

Abstract
An imbalance between activation and inhibition of the complement system has been implicated in the etiologies of numerous common diseases. Allotypic variants of a key complement fluid phase regulatory protein, complement factor H (CFH), are strongly associated with age-related macular degeneration (AMD), a leading cause of worldwide visual dysfunction, although its specific role in AMD pathogenesis is still not clear. CFH was isolated from individuals carrying combinations of two of the non-synonymous coding variants most strongly associated with AMD risk, V62/H402 (risk haplotype variants), I62/Y402 (non-risk haplotype variants), and V62/Y402. These proteins were used in two functional assays (cell surface- and fluid phase-based) measuring cofactor activity of CFH in the factor I-mediated cleavage of C3b. Though no variant-specific differences in the cofactor activity were detected, when heparan sulfate (HS) was added to these assays it accelerated the rate of C3b cleavage and this effect could be modulated by degree of HS sulfation. Bruch’s membrane/choroid, a site of tissue damage in AMD, contains high concentrations of glycosaminoglycans, including HS. Addition of human Bruch’s membrane/choroid to the fluid phase assay accelerated the C3b cleavage and this effect was lost after treatment of the tissue with heparinase III. Binding of CFH variants to Bruch’s membrane/choroid isolated from elderly, non-AMD donor eyes, was similar, as was the functional activity of bound CFH. These findings refine our understanding of interactions of HS and complement and support the hypothesis that these interactions play a role in the transition between normal aging and AMD in Bruch’s membrane/choroid.
Keywords: complement, age-related macular degeneration, retinal pigment epithelium
Introduction
Complement factor H (CFH) is an essential regulatory protein that plays a critical role in the homeostasis of the complement system in plasma and in the protection of host cells and tissues from damage by complement activation (1). A major biological function of CFH is its interaction with C3b (2). CFH binds to C3b, accelerating the decay of C3bBb, the complement alternative pathway C3-convertase. It also serves as a cofactor for factor I-mediated proteolytic inactivation of C3b (3). Once CFH binds C3b the serine protease factor I inactivates the C3b by cleaving the alpha chain of the C3b molecule, resulting in the formation of iC3b a major complement opsonic C3 fragment (4). Polyanionic molecules on cell surfaces, including sialic acid, heparan sulfate (HS), and other glycosaminoglycans (GAGs), can increase the affinity of CFH for surface bound C3b (5).
Variations in the complement factor H gene (CFH) are strongly associated with the risk of developing age-related macular degeneration (AMD), the leading cause of irreversible blindness in the Western world (6). These CFH variants account for up to 50% of the population attributable risk for AMD (7-10). Among the non-synonymous coding variants of CFH, two [V62 (residue 44 of the mature protein; rs800292) and H402 (384 in the mature protein; rs1061170)] are strongly associated with the major AMD risk haplotype.
The effects these amino acid substitutions have on CFH function and thus the mechanism underlying increased risk of developing AMD are poorly understood. It is reasonable to speculate that these amino acid substitutions alter the binding properties of CFH protein variants. This contention is supported by studies of recombinant fragments of CFH spanning these substitutions that show allotypic differences in binding of CFH to known associated molecules. The H402 variant is located in the short consensus repeat 7 (SCR7) region of CFH, a region that has been implicated in binding to polyanions, C-reactive protein (CRP) and the M-Protein of Streptococcus pyogenes (11-14). The I62V variant is located in SCR1, a domain within the N-terminal complement regulatory site that is responsible for cofactor activity of factor H in the factor I-mediated cleavage of C3b (1, 14-18).
AMD is characterized histopathologically by the progressive accumulation of diffuse basal deposits and focal deposits comprised of protein and lipid, known as drusen, located between the Bruch’s membrane/choroid complex and the basal surface of the retinal pigment epithelium (RPE). Bruch’s membrane is comprised of distinct sub lamina containing collagen and elastin-based fibrous connective tissue elements and a variety of proteoglycans, including HS-rich proteoglycans (19, 20). One allotypic variant of CFH associated with AMD risk, H402, differentially binds the glycosaminoglycan (GAG), heparin, in studies comparing recombinant fragments of CFH spanning SCR7 with H or Y at position 402 (21, 22). Differential binding of the H402 and Y402 variants of CFH to other GAGs and C-reactive protein has been observed (21-24).
These observations, combined with those showing the presence of abundant CFH and HS proteoglycans in the Bruch’s membrane/choroid complex led us to test the hypothesis that changes in the affinity of CFH for different GAGs may contribute to AMD pathobiology. We isolated full-length CFH from the plasma of individuals homozygous for combinations of two of the non-synonymous coding variants most strongly associated with the major AMD risk haplotype, V62/H402 (both risk variants), the major protective haplotype I62/Y402 (both non-risk variants), and V62/Y402. We employed these native forms of CFH in functional assays to analyze interactions with various purified or tissue-associated GAGs.
These studies were directed toward elucidating differences between the allotypic variants that might contribute to AMD development and progression. In addition, they addressed our understanding of CFH function in the normal tissue, which is necessary to appreciate functional deficits associated with disease.
Herein, we show that purified, full-length CFH isolated from plasma from individuals homozygous for the variant combinations I62/Y402, V62/Y402 and V62/H402, exhibit similar cofactor activity in the proteolytic cleavage of C3b by factor I in the assays employed. We further demonstrate that HS accelerates the rate of conversion of C3b to iC3b by factors H and I, that this conversion is sulfation dependent, and that the addition of isolated human Bruch’s membrane/choroid to components of the fluid phase assay can accelerate this conversion in a fashion similar to that of adding purified HS to the assay. In addition, both the V62/Y402 and V62/H402 variants of CFH bind to Bruch’s membrane/choroid tissue to the same extent and, once bound, they have nearly identical functional activities.
We hypothesize that one function of HS in normal Bruch’s membrane/choroid is to regulate the extent of complement activation by modulating the rate of conversion of C3b to iC3b by factors H and I. The acceleration of C3b decay could theoretically a) protect proximal RPE cells from the consequences of activated complement components and/or b) prevent complement-mediated clearance of debris, resulting in the build-up of sub-RPE deposits. Our data predicts that changes in Bruch’s membrane/choroid HS composition that occur with aging and disease will impact complement activation at this interface.
Materials and Methods
Plasma and tissue samples
Plasma was collected from research subjects at the Duke University Eye Center and The University of Iowa and purchased from the American Red Cross. Informed consent was obtained from all study participants, and all information was collected and protected according to standardized guidelines and in compliance with HIPAA regulations. Blood samples were collected into EDTA tubes, centrifuged within one hour and the plasma stored at -80°C. Human donor eyes with no history of ocular disease used in these studies were obtained from the North Carolina Eye Bank (Winston-Salem, NC) within up to 14 h of death (with an average procurement time of 7.5 h, Table I).
Table I.
Human donor eyes used for Bruch’s membrane analysis
| Donor | Age (y) | Gender | Death to Procurement (h:min) | Cause of Death |
|---|---|---|---|---|
| 1696-07-01 | 85 | F | 6:17 | Cancer - Lung |
| 1697-07-01 | 72 | M | 13:15 | Myocardial Infarction |
| 1698-07-01 | 51 | F | 7:50 | Sepsis |
| 0527-08-01 | 70 | F | 9:53 | Cardiac Arrest |
| 0924-08-03 | 68 | F | 9:25 | Renal Failure |
| 1082-08-01 | 35 | F | 5:57 | Seizures |
| 1086-08-01 | 80 | F | 5:05 | Pneumothorax |
| 1309-08-01 | 92 | M | 3:30 | Dementia |
| 1687-08-01 | 70 | M | 4:45 | Myocardial Infarction |
| 1546-09-01 | 83 | M | 8:50 | Cancer - Lung |
| 1744-09-01 | 83 | M | 4:30 | Ischemic Cardiomyopathy |
| 1880-09-01 | 79 | M | 23:30 | Cancer - Skin |
| 1881-09-01 | 84 | F | 4:25 | Ischemic Cardiomyopathy |
Buffers and reagents
Buffers employed include veronal-buffered saline (VBS); VBS with 0.15 mM calcium chloride, 1.0 mM magnesium chloride, and 0.1% gelatin (GVBS++); VBS with 0.1% gelatin (GVBS) plus 10 mM EDTA (EDTA-GVBS), 10% dextrose (w/v), with 2 mM magnesium chloride and 0.3 mM calcium chloride, and 0.2% gelatin (D5WG++), and 60% D5WG++ with 40% GVBS++ to give 60% DGVBS++. Human purified complement components C1, C2, C3, C3b, C4 and factors H and I were all obtained from CompTech, Inc (Tyler, TX). Sheep erythrocytes in Alsever’s solution were obtained from Lampire Biological Laboratories (Pipersville, PA). Mouse anti-human CFH MRC OX-24 and MRC OX-23 from Serotec Ltd. (Oxford, UK); polyclonal goat anti-human CFH (Quidel Corporation, San Diego, CA); CRP from Chemicon International (Temecula, CA); and HS from porcine intestine (HS-PI) from Celsus laboratories (Cincinnati, OH), trypsin (bovine pancreas), heparin (H-3393), HS from bovine kidney (HS-BK) and chondroitin sulfate (CS) A, B, and C (CS-A, CS-B, CS-C) were from Sigma (St. Louis, MO). Over-sulfated heparan sulfate, de-N-sulfated heparin, and fully de-O-sulfated heparin were from Neoparin, Inc. (San Leandro, CA). Rabbit anti-sheep hemolysin was prepared as previously described (25). 125I-0.1 mCi/μl (GE Healthcare, Piscataway, NJ), BioSpin 6 columns from Bio-Rad Laboratories, Inc. (Hercules, CA).
