

Abstract
We describe a fluorescence microscopy method, Co-Translational Activation by Cleavage (CoTrAC) to image the production of protein molecules in live cells with single-molecule precision without perturbing the protein's functionality. This method makes it possible to count the numbers of protein molecules produced in one cell during sequential, five-minute time windows. It requires a fluorescence microscope with laser excitation power density of ~0.5 to 1 kW/cm2, which is sufficiently sensitive to detect single fluorescent protein molecules in live cells. The fluorescent reporter used in this method consists of three parts: a membrane targeting sequence, a fast-maturing, yellow fluorescent protein and a protease recognition sequence. The reporter is translationally fused to the N-terminus of a protein of interest. Cells are grown on a temperature-controlled microscope stage. Every five minutes, fluorescent molecules within cells are imaged (and later counted by analyzing fluorescence images) and subsequently photobleached so that only newly translated proteins are counted in the next measurement.
Fluorescence images resulting from this method can be analyzed by detecting fluorescent spots in each image, assigning them to individual cells and then assigning cells to cell lineages. The number of proteins produced within a time window in a given cell is calculated by dividing the integrated fluorescence intensity of spots by the average intensity of single fluorescent molecules. We used this method to measure expression levels in the range of 0-45 molecules in single 5 min time windows. This method enabled us to measure noise in the expression of the λ repressor CI, and has many other potential applications in systems biology.
Keywords: Biophysics, Issue 73, Biochemistry, Genetics, Chemistry, Molecular Biology, Cellular Biology, Microbiology, Proteins, Single molecule, fluorescence protein, protein expression, cotranslational activation, CoTrAC, cell culture, fluorescent microscopy, imaging, translational activation, systems biology
Protocol
1. Strain Engineering Workflow
Insert sequences encoding (a) a membrane-localization sequence, (b) a fast-maturing fluorescent protein and (c) a protease recognition sequence N-terminal to and in frame with a protein of interest (e.g. a transcription factor). We used the membrane targeted Tsr-Venus reporter 2 and fused it to the protease recognition sequence Ubiquitin (Ub) to count the number of expressed bacteriophage λ repressor CI protein molecules. Details on how we constructed the fusion protein Tsr-Venus-Ub-CI, replacing the wild-type CI coding region and incorporating the construct into the E. coli chromosome using λ Red recombination 3, are described in detail in ref. 1.
Verify all modifications by sequencing.
Transform a plasmid encoding a protease specific to the recognition sequence used above into a strain expressing the fluorescent reporter and protein of interest in translational fusion. We used the protease Ubp1, which cleaves immediately after the C-terminal Ubiquitin residue 4.
2. Culture Cells and Prepare Sample
Pick one E. coli colony of interest from a freshly streaked agar plate into 1 ml M9 minimal media 5 supplemented with 1X MEM amino acids and appropriate antibiotics. Incubate in a shaker at desired temperature long enough to reach OD600 > 1.0 (optical density at 600 nm).
Dilute the culture into 1 ml fresh M9 medium with appropriate antibiotics to OD600 = 0.02. Incubate in a shaker at appropriate temperature. Initial cellular density can be increased if needed for low-temperature growth.
When the OD600 = 0.2-0.3 (at 37 °C with an initial cell culture at OD600 = 0.02, this will take 3-4 hr), start preparing the agarose gel pad (step 3).
The cells are ready for imaging when OD600 = 0.3-0.4.
Transfer 1 ml incubated culture into a 1.5 ml microcentrifuge tube, centrifuge at 10,000 x g for 1 min in a benchtop microcentrifuge.
Discard the supernatant and add 1 ml fresh M9 medium and resuspend the pellet gently.
Centrifuge at 10,000 x g for 1 min.
Repeat step 1.6 and 1.7. Discard the supernatant.
Gently resuspend in 1 ml M9 media. Cells can be directly used for microscope imaging or diluted 10- to 100-fold to ensure low cell densities for timelapse imaging. Note that low cell densities are important for extended timelapse imaging. The imaging chamber (step 4) is sealed during the experiment. Due to exponential growth of cells, high initial cell concentrations can significantly deplete oxygen within the chamber after prolonged growth in the gel pad, reducing fluorescent protein maturation and affecting cell growth.
3. Prepare the Agarose Solution
Weigh 10-20 mg low-melting-temperature agarose into a 1.5 ml microcentrifuge tube.
