis of asthma.
The transcription factor T-bet, which is known to transactivate the Ifnγ gene, was found to be down-regulated in asthmatic patients. In this paper, we observed that the PBMC of children with asthma have lower values of T-bet mRNA that coexist with relative higher levels of IL-6 mRNA. Further epigenetic studies are underway in these children to see if T-bet inhibits IL-6 at the promoter level or if pSTAT-3 downstream of IL-6 inhibits T-bet expression or both mechanism are present in asthma. In this paper we analyzed the effect of T-bet on IL-6 expression and the effect of IL-6 signal transduction in the presence and absence of T-bet in different settings of asthma. Here, we found that T-bet(−/−) mice, which have a more severe phenotype of asthma than wild-type mice, show an increased production of IL-6 in lung tissue as well as CD4+ T cells. In addition, downstream of IL-6, T-bet deficient lung CD4+ T cells show increased expression of Batf, the transcription factor not only orchestrating TFH cells and B cells in the B cell zone in the lymphatic organs, but also controlling TH17 cell development17,18,19. Moreover, we found that IL-6 does not control RORγt mRNA expression in lung CD4+ T cells. Our data suggest that T-bet inhibits the development of Th17 cells by inducing IL-21R and inhibiting IL-10 expression in lung CD4+ T cells in asthma.
We also found that OVA sensitization and challenge in T-bet deficient mice led to an up-regulation of IL-21, which can then bind to its receptor and further induce the interaction of BATF/IRF4 at the IL-10 and IL-17A promoters. This results in the hyperproliferation of TH2 and TH17 cells seen in asthmatic T-bet deficient mice. Because TH17 and Treg cells control each other, we went on to investigate CD4+CD25+Foxp3+ Treg cells. Although Treg numbers were decreased in both wild-type and T-bet(–/–) mice after allergen exposure, T-bet(–/–) asthmatic mice showed significantly lower numbers of Treg cells, which is probably secondary to the overproduction of IL-6 in these mice. However, the exact mechanism on how IL-6 controls T regulatory cells needs further investigation.
SIT is a successful therapy for allergic asthma and is accompanied by reduced IgE levels, inflammation and AHR2. However, the immunological changes present in this treatment are not clearly documented. To attempt to improve this understanding, we asked whether IL-6 regulation would be involved in this treatment. We found that SIT decreased not only IL-6, but also RORγt, BATF, IRF4 and IL-21R in the lungs of treated asthmatic T-bet(–/–) mice. These findings were accompanied by reduced effector TH17 cells, as well as IL-10 and IL-2 production.
We then asked whether blockade of the IL-6R would recapitulate the results seen after subcutaneous immunotherapy in T-bet deficient mice. We found that local anti-IL-6R antibody treatment had an even higher effect on the immunological markers inhibited by the SIT in the lung of T-bet deficient mice. Specifically, we observed a specific down-regulation of BATF, IL-21/IL-21R and IL-10 after anti-IL-6R antibody treatment. This resulted in a decreased differentiation of naïve T cells into TH2 cells. However, the down-regulation of IL-17A after anti-IL-6R antibody treatment was not statistically different from IgG treatment for yet unknown reasons. One possibility could relate to our findings showing that IL-6 induces BATF and not RORγt in the CD4+ T cells from T-bet deficient mice. Similarly to IL-17A also IRF4 was not significantly down-regulated by anti-IL-6R antibody treatment as compared to IgG. This is consistent with an IL-17A inducing role of IRF4 on IL-17A. In fact, it has been previously demonstrated that retrovirus over-expressing IRF4 induce IL-17A43. Moreover, IRF4 in conjunction with IL-6 induces Rorγt. Thus the residual IRF4 after anti-IL-6R antibody treatment might be responsible for the presence of IL-17A and RORγt. Thus it is possible that IL-17A modulation in asthma in the absence of T-bet requires both IL-6 and probably other not yet described factors that regulate RORγt such as IRF4. Further studies in this direction are underway in our laboratory. The anti-IL-6R treatment also led to a down-regulation of IL-21/IL-21R. IL-21 is an important cytokine that maintains the effector cells and has received attention because of its role in auto-immunity44. In contrast to IL-21R which is distributed to B, T, NKT cells and DCs, IL-21 is limited and support the effector T cells and NKT cells44. Finally we found a down-regulation of TH2 cytokines and IgE serum levels after anti IL-6R antibody treatment. It is possible that the IL-21/IL-21R reflects a decrease in Th2 cells as IL-21 has been described to be a Th2 cytokine45. These inhibitory effects on lymphocytes were also evident by the inhibition of the hyper-proliferation of CD4+ T cells and B cell maturation in the lung of anti-IL-6R treated mice.
