

Abstract
Hydrophilic interaction chromatography (HILIC) for separations of peptides has been employed infrequently, particularly considering that this technique was introduced over 20 years ago. The present manuscript describes a radical departure from the traditional HILIC elution approach, where separations are achieved via increasing salt (sodium perchlorate) gradients in the presence of high isocratic concentrations (>80%) of acetonitrile, denoted HILIC/SALT. This initial study compared to reversed-phase chromatography (RPC), HILIC and HILIC/SALT for the separation of mixtures of synthetic peptide standards varying in structure (amphipathic α-helix, random coil), length (10–26 residues), number of positively charged residues (+1 to +11) and hydrophilicity/hydrophobicity. Results showed a marked superiority of the HILIC/SALT approach compared to traditional HILIC and excellent complementarity to RPC for peptide separations. We believe these initial results offer a new dimension to HILIC, enabling it to transform from an occasional HPLC approach for peptide separations to a more generally applicable method.
Keywords: Hydrophilic interaction chromatography, (HILIC), HILIC/SALT, Peptides, De novo designed peptide standards
1. Introduction
Since the introduction of the term hydrophilic interaction chromatography (HILIC) by Alpert in 1990 [1] to distinguish it from traditional normal phase chromatography, HILIC has seen use in the separation and analysis of a wide range of solutes, including peptides [1–12]. In HILIC, analytes interact with a hydrophilic stationary phase and are eluted with a relatively hydrophobic binary eluent, with water the stronger eluting component (acetonitrile generally representing the water-miscible organic component). Thus, HILIC represents a complementary, orthogonal method to reversed-phase chromatography (RPC), the latter technique being the most widely used separative mode for peptides [13,14]. HILIC also has high solvent compatibility with mass spectroscopy (MS) in the electrospray ionization (ESI) mode [15,16].
Despite such reported applications, reports of employing HILIC for peptide separations are still relatively sparse, particularly considering that this technique was introduced over 20 years ago [1]. Even where HILIC separations are reported, such as its use in multidimensional/proteomics applications [9,17–22], the HILIC dimension is frequently not traditional HILIC, i.e., carried out in the absence of electrostatic interactions. Instead, such mixed-mode approaches as ZIC-HILIC are employed [17,18,20,21] or electrostatic interactions due to ionized silanols are observed [9,18]. Excellent recent reviews [23,24] highlight the point that the term “HILIC” is used even where the separations are actually mixed-mode (generally hydrophilic and electrostatic), either by design (e.g., zwitterionic ZIC-HILIC packings, ion-exchange packings) or through the presence of ionized silanols. The authors of Ref. [24] note that HILIC phases can be grouped into neutral polar or ionic surfaces. In our view, the term “HILIC mode” is more accurate for any separation involving hydrophilic interactions in combination with other packing properties such as charge, with “HILIC” referring only to “pure” HILIC separations.
When evaluating HILIC for the separation of peptides in 1991 [25], our laboratory could not demonstrate any significant advantage of HILIC for peptides at that time, either as a complement to RPC or as a separation technique in its own right. Granted that HILIC offered a niche approach for the separation of very polar peptides unable to be retained and resolved by RPC packings, but it was felt that its potential would be better realized for peptide separations if it could be utilized for separations of peptide mixtures containing peptides with a wide range of varying properties (charge, structure, length, etc.), with markedly different selectivities to RPC. Indeed, we developed such an approach by demonstrating that mixed-mode HILIC/cation-exchange chromatography (HILIX/CEX) [25] could rival RPC for peptide separations [26]. Briefly, HILIC/CEX combined the most advantageous aspects of two widely different separation mechanisms: a separation based on hydrophilicity/hydrophobicity differences between peptides overlaid on a separation based on net charge. A characteristic of HILIC/CEX separations is the presence of a high organic modifier concentration (generally acetonitrile) to promote hydrophilic interactions between the solute and the hydrophilic/charged CEX stationary phase. The effect of varying acetonitrile concentrations on selectivity of peptide separations on cation-exchange columns had previously been noted [27–29]. In addition, selectivity variations on anion-exchange columns in the presence of varying acetonitrile concentrations had also been reported [30]. However, our laboratory was the first to carry out systematic investigations into the potential of mixed/mode HILIC/CEX for peptide applications. Ref. [31] represents a comprehensive review of mixed-mode HILIC/CEX of peptides and proteins.
With the relative maturity of HILIC as a separative method, coupled with the improving selections of HILIC packing materials, we believed revisiting the potential of HILIC as a generally useful peptide separation tool was overdue. Specifically, we believed expanding the relatively narrow range of peptide elution options (decreasing acetonitrile gradient from a high starting concentration of acetonitrile) would help to achieve this goal. Although the retention mechanism of HILIC is not completely understood, it has been concluded that retention is affected by partitioning as well as adsorption, hydrogen bonding or a combination of these phenomena [32]. Such interactions are, thus, also likely to be present in mixed-mode HILIC/CEX and are overcome by increasing salt in the presence of 70–90% acetonitrile [31]. Thus, we decided to evaluate similar elution conditions for peptides from HILIC packings, i.e., a radical departure from the traditional decreasing organic modifier gradient, with this approach being denoted HILIC/SALT (an increasing linear salt gradient in the presence of high isocratic concentrations of acetonitrile).
HILIC retention behaviour of tryptic peptides on various HILIC packings has been examined [10], making the possibly intuitive conclusions that charged amino acid residues, such as His, Lys and Arg, promote retention in HILIC, whereas non-polar residues such as Phe, Trp, Ile and Leu prevent such interactions. HILIC peptide retention coefficients were derived with different ion-pairing reagents [4]. A systematic study of HILIC of peptides on a number of stationary phases has been carried out [11]. However, despite interesting results arising from such studies, they still had the common weakness of being carried out with mixtures of unrelated peptides of different sizes and compositions, making it difficult to delineate the influence of specific side-chain structure and overall peptide characteristics on HILIC retention behaviour. Hence, our favoured approach is to employ mixtures of well-defined de novo designed peptide standards (varying systematically in composition, sequence, number of positively charged residues, length and structure), not only to evaluate the effectiveness of HILIC/SALT specifically but also to offer potential insights into the mechanism of HILIC and HILIC/SALT.
2. Experimental
2.1. Materials
HPLC-grade water was prepared by an E-pure water purification system from Barnstead International (Dubuque, IA, USA). ACS-grade orthophosphoric acid and acetonitrile were obtained from Fisher Scientific (Fairlawn, NJ, USA). Trifluoroacetic acid (TFA) was obtained from Pierce (Rockford, IL, USA). Sodium perchlorate was obtained from MP Biomedicals (Solon, OH, USA).