Purification of CFH from plasma
CFH was isolated from 5 ml of plasma collected from individuals homozygous for the double variants I62/Y402, V62/Y402, and V62/H402 or from human plasma obtained from the American Red Cross in which these three homozygous double variants of CFH were identified using MALDI-mass spectrometry (26). The plasma was diluted to 20 ml with 10 mM phosphate buffer pH 7.4 and loaded onto a 1 ml HiTrap Heparin HP column (GE Healthcare) to capture the heparin binding proteins. Bound proteins were eluted in a NaCl gradient, concentrated and applied to a Superose 16/10 size exclusion FPLC column (GE Healthcare) and 50, 500 μl fractions were collected. Following analysis of the fractions by Western and on Coomassie stained gels, the fraction determined to have the highest concentration of CFH without any trace of the truncated CFH isoform, FHL1, was used in subsequent experiments.
CFH concentration determination
Plasma levels of CFH were determined by ELISA and by densitometry. Anti-CFH IgG fraction was prepared from a goat anti-CFH serum (Quidel Corporation) by ammonium sulfate precipitation. 96-well Nunc Maxisorp plates (eBioscience) were coated with 5 μg anti-CFH IgG fraction in 200 μl coating buffer (0.1 M sodium bicarbonate, PH 9.5) overnight at 4°C and blocked with 1% BSA/PBS for 1 h. Wells were washed 3 times with 0.05% Tween-20/PBS and incubated with diluted CFH samples for 1 h. After washes, monoclonal mouse anti-CFH antibody (OX 24, 1:500) (Serotec) was added for 1 h followed by a HRP-conjugated donkey anti-mouse secondary antibody (1:10000) (Jackson ImmunoResearch Laboratories) for 1 h. CFH was detected by using o-Phenylenediamine (OPD) substrate (Sigma). OD was read at 450 nm with a plate reader (Molecular Devices, Sunnyvale, CA). The ELISA detected CFH in a linear range at concentrations of 10 to 90 ng/ml.
An antibody-independent method for measuring purified CFH concentration was developed to ensure that the anti-CFH antibodies used in these quantitation assays did not preferentially recognize one variant over another. Non-reduced protein samples and five concentrations of standards ranging from 100-500 ng CFH (CompTech)/well were run on a 12% Bis-Tris Criterion XT Precast Gel (Bio-Rad). The gel was stained with Bio-Safe Coomassie Stain (Bio-Rad), imaged on an ImageScanner (GE Healthcare) and the band densities determined using ImageJ (NIH). The protein concentrations were determined from the standard curve.
Preparation of radiolabeled C3
50 μl of C3 (5 mg/ml) was labeled with 200 μCi of 125I using IODO-BEADS (Pierce), following the manufacturer’s protocol. Briefly, 50 μl PBS was added to two washed IODO-BEADS, followed by 2 μl of 125I, incubated for 5 min at room temperature, 50 μl of protein was added for 30 min at room temperature and a BioSpin 6 column used to remove any unbound radioactive iodine. The specific activity of the proteins ranged from 0.2 to 0.35μCi/μg.
Measurement of cofactor activity of CFH using a cell surface-based assay
C3b-coated sheep erythrocytes (EC3b) were prepared using the method of Gaither et al. (27). Sheep erythrocytes (E) were sensitized by incubation with rabbit anti-sheep hemolysin (EA) followed by the sequential addition of human complement components C1, C4, C2 and radiolabeled C3. C1 (5.6 μl of 2.13×106 units/ml) was incubated with 10 ml of EA (1.5 × 108 cells/ml in 60% DGVBS++) at 30°C for 15 min, washed, followed by C4 (10 μl of 2.86×107 units/ml), incubated at 37°C for 45 min, generating EAC14. C2 (3.1 μl of 7.5×106 units/ml) and 10 μl of 2.5 mg/ml 125I-C3 was added to 1.0 ml of the EAC14 cells (1.5×108 cells/ml) and incubated at 30°C for 45 min, and 37°C for 60 min, producing 125I-EAC43b referred to as 125I-EC3b(27). Purified human CFH, in the presence of limited factor I, was added to 50 μl 125I-EC3b cells and incubated at 37°C for 4 min, resulting in factor I-mediated cleavage of C3b to cell-bound iC3b. The amount of 125I-iC3b formed from 125I-C3b was determined quantitatively as 125I-C3c released by the addition of 0.25% trypsin. iC3b is trypsin sensitive undergoing cleavage to release C3c, whereas C3b resists trypsin cleavage. Radiolabeled C3d fragments (31 kDa) and uncleaved radiolabeled C3b remained cell-bound (28). The cells were centrifuged and the supernatant and cell pellets counted in a Packard Cobra II auto-gamma counter (Downers Grove, Il). The C3c cleaved and released into the supernatant was expressed as the percentage of counts in the supernatant compared to the total number of counts:
Measurement of cofactor activity of CFH by fluid-phase assay
350 ng purified, allotypic variant specific CFH and 35 ng factor I (CompTech) were diluted to 21.4 μl in EDTA-VBS and placed on ice. 585 ng C3b (CompTech Lot 22) was added in a volume of 3.6 μl cold EDTA-VBS and after mixing, the tubes were incubated for specific times at 37°C, recovered on ice and 18 μl removed to a tube containing 5μl 4X, XT sample buffer with reducing agent (BioRad). These samples were placed in a dry bath incubator at 105°C for 7 min and resolved on a 10% Bis-Tris Criterion XT gel with MOPS buffer. The gels were fixed immediately in 10% methanol/5% acetic acid for at least 30 min before silver staining. The gels were scanned using an ImageScanner and the band densities determined using MacBiophotonics ImageJ.
Addition of GAGs and C-reactive protein (CRP) to the CFH assays
The cell surface-based assay described above was modified by preincubating 250 ng purified CFH (either V62/H402 or I62/Y402) and 40 ng factor I with 95 μg heparin, HS-PI or HS-BK, CS-A, CS-B, or CS-C and 47.5 μg CRP in a total volume of 42 μl EDTA-GVBS buffered solution for 30 min at room temperature followed by 50 μl of cells (EAC43b*) at a concentration 1.5×108 cells/ml. Reactions were incubated at 37°C for 5 min before stopping them with the addition of ice cold buffer. The cells were centrifuged, the supernatant removed, and trypsin was added. The reaction was completed as described above.
Fluid-phase cofactor activity assays were performed by preincubating 350 ng CFH, factor I (35 ng) and 50 μg heparin HS-PI, or HS-BK for 30 min at room temperature before ice cold C3b (585 ng) was added and immediately incubated at 37°C for increasing times. Tubes were placed on ice and 18 μl of the 25 μl total volume was added to 5 μl reducing sample buffer. The tubes were boiled and samples loaded to a 10% Bis-Tris Criterion XT gel and processed as described above.
Additional fluid-phase cofactor assays were performed in the presence of chemically-modified GAGs. Over-sulfated heparan sulfate or fully de-O-sulfated heparin or de-N-sulfated heparin (146 μg each) were added to CFH (390 ng), factor I (200 ng), and C3b (3.4 μg) in a total volume of 70 μl EDTA-VBS at 37°C. Ten μl aliquots were removed at 0, 2, 4, 8 and 20 min, added to 5 μl of reducing buffer, boiled at 105°C for 7 min, and separated on a 10% Bis-Tris XT Criterion gel that was silver stained.