Taken together, these data demonstrated a multi target immune-regulatory effect of anti-IL-6R antibody immunotherapy that works by inhibiting the main cellular components of allergic asthma in the absence of T-bet. This therapy results in reduction of TH2 and markers of TH17 cells, inflammation and IgE production. We found a central regulatory role of BATF-IRF4 complex downstream of the IL-6R in mediating this effect because controlling both TH2, TH17 and B cells that is important for the development of new immune-therapeutical strategies for allergic asthma.
Methods
Human studies
Isolation of PBMCs
Heparinized blood was transferred to a 15 ml sterile tube and diluted with an equal volume of PBS, inverted and carefully stratified on Ficoll-Hypaque. After centrifugation, the layer of peripheral blood mononuclear cells (PBMCs), between plasma and Ficoll, was carefully transferred into a new 15 ml tube. On the one hand the RNA was isolated by using PeqGold RNA Pure according to the manufactureŕs protocol (PeqLab, Erlangen, Germany). On the other hand RNA was isolated with an AllPrep DNA/RNA Mini Kit (Quiagen, Hilden, Germany). Quantitative real time PCR was performed as described below. This study was approved by the ethics committee of the Friedrich-Alexander University Erlangen-Nürnberg, Germany (Re-No 4435). Informed consent was obtained from the parents of the all human participants (children) of this study.
Mice
Wild-type and T-bet(−/−) mice (the latter generous gift from Prof Laurie Glimcher) were on a Balb/c genetic background. The experiments were performed with male mice at the age of 6–8 weeks. The animals were bread in the animal facility adjacent to our institute with temperature control and had free access to food and water. All experiments were performed with approved licences (23-177-07/G09-1-008 from ethical review board Rheinland-Pfalz and 54-2532.1-2/10 from government of Mittelfranken, Bavaria).
Murine model of allergic asthma and antibody treatment
Wild-type mice and T-bet(−/−) mice received an intraperitoneal injection of 100 μg OVA (Calbiochem, San Diego, CA) complexed with 10% alum (Sigma Aldrich, Steinheim, Germany) on day 0. On days 7, 8 and 9 the animals were treated with OVA or PBS intranasally (2 mg OVA/ml PBS in solution). Some of the mice also received 75 μg/day α-mIL-6R monoclonal antibody MR16-1 (Chugai Pharmaceuticals) or rat IgG control (R&D Systems, Wiesbaden) intranasally about 30 minutes before the allergen challenge on days 7 and 8. On day 10 the mice were sacrificed to isolate lung cells as described below.
Murine model of immunotherapy
The immunotherapy model was modified from a previously described protocol46. Briefly, on days 0 and 2 T-bet(−/−) mice were intraperitoneally immunized with 100 μg Ovalbumin (Calbiochem, San Diego, CA) complexed with 10% alum (Sigma Aldrich, Steinheim, Germany) or PBS. The immunotherapy group received 1000 μg OVA subcutaneously on days 19, 21 and 23. On days 33, 34 and 35 the mice received OVA intranasally (2 mg OVA/ml PBS in solution) and on day 36 the mice were sacrificed.
Collection and analysis of the BAL
BALF of the right lung was performed 24 h after the last allergen challenge with 0.8 ml saline for two times. BALF was collected and centrifuged at 1500 rpm for 5 min. The supernatants were frozen and subsequently analyzed by ELISA. The cell pellets were resuspended in 1 ml PBS and an aliquot was stained with trypan blue solution and cells were counted using a Neubauer chamber. Eosinophils were detected by fluorescence-activated cell sorting (FACS) analysis. The cell surface staining was performed with antibodies against CD3-Fitc (eBioscience, Frankfurt, Germany), GR-1-Pe (BD Bioscience, Heidelberg, Germany), CD45R-PeCy5.5 (eBioscience, Frankfurt, Germany) and CCR3-APC (BD Bioscience, Heidelberg, Germany) for 30 min at 4°C.
Histological analysis
Lung tissues were analyzed by using paraffin-embedded tissue slices for histology. After staining with Hematoxylin/Eosin peribronchial and perivascular inflammation was assessed by a pathologist blinded to the experimental group assignments of the individual lungs. Inflammation was graded by using a semi-quantitative scoring system with a range pending between 1 (mild) and 4 (severe) as described before37.