2.2. Peptide synthesis and purification
Synthesis of the amphipathic α-helical peptides D1–4 and D12–D14 peptides (Table 2) was carried out by standard solid-phase peptide synthesis methodology using t-butyloxy-carbonyl (t-Boc) chemistry and 4-methylbenzhydrylamine resin (substitution level 0.97 mmol/g) followed by cleavage of the peptide from the resin. The coupling procedure was catalyzed by 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/hydroxybenzotriazole (HOBt) (5-fold excess to amino groups on the resin) in dimethyl-formamide (DMF)/dichloromethane (DCM) (1:1) with N,N-diisopropylethylamine (DIPEA) (2-fold excess to amino groups on the resin) for 1–4 h. The couplings were checked for completion by the Kaiser ninhydrin test and the couplings were repeated if necessary to obtain a negative result for the free amines. The deprotection procedure (removal of Boc protecting group) was carried out by treatment of the resin with TFA/DCM (1:1) for 10–15 min, then washed with DCM three times and DMF three times. Before the next coupling step, the resin should be neutralized with 10% DIPEA in DCM. The above coupling, deprotection, neutralization and wash steps were repeated for 25 cycles to complete the synthesis of the 26-residues peptide. The N-terminal α-amino group was acetylated with acetic anhydride in the presence of DIPEA in DCM for 10–15 min. The peptide resin was dried under vacuum and stirred with anhydrous hydrogen fluoride (HF) with an appropriate scavenger such as anisole for 1 h between 0 °C and 4 °C. Following removal of HF under reduced pressure, the resin was washed with ethyl ether. The cleaved peptide was extracted from the resin by washing the resin with acetonitrile/water (1:1, with 0.2% TFA) and the solution lyophilized to obtain the crude peptide. The remaining peptides were synthesized by similar protocols. Crude peptides were then purified by preparative RPC as described previously [33]. Peptide standards denoted S1–S5 (Table 1) are available from Pierce.
Table 2.
Sequences of synthetic amphipathic α-helical peptides.
| Series | Peptide Name | Peptide Sequence | Net Charge pH 2 | Number of Ala to Leu Substitutions |
|---|---|---|---|---|
| LX | LS7 | Ac–ELEKLLSELEKLLKELEK–amide | +4 | – |
| LT7 | Ac–ELEKLLTELEKLLKELEK–amide | +4 | – | |
| LV7 | Ac–ELEKLLVELEKLLKELEK–amide | +4 | – | |
|
| ||||
| D1–D4 | D1 | Ac–KWKSFLKTFKSAKKTVLHTALKAISS–amide | +8 | 0 |
| D2 | Ac–KWKSFLKTFKSAKKTVLHTLLKAISS–amide | +8 | 1 | |
| D3 | Ac–KWKSFLKTFKSLKKTVLHTLLKAISS–amide | +8 | 2 | |
| D4 | Ac–KWKSFLKTFKSLKKTVLHTLLKLISS–amide | +8 | 3 | |
|
| ||||
| D12–D14 | D14 | Ac–KWKSFLKTFSKAKKKKLKTLLKAISK–amide | +11 | 0 |
| D13 | Ac–KWKSFLKTFSKLKKKKLKTLLKAISK–amide | +11 | 1 | |
| D12 | Ac–KWKSFLKTFSKLKKKKLKTLLKLISK–amide | +11 | 2 | |
Ac denotes Nα-acetyl; amide denotes Cα-amide. Variations in hydrophobicity of the peptide analogues are indicated in bold. D1–D4 and D12–D14 are α-helical antimicrobial peptides [33].
Table 1.
Sequences of synthetic random-coil peptide standards.
| Series | Peptide Standard | Peptide Sequence | Net Charge pH 2 | Change in Carbon atom Content |
|---|---|---|---|---|
| S1–S5 | S1 | R–G–A–G–G–L–G–L–G–K–amide | +3 | 1 |
| S2 | Ac–R–G–G–G–G–L–G–L–G–K–amide | +2 | 0 | |
| S3 | Ac–R–G–A–G–G–L–G–L–G–K–amide | +2 | 1 | |
| S4 | Ac–R–G–V–G–G–L–G–L–G–K–amide | +2 | 2 | |
| S5 | Ac–R–G–V–V–G–L–G–L–G–K–amide | +2 | 3 | |
|
| ||||
| RX1–4 | RX1 | Ac–G–G–G–L–G–G–A–G–G–L–K–amide | +1 | – |
| RX2 | Ac–K–Y–G–L–G–G–A–G–G–L–K–amide | +2 | – | |
| RX3 | Ac–G–G–A–L–K–A–L–K–G–L–K–amide | +3 | – | |
| RX4 | Ac–K–Y–A–L–K–A–L–K–G–L–K–amide | +4 | – | |
|
| ||||
| HILIC 3 | 3a | G–G–G–G–K–L–G–L–G–K–amide | +3 | 0 |
| 3b | G–G–A–G–K–L–G–L–G–K–amide | +3 | 1 | |
| 3c | G–G–A–A–K–L–G–L–G–K–amide | +3 | 2 | |
| 3d | G–G–V–G–K–L–G–L–G–K–amide | +3 | 3 | |
| 3e | G–G–V–A–K–L–G–L–G–K–amide | +3 | 4 | |
| 3f | G–G–I–G–K–L–G–L–G–K–amide | +3 | 4 | |
| 3g | G–G–I–A–K–L–G–L–G–K–amide | +3 | 5 | |
| 3h | G–G–I–V–K–L–G–L–G–K–amide | +3 | 7 | |
| 3i | G–G–I–I–K–L–G–L–G–K–amide | +3 | 8 | |
Ac denotes Nα-acetyl; amide denotes Cα-amide. Variations in hydrophobicity of the peptide analogues for S1–S5 and HILIC 3 standards are indicated in bold. For the RX1 to RX4 standards the Lys residues are bolded.
2.3. Instrumentation
All HPLC runs were carried out on an Agilent 1100 Series liquid chromatograph, which included a G311A quaternary pump (Agilent Technologies, Foster City, CA, USA).
2.4. Columns
RPC runs were carried out on a Halo Peptide ES-C18 columns (100 mm × 2.1 mm I.D.; 2.7 μm particle size). HILIC and HILIC/SALT runs were carried out on a Halo Penta-HILIC column (100 mm × 2.1 mm I.D.; 2.7 μm particle size). The columns were donated by Advanced Materials Technology, Inc. (Wilmington, DE, USA). These superficially porous packings are based on fused core technology [34], where a porous silica layer is fused to a solid silica particle [34–39]. The Halo packings are comprised of a 0.5 μm porous (90 Å ° pore size) silica layer fused to a solid core silica particle of 1.7 μm diameter. A schematic representation of the poly-hydroxylic Penta-HILIC packing is shown in Fig. 1.
Fig. 1.
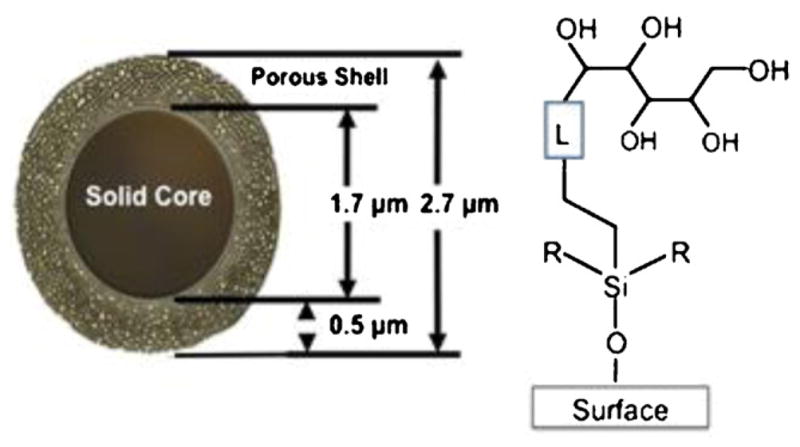
HILIC particles have a solid core of 1.7 μm diameter with a porous shell of 0.5 μm surrounding the solid core to give a particle of 2.7 μm in diameter. The pentan-OL is linked to the silica surface to prepare a hydrophilic interaction chromatography (HILIC) column.
2.5. HPLC conditions
RPC: linear AB (increasing acetonitrile) gradient at a flow-rate of 0.3 ml/min and a temperature of 30 °C, where eluent A is 0.2% aq. H3PO4 or 0.2% TFA and eluent B is 0.2% H3PO4 or 0.18% TFA in acetonitrile. The pH values of a 100% aqueous solution of 0.2% H3PO4 are in the region of pH 2, whereas the actual value in the presence of high concentrations of acetonitrile is difficult to determine. However, we retain the description of pH 2 for HILIC and HILIC/SALT mobile phases for the sake of simplicity.
HILIC: linear AB (decreasing acetonitrile) gradient from starting conditions of varying levels of acetonitrile concentrations, following 5-min isocratic elution with these starting conditions, at a flow-rate of 0.3 ml/min and temperature of 30 °C, where eluent A is 0.2% H3PO4 in acetonitrile and eluent B is 0.2% aq. H3PO4.
HILIC/SALT: HILIC/SALT, linear (increasing NaClO4) gradient in the presence of an isocratic concentration of acetonitrile containing 0.2% H3PO4, following 5-min isocratic elution in the absence of salt, at a flow-rate of 0.3 ml/min and a temperature of 30 °C.
The solvent systems employed on the quaternary chromatography system for the HILIC and HILIC/SALT runs were: eluent A = 0.2% H3PO4 in acetonitrile; eluent B = 0.2% aq. H3PO4; eluent C = 300 mM NaClO4 in 90% aq. acetonitrile containing 0.2% H3PO4; eluent D = 300 mM NaCl04 in 0.2% aq. H3PO4. The instrument was then programmed to utilize various combinations of these four solvents for the specific HILIC and HILIC/SALT run conditions. All chromatograms shown can be achieved with linear AB binary gradients. However, the quaternary eluent system allows complete flexibility for method development where the same eluents can be used for all runs rather than having the necessity of preparing more eluents constantly for different mobile phase conditions when using a binary system.
3. Results and discussion
3.1. Development of novel HILIC/SALT separations of peptides
3.1.1. Principles of HILIC versus RPC
As noted above, the term HILIC was originally introduced to describe separations based on solute hydrophilicity [1]. Thus, separations by HILIC, in a manner similar to normal phase chromatography (to which it is related), depends on hydrophilic interactions between the solutes and a hydrophilic stationary phase, i.e., solutes are eluted in order of increasing hydrophilicity (decreasing hydrophobicity). This is, of course, in direct contrast to RPC, where solutes are eluted from a hydrophobic stationary phase in order of increasing hydrophobicity (decreasing hydrophilicity). Unlike small molecules, peptides (and proteins) are considered to favour an adsorption/desorption mechanism during RPC, i.e., only limited partitioning between the mobile and stationary phases, due to multisite binding with the hydrophobic packing [40,41]. Thus, peptide separations during RPC are mainly effected by a linear increasing gradient of an organic modifier (generally acetonitrile). HILIC, in contrast, is generally carried out in starting conditions of high organic solvent concentration (typically >70% acetonitrile). It has been noted that the presence of highly polar functionalities (such as the hydroxyl groups on the present HILIC packing; Fig. 1) in HILIC packings attracts water molecules from the mobile phase solvent to form water-enriched layers on the stationary phase surface [1,32]. This water layer then allows analytes to partition between this enriched water layer and the relatively less polar mobile phase. Interestingly, as far back as 1951 [42], it was described that hydrophilic stationary phases such as ion-exchange resins contained a water-enriched layer on the surface. In fact, the mechanism of HILIC separations is viewed as quite complex with retention being affected by partitioning as well as adsorption, hydrogen bonding or a mixture of such processes [32]. Increasing the concentration of water in the eluent (i.e., a decreasing gradient of organic modifier) will, of course, disrupt hydrogen bonding interactions, if present, as well as encourage partitioning of the polar solutes into the mobile phase as the relative polarity of the latter increases. Whatever the actual mechanism(s), the result will be the same, i.e., elution from the HILIC packing in order of increasing solute hydrophilicity (decreasing concentration of organic modifier, e.g., acetonitrile).
3.1.2. HILIC/SALT
An alternative approach to disrupting interactions with the hydrophilic stationary phase induced by a high background concentration of organic modifier is to effect peptide elution by a linear increasing salt gradient in the presence of a constant (isocratic) acetonitrile concentration (above 70%). We believed that such an approach would result in elution of the peptide and would be achieved in an analogous manner to that of traditional decreasing acetonitrile concentration (i.e., disruption of any hydrogen bonding between solute and enriched water layer; increased polarity of mobile phase relative to stationary phase), albeit with the potential of enhanced flexibility and effectiveness for resolution of peptide mixtures. Although Alpert [1] suggested that gradient elution in HILIC could be accomplished by “increasing the polarity of the mobile phase” through an increasing salt gradient in the presence of high concentrations of acetonitrile, the present study represents the first sustained effort to investigate the potential value of such an approach in order to widen the scope of HILIC for peptide separations.
3.1.3. HILIC and HILIC/SALT conditions
In these initial studies, peptide mixtures were run in mobile phases based on 0.2% aq. H3PO4 for both HILIC and HILIC/SALT. The hydrophilic phosphate anion [43–45], on interacting with positively charged peptide groups, maximizes the overall hydrophilicity of the peptides relative to the use of more hydrophobic anionic ion-pairing reagents such as TFA [43–45], thereby maximizing their interaction with the hydrophilic stationary phase. In addition, the resulting low pH (pH 2) will also simplify interpretation of the peptide elution profiles (1) by suppressing any ionization of free, underivatized silanol groups, if present, which may otherwise interact with positively charged peptide groups and (2) by suppressing ionization of acidic side-chains (specifically glutamic acid in the present study), thereby maximizing the basic, positively charged character of the peptides. The Penta-HILIC column employed for this study is known to exhibit very limited silanol-derived ionic characteristics, compared with bare silica materials, as evidenced by low ionic strength dependence on peak shape and retention of small basic organic molecule HILIC separations (unpublished data). In addition, we carried out a rigorous investigation of potential ionized silanol effects on the Penta-HILIC packing, even at low pH, with highly positively charged peptides denoted RX1–4 and D14 (sequences shown in Tables 1 and 2, respectively). The random coil peptides RX1–4 (Table 1) were originally introduced as silanol monitoring standards for RPC columns, with the +4 RX4 peptide having a charge density of +0.36 (charge/residue) being particularly sensitive to ionized silanols [46]. These peptides were run on the HILIC column under HILIC/SALT conditions in the presence of isocratic acetonitrile concentrations of 0%, 10%, 20%, 30%, 40%, 50%, 60%, 70% and 80%. No retention of the peptides was observed from 0 to 70% acetonitrile, with only very weak retention seen at 80% acetonitrile. Although these results were good indicators of the lack of electrostatic interactions on the Penta-HILIC, we now took this probe of silanol activity a step further by carrying out the same runs with a highly positively charged peptide denoted D14 (sequence shown in Table 2). This peptide contains eleven positively charged lysine residues, resulting in an overall charge density of +11 per 26 residues (+0.42 charge/residue). This amphipathic α-helical peptide has eight lysine residues located in the 14-residue polar face (Fig. 8), i.e., a high positive charge density of +0.57 (charge/residue) on this polar preferred binding domain. No retention of the peptide was observed in the presence of isocratic acetonitrile concentrations of 0%, 10%, 20%, 30%, 40%, 50% and 60% in 0.2% aq. H3PO4, with only very weak retention seen at 70% acetonitrile and the peptide being eluted prior to the salt gradient. Our results clearly show that electrostatic interactions due to ionized silanols are not a factor in the mechanism of HILIC or HILIC/SALT on the Penta-HILIC column.
Fig. 8.

Helical net representations of synthetic amphipathic α-helical peptide standards. Residues comprising the non-polar face are circled, with lines between non-polar residues denoting i to i + 3 and i to i + 4 hydrophobic interactions between large hydrophobes along the helix. The shaded residues denote substitutions made in the “native” peptides: D1 in the +8 series D1–D4 and D14 in the +11 series D12–D14. Sequences of the peptides are shown in Table 2.
For HILIC/SALT, NaClO4 was employed as the salt for elution of peptides from the HILIC column because of its excellent solubility in aqueous solution even in the presence of high concentrations of organic modifier [26,47]. NaClO4 is also a strong chaotropic reagent, i.e., disruptor of water structure [47].
3.1.4. Synthetic peptide standards
Our extensive work in the de novo design and synthesis of peptide standards for monitoring HPLC column performance and aiding the development of novel HPLC protocols [13,14,46,48–53] is a result of our premise that peptide solutes are best suited for monitoring peptide behaviour during HPLC, since it is preferable to use standards that are structurally similar to the sample of interest [52]. This is particularly pertinent to RPC considering, as noted above, the fundamentally different mechanism of interaction of peptides with hydrophobic stationary phases compared to small molecules. In addition, designed peptide standards avoid the disadvantages of the use of random mixtures of native peptides which is generally not informative, since if an improvement in selectivity is observed the reason may be unclear due to this random nature where size, amino acid composition, conformation and sequences of the peptides in the mixture may vary considerably. Hence, our preferred approach has always been the design and synthesis of peptide standards with minimal sequence variation to probe column selectivity for the separation of peptide mixtures.
In this initial assessment of the potential of HILIC/SALT, we chose to utilize six mixtures of synthetic peptide standards encompassing a range of structure (α-helix, random coil), length (10–26 residues), net number of positively charged residues (+1 to +11 at pH 2) and hydrophilicity/hydrophobicity variations within the six peptide mixtures. Sequences of the three sets of random coil peptide standards and three sets of α-helical peptide standards are shown in Tables 1 and 2, respectively.
3.2. HILIC/SALT separation of random coil peptide standards
3.2.1. S1–S5 standards
The peptide standard mixture S1–S5 (Table 1) was originally introduced for monitoring RPC column performance over 35 years ago by this laboratory [48] and have been employed extensively since that time [13,25]. The peptides increase in hydrophobicity (decrease in hydrophilicity from S2 to S5, Table 1). These peptides also contain two positively charged residues with no potentially negatively charged residues present. The sequence of S1 is identical to that of S3, albeit with a free (unblocked) α-amino group and a resulting +3 charge. The presence of Gly residues (a helix-disrupting residue) throughout the sequence ensures the peptides are unstructured, even in the presence of the helix-inducing conditions of RPC (hydrophobic stationary phase; acetonitrile in mobile phase) and HILIC or HILIC/SALT (high acetonitrile concentration in the mobile phase) [54,55].
From Fig. 2A, the peptides are eluted, as expected, in order of increasing hydrophobicity with 0.2% aq. H3PO4 employed as the counterion (for a direct comparison with HILIC and HILIC/SALT, where 0.2% H3PO4 is also used).
Fig. 2.

Separation of S1–S5 synthetic random coil peptide standards by RPC (panel A), HILIC (panel B) and HILIC/SALT (panel C). Conditions: RPC, linear AB (increasing acetonitrile) gradient of 1% acetonitrile/min from 2% aq. acetonitrile; HILIC, linear AB (decreasing acetonitrile) gradient of −0.25% acetonitrile/min from starting conditions of 90% aq. acetonitrile/0.2% H3PO4, following 5-min isocratic elution with these starting conditions; HILIC/SALT, linear (increasing salt) gradient of 2 mM NaClO4/min in the presence of 90% aq. acetonitrile/0.2% H3PO4, following 5-min isocratic elution in starting conditions of 90% aq. acetonitrile/0.2% H3PO4, Sequences of peptides S1–S5 shown in Table 1.
Fig. 2B demonstrates a traditional example of HILIC compared to the RPC run of Fig. 2A, where the elution order of S1–S5 is completely reversed relative to RPC, i.e., in order of increasing hydrophilicity.
From Fig. 2C, the HILIC/SALT separation results in an order of peptide elution identical to the HILIC run (Fig. 2B) but with a significantly superior elution range from S1 to S5 of 41.6 min compared to HILIC (25.7 min elution range) within a similar run time. In addition, the decreasing acetonitrile gradient to achieve the HILIC separation was necessarily extremely shallow (−0.25% acetonitrile/min.)
3.2.2. RX1–RX4 standards
In addition to monitoring RPC columns [13,46], the RX1–4 peptide standard mixture (Table 1) was also originally designed for assessing cation-exchange columns [13,50], with the presence of Gly residues throughout the sequence again ensuring no α-helical structure. The standards exhibit +1 to +4 net charges from RX1–4, respectively, with no acidic, potentially negatively charged residues present. The presence of Tyr residues in peptides RX2 and RX4 allows detection at 280 nm in addition to peptide bond absorbance at 210 nm.
From Fig. 3A, the RPC elution order of the standards is RX1 < RX2 < RX3 < RX4 with TFA as the counterion. The results obtained with the more hydrophilic H3PO4 were different from the more hydrophobic TFA ion-pairing reagent with RX2 and, especially, RX1 being eluted very early with subsequent peak broadening (results not shown). The elution order was essentially the same as obtained with TFA but with RX3 and RX4 being coeluted.
Fig. 3.

Separation of RX1–4 synthetic random coil peptide standards by RPC (panel A) and HILIC/SALT (panel B). Conditions: RPC, linear AB (increasing acetonitrile) gradient of 1% acetonitrile/min from 2% aq. acetonitrile; HILIC/SALT, linear (increasing salt) gradient of 2 mM NaClO4/min in the presence of 90% aq. acetonitrile/0.2% H3PO4, following 5-min isocratic elution in starting conditions of 90% aq. acetonitrile/0.2% H3PO4. Sequences of peptides shown in Table 1.
Fig. 3B now shows the HILIC/SALT separation of the four peptides, with the striking observation that the elution order of RX1–4 is identical to that of RPC (Fig. 3A), i.e., not a simple reversal of elution order as was observed for S1–S5 (Fig. 2). Note that the elution order of the peptides in HILIC/SALT was identical to that obtained in traditional HILIC (not shown), albeit the resolution obtained with HILIC/SALT was again superior to that of HILIC within a similar run time. Thus, the value of the RX1–4 standards lies in their highlighting that, as noted above, HILIC is not necessarily a simple reversal of RPC; rather, both methods depend on differences in overall expressed peptide hydrophilicity/hydrophobicity under the particular mobile phase conditions employed.
Alpert noted that charged basic groups in a solute lead to pronounced hydrophilicity and retention [1]. Also, as seen in Fig. 2, the +3 peptide (S1) was eluted later than the +2 peptides (S2–S5) in HILIC (Fig. 2B) and HILIC/SALT (Fig. 2C) compared to RPC (Fig. 2A). This is not surprising given the inherent hydrophilicity of a charged group. However, the results shown in Fig. 3 clearly demonstrate that the number of positively charged groups on the solute (peptides in this case) and the contribution of charge to overall peptide hydrophilicity is magnified in HILIC and HILIC/SALT compared to RPC. This leads to an even more potentially interesting complementarity to RPC instead of just simple elution reversal for specific peptide mixtures.
3.2.3. HILIC 3 standards
The HILIC 3 mixture is composed of nine peptides of identical charge (+3), with only a subtle hydrophobicity variation between adjacent peptides – generally just a single CH2 group change from peptide to peptide (Table 1) [31,51]. The only exception is an even more subtle hydrophobicity change between peptides 3e/3f which have the same number of carbon atoms and the same mass (the Val-Ala sequence is changed to an Ile-Gly sequence). Thus, this peptide mixture represents a particularly potent test of RPC and HILIC column selectivity. Once more, the presence of Gly residues throughout the sequence of these standards ensures the absence of any secondary structure.
Fig. 4 compares RPC and HILIC of the nine-peptide mixture. RPC in the presence of TFA (Fig. 4A) and H3PO4 (Fig. 4B) is able to separate the nine peptides, albeit not quite to baseline for the d/e and f/g peptide pairs. Indeed, the separations are similar for the two ion-pairing reagent systems, the main difference being the earlier elution of the peptides in the presence of the more hydrophilic H3PO4 compared to TFA. Fig. 4C shows the HILIC separation of the nine peptides with 90% starting concentration of acetonitrile. All nine peptides are seen, with a straightforward reversal of peptide elution compared to RPC (Fig. 4A and B). A decreasing shallow gradient of −0.25% acetonitrile/min was required to achieve this separation, which nonetheless demonstrated the similar ability of HILIC compared to RPC to resolve a challenging peptide mixture. Fig. 4D now demonstrates how modifying the starting acetonitrile concentration (i.e., a decrease to 85%) may achieve the same satisfactory separative result within a shorter run time: tR peptide i − a = 11.8 min for 90% run (top right) and 11.9 min for 85% run (bottom right), with a 20 min decrease in run time for the latter run. It is interesting to note that the peak broadening of the peptides during HILIC (Fig. 4C and D) compared to RPC (Fig. 4A and B), which increases with increasing hydrophilicity of the peptides, is eliminated during HILIC/SALT elution (Fig. 5).
Fig. 4.

Separation of +3 synthetic random coil peptide standards by RPC (panels A and B) and HILIC (panels C and D). Conditions: RPC, linear AB (increasing acetonitrile) gradient of 1% acetonitrile/min from 2% aq. acetonitrile, with TFA (panel A) or H3PO4 (panel B) as the ion-pairing reagent; HILIC, linear AB (decreasing acetonitrile) gradient of −0.25% acetonitrile/min from starting conditions of 90% aq. acetonitrile/0.2% H3PO4 or 85% aq. acetonitrile/0.2% H3PO4, following 5-min isocratic elution with these starting conditions. Sequences of peptides a–i are shown in Table 1.
Fig. 5.

HILIC/SALT separation of +3 synthetic random coil peptide standards. Conditions: linear (increasing salt) gradient of 1 mM NaClO4/min (panel A) or 2 mM NaClO4/min (panels B and C) in the presence of 90% (panels A and B) or 85% (panel C) aq. acetonitrile/0.2% H3PO4, following 5-min isocratic elution in starting conditions of 90% aq. acetonitrile/0.2% H3PO4 (panels A and B) or 85% aq. acetonitrile/0.2% H3PO4 (panel C). Sequences of peptides a–i shown in Table 1.
Fig. 5 illustrates the effect of varying run conditions on the HILIC/SALT separation of the HILIC 3 standards. Thus, at a gradient rate of 1 mM salt/min and in the presence of 90% acetonitrile (Fig. 5A), the peptides exhibit a separation range (from peptide i to a) of 67.4 min. From Fig. 5B, increasing the gradient to 2 mM salt/min still maintained a separation range of 37.8 min (in a significantly decreased run time compared to the 1 mM/min run) compared to both the RPC and HILIC runs shown in Fig. 4. Note also that the majority of the nine peptides in the HILIC/SALT runs were resolved to baseline with the interesting, and sole, exception of the e/f peptide pair, i.e., the two peptides with identical carbon content. This particular peptide pair was seen to be resolved by both RPC and HILIC (Fig. 4). However, partial resolution of the e/f peptide pair was achieved when lowering the acetonitrile concentration to 85%, whilst maintaining a 2 mM salt/min gradient (Fig. 5C), showing the importance of the interplay between the salt gradient rate and the isocratic concentration of acetonitrile. Finally, as noted above, the improvement in peak shape of these peptides during the HILIC/SALT runs in Fig. 5 compared to HILIC (Fig. 4C and D) is clear, i.e., elution with a salt gradient in the presence of a high acetonitrile concentration (Fig. 5) results in improved peak shape compared to elution by a decreasing acetonitrile gradient (Fig. 4C and D).
3.3. Separation of amphipathic α-helical peptide standards
From Table 2, all three sets of peptide standards are amphipathic α-helical peptides, i.e., with non-polar and polar faces. Thus, the non-polar face represents a preferred binding domain for RPC [55], whereas the polar face could represent a preferred binding interaction domain for HILIC, as has previously been demonstrated for mixed-mode HILIC/CEX [31,56]. It has previously been shown that high concentrations of organic modifiers such as acetonitrile can induce helix formation in a potentially helical peptide [54,55]. Thus, under characteristic conditions of RPC (increasing acetonitrile concentration in mobile phase plus the hydrophobic environment of the stationary phase) and HILIC or HILIC/SALT (high concentrations of acetonitrile), the model peptides shown in Table 2 would be expected to be α-helical, allowing interaction of the hydrophobic face or hydrophilic face with the RPC and HILIC matrices, respectively. The peptides shown in Table 2 offer interesting insights into the differences and resolving capabilities of RPC, HILIC and HILIC/SALT for such peptides, differing as they do in the type and position of residue substitutions: either in the polar face (LX7 peptides) or in the non-polar face (D1–D4 and D12–D14 peptides).
3.3.1. LX7 standards
Fig. 6 shows helical wheel (left) and helical net (right) representations of the LX7 sequence, where position X, in the polar face, is substituted by Ser (LS7), Thr (LT7) or Val (LV7) [56]. Serine and threonine are classed as polar amino acids with the latter being slightly less polar than the former; in contrast, valine is classed as a significantly non-polar amino acid [57]. All four peptides have the same net charge of +4 at pH 2.
Fig. 6.

Helical wheel (left) and helical net (right) representations of synthetic amphipathic α-helical LX7 peptides, where position X7, in the polar face of the helix, is substituted by valine (LV7), serine (LS7) or threonine (LT7). The radius of the α-helix is taken as 2.5 Å with 3.6 residues per turn, a residue translation of 1.5 Å and thus a pitch of 5.4 Å. The Lys residues in the polar face are shaded. Sequences of peptides shown in Table 2.
From Fig. 7, RPC was, not surprisingly, completely unable to separate the three peptides in the presence of TFA (Fig. 7A) or H3PO4 (Fig. 7B). Due to the preferential binding of the non-polar face to the RPC stationary phase, this mode of HPLC cannot differentiate between peptides substituted in the polar face, even when the substitutions vary widely in polarity, i.e., from polar amino acids Ser and Thr to the hydrophobic Val.
Fig. 7.

Separation of a mixture of synthetic amphipathic α-helical peptide standards (LV7, LT7, LS7) by RPC (panels A and B), HILIC (panel C), isocratic HILIC (panel D) and HILIC/SALT (panel E). Conditions: RPC, linear AB (increasing acetonitrile) gradient of 1% acetonitrile/min from 2% aq. acetonitrile, with TFA (panel A) or H3PO4 (panel B) as the ion-pairing reagent; isocratic HILIC, 81% aq. acetonitrile/0.2% H3PO4; HILIC, linear AB (decreasing acetonitrile) gradient of −0.25% acetonitrile/min from starting conditions of 90% aq. acetonitrile/0.2% H3PO4, following 5-min isocratic elution with these starting conditions; HILIC/SALT, linear (increasing salt) gradient of 2 mM NaClO4/min in the presence of 90% aq. acetonitrile/0.2% H3PO4, following 5-min isocratic elution in starting conditions of 90% aq. acetonitrile/0.2% H3PO4. Sequences of peptides shown in Table 1 and Fig. 6.
Some success is seen in separating the three peptides by HILIC (Fig. 7C), with the peptides being eluted in order of increasing hydrophilicity: LV7 < LT7 < LS7. However, the range of separation from LV7 to LS7 was just 2.5 min, even with a shallow decreasing acetonitrile gradient of −0.25%/min. Isocratic HILIC elution in the presence of 81% acetonitrile produced the profile shown in Fig. 7D. Although the separation showed an improvement over that of HILIC in gradient mode (Fig. 7C), this was at the expense of extensive peak broadening under isocratic conditions (Fig. 7D). The most dramatic separation is achieved by HILIC/SALT (Fig. 7E), with a range of separation between first and last eluted peaks of 19 min under the conditions employed (90% isocratic acetonitrile; 2 mM salt/min), as well as excellent peak shape compared to isocratic HILIC elution (Fig. 7D). It should also be noted that, in an analogous manner to isocratic RPC of peptides [40], the isocratic separation of the three peptides was extremely sensitive to acetonitrile concentration and optimization (in this case, satisfactory separation within a similar run to the HILIC/SALT run in Fig. 7E) took several attempts to achieve. This narrow “partitioning window” for RPC of peptides is well known [40] and also appears to be a factor in isocratic HILIC elution. The HILIC and HILIC/SALT results support the premise of preferred interaction of the polar face of the amphipathic α-helical peptides with the HILIC stationary phase. Further, the HILIC/SALT approach appears to enhance hydrophilic/hydrophobic differences between substituted residues in the polar face of the peptides, as evidenced by the much more dramatic separation of the peptides by HILIC/SALT compared to traditional HILIC.
3.3.2. D1–D4 and D12–D14 standards
Fig. 8 shows helical net representations of the +8 series (D1–D4; Table 2) and +11 series (D12–D14; Table 2) amphipathic α-helical peptide standards [33]. In both series, Ala to Leu substitutions are being made in the more non-polar face of the peptide, increasing the overall hydrophobicity of the peptides (D1 < D2 < D3 < D4 and D14 < D13 < D12). Concomitant with these substitutions of the small Ala hydrophobe with the more bulky Leu hydrophobe is an increase of hydrophobic interactions (i to i + 3, denoting a residue at position i interacting with a residue three positions away in the sequence; i to i + 4, denoting a residue at position i interacting with a residue four positions away in the sequence) between non-polar residues on the non-polar face.
Fig. 9A shows the RPC elution profile of the combined D1–D4 and D12–D14 peptide mixtures. Despite some overlap between the two peptide mixtures (specifically, D1 is eluted just prior to D12), the peptide components of the individual peptide mixtures are seen to be resolved in order of increasing hydrophobicity.
Fig. 9.

Separation of a mixture of synthetic α-helical peptides (D1–D4, D12–D14) by RPC (panel A), HILIC (panel B) and HILIC/SALT (panels C and D). Conditions: RPC, linear AB (increasing acetonitrile) gradient of 1% acetonitrile/min from 2% acetonitrile; HILIC, linear AB (decreasing acetonitrile) gradient of −0.5% acetonitrile/min from starting conditions of 90% aq. acetonitrile/0.2% H3PO4, following 5-min isocratic elution with these starting conditions; HILIC/SALT, linear (increasing salt) gradient of 2 mM NaClO4/min (panel C) or 1 mM NaClO4/min (panel D) in the presence of 85% aq. acetonitrile/0.2% H3PO4 (panel C) or 80% aq. acetonitrile/0.2% H3PO4 (panel D), following 5-min isocratic elution in starting conditions of 85% aq. acetonitrile/0.2% H3PO4 (panel C) or 80% aq. acetonitrile/0.2% H3PO4 (panel D). Sequences of peptides shown in Table 2 and Fig. 8.
From Fig. 9B, HILIC is only able to achieve a very limited resolution of the peptides, with the D1–4 (+8) peptide series being separated from the D12–D14 (+11) series but no resolution of peptides within these groups. Thus, the overall more highly charged +11 peptides are being eluted later than the +8 peptides. However, clearly HILIC cannot differentiate well the differences in hydrophilicity/hydrophobicity of the peptides within these groups where Ala to Leu substitutions were made on the non-polar face of the helix.
In contrast to HILIC, the HILIC/SALT approach produced a dramatic improvement in peptide separation, with both peptide series resolved to near baseline in the presence of 85% acetonitrile and a salt gradient of 2 mM/min (Fig. 9C). Within the two peptide series, the peptides are eluted in reversed order to that of RPC (Fig. 9A), albeit with much improved separation between the two peptide groups. This is a particularly significant observation when one considers that Ala to Leu substitutions are being made on the non-polar face of the α-helical peptides. Thus, whilst the polar face represents a preferred region of interaction with the HILIC stationary phase, the HILIC/SALT elution approach also appears to be sensitive to overall peptide hydrophilicity/hydrophobicity (also indicated in Fig. 7 for the LX7 peptides) in a more significant manner than traditional HILIC (Fig. 9B). Fig. 9D again demonstrates how the HILIC/SALT approach may be manipulated to maintain excellent separations within a lesser run time, i.e., decreasing the isocratic acetonitrile concentration to 80% and the salt gradient to 1 mM/min.
4. Conclusions
This study introduces a new dimension to traditional HILIC for the separation of peptides, termed HILIC/SALT. This approach is characterized by the employment of increasing salt (NaClO4) gradients in the presence of high isocratic acetonitrile (80–90%) concentrations to elute peptides in contrast to the traditional decreasing acetonitrile gradient elution method. This initial study applied RPC, HILIC and HILIC/SALT to the separation of mixtures of synthetic peptide standards varying in structure (amphipathic α-helix, random coil), length (10–26 residues), charge (+1 to +11) and hydrophilicity/hydrophobicity. Results showed consistent and marked superiority of the HILIC/SALT approach for separation of these peptide standards compared to traditional HILIC, offering a flexible and excellent complement to RPC for peptide separations, including a role as a component of multicolumn HPLC. Current efforts are directed to expanding the range of mobile phase modifiers that could be used for HILIC/SALT, perhaps to include more MS tolerant conditions. In addition, the ability of HILIC/SALT to separate more complex peptide mixtures, such as those resulting from protein digests, as well as its utility for highly polar peptides not retained by RPC columns will be investigated. Nevertheless, these initial results indicate an excellent potential for HILIC packings using HILIC/SALT to enter the mainstream of approaches to peptide separations.
Acknowledgments
This work was supported by an NIH Grant (RO1 GM61855) to Robert S. Hodges and the John Stewart Chair in Peptide Chemistry to Robert S. Hodges and an NIH Grant (GM093747) to Advanced Materials Technology.
References
- 1.Alpert AJ. J Chromatogr. 1990;499:177. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 2.Oyler AR, Armstrong BL, Cha JY, Zhou MX. J Chromatogr A. 1996;724:378. [Google Scholar]
- 3.Yoshida T. Anal Chem. 1997;69:3038. doi: 10.1021/ac9702204. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida T. J Chromatogr A. 1998;808:105. doi: 10.1016/s0021-9673(98)00092-2. [DOI] [PubMed] [Google Scholar]
- 5.Schichtherle-Cerny H, Affotler M, Cerny C. Anal Chem. 2003;75:2349. doi: 10.1021/ac026313p. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida T. J Biochem Biophys Methods. 2004;60:265. doi: 10.1016/j.jbbm.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Wuhrer M, Koeleman CAM, Hokke CH, Deelder AM. Int J Mass Spectrom. 2004;232:51. [Google Scholar]
- 8.Wuhrer M, Koeleman CAM, Hokke CH, Deelder AM. Anal Chem. 2005;77:886. doi: 10.1021/ac048619x. [DOI] [PubMed] [Google Scholar]
- 9.Gilar M, Olivova P, Daly AE, Gebler JC. Anal Chem. 2005;77:6426. doi: 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- 10.Gilar M, Jaworski A. J Chromatogr A. 2011;1218:8890. doi: 10.1016/j.chroma.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Dorpe SV, Vergote V, Pezeshki A, Burvenich C, Peremans K, Spiegeleer BD. J Sep Sci. 2010;33:728. doi: 10.1002/jssc.200900476. [DOI] [PubMed] [Google Scholar]
- 12.Harscoat-Schiavo C, Nioi C, Ronat-Heit E, Paris C, Vanderesse R, Fournier F, Marc I. Anal Bioanal Chem. 2012;403:1939. doi: 10.1007/s00216-012-5987-6. [DOI] [PubMed] [Google Scholar]
- 13.Mant CT, Hodges RS, editors. HPLC of Peptides and Proteins: Separation, Analysis and Conformation. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 14.Mant CT, Chen Y, Yan Z, Popa TV, Kovacs JM, Mills JB, Tripet BP, Hodges RS. In: Methods in Molecular Biology. Fields G, editor. Humana Press; Totowa, NJ, USA: 2007. p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen HP, Schug KA. J Sep Sci. 2008;31:1465. doi: 10.1002/jssc.200700630. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, Boysen RI, Hearn MTW. J Chromatogr A. 2009;1216:5518. doi: 10.1016/j.chroma.2009.05.085. [DOI] [PubMed] [Google Scholar]
- 17.Boersema PJ, Divecha N, Heck AJR, Mohammed S. J Proteome Res. 2007;6:937. doi: 10.1021/pr060589m. [DOI] [PubMed] [Google Scholar]
- 18.Boersema PJ, Mohammed S, Heck AJR. Anal Bioanal Chem. 2008;391:151. doi: 10.1007/s00216-008-1865-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ow SY, Salim M, Noirel J, Evans C, Wright PC. Proteomics. 2011;11:2341. doi: 10.1002/pmic.201000752. [DOI] [PubMed] [Google Scholar]
- 20.Di Palma S, Boersema PJ, Heck AJR, Mohammed S. Anal Chem. 2011;83:3440. doi: 10.1021/ac103312e. [DOI] [PubMed] [Google Scholar]
- 21.Di Palma S, Raijmakers R, Heck AJR, Mohammed S. Anal Chem. 2011;83:8352. doi: 10.1021/ac2018074. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Y, Kong RPW, Li G, Lam MPY, Law CH, Lee SMY, Lam HC, Chu IK. J Sep Sci. 2012;35:1755. doi: 10.1002/jssc.201200054. [DOI] [PubMed] [Google Scholar]
- 23.Jandera P. Anal Chim Acta. 2011;692:1. doi: 10.1016/j.aca.2011.02.047. [DOI] [PubMed] [Google Scholar]
- 24.Buszewski B, Noga S. Anal Bioanal Chem. 2012;402:231. doi: 10.1007/s00216-011-5308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu BY, Mant CT, Hodges RS. J Chromatogr. 1991;548:13. doi: 10.1016/s0021-9673(01)88590-3. [DOI] [PubMed] [Google Scholar]
- 26.Zhu BY, Mant CT, Hodges RS. J Chromatogr. 1992;594:75. [Google Scholar]
- 27.Stenman UH, Laatikainen T, Salminen K, Huhtala ML, Leppaluoto J. J Chromatogr. 1984;297:399. doi: 10.1016/s0021-9673(01)89061-0. [DOI] [PubMed] [Google Scholar]
- 28.Crimmins DL, Gorka J, Thoma RS, Schwartz BD. J Chromatogr. 1988;443:63. doi: 10.1016/s0021-9673(00)94783-6. [DOI] [PubMed] [Google Scholar]
- 29.Alpert AJ, Andrews PC. J Chromatogr. 1988;443:85. doi: 10.1016/s0021-9673(00)94785-x. [DOI] [PubMed] [Google Scholar]
- 30.Dizdaroglu M. J Chromatogr. 1985;334:49. [Google Scholar]
- 31.Mant CT, Hodges RS. J Sep Sci. 2008;31:2754. doi: 10.1002/jssc.200800243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hemstrom P, Irgum K. J Sep Sci. 2006;29:1784. doi: 10.1002/jssc.200600199. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Z, Vasil AI, Gera L, Vasil ML, Hodges RS. Chem Biol Drug Design. 2011;77:225. doi: 10.1111/j.1747-0285.2011.01086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Stefano JJ, Langlois TJ, Kirkland JJ. J Chromatogr Sci. 2008;46:254. doi: 10.1093/chromsci/46.3.254. [DOI] [PubMed] [Google Scholar]
- 35.Kirkland JJ, Langlois TJ. Am Lab. 2007;39:18. [Google Scholar]
- 36.Cunliffe JM, Maloney TD. J Sep Sci. 2007;30:3104. doi: 10.1002/jssc.200700260. [DOI] [PubMed] [Google Scholar]
- 37.Marchetti N, Cavazzini A, Gritti F, Guiochon G. J Chromatogr A. 2007;1163:203. doi: 10.1016/j.chroma.2007.06.046. [DOI] [PubMed] [Google Scholar]
- 38.Ruta J, Guillarme D, Rudaz S, Veuthey JL. J Sep Sci. 2010;33:1. doi: 10.1002/jssc.201000023. [DOI] [PubMed] [Google Scholar]
- 39.Schuster SA, Boyes BE, Wagner BM, Kirkland JJ. J Chromatogr A. 2012;1228:232. doi: 10.1016/j.chroma.2011.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mant CT, Burke TWL, Hodges RS. Chromatographia. 1987;24:565. [Google Scholar]
- 41.Mahoney WC. Biochim Biophys Acta. 1982;704:284. doi: 10.1016/0167-4838(82)90158-3. [DOI] [PubMed] [Google Scholar]
- 42.Gregor HP, Collins FC, Pope M. J Colloid Sci. 1951;6:304. [Google Scholar]
- 43.Guo D, Mant CT, Hodges RS. J Chromatogr. 1987;386:205. doi: 10.1016/s0021-9673(01)94598-4. [DOI] [PubMed] [Google Scholar]
- 44.Shibue M, Mant CT, Hodges RS. J Chromatogr A. 2005;1080:58. doi: 10.1016/j.chroma.2005.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shibue M, Mant CT, Hodges RS. J Chromatogr A. 2005;1080:68. doi: 10.1016/j.chroma.2005.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mant CT, Hodges RS. Chromatographia. 1987;24:805. [Google Scholar]
- 47.Sereda TJ, Mant CT, Hodges RS. J Chromatogr A. 1997;776:153. doi: 10.1016/s0021-9673(97)00150-7. [DOI] [PubMed] [Google Scholar]
- 48.Mant CT, Hodges RS, Magazine LC. Liq Chromatogr HPLC. 1986;4:250. [Google Scholar]
- 49.Mant CT, Parker JMR, Hodges RS. J Chromatogr. 1987;397:99. doi: 10.1016/s0021-9673(01)84993-1. [DOI] [PubMed] [Google Scholar]
- 50.Mant CT, Burke TWL, Black JA, Hodges RS. J Chromatogr. 1988;458:193. doi: 10.1016/s0021-9673(00)90564-8. [DOI] [PubMed] [Google Scholar]
- 51.Mant CT, Hodges RS. J Sep Sci. 2008;31:1573. doi: 10.1002/jssc.200700619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mant CT, Cepeniene D, Hodges RS. J Sep Sci. 2010;33:3005. doi: 10.1002/jssc.201000518. [DOI] [PubMed] [Google Scholar]
- 53.Mant CT, Hodges RS. J Chromatogr A. 2012;1230:30. doi: 10.1016/j.chroma.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lau SYM, Taneja AK, Hodges RS. J Chromatogr. 1984;317:129. [Google Scholar]
- 55.Zhou NE, Mant CT, Hodges RS. Peptide Res. 1990;3:8. [PubMed] [Google Scholar]
- 56.Mant CT, Litowski JR, Hodges RS. J Chromatogr A. 1998;816:65. doi: 10.1016/s0021-9673(98)00507-x. [DOI] [PubMed] [Google Scholar]
- 57.Kovacs JM, Mant CT, Hodges RS. Biopolymers (Pept Sci) 2006;84:298. doi: 10.1002/bip.20417. [DOI] [PMC free article] [PubMed] [Google Scholar]