Add appropriate volume M9 minimal medium without antibiotics to make a 3% agarose solution.
Heat the agarose solution for 30 min at 70 °C to melt, inverting the tube to ensure that the solution is completely melted and homogenous. The gel pad can be poured at this point (step 4) or the temperature can be decreased to 50 °C and held for several hours for later use.
4. Prepare Gel Pad on the Chamber
About 50 min before imaging, rinse a Microaqueduct slide with water.
Rinse one cleaned cover glass (using a stringent cleaning method such as that described in 6) and Microaqueduct slide with sterile water and dry by blowing with compressed air.
Place the rubber gasket on the Microaqueduct slide so that it covers the inlets and outlets on the glass side of the Microaqueduct slide (this can be modified for applications in which media is perfused through the sample). Apply 50 μl agarose solution (step 3) to the center of the Microaqueduct slide.
Top the agarose solution with the cleaned, dry cover glass.
Let the agarose solution stand at room temperature for 30 min.
Meanwhile, prepare a cell sample (step 2).
Carefully peel off the cover glass from the top of gel pad. Add 1.0 μl washed cell culture to the top of the gel pad. Wait for ~2 min for the culture to be absorbed by the gel pad. It is important not to wait so long that the gel pad overdries, but long enough that cells are properly adhered to the gel pad. The Ideal waiting time will vary with temperature and humidity.
While waiting, dry another precleaned cover glass.
Cover the sample with the new cover glass ensuring that the cover glass and Microaqueduct slide are well aligned.
Assemble the temperature controlled growth chamber following the manufacturer's instructions. It can now be used for imaging on microscope (step 5).
5. Imaging and Acquisition of Timelapse Movies
Turn on the laser and microscope following manufacturer's instructions (e.g. the laser may need to warmed up for ~0.5 hr before the experiment).
Lock the assembled chamber to the stage insert on the microscope. Set the temperature of the growth chamber and the objective heater at 37 °C or other desired growth temperature.
If imaging above room temperature, the focus or gel pad will usually drift significantly for ~15 min after the initial temperature shift. In practice, use this time to find regions of interest and modify imaging scripts as necessary for an experiment.
Find cells on the microscope and store the position of each cell after centering it within a predefined and aligned imaging region. More than one cell can be imaged in each image acquisition time window. For timelapse imaging over many generations, ensure that imaged cells are initially separated from other cells by at least a few hundred μm so that other colonies will not enter the imaging region during growth.
Adjust laser excitation intensity so that almost all fluorescent molecules photobleach within 6 exposures (Figure 5).
Using an automated imaging script/journal, acquire timelapse data using the algorithm described in the experimental workflow in Figure 3.
Representative Results
Typical results from a CoTrAC experiment tracking the production of the λ repressor CI are shown in Figures 4 and 5. In this experiment, 12 colonies were imaged at 5 min intervals. At each time point, the colony was first autofocused and centralized within the imaging region. Next, the centered/focused position was stored and a brightfield image was acquired. The stage was then translocated by ~0.5 μm along the z-axis to move from the brightfield focal plane to the plane bisecting the cells (without phase-contrast or differential interference contrast (DIC) optics, semi-transparent objects can be imaged at slightly out-of-focus planes 8). Finally, six images were acquired at 1 sec intervals, each with a 100 msec exposure (Figure 5 shows one such time point). This was repeated for all cells at each time point. Because almost all Venus molecules are photobleached at each timepoint, the fluorescent spots in Figures 4 and 5 correspond to Venus molecules that matured (became fluorescent) within the past 5 min. Venus molecules mature quickly in vivo with a half-life of ~2-7 min 1, 2, 7 and cleavage by Ubp1 is very efficient 4, 9. Therefore, the number of protein molecules produced since the last measurement can be accurately inferred from the number of Venus molecules counted on the membrane.
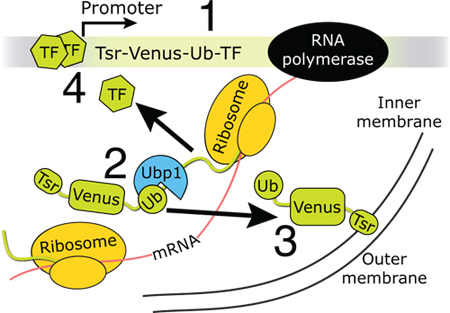 Figure 1. Using CoTrAC to count the expression of single transcription factor molecules.(1) A reporter including the membrane-localization sequence Tsr, the fast-maturing YFP Venus, and Ubiquitin (Ub) is expressed in translational fusion with a transcription factor from a promoter regulated by the transcription factor. (2) The protease Ubp1 co-translationally cleaves the nascent polypeptide after the C-terminal Ub residue. (3) The Tsr-Venus-Ub reporter localizes to the membrane where it can be counted at the single-molecule level. (4) The transcription factor can regulate its own expression. Method cartoon.
Figure 1. Using CoTrAC to count the expression of single transcription factor molecules.(1) A reporter including the membrane-localization sequence Tsr, the fast-maturing YFP Venus, and Ubiquitin (Ub) is expressed in translational fusion with a transcription factor from a promoter regulated by the transcription factor. (2) The protease Ubp1 co-translationally cleaves the nascent polypeptide after the C-terminal Ub residue. (3) The Tsr-Venus-Ub reporter localizes to the membrane where it can be counted at the single-molecule level. (4) The transcription factor can regulate its own expression. Method cartoon.
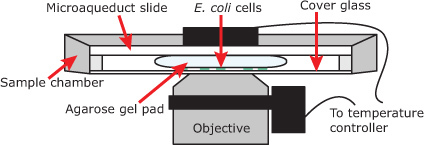 Figure 2. Schematic of sample chamber used in CoTrAC experiments. Cells are placed between agarose gel pad and cover glass. Microaqueduct slide and objective are held at a fixed temperature using an electronic controller. Sample preparation.
Figure 2. Schematic of sample chamber used in CoTrAC experiments. Cells are placed between agarose gel pad and cover glass. Microaqueduct slide and objective are held at a fixed temperature using an electronic controller. Sample preparation.
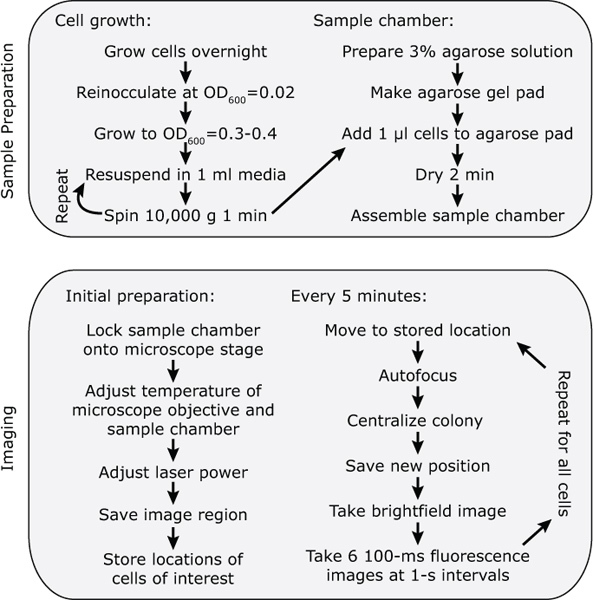 Figure 3. Workflow of a typical CoTrAC experiment. Experimental workflow.
Figure 3. Workflow of a typical CoTrAC experiment. Experimental workflow.
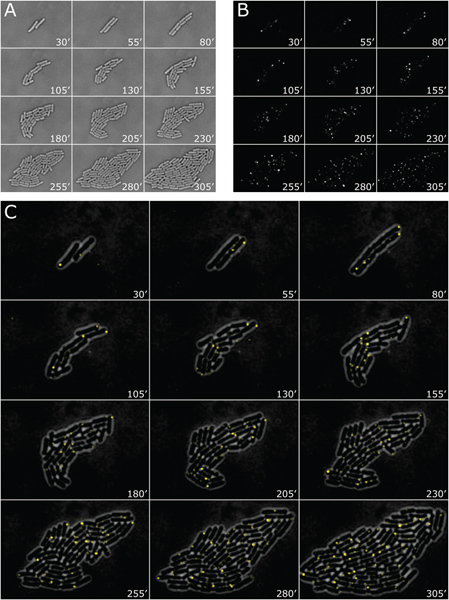 Figure 4. Typical timelapse data from a CoTrAC experiment. Images from a single colony are shown at 25 min intervals. In this experiment, data were acquired every 5 min from 12 different colonies. (A) Brightfield images. (B) Venus (YFP) fluorescence image (average background subtracted to account for change in background of entire imaging field over time). (C) Overlay image shows pole localization of Tsr-Venus-Ub reporter molecules and heterogeneity in the number of molecules produced in 5 min time windows in different cells (brightfield image is inverted and Venus image is bandpass filtered). Click here to view larger figure.
Figure 4. Typical timelapse data from a CoTrAC experiment. Images from a single colony are shown at 25 min intervals. In this experiment, data were acquired every 5 min from 12 different colonies. (A) Brightfield images. (B) Venus (YFP) fluorescence image (average background subtracted to account for change in background of entire imaging field over time). (C) Overlay image shows pole localization of Tsr-Venus-Ub reporter molecules and heterogeneity in the number of molecules produced in 5 min time windows in different cells (brightfield image is inverted and Venus image is bandpass filtered). Click here to view larger figure.
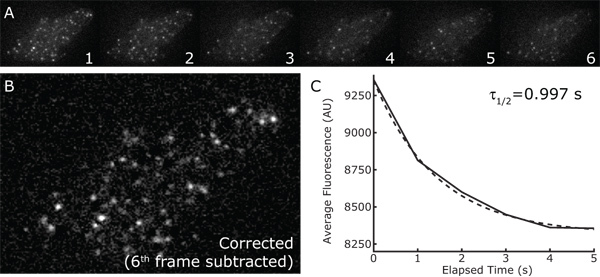 Figure 5. Typical data from a single time point in a CoTrAC experiment.(A) Venus (YFP) fluorescence images. Six 100 msec exposures were acquired at the beginning of each 5 min time interval. Each of the 6 exposures was separated by 900 msec to allow time for transiently dark Venus molecules that had blinked off to become fluorescent. (B) For analysis, image 6 is subtracted from image 1 to correct for unbleached molecules and autofluorescence background. Spots in this image are localized to specific cell lineages and quantified by their integrated fluorescence intensity. (C) The average integrated fluorescence of the colony in A is plotted over 6 images (solid line). Fitting this line to an exponential decay plus a constant offset (dashed line) gives a decay half-time of 1 sec. In this experiment, Venus molecules photobleach much more quickly than cellular autofluorescence, so this means that approximately half of the total number of Venus molecules are photobleached in each frame. Photobleaching progress at a single time point.
Figure 5. Typical data from a single time point in a CoTrAC experiment.(A) Venus (YFP) fluorescence images. Six 100 msec exposures were acquired at the beginning of each 5 min time interval. Each of the 6 exposures was separated by 900 msec to allow time for transiently dark Venus molecules that had blinked off to become fluorescent. (B) For analysis, image 6 is subtracted from image 1 to correct for unbleached molecules and autofluorescence background. Spots in this image are localized to specific cell lineages and quantified by their integrated fluorescence intensity. (C) The average integrated fluorescence of the colony in A is plotted over 6 images (solid line). Fitting this line to an exponential decay plus a constant offset (dashed line) gives a decay half-time of 1 sec. In this experiment, Venus molecules photobleach much more quickly than cellular autofluorescence, so this means that approximately half of the total number of Venus molecules are photobleached in each frame. Photobleaching progress at a single time point.
Discussion
The CoTrAC method can be generalized to measure the production of other proteins where conventional N- or C-terminal fluorescent protein fusions may disrupt protein activity. The CoTrAC strategy has three unique advantages over current methods. First, co-translational fusion ensures that one molecule of the fluorescent reporter is produced for each molecule of the protein of interest, allowing accurate counting of protein production in real time. Second, the membrane-targeted reporter Tsr-Venus enables single-molecule detection of fluorescent reporters, enabling the detection of proteins expressed at very low levels. Third and most importantly, the co-translational cleavage of the fluorescent reporter allows the protein of interest to retain its near-wild-type sequence and function normally-using Ubiquitin as a protease-recognition sequence in E. coli, target proteins only differ from their wild-type peptide sequences in that the first N-terminal N-formyl-methionine is changed to methionine.
When using the CoTrAC method, It is important to keep in mind that the CoTrAC method measures the number of protein molecules produced at each 5-min time window, which is distinct from the measurement of the number of protein molecules present per cell (i.e. the protein concentration). Most single-cell fluorescence gene expression assays measure protein concentration, which is the balanced result of protein production and degradation. Therefore, the degradation rate of the protein of interest must be considered carefully if the concentration measurement is used to infer protein production information. Furthermore, protein concentration is subject to fluctuations caused by partitioning of the protein between two daughter cells at cell division, which may be significant for low-expression-level proteins and mistakenly interpreted as arising from protein production dynamics. The CoTrAC method can be modified to measure protein concentration by not photobleaching newly produced reporter molecules, but this must be done with extra scrutiny. One must verify that the degradation rates of the fluorescent reporter and the protein of interest are similar, and that they are both similarly partitioned into two daughter cells upon cell division, so that the concentration of the fluorescent reporter reflects that of the protein of interest.
We note that while the CoTrAC does not alter the protein sequence, the addition of the reporter gene changes the mRNA sequence. Therefore, transcription and translation dynamics and/or mRNA transcript degradation rates may be perturbed. If the CoTrAC method is used to observe protein production in the wild-type context, it is important to compare expression levels of the target protein both in its wild-type sequence and with the CoTrAC fluorescent fusion tag (e.g. in ref. 1 we compared CI expression in our CoTrAC construct to that in a wild-type lysogen). Below we discuss considerations necessary for generalizing the CoTrAC method.
First, we discuss the possibility of using sequences other than Tsr-Venus-Ub. The general requirements are: (1) the reporter must be localized to some position within the cell so that it does not diffuse quickly (with Tsr, reporters are at the cell poles1). This is necessary for single-molecule detection of a fluorescent protein molecule as the signal from a quickly diffusing fluorescent protein molecule will usually not be detectable above a cell's autofluorescent background; (2), the fluorescent protein must be sufficiently bright to be detected on the single-molecule level and must mature (become fluorescent) quickly enough for the desired application (Venus is the fastest-maturing fluorescent protein to date 7); (3), the fluorescent protein must be photobleached after detection so that newly fluorescent proteins can be detected at subsequent time points (fluorescent proteins that are overly resistant to photobleaching are undesirable as excessive laser exposure can cause phototoxicity artifacts); (4), the protease recognition sequence cannot leave any residual amino acids on the protein of interest that will perturb its functionality (Ubp1 cleaves immediately after the carboxy-terminal Ub residue, leaving no extra residues at all).
Second, one must verify that the reporter is properly expressed and cleaved. Proteolytic cleavage separating the reporter from the protein of interest must be efficient; cleavage efficiency can be measured using Western blots against the reporter, the protein of interest or both (antibodies are commercially available for many fluorescent proteins such as the anti-GFP antibody JL-8 from Clontech, which binds Venus in Western blot). One should ensure that no uncleaved fusion protein is detected in blots against lysates of cells expressing the fusion protein at higher levels than those anticipated in CoTrAC experiments. Also, the reporter and protein of interest must be produced at a 1:1 ratio after cleavage. This can be checked by measuring the intensities of the cleaved bands in anti-reporter and anti-protein-of-interest Western blots normalized by the intensities of the uncleaved bands in strains lacking protease. If reporter and protein of interest are present at a 1:1 ratio after cleavage, then:
Note that since Western blots measure protein concentration, the resulting ratio will deviate from 1:1 if the reporter and the protein of interest exhibit different degradation rates after cleavage.
Third, one should verify that the protein of interest exhibits its expected functionality after cleavage. The presence of the large reporter construct fusion and/or localization to the cell membrane is expected to inactivate most transcription factors (the λ repressor CI was completely inactive without Ubp1 expression in our experiments1). Transformation of the protease-expressing plasmid should activate the protein of interest by liberating the protein from the bulky translational fusion tag. This phenomenon gives rise to other possible CoTrAC applications. Cleavage at the Ub carboxy terminus by Ubp1 has been used to expose non-canonical amino-terminal amino acids to elucidate the bacterial N-end rule of protein degradation 9. If fusion to a large, membrane-associated reporter inactivates a transcription factor (or other protein such as an enzyme), its activity could be quickly recovered by inducing protease expression. CoTrAC could be used to very rapidly "turn on" a preexisting pool of transcription factors by removing/adding an inhibitor/co-activator or otherwise initiating protease expression or activity. Lastly, expression of the fluorescent reporter may perturb cell physiology and it should be ensured that properties such as cell cycle time are unaffected; in the experiment described in ref. 1, we used the Tsr protein as a membrane-localization sequence because Tsr is already a highly expressed membrane protein and a small increase in Tsr expression was not expected to affect cell behavior.
Fourth, when a reporter sequence other than the specifically discussed Tsr-Venus-Ub is used, the corresponding imaging protocol should also be modified as needed. For example, different fluorescent proteins will require different illumination protocols to achieve a good balance between efficiently detecting fluorescent protein molecules, phototoxicity from laser exposure and photobleaching. Ideally, laser excitation should be at the absorbance peak of the chosen fluorescent protein; off-peak excitation requires higher excitation intensities (and thus higher phototoxicity and autofluorescence background) to achieve the same fluorescent protein emission and photobleaching characteristics. Also, the imaging frequency can be modified to be slower or faster than 5 min depending on the tolerance of the cells to the phototoxicity of laser exposure. In our experiments, 12 separate E. coli colonies were imaged every 5 min and followed for 7-8 cell cycles before observing substantial phototoxicity defects 1.
Finally, after time-lapse movies of protein production are acquired in a CoTrAC experiment (examples shown in Figures 4 and 5), fluorescent protein expression needs to be quantified by detecting fluorescent spots corresponding to one or more molecules, assigning molecules to individual cells and tracking cell lineages. We have described a custom Matlab routine to study λ repressor CI production 1. Briefly, one must first identify outlines of individual cells and time points corresponding to cell division in brightfield images while tracking cell lineages through subsequent image frames. We found that a 5 min time interval was sufficiently short that a cell in one frame usually corresponded to the cell in the previous frame with which it shared the most pixels in a segmented image. In cases where occasional large shifts of colonies between two subsequent frames were observed, manual assignment of individual cells to their lineages was required. Next, one needs to identify fluorescent spots and quantify their intensity. We found that fluorescent spots could be identified by (1) bandpass filtering the fluorescence image to remove low-frequency fluorescence background (e.g. autofluorescence) and high-frequency noise, (2) thresholding the image based on fluorescence intensity and (3) identifying objects larger than a few pixels in the thresholded image. The objects in the thresholded image were fit to a 2-dimensional Gaussian function plus a constant background, and the integrated intensity of the spot was taken as the product of the fit amplitude and variance. In our experiments, multiple Tsr-Venus-Ub reporters often co-localized at cell poles, creating spots that were brighter than those from single Tsr-Venus molecules. The number of Venus molecules within one spot was quantified by dividing the integrated intensity of the spot by the fluorescence intensity of a single Venus molecule, which was quantified by using a low-expression-level strain in which spots rarely contain more than one fluorescent protein molecule. This measurement can be further supplemented with in vitro measurements of single fluorescent protein molecules adhered to a glass slide. We have found that fluorescent protein properties are largely similar in in vivo and in vitro systems with similar buffer conditions.
Disclosures
We have no disclosures.
Acknowledgments
The plasmid pCG001 expressing Ubp1 was kindly provided by Rohan Baker at the John Curtin School of Medical Research. This work was funded by March of Dimes Research Grant 1-FY2011, March of Dimes Basil O'Connor Starter Scholar Research Award #5-FY20 and NSF CAREER award 0746796.
References
- Hensel Z, Feng HD, Han B, Hatem C, Wang J, Xiao J. Stochastic expression dynamics of transcription factor revealed by single-molecule noise analysis. Nat. Struct. Mol. Biol. 2012;19:797–802. doi: 10.1038/nsmb.2336. [DOI] [PubMed] [Google Scholar]
- Yu J, Xiao J, Ren X, Lao K, Xie XS. Probing Gene Expression in Live Cells, One Protein Molecule at a Time. Science. 2006;311:1600–1603. doi: 10.1126/science.1119623. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Nat. Acad. Sci. U.S.A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias JW, Varshavsky A. Cloning and Functional Analysis of the Ubiquitin-specific Protease Gene UBP1 of Saccharomyces cerevisiae. J. Biol. Chem. 1991;266:12021–12028. [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. ed. 3. New York: Cold Spring Harbor Laboratory Press; 2001. p. A2.2. [Google Scholar]
- Xiao J, Elf J, Li G, Yu J, Xie XS. Imaging gene expression in living cells at the single-molecule level. Single Molecules: a laboratory manual. 2007. pp. 149–169.
- Nagai T, et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Stagaman G, Forsyth J. Bright-field microscopy of semitransparent objects. J. Opt. Soc. Am. A. 1988;5:648–659. doi: 10.1364/josaa.5.000648. [DOI] [PubMed] [Google Scholar]
- Tobias JW, Shrader TE, Rocap G, Varshavsky A. The N-end rule in bacteria. Science. 1991;254:1374–137. doi: 10.1126/science.1962196. [DOI] [PubMed] [Google Scholar]