Isolation of lung CD4+ T cells
CD4+ T cells were purified as previously described47 from isolated lung cell suspension by incubation with anti-mouse CD4 L3T4 microbeads and positively sorted in a magnetic cell sorter system according to the manufactoreŕs protocol (MACS; Miltenyi Biotech, Germany). CD4+ T cells were cultured in RPMI with plate-bound α-CD3 and soluble α-CD28 for 24 h in a density of 106 cells/ml. Afterwards ELISA or RNA isolation was performed.
Flow cytometric analysis and intracellular staining
Total lung cells were stained with anti-CD4-Alexa488, anti-CD25-PerCp-Cy5.5 (BD Bioscience, Heidelberg, Germany), anti-CD19-Fitc and anti-IL-6R-Pe (eBioscience, Frankfurt, Germany) antibodies for 30 min at 4°C and washed once before measuring. For intracellular staining with IL-10 or IL-6 cells were cultured in RPMI medium at a concentration of 2 × 106 cells/ml over night with plate-bound α-CD3 (2 μg/ml) and soluble α-CD28 (2 μg/ml). The next day cells were stimulated with PMA (1 ng/ml, Sigma-Aldrich, Steinheim, Germany), Ionomycin (1 μM, Sigma-Aldrich, Steinheim, Germany) and GolgiStop (BD Bioscience, Wiesbaden, Germany). After centrifugation the cells were incubated with surface markers for 30 min at 4°C. Afterwards the cells were fixed with fixation/permeabilization solution (eBioscience, Frankfurt, Germany) for 35 min at 4°C and then washed with permeabilization buffer (eBioscience, Frankfurt, Germany). Subsequently, the cells were incubated with antibodies against IL-10 conjugated with APC (BD Bioscience, Wiesbaden, Germany) or IL-6 conjugated with Pe (eBioscience, Frankfurt, Germany) for 30 min at 4°C in permeabilization buffer and washed once. The samples were acquired by using a FACS-Calibur and analyzed with FlowJo.
ELISA
Mouse IL-2, IL-4, IL-5, IL-10 and total IgE were detected by using OptEIA™ sandwich ELISA kits from BD Bioscience (Heidelberg, Germany). Mouse IL-13 and IL-17 were detected by using a Duoset™ sandwich ELISA kit from R&D Systems (Wiesbaden, Germany).
RNA isolation and quantitative real time-PCR
Total lung tissue was homogenized and total RNA was then extracted by using PeqGold RNA Pure according to the manifacturer's protocol (PeqLab, Erlangen, Germany). Cells were directly resuspended in PeqGold RNA Pure for RNA isolation. RNA (1 μg) was then reverse transcribed using the first strand cDNA synthesis kit for RT-PCR (MBI Fermentas, Sat. Leon-Rot, Germany). The resulting template-cDNA was amplified by quantitative real-time PCR using SsoFast EvaGreen Supermix (Bio-Rad Laboratories, München, Germany). The qPCR was performed with a cycle of 2 min 98°C, 50 cycles at 5 s 95°C, 10 s 60°C, followed by 5 s 65°C and 5 s 95°C in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories). The primers and sequences used for mouse were as follows: mRORγT (5′-GTG TGC TGT CCT GGG CTA CC-3′, 5′-AGC CCT TGC ACC CCT CAC AG-3′), mBATF (5′-GTT CTG TTT CTC CAG GTC C-3′, 5′-GAA GAA TCG CAT CGC TGC-3′), mIRF4 (5′-ACGCTGCCCTCTTCAAGGCTT-3′, 5′-TGGCTCCTCTCGACCAATTCC-3′), mc-maf (5′-AGC AGT TGG TGA CCA TGT CG-3′, 5′-TGG AGA TCT CCT GCT TGA GG-3′), mIL21 (5′-CAG GAG GGG AGG AAA GAA AC-3′, 5′-GGG AAT CTT CTC GGA TCC TC-3′), mIL-21R (5′-CTC CCC CCT TGA ACG TGA CT-3′, 5′-TTG CCC CTC AGC ACG TAG TT-3′). The mRNA of the genes of interest was normalized using the mRNA levels of the housekeeping gene HPRT (5′-GCC CCA AAA TGG TTA AGG TT-3′, 5′-TTG CGC TCA TCT TAG GCT TT-3′). For human analyses the following primers and sequences were used: hHPRT (5′- TGA CAC TGG CAA AAC AAT GCA-3′, 5′- GGT CCT TTT CAC CAG CAA GCT-3′), hIL-6 (5′-CAA GAC TGA ACA CCG ACT AAG-3′, 5′- CTG GCT CTG AAA CAA AGG AT-3′) and hT-bet (5′-CAG AAT GCC GAG ATT ACT CAG-3′, 5′- GGT TGG GTA GGA GAG GAG AG-3′).