Preparation of Bruch’s membrane/choroid punches
Human eyes were treated as described previously by Hewitt et al. (29). Briefly, a cut was made around the ora serrata to remove the anterior chamber. The vitreous and retina were removed from the posterior eye cup exposing the RPE. The RPE was gently rubbed off and removed in PBS using a smooth, rounded glass rod. 6 mm diameter punches were taken through Bruch’s membrane, choroid and sclera using a trephine punch. The Bruch’s/choroid complex was then gently peeled away from the sclera. When punches from a pair of eyes were used (e.g. when the reaction rate was measured for C3b proteolytic cleavage with three types of CFH and with or without heparinase III treatment), the punches were selected from equivalent regions of the eye (e.g. mid-periphery). These punches were incubated in cold, distilled water containing protease inhibitors (0.1 M 6-aminohexanoic acid and 5 mM benzamidine hydrochloride) overnight and the next day tissue punches were washed by gently shaking in PBS and touching to filter paper to remove the loose fluffy choroid. The resulting semi-transparent membranes were fully suspended in the buffers during the process, thus exposing a large surface area. Four 6 mm punches were treated with heparinase III in 100 μl PBS for 2.5 h at 37°C. Heparinase III acts solely on HS by cleaving sulfated polysaccharide chains containing 1-4 linkages between hexosamines and glucuronic acid residues. Heparinase III from both Sigma (0.002 I.U. per four 6 mm punches) and IBEX (0.008 I.U. per four 6 mm punches) were used to treat these membranes. After enzyme treatment the membranes were washed thoroughly in cold EDTA-VBS buffer and added, on ice, to the reaction tube containing 1 μg CFH, and 260 ng factor I made up to 180 μl with EDTA-VBS. C3b (4.4 μg) was added and the tubes incubated at 37°C. 18 μl samples are removed at various time points and added to 6 μl reducing 4X sample buffer, the tubes are boiled and the samples resolved on a 10% Bis-Tris Criterion XT gel with MOPS buffer.
Adding CFH to Bruch’s membrane/choroid tissue
Six mm diameter punches of human Bruch’s membrane/choroid were isolated from the macula and peripheral retina as described in previous paragraph. As many eight punches were taken from the far peripheral retina just inside the ora serrata and up to six from around the optic nerve of a single eye, including one over the macula. One washed punch was added to 20 μl 10 mM phosphate (PO4) buffer with or without 700 ng of either the V62/H402 or V62/Y402 variant of CFH. This was slowly rocked at room temperature for 2.5 h, the punch was carefully removed, touched to filter paper, and added to 1 ml of 10 mM PO4 buffer. The punch slowly floated through the buffer and was gently agitated for 5 min, then the punch was removed from the buffer, touched to a piece of filter paper and added to 35 μl of XT sample buffer. The sample was vortexed vigorously for 30 seconds, allowed to sit at room temp for 10 min, vortexed again, spun down and the supernatant removed. 18 μl of the supernatant was added to 6 μl reducing 4X sample buffer, the tubes are boiled and the samples resolved on a 10% Bis-Tris Criterion XT gel with MOPS buffer. The gel was transferred to nitrocellulose and probed with mouse monoclonal anti-human CFH (OX 24, Serotec).
Functional assay using Bruch’s membrane/choroid tissue pre-incubated with CFH
Six mm diameter punches of Bruch’s membrane/choroid tissue were pre-incubated with 700 ng of either the V62/H402 or V62/Y402 variant of CFH and washed as described in the previous section. A single 6 mm tissue punch with CFH was added to 79 μl VBS/EDTA with 2.61 μl 0.1 mg/ml factor I. 2.2 μl of 1 mg/ml C3b and the tubes incubated at 37°C. 9 μl of the reaction mixture was removed at 0, 2, 4, 8, 16 and 24 min, and added to 12 μl reducing sample buffer on ice. The tubes were heated to 105°C for 7 min, and the sample added to 10% Bis-tris Criterion XT gels. The resultant gel was silver-stained and the density of the bands measured using ImageJ software.
Electron microscopic characterization of changes in Bruch’s membrane/choroid following heparinase III treatment
Electron microscopy was used to visualize the Bruch’s membrane/choroid punches following treatment with heparinase III. One 6 mm punch was isolated as described above and fixed in 4% paraformaldehyde for 30 min, followed by 1% paraformaldehyde for 1 h, then washed and stored at 4°C in PBS. The punch was cut into sections and either left in PBS or treated with four times the concentration of heparinase III used in the assay due to fixation of this tissue. The tissue was stained with Cupromeronic Blue, which selectively stains sulfated proteoglycans at a critical electrolyte concentration (30). Briefly, tissue was rinsed several times in a rinsing solution [25 mM sodium acetate (Fisher Scientific), 0.2 M magnesium chloride (MgCl, EM Science), pH5.7], before incubating in staining solution [0.2M MgCl, 2.5% glutaraldehyde (Electron Microscopy Sciences), 0.05% Cupromeronic Blue (United States Biological), 25 mM sodium acetate, pH5.7] overnight in the dark at room temperature. The tissue was rinsed again, incubated in 0.5% ammonium tungstate (Sigma) for 1 h, dehydrated in a graded ethanol series followed by propylene oxide, infiltrated and embedded with a mixture of Epon and Spurr resins, and polymerized at 60°C for 36-48 h. Thin sections of ~60 nm were cut, mounted on copper grids, counterstained with uranyl acetate, and examined on a Tecnai G2 TWIN transmission electron microscope (FEI).
Data and Statistical analyses
Statistical analysis of the cell-based assay was restricted to comparison of CFH allotypes within the same experiment, largely because of differences between each batch of the EC3b cell preparations. All three allotypes were always isolated at the same time, which resulted in little inter-experiment variation. The purified human CFH was never frozen; therefore statistical analysis of the activity of the CFH allotypes in the cell-based assay was based on data from within the same assay. A t-test comparing a difference with the standard error of the difference was performed using an equation for nonlinear regression and comparing two curves (GraphPad Software,Inc.; curvefit.com). In this case the standard error is reported by nonlinear regression. The following equation was used to determine t: t = (B1-B2)/√(SE12 + SE22). Where B1 and B2 are the EC50 calculated by graphing (using SigmaPlot 9) the % cpm in the supernatant (a measure of C3c release) against concentration of CFH (Fig. 1A) or time of incubation at 37°C (data not shown) and fitted to the equation: f = a*x/(b+x). The two-tailed P value was calculated from t using the tdist (t, degrees of freedom, 2).
FIGURE 1.
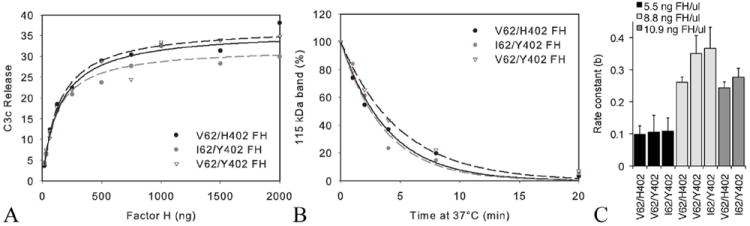
(A) Cofactor activities of allotypic variants of purified CFH (FH). In this graph of a representative assay, C3c release was measured (% cpm in supernatant) after 4 min at 37°C in cell-based assays of CFH cofactor activity with increasing concentrations of CFH (I62/Y402, V62/Y402 or V62/H402), factor I and 125I-EC3b cells. (B) Cleavage of C3b was measured in the fluid-phase assay using the I62/Y402, V62/Y402 or V62/H402 variants of purified CFH. Decay of the 115 kDa band (C3bα′) was determined by densitometry using ImageJ, expressed as a % of the 115 kDa band at time zero and plotted using SigmaPlot. (C) The rate constant for the exponential decay was measured using the equation y=a*exp(-b*x). There was no statistically significant difference in the rate constants between variants of CFH used. This held true over a range of CFH concentrations tested (5.5, 8.8 and 10.9 ng FH/μl) (p>0.07 for all comparisons of the different variants). [5.5 ng FH/μl], n=7; [8.8 ng FH/μl], n=3; and [10.9 ng FH/μl], n=5.
In the fluid phase assay, the percent of the 115 kDa band of C3b was expressed by the equation: f = a*exp(-b*x). The differences in the rate constant ‘b’ due to changes in the experimental conditions (CFH variant used, and/or addition of GAGs or other biological molecules added to the reaction) were analyzed using a Wilcoxon two sample test (SigmaPlot Version 9, SYSTAT software). This test compared the rate constants calculated for each set of data produced under identical assay conditions. Differences with p < 0.05 were considered statistically significant. A similar analysis was performed when comparing C3c release (Fig. 2) and band densities (Fig. 6).
FIGURE 2.
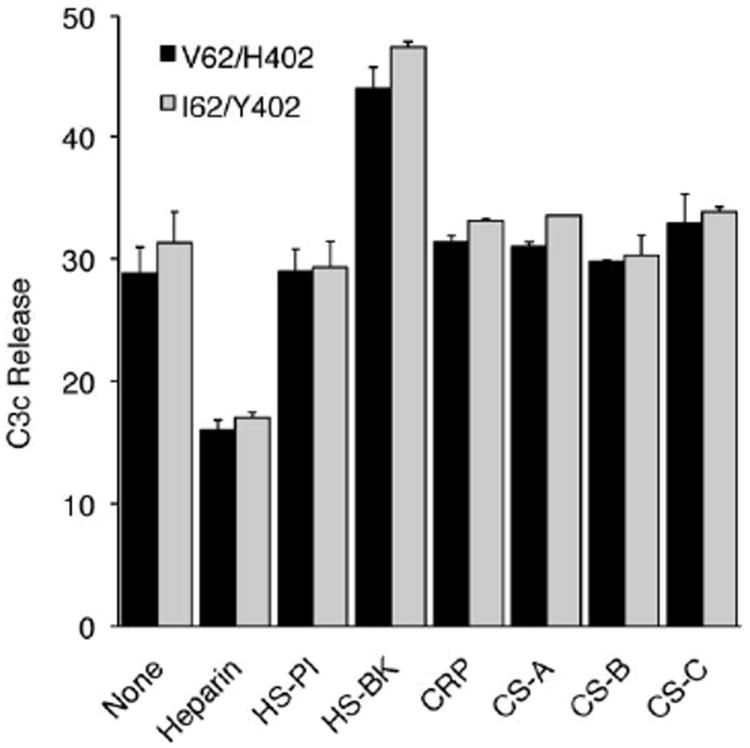
The effect of various molecules on cofactor activity of allotypic variants of purified CFH in the cell surface-based assay. CFH, factor I and heparin, heparan sulfate (HS) isolated from porcine intestinal mucosa (HS-PI), HS isolated from bovine kidney (HS-BK), chondroitin sulfate (CS)-A, CS-B, CS-C or CRP were preincubated at room temperature before 125I- EC3b cells were added and incubated for at 37°C (mean ± SD). Differences in between the allotypic variants are not statistically significant, but differences between the effect of heparin and HS-BK on the modulation of the co-factor activity are. Heparin decreased C3c release (***, p< 0.001) and HS-BK increased C3c release (p< 0.001). Data depicted are representative of two independent experiments, each done in triplicate.
FIGURE 6.
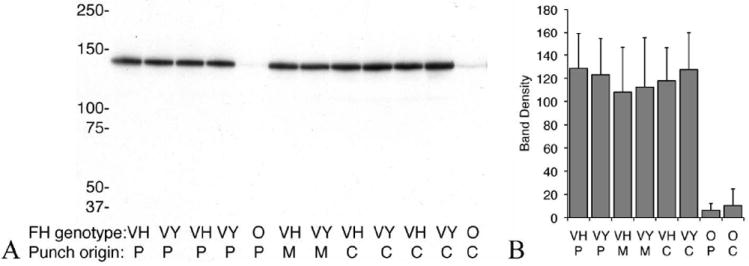
CFH binding to Bruch’s membrane/choroid. Immunoblot of CFH variant binding to a single 6 mm Bruch’s membrane/choroid punches isolated from different regions of the human eye. Peripheral (P), central (C) or macula (M) punches were incubated with buffer alone (O) or in buffer with V62/Y402 (VY) or V62/H402 (VH) variants of CFH. Following incubation the punches were washed thoroughly and added to 35 μl of XT sample buffer. The sample was vortexed vigorously for 30 seconds, left at room temperature for 10 min, vortexed again, spun down and supernatant was run on a gel as previously described. The gel was transferred to nitrocellulose and probed with mouse monoclonal anti-human CFH (Serotec, OX24). Immunoreactive proteins were visualized with ECL and quantified by densitometry, n=18 for both VH and VY on peripheral punches, n=13 for both on central punches and n=3 for macular punches. (B) No statistical difference in the amount of each added variant of CFH bound to the Bruch’s membrane/choroid tissue was detected (p> 0.07), no matter where the punches were taken from. Very little endogenous CFH is detected in the punches with no added CFH.
Results
Cofactor activities of three allotypic variants of CFH are the same when measured using cell surface- or fluid-phase-based functional assays of the factor I-mediated proteolytic cleavage of C3b
Full-length CFH was purified from plasma samples of individuals homozygous for three combinations of risk or normal alleles of both non-synonymous coding variants associated with AMD (V62/H402-both risk variants, I62/Y402- both non-risk variants, and V62/Y402). These CFH preparations were used in a cell surface-based functional assay performed many times using nine different sets of preparations of CFH (two I62/Y402 donors; two V62/Y402 donors and five V62/H402 donors). The assay conditions were optimized to detect variations in the rate of the reaction in response to small changes in CFH concentration. There were significant batch-to-batch variation in the EC3b cells used in this assay, therefore the reaction rates were compared intra-assay but not inter-assay (Fig. 1A). The rates of C3b cleavage were similar for each CFH variant tested (Fig. 1A, p> 0.5). Based on pair-wise comparisons of the rate curves obtained with each allotypic variant there was no statistically significant difference between any of the variants: I62/Y402 vs. V62/Y402, p= 0.43; V62/Y402 vs. V62/H402, p= 0.84; and I62/Y402 vs. V62/H402, p= 0.44.
The cofactor activity of these CFH preparations was also measured using a fluid-phase assay and again there was no statistically significant difference among the variants of CFH tested (Fig. 1B,C). In Figure 1B, a representative graph of the decrease of the 115 kDa band of C3 against time of incubation at 37°C for the 3 allotypic variants, is shown.
Effect of heparin, HS, chondroitin sulfate (CS)-A, -B and -C and CRP on the functional cofactor activity of CFH
CFH binds a number of different molecules that have been shown to modulate its activity (31). The SCR7 domain containing the H402 variant interacts with heparin, CRP and the M-protein of Streptococcus pyogenes (1, 12, 13). SCR7 and 20 function in heparin and polyanion recognition (2, 23, 32-37). The effect of various polyanions on the cofactor activities of allotypic variants of CFH was analyzed in the cell surface-based assay. Different polyanions from various sources were preincubated with each of the forms of CFH and factor I prior to analysis of cofactor activity as follows: purified CFH (V62/H402 or V62/Y402), and factor I were preincubated with either heparin, HS [porcine intestinal mucosa (HS-PI) and bovine kidney (HS-BK)], CS-A, CS-B, CS-C or CRP. As summarized in Fig. 2, two molecules from this series had a significant effect on the proteolytic breakdown of C3b: (1) heparin significantly decreased the rate of C3c release (p< 0.001), and (2) HS-BK significantly increased the rate of C3c release (p< 0.001). HS-PI, CS-A, CS-B, CS-C and CRP had no effect, nor were any CFH allotypic variant-associated differences detected.
The effects of heparin and HS-BK on the CFH cofactor activity were further analyzed using the fluid-phase assay. The rate of decay of C3bα ′ and the increase in the 68 kDa band showed the same trends detected by the cell surface-based assay in which heparin slowed the reaction. HS-BK accelerated it and there were no differences associated with the allotypic variants of CFH used (Fig. 3A).
FIGURE 3.
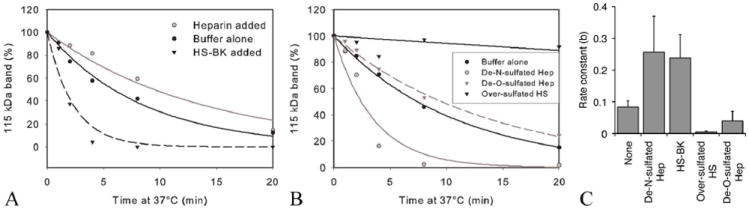
The rate of C3b cleavage is reduced by highly sulfated GAGs (heparin and over-sulfated HS) and accelerated by less N-sulfated GAGs (HS-BK and de-N-sulfated heparin). (A) Cleavage of C3b was measured in the fluid-phase assay using purified CFH with heparin (Hep) or HS-BK added. CFH (390 ng) and Factor I (200 ng) with or without 146 μg of a specific GAG were mixed and added to C3b (3.4 μg) in a total volume of 70 μl EDTA-VBS. At various time points, 10μl aliquots were removed and proteins separated on a gel as previously described. Decay of the 115 kDa band (C3bα′) was determined by densitometry using ImageJ, expressed as a % of the 115 kDa band at time zero and plotted using SigmaPlot. (B) Cleavage of C3b measured in the fluid-phase assay using purified CFH with sulfate-modified heparin (de-N- or de-O-sulfated) or over-sulfated HS added. (C) The rate of the fluid phase reaction is greatly reduced by over-sulfated HS (***, p< 0.001) and to a lesser extent heparin and de-O-sulfated heparin (**, p< 0.01) whereas HS-BK and de-N-sulfated heparin significantly increase the rate of this reaction (p< 0.001). Similar results were obtained using V62/Y402 or V62/H402 (data not shown). None (no added GAG), n=18; de-N-sulfated heparin, n=18; HS-BK, n=6; over-sulfated HS, n=9; de-O-sulfated heparin, n=6
The sulfation pattern of GAGs is the key to their capacity to modulate the rate of proteolytic cleavage of C3b by factors H and I
We hypothesized that the differences in the modulatory effect of heparin, HS-PI and HS-BK derive from this relative sulfation. Each of the purified GAGs tested on our assays was a heterogeneous mixture of molecules with varied molecular weights and sulfation patterns. Previous studies of HS isolated from bovine kidney (HS-BK) showed that it is less sulfated than the HS isolated from porcine intestinal mucosa (HS-PI) or heparin, which is the most highly sulfated of these GAGs (38, 39).
The Azadi laboratory, at the Complex Carbohydrate Research Center of the University of Georgia, Athens, GA, carried out disaccharide analysis of the HS-BK, HS-PI, and heparin used in this study. The percentage of disaccharides with N-sulfation and O-sulfation in the heparin and the two HS samples are shown in Table II (40). The overall extent of O-sulfation is very similar in the HS samples whereas the amount of N-sulfation in the HS-BK is markedly less than the HS-PI potentially accounting for why HS-BK accelerates the C3b cleavage reaction compared to HS-PI. Heparin was confirmed to be the most highly sulfated of all these GAGs.
Table II.
Disaccharide analysis of Heparin and HSs used in C3b cleavage assays
| Disaccharidea
|
|||
|---|---|---|---|
| Nomenclatureb (sulfation) | Heparin | HS-PI | HS-BK |
| D0A0 | 5.55 | 43.25 | 60.22 |
| D0S0 (N) | 3.38 | 22.57 | 8.05 |
| D0A6 (O) | 6.30 | 14.27 | 20.39 |
| D2A0 (O) | 2.61 | 0.61 | 0.71 |
| D0S6 (N + O) | 13.44 | 10.60 | 5.00 |
| D2S0 (N + O) | 4.97 | 3.42 | 2.22 |
| D2A6 (O) | 3.14 | 0.29 | 0.56 |
| D2S6 (N + O) | 60.60 | 5.00 | 2.86 |
| total N-sulfated (N) disaccharides | 82.39 | 41.59 | 18.13 |
| total O-sulfated (O) disaccharides | 91.06 | 34.19 | 31.74 |
GAG samples were digested with a heparinase mixture. Disaccharides were analyzed by HPLC. Values are represented as percent nanogram of total HS
Disaccharide nomenclature is according to Lawrence et al. REF. D0A0, unsulfated Δ4,5-uronic acid-N-acetylglucosamine; D0S0, monosulfated Δ4,5-uronic acid-N-sulfated glucosamine; D0A6, monosulfated Δ4,5-uronic acid-N-acetylglucosamine-6-O-sulfate; D2A0, 2-O-sulfated Δ4,5-uronic acid-N-acetylglucosamine; D0S6, disulfated Δ4,5-uronic acid-N-sulfoglucosamine-6-O-sulfate; D2S0, disulfated 2-O-sulfated Δ4,5-uronic acid-N-sulfoglucosamine; D2A6, 2-O-sulfated Δ4,5-uronic acid- N-acetylglucosamine-6-O-sulfate; D2S6, trisulfated 2-O-sulfated Δ4,5-uronic acid-N-sulfoglucosamine-6-O-sulfate.
To confirm that the extent of sulfation of the GAGs present in the fluid phase reaction is responsible for modulating the rate of C3b cleavage by factors H and I, de-N-sulfated heparin, de-O-sulfated heparin or over-O-sulfated HS were used in the assay (Figure 3B,C). Addition of the least sulfated modified GAG, de-N-sulfated heparin, mimicked the effect of HS-BK, increasing the rate of cleavage of C3b (p< 0.001). The addition of a highly sulfated GAG (over-O-sulfated HS), similar to heparin, slowed the fluid phase reaction to an even greater extent than heparin (p< 0.001). Together, these results support the hypothesis that the degree of sulfation of these different glycosaminoglycans accounts – at least in part – for the differences in their ability to modulate complement activation through C3b cleavage. When de-O-sulfated heparin was used in the reaction there was a decrease in the rate of C3b cleavage compared to the rate in the absence of GAGs (p< 0.05). This effect on the rate constant was less pronounced than the decrease observed in the presence of over-sulfated HS. These data indicate that it is not only the extent, but the position of sulfation that controls modulation of the reaction by GAGs.
The rate of C3b cleavage is accelerated in the presence of human Bruch’s membrane/choroid punches
An increase in the rate of proteolytic cleavage of C3b was measured when Bruch’s membrane/choroid punches were added to the fluid phase assay. This was repeated using two allotypic variants of purified CFH ((V62/H402 and I62/Y402), which both showed a similar rate increase in the presence of the tissue (Fig. 4A). The rate constant for the reactions of the CFH and I mediated proteolytic cleavage of the 115 kDa band of C3b increased significantly (p< 0.05) in the presence of Bruch’s membrane/choroid punches (four 6 mm punches per reaction), but in an allotype independent manner (Fig. 4B). In order to establish that this effect was not due to the presence of endogenous CFH within the tissue punches, four pooled punches were analyzed for the presence of CFH revealing a maximal increase in the total amount of CFH of only 5%, which was not sufficient to increase the rate of the reaction to the extent we had measured (data not shown). To assess whether this increase in the rate of the reaction could be attributed to the HS in the tissue, heparinase III was used to pre-treat the Bruch’s membrane/choroid punches. Heparinase III pretreatment of the punches abolished the increased reaction rate over the normal rate of the reaction (Fig.5A). Collectively, these results suggest that the increase in the rate of C3b cleavage upon addition of human Bruch’s membrane/choroid tissue is due to HS in the tissue and not to other extracellular matrix components present in the tissue or endogenous CFH.
FIGURE 4.
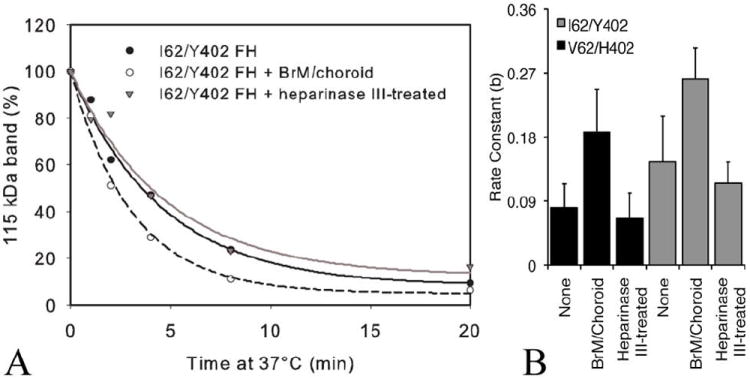
Addition of human Bruch’s membrane/choroid punches increases the rate of cleavage of C3b by factor I and variants of CFH. (A) Cleavage of C3b was measured in the presence of four 6 mm diameter punches of Bruch’s membrane/choroid from the peripheral (two) and central (two) regions of posterior eyecups from a pair of human eyes (68-year-old donor) per reaction. Punches (four treated or four untreated with heparinase III) were added to CFH and factor I in ice cold EDTA-VBS. C3b was added, and the tubes placed at 37°C. The control tube had no punches added. At various times 18 μl was removed and run on a gel as previously described. Representative results from one isoform, I62/Y402, are depicted. (B) I62/Y402 and V62/H402 CFH both showed statistically significant increases in the rate constant in the presence of tissue (p< 0.01; n=5 each). There were no statistically significant differences between the variants of CFH alone compared to reactions with tissue pre-treated with heparanase III V62/H402). Data depicted are representative of 5 independent experiments from 5 pairs of donor eyes.
FIGURE 5.
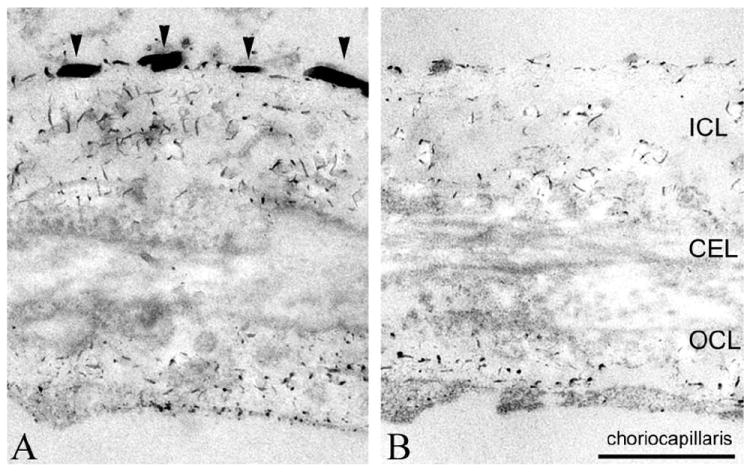
Electron micrograph of human Bruch’s membrane/choroid stained for proteoglycans using Cupromeronic Blue. (A) HS [arrowheads in inner collagenous layer (ICL)], chondroitin sulfate and dermatan sulfate are stained, and can be distinguished based on their length/diameter and locations (Call and Hollyfield, 1990). (B) The HS associated with the basal lamina of pigment epithelium and choriocapillaris has disappeared following heparinase III treatment. CEL, central elastin layer; OCL, outer collagen layer. Scale bar = 500 nm.
Electron microscopy revealed a marked loss of HS in Bruch’s membrane/choroid upon treatment with heparinase III. Significantly more HS was detected in human Bruch’s membrane/choroid stained with Cupromeronic Blue compared to Bruch’s membrane/choroid following heparinase III treatment (Fig. 5B,C). Call and Hollyfield (19) described similar filaments containing HS that could be eliminated with nitrous acid.
V62/Y402 and V62/H402 CFH bind equally to Bruch’s membrane/choroid tissue
Although no statistically significant changes in the cofactor activity of the allotypic variants of CFH were detected by the above assays, we hypothesized that even if the activity is the same, a difference in the local concentration (Bruch’s membrane/choroid-associated CFH) of each allotype of CFH might explain the genotypic “risk” of developing AMD. We therefore sought to determine if variants of CFH (Y402 and H402) differentially bound to and/or were retained on the GAGs and other polyanions on Bruch’s membrane/choroid tissue. Both allotypes showed statistically similar binding to the tissue, independent of the region of the eye from which the tissue punch was isolated (peripheral, central, or macular, Fig. 6).
Furthermore, the ability of the exogenous CFH bound to the Bruch’s membrane/choroid punch to act as a cofactor in the proteolytic cleavage of C3b was tested and both V62/Y402 and V62/H402 behaved similarly. This was similar for punches taken either centrally, peripherally, or from the macular region (Fig. 7).
FIGURE 7.
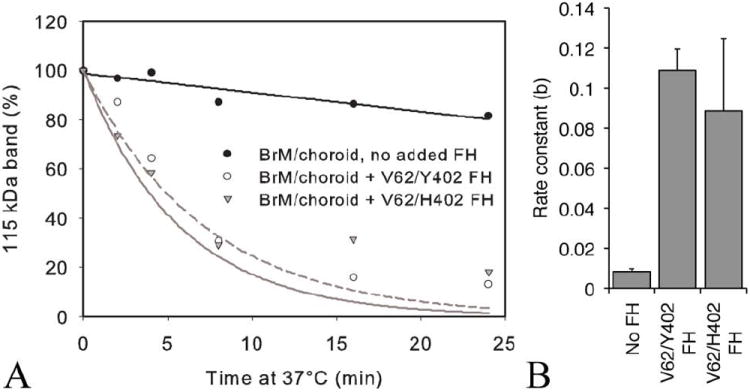
Functional cofactor activity of CFH variant prebound to a single 6 mm Bruch’s membrane/choroid punch. (A) Cleavage of C3b measured in the fluid-phase assay using one Bruch’s membrane/choroid 6 mm punch preincubated with or without CFH (V62/Y402 or V62/H402) and washed, then added to VBS/EDTA, factor I and C3b. Aliquots were removed at 0, 2, 4, 8, 16, and 24 min and these samples were boiled and run with reducing buffer on 10% Bis-tris Criterion XT gels with MOPS buffer. After silver staining, the gel was scanned and the density of the 115 kDa band measured with ImageJ software, and SigmaPlot 9 used to create the graphs. The example shown is representative of 6 separate experiments. (B) Rate constants showed no significant differences between CFH variants independent of region tissue punch was taken from (i.e. periphery (as shown) or central) (p> 0.2).
Discussion
The effects of the CFH amino acid variants associated with increased risk of developing AMD are poorly understood, although they are located within regions of the protein known to function either in the cofactor activity of CFH in the factor I-mediated cleavage of C3b [V62 in SCR1,(1, 14-18)] and in binding to polyanions [H402 in SCR7,(12, 14)]. To further refine our understanding of these amino acid substitutions, two assays to measure the cofactor activity of CFH in the factor I-mediated proteolytic cleavage of C3b were optimized to examine the activity of allotypic variants of CFH alone and in the presence of specific GAGs. Interestingly, these two assays gave essentially the same results. Allotypic variants of CFH showed similar levels of activity in these assays. Moreover, all assays used full-length CFH purified from the plasma samples of individuals with specific CFH genotypes, suggesting that any modifications that may have occurred in human blood does not appear to alter the results.
Importantly, the addition of specific GAGs to both the fluid-phase and cell-based assays altered the rate of C3b conversion to iC3b. Chondroitin sulfate-A, -B, or -C had no effect on the rate of the reaction, but heparin and HS modulated the cleavage rate. HS-BK accelerated the rate, HS-PI had no effect, and heparin decreased the rate. Studies using de-N-sulfated heparin, de-O-sulfated heparin, and over-sulfated HS indicated that both the position and extent of sulfation of the GAG are critical in determining its effect on the reaction rate. Over-O-sulfated HS slowed the C3b being converted to the inactive iC3b to a greater extent than the highly sulfated heparin used in the initial assays. This would increase the half-life of C3b and lead to complement activation in vivo. The presence of de-N-sulfated heparin in the fluid-phase reaction accelerated C3b cleavage in a manner similar to that of the poorly N-sulfated HS-BK, which would inhibit complement activation. Interestingly, de-O-sulfated heparin still decreased the rate of C3b cleavage, whereas de-N-sulfated heparin increased the rate, supporting a role for N-sulfation rather than O-sulfation being the important modification in accelerating this reaction. There are several possible explanations for these observed effects. For example, treatment of the reaction components (C3b, CFH or factor I) with the less N-sulfated HSs (e.g. HS-BK or de-N-sulfated heparin) might increase the affinity of the proteins in the reaction; or treatment of CFH with these GAGs might enhance the polyanion-induced self-association of CFH, as described by Pangburn et al. (41); and finally, these GAGs could act as bridges for bringing the components of the reaction together more efficiently. These results support mechanisms by which GAGs influence C3b degradation.
HS proteoglycans are expressed and secreted by most mammalian cells and are distributed on both cell surfaces and in the extracellular matrix. The HS composition of specific organs and tissues varies widely, with the structural makeup changing in response to extracellular signaling. Age- and disease-dependent changes have been documented in many tissues and extracellular matrices, including Bruch’s membrane (38, 42, 43). To test the hypothesis that HS modulates complement activation in the eye where AMD manifests, we added Bruch’s membrane/choroid tissue, which is rich in HS proteoglycans, to the fluid-phase assay. This tissue was isolated from elderly human donor eyes. The cleavage reaction was accelerated indicating that the addition of tissue behaved in the same way as the addition of HS-BK and de-N-sulfated heparin. Furthermore, this effect could be reversed by pretreatment of the tissue with heparinase III. These observations provide supporting evidence that the effect observed was due to the presence of HS in the tissue.
Although our assays did not reveal allotypic variations in binding of CFH to components in Bruch’s membrane, it remains possible that subtle regional GAG changes might result in local differential activity of the variants but this was masked by the amount and complexity of tissue required to perform these assays. In addition to its cofactor activity, CFH performs decay-accelerating functions by removing Bb from the convertase and by preventing the initial binding of factor B to C3b, a function of CFH that was not addressed in this current study. It has not yet been determined if it is this function of CFH that is associated with the “risk” of developing AMD.
To date, all studies of documented differences between the allotypic variants of CFH have employed recombinant fragments of CFH and not the full-length molecule. Recombinant fragments of the allotypic variants of CFH differ in their binding affinities for GAGs (21-24), and more specifically, in their binding to Bruch’s membrane (44). Clark et al. (44) examined the binding of recombinant fragments of CFH composed of SCRs 6 to 8 containing the Y402 or H402 variant to cryosections of human macula and found that the H402 form bound less strongly. When full-length CFH was employed in a study investigating binding to heparin, differential binding was no longer detected, a result similar to that obtained in our study (21). It is intriguing that full-length CFH binding to isolated Bruch’s membrane/choroid did not show any difference regardless of which allotypic variant of CFH was applied. This contradicts the currently held view in the AMD field that the effect of the CFH variants associated with risk of AMD is due, at least in part, to differential binding of these variants to purified GAGs or proteoglycans in Bruch’s membrane/choroid. Since our studies analyzed the binding and activity of full-length forms of CFH variants, they may be more reflective of the in vivo behavior of CFH. Our results support the hypothesis that modulators of CFH function, such as HS, contribute to CFH-associated risk in AMD.
There are interesting parallels between the extracellular matrix of the eye, Bruch’s membrane, and the kidney’s glomerular extracellular matrix. Both the ocular Bruch’s membrane and the renal glomerular basement membrane are particularly vulnerable to damage from complement activation and dysregulation in a variety of complement-related diseases, including membranoproliferative glomerulonephritis or dense deposit disease (45). Both structures share similar features. For example, they both form a HS-rich barrier between fenestrated vasculature with high blood flow and a layer of highly metabolically active cells: the RPE cells juxtaposed to Bruch’s membrane and the podocytes juxtaposed to the glomerular basement membrane. These two extracellular matrix ‘membranes’ appear uniquely sensitive to the dysfunction induced by the deposition of C3 cleavage products and subsequent damage, in individuals with specific CFH genotypes leading to its dysfunction (10, 45-49).
It appears that CFH may play a critical role in normal conditions in preventing complement activation in these tissues, which notably lack endogenous membrane-bound complement regulators (50). We hypothesize that the GAGs in these tissues play a role in modulating the activation of complement. Age-related changes in the composition of the HS may alter local complement activation including production of C5b-9 and recruitment of macrophages resulting in local damage to the tissues in these regions.
Acknowledgments
We thank Drs. Margaret Pericak-Vance (University of Miami) and Jonathan Haines (Vanderbilt University) whose grant (NEI EY012118) funded collection of a portion of the samples used in this work. We gratefully acknowledge Dr. Lincoln Johnson for helpful comments and discussions, Dr. Nikolai Skiba for performing MALDI analysis of CFH and the excellent technical assistance of Marybeth Groelle and Lisa Hancox.
This work was supported by National Institutes of Health (NIH) Grants R01 EY019038 (CBR), P30 EY005722 (Duke Eye Center) and R24 EY017404 (GSH); and by Research to Prevent Blindness, Inc. (RPB) Core Grants to the Duke Eye Center and John A. Moran Eye Center, RPB Special Scholars Award (CBR), The Foundation Fighting Blindness (CBR), The Ruth and Milton Steinbach Fund (CBR), and Macular Vision Research Foundation (CBR). The disaccharide analysis of the GAGs performed by the Azadi laboratory was supported by NIH 5-P41-RR05351 to the Complex Carbohydrate Research Center.
Footnotes
Abbreviations used in the paper: CFH, complement factor H protein; CFH, CFH gene; AMD, age-related macular degeneration; HS, heparan sulfate; SCR, short consensus repeat; GAG, glycosaminoglycan; CRP, C-reactive protein; RPE, retinal pigmented epithelium; GVBS, gelatin-veronal-buffered saline; HS-PI, HS from porcine intestine; HS-BK, HS from bovine kidney; CS-A, CS-B, CS-C, chondroitin sulfate (CS) A, B, and C
References
- 1.Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Molecular immunology. 2004;41:355–367. doi: 10.1016/j.molimm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Sharma AK, Pangburn MK. Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10996–11001. doi: 10.1073/pnas.93.20.10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiScipio RG. Ultrastructures and interactions of complement factors H and I. J Immunol. 1992;149:2592–2599. [PubMed] [Google Scholar]
- 4.Soames CJ, Sim RB. Interactions between human complement components factor H, factor I and C3b. The Biochemical journal. 1997;326(Pt 2):553–561. doi: 10.1042/bj3260553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:3982–3986. doi: 10.1073/pnas.87.10.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein BE, Klein R, Lee KE. Incidence of age-related cataract over a 10-year interval: the Beaver Dam Eye Study. Ophthalmology. 2002;109:2052–2057. doi: 10.1016/s0161-6420(02)01249-6. [DOI] [PubMed] [Google Scholar]
- 7.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science (New York, N Y) 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science (New York, N Y) 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 9.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science (New York, N Y) 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 10.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blackmore TK, Sadlon TA, Ward HM, Lublin DM, Gordon DL. Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J Immunol. 1996;157:5422–5427. [PubMed] [Google Scholar]
- 12.Giannakis E, Jokiranta TS, Male DA, Ranganathan S, Ormsby RJ, Fischetti VA, Mold C, Gordon DL. A common site within factor H SCR 7 responsible for binding heparin, C-reactive protein and streptococcal M protein. European journal of immunology. 2003;33:962–969. doi: 10.1002/eji.200323541. [DOI] [PubMed] [Google Scholar]
- 13.Jarva H, Jokiranta TS, Hellwage J, Zipfel PF, Meri S. Regulation of complement activation by C-reactive protein: targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8-11. J Immunol. 1999;163:3957–3962. [PubMed] [Google Scholar]
- 14.Schmidt CQ, Herbert AP, Kavanagh D, Gandy C, Fenton CJ, Blaum BS, Lyon M, Uhrin D, Barlow PN. A new map of glycosaminoglycan and C3b binding sites on factor H. J Immunol. 2008;181:2610–2619. doi: 10.4049/jimmunol.181.4.2610. [DOI] [PubMed] [Google Scholar]
- 15.Alsenz J, Lambris JD, Schulz TF, Dierich MP. Localization of the complement-component-C3b-binding site and the cofactor activity for factor I in the 38kDa tryptic fragment of factor H. The Biochemical journal. 1984;224:389–398. doi: 10.1042/bj2240389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM. Identification of complement regulatory domains in human factor H. J Immunol. 1995;155:348–356. [PubMed] [Google Scholar]
- 17.Jokiranta TS, Zipfel PF, Hakulinen J, Kuhn S, Pangburn MK, Tamerius JD, Meri S. Analysis of the recognition mechanism of the alternative pathway of complement by monoclonal anti-factor H antibodies: evidence for multiple interactions between H and surface bound C3b. FEBS Lett. 1996;393:297–302. doi: 10.1016/0014-5793(96)00905-2. [DOI] [PubMed] [Google Scholar]
- 18.Kuhn S, Skerka C, Zipfel PF. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J Immunol. 1995;155:5663–5670. [PubMed] [Google Scholar]
- 19.Call TW, Hollyfield JG. Sulfated proteoglycans in Bruch’s membrane of the human eye: localization and characterization using cupromeronic blue. Experimental eye research. 1990;51:451–462. doi: 10.1016/0014-4835(90)90158-q. [DOI] [PubMed] [Google Scholar]
- 20.Hogan MJ, Alvarado JA, Weddell JE. Histology of the Human Eye: An Atlas and Textbook. W B Sanders Company; Philadelphia: 1971. [Google Scholar]
- 21.Clark SJ, Higman VA, Mulloy B, Perkins SJ, Lea SM, Sim RB, Day AJ. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. The Journal of biological chemistry. 2006;281:24713–24720. doi: 10.1074/jbc.M605083200. [DOI] [PubMed] [Google Scholar]
- 22.Herbert AP, Deakin JA, Schmidt CQ, Blaum BS, Egan C, Ferreira VP, Pangburn MK, Lyon M, Uhrin D, Barlow PN. Structure shows that a glycosaminoglycan and protein recognition site in factor H is perturbed by age-related macular degeneration-linked single nucleotide polymorphism. The Journal of biological chemistry. 2007;282:18960–18968. doi: 10.1074/jbc.M609636200. [DOI] [PubMed] [Google Scholar]
- 23.Ormsby RJ, Jokiranta TS, Duthy TG, Griggs KM, Sadlon TA, Giannakis E, Gordon DL. Localization of the third heparin-binding site in the human complement regulator factor H1. Molecular immunology. 2006;43:1624–1632. doi: 10.1016/j.molimm.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 24.Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, Jowitt TA, Clark SJ, Tarelli E, Uhrin D, Barlow PN, Sim RB, Day AJ, Lea SM. Structural basis for complement factor H linked age-related macular degeneration. The Journal of experimental medicine. 2007;204:2277–2283. doi: 10.1084/jem.20071069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayer MM. Complement and complement fixation. In: Kabat EA, Mayer MM, editors. Experimental Immunochemistry. Charles C Thomas; Springfield: 1961. p. 133. [Google Scholar]
- 26.Kelly U, Rickman CB, Postel EA, Hauser MA, Hageman GS, Arshavsky VY, Skiba NP. Rapid and sensitive method for detection of Y402, H402, I62, and V62 variants of complement factor H in human plasma samples using mass spectrometry. Investigative ophthalmology & visual science. 2009;50:1540–1545. doi: 10.1167/iovs.08-2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaither TA, Hammer CH, Frank MM. Studies of the molecular mechanisms of C3b inactivation and a simplified assay of beta 1H and the C3b inactivator (C3bINA) J Immunol. 1979;123:1195–1204. [PubMed] [Google Scholar]
- 28.Reid KB, Nolan KF, Lijnen HR, Collen D. Proteolytic enzymes in coagulation, fibrinolysis, and complement activation. Introduction. Methods Enzymol. 1993;223:1–9. doi: 10.1016/0076-6879(93)23034-k. [DOI] [PubMed] [Google Scholar]
- 29.Hewitt AT, Nakazawa K, Newsome DA. Analysis of newly synthesized Bruch’s membrane proteoglycans. Investigative ophthalmology & visual science. 1989;30:478–486. [PubMed] [Google Scholar]
- 30.Scott JE. Collagen--proteoglycan interactions. Localization of proteoglycans in tendon by electron microscopy. The Biochemical journal. 1980;187:887–891. doi: 10.1042/bj1870887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pangburn MK. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology. 2000;49:149–157. doi: 10.1016/s0162-3109(00)80300-8. [DOI] [PubMed] [Google Scholar]
- 32.Blackmore TK, Hellwage J, Sadlon TA, Higgs N, Zipfel PF, Ward HM, Gordon DL. Identification of the second heparin-binding domain in human complement factor H. J Immunol. 1998;160:3342–3348. [PubMed] [Google Scholar]
- 33.Jokiranta TS, Hellwage J, Koistinen V, Zipfel PF, Meri S. Each of the three binding sites on complement factor H interacts with a distinct site on C3b. The Journal of biological chemistry. 2000;275:27657–27662. doi: 10.1074/jbc.M002903200. [DOI] [PubMed] [Google Scholar]
- 34.Oppermann M, Manuelian T, Jozsi M, Brandt E, Jokiranta TS, Heinen S, Meri S, Skerka C, Gotze O, Zipfel PF. The C-terminus of complement regulator Factor H mediates target recognition: evidence for a compact conformation of the native protein. Clinical and experimental immunology. 2006;144:342–352. doi: 10.1111/j.1365-2249.2006.03071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferreira VP, Herbert AP, Hocking HG, Barlow PN, Pangburn MK. Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol. 2006;177:6308–6316. doi: 10.4049/jimmunol.177.9.6308. [DOI] [PubMed] [Google Scholar]
- 36.Pangburn MK, Pangburn KL, Koistinen V, Meri S, Sharma AK. Molecular mechanisms of target recognition in an innate immune system: interactions among factor H, C3b, and target in the alternative pathway of human complement. J Immunol. 2000;164:4742–4751. doi: 10.4049/jimmunol.164.9.4742. [DOI] [PubMed] [Google Scholar]
- 37.Herbert AP, Uhrin D, Lyon M, Pangburn MK, Barlow PN. Disease-associated sequence variations congregate in a polyanion recognition patch on human factor H revealed in three-dimensional structure. The Journal of biological chemistry. 2006;281:16512–16520. doi: 10.1074/jbc.M513611200. [DOI] [PubMed] [Google Scholar]
- 38.Rabenstein DL. Heparin and heparan sulfate: structure and function. Natural product reports. 2002;19:312–331. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 39.Wei Z, Lyon M, Gallagher JT. Distinct substrate specificities of bacterial heparinases against N-unsubstituted glucosamine residues in heparan sulfate. The Journal of biological chemistry. 2005;280:15742–15748. doi: 10.1074/jbc.M501102200. [DOI] [PubMed] [Google Scholar]
- 40.Lawrence R, Lu H, Rosenberg RD, Esko JD, Zhang L. Disaccharide structure code for the easy representation of constituent oligosaccharides from glycosaminoglycans. Nat Methods. 2008;5:291–292. doi: 10.1038/nmeth0408-291. [DOI] [PubMed] [Google Scholar]
- 41.Pangburn MK, Rawal N, Cortes C, Alam MN, Ferreira VP, Atkinson MA. Polyanion-induced self-association of complement factor H. J Immunol. 2009;182:1061–1068. doi: 10.4049/jimmunol.182.2.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadanaka S, Kitagawa H. Heparan sulphate biosynthesis and disease. J Biochem. 2008;144:7–14. doi: 10.1093/jb/mvn040. [DOI] [PubMed] [Google Scholar]
- 43.Verdugo ME, Ray J. Age-related increase in activity of specific lysosomal enzymes in the human retinal pigment epithelium. Experimental eye research. 1997;65:231–240. doi: 10.1006/exer.1997.0325. [DOI] [PubMed] [Google Scholar]
- 44.Clark SJ, Sim RB, Day AJ, Bishop PN. Differential Binding of the 402Y and AMD-Associated 402H Variants of Complement Factor H. Association for Research in Vision and Ophthalmology. 2008 Abstract 5159. [Google Scholar]
- 45.Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, Lambris JD, Lanning L, Lutz HU, Meri S, Rose NR, Salant DJ, Sethi S, Smith RJ, Smoyer W, Tully HF, Tully SP, Walker P, Welsh M, Wurzner R, Zipfel PF. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol. 2005;16:1392–1403. doi: 10.1681/ASN.2005010078. [DOI] [PubMed] [Google Scholar]
- 46.Duvall-Young J, MacDonald MK, McKechnie NM. Fundus changes in (type II) mesangiocapillary glomerulonephritis simulating drusen: a histopathological report. Br J Ophthalmol. 1989;73:297–302. doi: 10.1136/bjo.73.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saunders RE, Goodship TH, Zipfel PF, Perkins SJ. An interactive web database of factor H-associated hemolytic uremic syndrome mutations: insights into the structural consequences of disease-associated mutations. Hum Mutat. 2006;27:21–30. doi: 10.1002/humu.20268. [DOI] [PubMed] [Google Scholar]
- 48.Rose KL, Paixao-Cavalcante D, Fish J, Manderson AP, Malik TH, Bygrave AE, Lin T, Sacks SH, Walport MJ, Cook HT, Botto M, Pickering MC. Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice. The Journal of clinical investigation. 2008;118:608–618. doi: 10.1172/JCI32525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boon CJ, van de Kar NC, Klevering BJ, Keunen JE, Cremers FP, Klaver CC, Hoyng CB, Daha MR, den Hollander AI. The spectrum of phenotypes caused by variants in the CFH gene. Molecular immunology. 2009;46:1573–1594. doi: 10.1016/j.molimm.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 50.Kavanagh D, Richards A, Atkinson J. Complement regulatory genes and hemolytic uremic syndromes. Annu Rev Med. 2008;59:293–309. doi: 10.1146/annurev.med.59.060106.185110. [DOI] [PubMed] [Google Scholar]