Statistical analysis
Differences were evaluated for significance by the Student's two-tailed t test for parametric data. * p ≤ 0,05, ** p ≤ 0,01, *** p ≤ 0,001. Data are given as mean values ± s.e.m.
Author Contributions
S.K. and S.F. initiated and completed the project. S.F. and S.K. designed the experiments. S.K. performed the experiments. S.F. wrote the manuscript. S.K. and S.F analyzed the data. H.A.L. and R.R. did the histology and histological analysis. S.R. and C.Ü. contributed to many experiments. S.M. did the SIT experiments and S.M and A.G. did the experiments on peripheral blood cell analysis of children. C.R. and T.Z. took care of the clinical part of the study.
Supplementary Material
supplementary data
Acknowledgments
The authors thank Prof Laurie Glimcher, Harvard Medical School (Boston, MA, USA) for providing T-bet(−/−) mice and Chugai Pharmaceuticals for providing us with α-mIL-6R monoclonal antibody MR16-1. Moreover we thank Sonja Trump, Daniel Engelbrecht, Evelin Muschiol, Ines Yawa and Lena Schramm for their technical assistance. This work was supported by an SFB 643 (Strategien der Zellulären Immunintervention; Friedrich-Alexander Universität Erlangen-Nürnberg) grant and from the European grant PreDicta (Post-infectious immune reprogramming and its association with persistence and chronicity of respiratory allergic diseases).
References
- Holgate S. T. & Polosa R. Treatment strategies for allergy and asthma. Nat Rev Immunol 8, 218–230 (2008). [DOI] [PubMed] [Google Scholar]
- Akdis C. A. Therapies for allergic inflammation: refining strategies to induce tolerance. Nat Med 18, 736–749 (2012). [DOI] [PubMed] [Google Scholar]
- Akdis M. & Akdis C. A. Therapeutic manipulation of immune tolerance in allergic disease. Nat Rev Drug Discov 8, 645–660 (2009). [DOI] [PubMed] [Google Scholar]
- Durham S. R. et al. Long-term clinical efficacy in grass pollen-induced rhinoconjunctivitis after treatment with SQ-standardized grass allergy immunotherapy tablet. J Allergy Clin Immunol 125, 131–138 e131–137 (2010). [DOI] [PubMed] [Google Scholar]
- Fujita H., Soyka M. B., Akdis M. & Akdis C. A. Mechanisms of allergen-specific immunotherapy. Clin Transl Allergy 2, 2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xepapadaki P. & Papadopoulos N. G. Childhood asthma and infection: virus-induced exacerbations as determinants and modifiers. Eur Respir J 36, 438–445 (2010). [DOI] [PubMed] [Google Scholar]
- Dienz O. & Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol 130, 27–33 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006). [DOI] [PubMed] [Google Scholar]
- He R., Oyoshi M. K., Jin H. & Geha R. S. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci U S A 104, 15817–15822 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley L. et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 181, 4089–4097 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R. H. et al. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med 180, 720–730 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V. et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat Immunol 12, 96–104 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant D. M., Gaffen S. L., Riesenfeld E. P., Irvin C. G. & Metzger D. W. Development of allergen-induced airway inflammation in the absence of T-bet regulation is dependent on IL-17. J Immunol 183, 5293–5300 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotto S. et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science 295, 336–338 (2002). [DOI] [PubMed] [Google Scholar]
- Doganci A. et al. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest 115, 313–325 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotto S. et al. Local blockade of IL-6R signaling induces lung CD4+ T cell apoptosis in a murine model of asthma via regulatory T cells. Int Immunol 19, 685–693 (2007). [DOI] [PubMed] [Google Scholar]
- Betz B. C. et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med 207, 933–942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ise W. et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol 12, 536–543 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G. J. & Dong C. BATF: bringing (in) another Th17-regulating factor. J Mol Cell Biol 1, 66–68 (2009). [DOI] [PubMed] [Google Scholar]
- Schraml B. U. et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460, 405–409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M. et al. A validated regulatory network for th17 cell specification. Cell 151, 289–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E. et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338, 975–980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P. et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munthe-Kaas M. C. et al. T cell-specific T-box transcription factor haplotype is associated with allergic asthma in children. J Allergy Clin Immunol 121, 51–56 (2008). [DOI] [PubMed] [Google Scholar]
- Broide D. H. et al. Cytokines in symptomatic asthma airways. J Allergy Clin Immunol 89, 958–967 (1992). [DOI] [PubMed] [Google Scholar]
- Marini M., Vittori E., Hollemborg J. & Mattoli S. Expression of the potent inflammatory cytokines, granulocyte-macrophage-colony-stimulating factor and interleukin-6 and interleukin-8, in bronchial epithelial cells of patients with asthma. J Allergy Clin Immunol 89, 1001–1009 (1992). [DOI] [PubMed] [Google Scholar]
- Yokoyama A. et al. Circulating interleukin-6 levels in patients with bronchial asthma. Am J Respir Crit Care Med 151, 1354–1358 (1995). [DOI] [PubMed] [Google Scholar]
- Zhou L. et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8, 967–974 (2007). [DOI] [PubMed] [Google Scholar]
- Aujla S. J. & Alcorn J. F. T(H)17 cells in asthma and inflammation. Biochim Biophys Acta 1810, 1066–1079 (2011). [DOI] [PubMed] [Google Scholar]
- Ivanov, II,. Zhou L. & Littman D. R. Transcriptional regulation of Th17 cell differentiation. Semin Immunol 19, 409–417 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustle A. et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol 8, 958–966 (2007). [DOI] [PubMed] [Google Scholar]
- Huber M. et al. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc Natl Acad Sci U S A 105, 20846–20851 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt V. et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 33, 192–202 (2010). [DOI] [PubMed] [Google Scholar]
- Huber S. et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. et al. c-Maf regulates IL-10 expression during Th17 polarization. J Immunol 182, 6226–6236 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotto S. et al. Asthmatic changes in mice lacking T-bet are mediated by IL-13. Int Immunol 17, 993–1007 (2005). [DOI] [PubMed] [Google Scholar]
- Doganci A. et al. IL-2 receptor beta-chain signaling controls immunosuppressive CD4+ T cells in the draining lymph nodes and lung during allergic airway inflammation in vivo. J Immunol 181, 1917–1926 (2008). [DOI] [PubMed] [Google Scholar]
- Ellyard J. I. & Vinuesa C. G. A BATF-ling connection between B cells and follicular helper T cells. Nat Immunol 12, 519–520 (2011). [DOI] [PubMed] [Google Scholar]
- Yoshizaki K. et al. Isolation and characterization of B cell differentiation factor (BCDF) secreted from a human B lymphoblastoid cell line. J Immunol 132, 2948–2954 (1984). [PubMed] [Google Scholar]
- Hiramatsu Y. et al. c-Maf activates the promoter and enhancer of the IL-21 gene, and TGF-beta inhibits c-Maf-induced IL-21 production in CD4+ T cells. J Leukoc Biol 87, 703–712 (2010). [DOI] [PubMed] [Google Scholar]
- Yang Y., Ochando J., Yopp A., Bromberg J. S. & Ding Y. IL-6 plays a unique role in initiating c-Maf expression during early stage of CD4 T cell activation. J Immunol 174, 2720–2729 (2005). [DOI] [PubMed] [Google Scholar]
- Neurath M. F. & Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev 22, 83–89 (2011). [DOI] [PubMed] [Google Scholar]
- Mudter J. et al. IRF4 regulates IL-17A promoter activity and controls RORgammat-dependent Th17 colitis in vivo. Inflamm Bowel Dis 17, 1343–1358 (2011). [DOI] [PubMed] [Google Scholar]
- Spolski R. & Leonard W. J. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol 26, 57–79 (2008). [DOI] [PubMed] [Google Scholar]
- Pesce J. et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest 116, 2044–2055 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taher Y. A., van Esch B. C., Hofman G. A., Henricks P. A. & van Oosterhout A. J. 1alpha, 25-dihydroxyvitamin D3 potentiates the beneficial effects of allergen immunotherapy in a mouse model of allergic asthma: role for IL-10 and TGF-beta. J Immunol 180, 5211–5221 (2008). [DOI] [PubMed] [Google Scholar]
- Sauer K. A., Scholtes P., Karwot R. & Finotto S. Isolation of CD4+ T cells from murine lungs: a method to analyze ongoing immune responses in the lung. Nat Protoc 1, 2870–2875 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary data