

Pheochromocytomas (PCCs) and paragangliomas (PGLs) are rare catecholamine-secreting tumors derived from chromaffin cells originating in the neural crest. This review discusses the currently available diagnostic and therapeutic options for patients with malignant PCCs and PGLs and details the molecular rationale and clinical evidence for novel and emerging diagnostic and therapeutic strategies.
Keywords: Pheochromocytoma, Paraganglioma, Malignant, Therapeutics, Genetic mutation
Learning Objectives
Discuss the advances in molecular genetics which have uncovered new hereditary and germline mutations contributing to the development of pheochromocytoma and paraganglioma and identify the genotype/phenotype patterns which facilitate more accurate determination of malignant potential.
Describe the current imaging modalities used in the diagnosis of pheochromocytoma and paraganglioma and evaluate the efficacy of functional imaging modalities according to tumor genotype.
Evaluate the current preclinical molecular research contributing to the selection of targeted therapies for malignant pheochromocytoma and paraganglioma.
Abstract
Pheochromocytomas (PCCs) and paragangliomas (PGLs) are rare catecholamine-secreting tumors derived from chromaffin cells originating in the neural crest. These tumors represent a significant diagnostic and therapeutic challenge because the diagnosis of malignancy is frequently made in retrospect by the development of metastatic or recurrent disease. Complete surgical resection offers the only potential for cure; however, recurrence can occur even after apparently successful resection of the primary tumor. The prognosis for malignant disease is poor because traditional treatment modalities have been limited. The last decade has witnessed exciting discoveries in the study of PCCs and PGLs; advances in molecular genetics have uncovered hereditary and germline mutations of at least 10 genes that contribute to the development of these tumors, and increasing knowledge of genotype-phenotype interactions has facilitated more accurate determination of malignant potential. Elucidating the molecular mechanisms responsible for malignant transformation in these tumors has opened avenues of investigation into targeted therapeutics that show promising results. There have also been significant advances in functional and radiological imaging and in the surgical approach to adrenalectomy, which remains the mainstay of treatment for PCC. In this review, we discuss the currently available diagnostic and therapeutic options for patients with malignant PCCs and PGLs and detail the molecular rationale and clinical evidence for novel and emerging diagnostic and therapeutic strategies.
Implications for Practice:
Malignant pheochromocytoma and paraganglioma represent a significant management challenge. The diagnosis of malignancy is frequently made in retrospect and traditional treatment modalities have been limited. Recent exciting advances in molecular genetics have uncovered hereditary and germline mutations of at least 10 genes that contribute to the development of these tumors. Increasing knowledge of genotype-phenotype interactions facilitates more accurate determination of malignant potential and has prompted investigation into targeted therapeutics with promising results. There have also been significant advances in functional and radiological imaging and in the surgical approach to adrenalectomy, which remains the mainstay of treatment for pheochromocytoma. Due to the rarity of this tumor, large-scale clinical studies that would progress clinical practice are rare, and the requirement for international collaboration is crucial. The need for large-scale international multicenter studies to effectively exploit the molecular and genetic knowledge gained in this area is highlighted in this review.
Introduction
Pheochromocytomas (PCCs) and paragangliomas (PGLs) are rare catecholamine-secreting tumors derived from chromaffin cells originating in the neural crest [1]. These tumors can occur in any anatomical location of sympathetic/parasympathetic nervous tissue. Since their original description in 1886, they have had many designations, including pheochromocytoma, chaemodectoma, glomus tumors, and paragangliomas. The World Health Organization (WHO) has recently recommended that the term pheochromocytoma be reserved for intra-adrenal tumors, with all others defined as sympathetic or parasympathetic paragangliomas further categorized by site (pelvis, abdomen, mediastinum/thorax, head and neck) to ensure consistency in research [1]. Their incidence has been reported as 0.4–9.5 per million for PCCs [1–5] and 1.5 per million for PGLs [6, 7]. PCCs/PGLs are present in 0.1%–1% of patients with hypertension [8, 9], whereas undiagnosed lesions account for approximately 5% of adrenal incidentalomas [10]. There is a peak incidence in the third and fourth decades of life, with equal incidence in men and women [11].
The classic symptoms experienced by patients with secretory PCCs and sympathetic PGLs result from excessive circulating catecholamines. Symptoms, including headaches, palpitations, diaphoresis, and anxiety, are typically intermittent in nature. Up to 21% of PCC may be asymptomatic [12, 13], which may occur due to desensitization of the cardiovascular system to high circulating catecholamine levels [14]. Interestingly, symptoms are more likely to occur in women [15]. If not treated, this catecholamine excess can result in a hypertensive crisis, which can lead to stroke or fatality. Parasympathetic PGLs are a distinct subgroup predominantly found in the head and neck region, 95% of which are nonsecretory. These PGLs present in an alternative fashion that may be dictated by their specific location, such as tinnitus and hearing loss in patients with tympanic PGLs or cranial nerve deficits in those with jugular PGLs [16, 17].
Some PCCs and PGLs may be malignant. The true incidence of malignancy is difficult to accurately determine; it has traditionally been cited as 10% [1], but it may range from 2.4% to 50%, depending on the definition of malignancy used and the specific population in question [18–20]. One of the main diagnostic challenges has been the recognition of malignant potential. Malignant PCCs/PGLs have been described as those that exhibit local invasion, have metastasized, or have recurred [21–23]. WHO currently defines malignant disease only by the presence of metastases; therefore, the diagnosis is often made only in retrospect.
The overall 5-year survival for patients with PCCs is approximately 89% [24]. It is worse for patients with malignant PCCs/PGLs, with 5-year survival rates varying between 20% and 70% [16, 25–27]. Patients with visceral metastatic disease have a worse prognosis than those with skeletal metastases [28]. Metastatic disease is the main factor associated with decreased survival; because it carries such a poor prognosis [24], accurately distinguishing benign from malignant tumors at the time of diagnosis may ensure appropriate treatment and adequate follow-up.
This review focuses on recent developments in the diagnosis of malignant PCCs and PGLs, encompassing biochemical, radiological, histological, and molecular analysis. In addition, newer treatment modalities and advances in individual targeted therapies will be discussed.
Genetic Mutations in Malignant Pheochromocytomas and Paragangliomas
Recent advances in malignant PCCs and PGLs can be largely attributed to the discovery of novel genetic mutations and the recognition that at least 30% have a genetic origin and are derived from a spectrum of at least ten germline mutations (Table 1) [29, 30]. Approximately 10% are associated with familial syndromes that have an autosomal dominant inheritance, including multiple endocrine neoplasia (MEN) 2A and 2B, Von Hippel Lindau syndrome (VHL), and neurofibromatosis type 1 (NF-1; Table 1) [31, 32]. Furthermore, up to 25% of apparently sporadic cases result from germline loss-of-function mutation in the genes encoding the subunits A[F2], B, C, and D of succinate dehyrdogenase (SDH) [11, 33–35]. Finally, novel genes such as transmembrane protein 127 (TMEM127) [36], kinesin family member 1B (KIF1Bβ) [37], EGL nine homolog 1 (EGLN1/PDH2) [38], MYC-associated factor X (MAX) [39], and hypoxia inducible factor 2α (EPAS/HIF2A) [40] have also been recently implicated in the development of PCCs/PGLs.
Table 1.
Genetic mutations associated with pheochromocytomas and paragangliomas
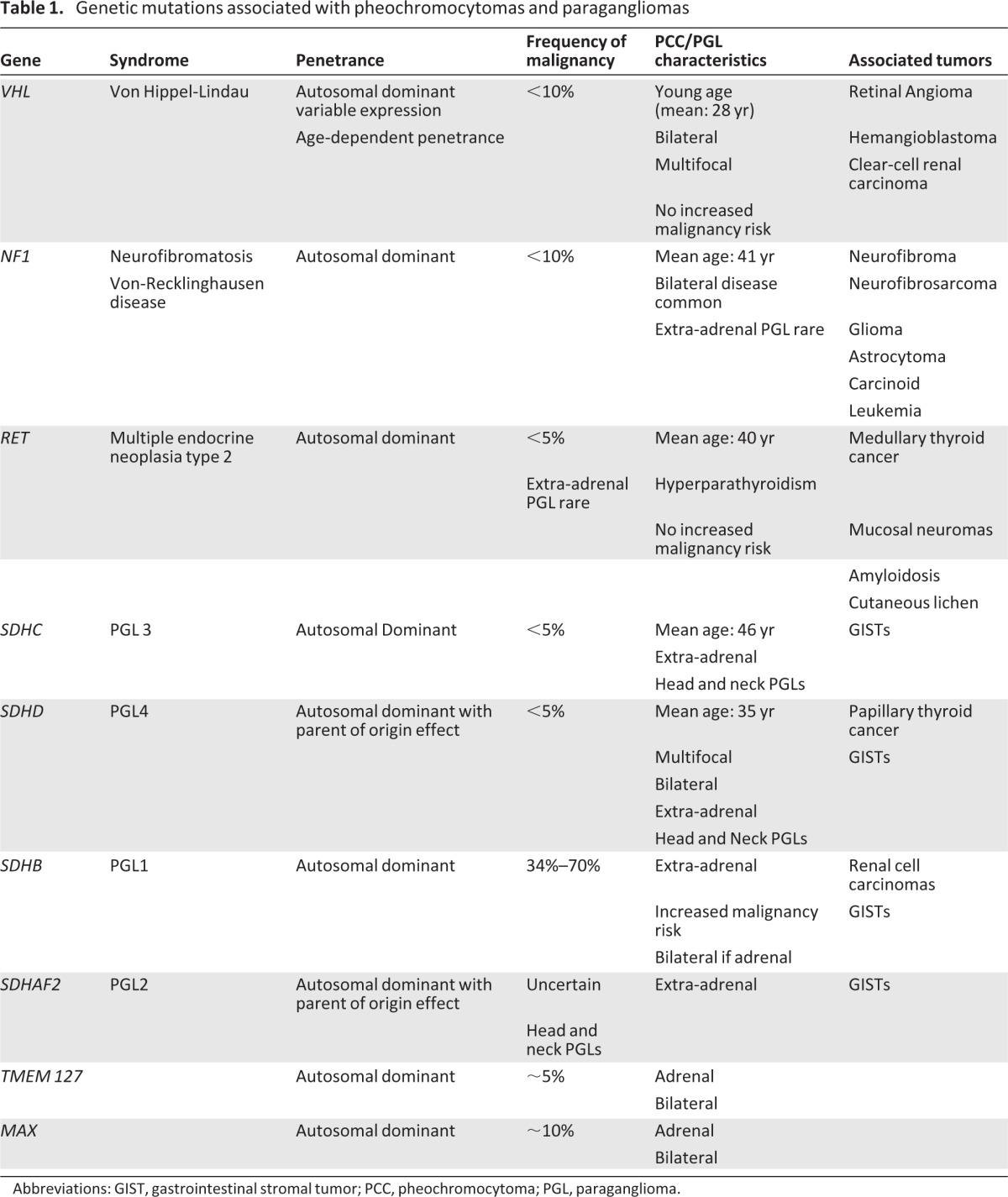
Abbreviations: GIST, gastrointestinal stromal tumor; PCC, pheochromocytoma; PGL, paraganglioma.
Considering all patients with SDH mutations, the subunit affected dictates the clinical features of disease (Table 1), and the likelihood of malignancy is strongly influenced by the underlying genetic aberration. SDHA mutations, initially described in autosomal recessively inherited juvenile encephalopathy [41], were thought to be absent from patients with PCCs/PGLs. However, more recent reports implicate heterozygous germline mutations in SDHA in PCCs/PGLs and head and neck PGLs [42, 43]. Succinte dehydrogenase complex assembly factor 2 (SDHAF2) encodes an evolutionarily highly conserved flavin-adenine dinucleotide factor [44] and is involved in the flavination of SDHA. The loss of SDHAF2 results in loss of SDH function with reduced stability of the SDH complex and a reduction in the subunit expression. It has been subsequently demonstrated that a missense mutation in the conserved region of SDHAF2 [c.232G>A (p.Gly78Arg)] is associated with head and neck PGLs [45, 46].
SDHB mutations are present in approximately 1.7%–6.7% of patients with apparently sporadic PCCs [30]. Although initially thought to have a high clinical penetrance, increased testing of these cases indicates a penetrance of 25%–40% [47]. SDHB mutations exhibit the highest frequency of malignancy. Approximately 20% of mutation carriers will develop malignant disease and up to 50% of patients with a malignant PCCs/PGLs harbor a germline SDHB mutation [20, 48–51]. In addition to PCCs and PGLs, SDHB mutations are also associated with renal cell carcinoma, which can have an aggressive phenotype in young patients [49, 52]. It has been recommended that patients with SDHB mutations be offered surveillance screening for renal cell carcinoma [53].
SDHD and SDHC mutations were initially described in head and neck PGLs but have since been reported in adrenal PCCs and PGLs at other sites [35, 49, 54–55]. The delineation of specific SDH subunit mutations has led to an improved understanding of disease associations. PCCs/PGLs can be found in association with gastrointestinal stromal tumors (GISTs) and pulmonary chondroma in the Carney triad [56], whereas SDHB and SDHC mutations are present in approximately 12% of patients with GISTs without PDGFRA receptor mutations [57, 58].
In addition to syndromic and familial mutations, novel genes associated with pheochromocytoma have recently been described. TMEM127 is a tumor suppressor gene located on chromosome 2q11 and has been linked to a clinical phenotype of adrenal PCCs [36]. The function of TMEM127 is thought to involve protein trafficking and it has been shown to aberrantly activate the MTOR signaling pathway [36, 59, 60]. TMEM127 mutations are predominantly associated with pheochromocytomas that are frequently bilateral and typically carry a low risk of malignancy (2%) [36, 60].
KIF1Bβ is a tumor suppressor gene located on chromosome 1p36.22 that is required for neuronal apoptosis and is frequently deleted in neural crest-derived tumors. Schlisio et al. reported two distinct KIF1Bβ mutations in patients with PCCs [37]; in these cases, the PCCs were bilateral with no evidence of metastases. Germline KIF1Bβ mutations also have been reported in association with other tumors, including neuroblastoma and lung adenocarcinoma [61].
Dysregulation of hypoxia-inducible factor (HIF) transcription factors has been investigated in relation to PCCs and PGLs; a germline EGLN1/PDH2 mutation was reported in association with congenital erythrocytosis and recurrent extra-adrenal PGLs [38]. However, a mutation analysis of EGLN1/PDH2, EGLN2/PDH1 and EGLN3/PDH3 in 82 patients with features of inherited PGLs detected no pathogenic mutations, suggesting that mutations in these genes may not be a frequent cause of inherited PCCs/PGLs [62]. Other novel mutations of genes in the hypoxia sensing pathway have been reported in association with PCCs/PGLs; somatic gain-of-function mutations in EPAS1/HIF-2A have been reported in two patients with polycythemia and PGLs [40]. Lorenzo et al. have also reported a novel inherited germline HIF2A mutation in a polycythemic patient who developed PGL [63].
Another recently described gene implicated in hereditary pheochromocytoma is MAX [39]. MAX protein is a cofactor of the proto-oncogene MYC and is a key component of the MYC-MAX-MXD1 network that regulates cell proliferation and differentiation, exhibiting crosstalk with the mammalian target of rapamycin (mTOR) pathway. Burchinon et al. [64] reported a multicentre series of 1,694 patients with PCCs/PGLs in whom MAX was sequenced. They ascertained that MAX germline mutations are present in 1.12% of cases of PCC/PGL in patients with no other known mutation. Burchinon et al. recommended that MAX should now be considered in the genetic workup of patients with PCCs/PGLs. The identification of TMEM127 and MAX emphasize the likelihood that there are multiple pathways implicated in the development of PCC/PGL.
The correlation between genetic mutations and biochemical phenotype in hereditary PCCs/PGLs may provide an opportunity for more streamlined genetic testing and earlier identification of malignant potential. The biochemical phenotype may have the potential to determine which of the genetic mutations is more likely in patients without a known familial mutation and could therefore also be used to rationalize genetic testing.
Uncovering the genetic complexity of these tumors provides invaluable insight into the factors that are associated with and drive malignant progression. This knowledge is the foundation for progress in the diagnosis and treatment of malignant PCCs/PGLs.
Diagnosis of Malignant PCCs/PGLs
Biochemistry
Traditional diagnostic testing for PCCs/PGLs has focused on catecholamine metabolism. It has been accepted that measurements of metanephrines (O-methylated metabolites of catecholamines) are superior diagnostic markers to both the parent catecholamines and other metabolites, including vanillymandelic acid [65–67]. Therefore, it is recommended that standard initial testing for PCCs/PGLs should include measurements of plasma-free and/or urinary-fractionated metanephrines [28]. Plasma-free metanephrines in particular have demonstrated high diagnostic sensitivity [68–70], but there is no clear evidence suggesting they are more accurate than urinary-free or urinary-deconjugated metanephrines. The main diagnostic challenge in PCCs/PGLs is recognition of malignant disease. Interest has focused on the possibility that biochemical information may help elucidate future malignant potential.
Genotype/Mutation Correlations
The pattern of metabolite secretion may be used to guide the identification of specific genetic mutations [71, 72]. A study by Eisenhofer et al. [72] measured free plasma concentrations of O-methylated metabolites in 173 patients with hereditary PCCs/PGLs; they demonstrated that patients with NF-1 and MEN-2 could be discriminated from those with VHL and SDH mutations in 99% of cases by the combination of normetanephrine and metanephrine, whereas measurements of plasma methoxytyramine further discriminated those patients with SDH mutations in 78% of cases. The authors found that the biochemical profile in patients with MEN-2 and NF-1 was characterized by increased plasma metanephrine concentrations (increased epinephrine production). In contrast, patients with VHL demonstrated solitary increases in normetanephrine, whereas 70% of patients with an SDH mutation demonstrated additional or solitary increases in methoxytyramine (dopamine production). More recently, it was reported that PCCs/PGLs associated with MAX mutations are characterized by substantial increases in normetanephrine and may be associated with normal or minor increases in metanephrine [64].
The correlation between genetic mutations and biochemical phenotype in hereditary PCCs/PGLs may provide an opportunity for more streamlined genetic testing and earlier identification of malignant potential. The biochemical phenotype may have the potential to determine which of the genetic mutations is more likely in patients without a known familial mutation and could therefore also be used to rationalize genetic testing.
Biochemical Markers of Malignancy
The search for biochemical markers of malignancy has been the focus of many researchers in the last decade but continues to remain elusive. Catecholamines (urinary and plasma), chromogranin, and novel markers, such as methoxytyramine, have been explored. However, accurate markers remain difficult to identify.
PCCs/PGLs are diagnosed biochemically by the measurement of plasma or 24-hour urinary excretion of catecholamine metabolites [28]. However, there is evidence that malignant PCCs may exhibit enzyme deficiencies that inhibit catecholamine metabolism, resulting in secretion of more premature catecholamines. High dopamine levels have been shown in malignant PCCs resulting from decreased expression of dopamine-β-hydroxylase [73]. Furthermore, high levels of dopamine secretion are associated with malignancy and a shorter metastasis-free interval in patients with PCCs/PGLs [74–76].
John et al. demonstrated that patients with high preoperative 24-hour urinary dopamine levels have an increased likelihood of having a malignant PCC [77], proposing this as a potential preoperative marker of malignancy. However, in an analysis of 120 patients with PCCs, Januszewicz et al. demonstrated that, despite increased urinary dopamine excretion in all patients with malignant PCCs, high levels were also observed in a subset of patients with benign tumors [78]. These findings are supported by a recent report by Zelinka et al. [76], who analyzed the dimension and biochemical profile of 41 metastatic and 108 benign PCCs; they found no difference in dopamine secretion between benign and malignant tumors, indicating that measurement of dopamine secretion does not have satisfactory discriminatory potential as a marker of malignancy.
Rao et al. [79] evaluated the diagnostic and prognostic value of chromogranin A in patients with PCCs/PGLs. They demonstrated that levels rose significantly between normal controls, benign pheochromocytomas, and malignant pheochromocytomas. These results, demonstrated in a small group of patients, have failed to be replicated. Although elevated levels of many granin-derived peptides are found in PCCs/PGLs, they rarely help to discriminate between benign and malignant tumors [80]. Recently, EM66, a secretogranin II-derived peptide present in chromaffin cells, has shown the most promise in early studies in distinguishing between benign and malignant disease [81, 82]. Malignant disease demonstrated lower gene expression and protein transcription, but this has yet to translate into a measurable circulating biomarker.
Eisenhofer et al. [83] explored the utility of catecholamine and metabolite measurements as biomarkers for malignant PCCs/PGLs in a cohort of 365 patients, 105 of whom had metastases. Plasma methoxytyramine, the O-methylated metabolite of dopamine demonstrated an almost fivefold increase in the presence of metastatic disease. Furthermore, although increased plasma methoxytyramine was associated with SDHB mutations and extra-adrenal disease, it remained predictive of malignancy in the absence of SDHB mutations. Consideration should therefore be given to measuring plasma methoxytyramine in all patients with SDH mutations and patients in whom malignancy is suspected. However, long-term follow-up studies will be required to establish the prognostic utility of this biomarker.
Radiology
The role of radiological imaging in PCCs and PGLs is to localize the primary tumor, evaluate for multifocal or metastatic disease, and determine management strategies in patients with metastatic disease. Computed tomography (CT) and magnetic resonance imaging (MRI) are sufficiently sensitive to localize the primary tumor, with sensitivities of 98%–100% for adrenal PCCs; MRI is more sensitive than CT for extra-adrenal PGLs (93% vs. 90%) [84]. However, these modalities are limited by lower specificity (approximately 70%) due to the high incidence of adrenal incidentalomas [85]. Functional imaging is often required to evaluate the extent of disease and to accurately stage patients. Increased knowledge of the molecular/genetic basis of the malignant disease has affected the approach to radiological investigation. The reported sensitivities of functional imaging of PCCs/PGLs may vary depending on the patient population involved and associated underlying genetic mutations [86, 87].
Metaiodobenzylguanidine Scintigraphy
Chromaffin cells express human norepinephrine transporters; via the latter, the catecholamine precursor metaiodobenzylguanidine (MIBG) is transported into the cells and stored in cytoplasmic granules by vesicular monoamine transporters [88]. Thus, I131/I123 MIBG has been used for the last two decades to image neuroendocrine tumors [89] because uptake reflects adrenergic innervation or catecholamine excretion. MIBG scintigraphy has a sensitivity and specificity of 94% (95% CI: 91%–97%) and 92% (95% CI: 87%–98%), respectively [90]. Although the sensitivity of I123 MIBG is superior to I131 for the detection of metastases, the overall sensitivity is decreased in malignancy [91]. This reduced sensitivity may result from a decreased expression of noradrenaline transporters in malignant PCCs/PGLs, dedifferentiation, or genotype/phenotype differences. VHL-associated PCCs/PGLs are also more likely to be negative on MIGB imaging, but this is thought to be due to a lower noradrenaline transporter expression [92]. Similarly, patients with SDHB mutations also have a high rate of false-negative MIBG imaging results because of an absence of its intracellular transporter [93]. False-negative rates of 60% I123 MIBG for have been reported for patients with hereditary PCCs/PGLs compared to 6% in patients with sporadic PCCs/PGLs [94]. For this reason, alternative imaging modalities should be considered for patients with known mutations in whom malignancy is suspected.
Positron Emission Tomography
Positron emission tomography (PET) has recently been used for the localization of PCCs/PGLs, particularly in patients for whom MIBG scanning is negative. Based on the measurement of biologically active, tracer-labeled molecules, this functional imaging modality is in widespread use in oncology. The most commonly used PET reagent is 18F-fluoro-2-deoxy-d-glucose (FDG), a nonspecific tracer that enters the cell via glucose transporters and undergoes phosphorylation to become 18F-FD-6P. The accumulation of this reagent is an index of increased glucose metabolism and is seen in malignant and inflammatory tissue. Recently, more specific PET reagents (involved in the metabolism of catecholamines) have been evaluated for the investigation of PCCs/PGLs.
Shulkin et al. [95] was the first to report the advantages of 18F-FDG imaging in PCCs/PGLs that do not accumulate MIBG. The uptake of 18F-FDG is not related to tumor secretory status; its superiority in malignant PCCs that do not accumulate I131 or I123 MIBG has been confirmed [87, 96]. For patients with SDHB-associated PCCs/PGLs, 18F-FDG PET is superior to I131-MIBG, I123-MIBG, 111In-pentetreotide, and 18F-FDA in detecting metastatic lesions, with a reported sensitivity approaching 100% [96]. The high sensitivity of 18F-FDG PET in SDHB-mutated tumors may be partially explained by loss of function of the SDH complex, which is involved in energy-producing metabolic processes (tricyclic acid cycle and oxidative phosphorylation). This impaired energy production may cause the cells to switch to glycolysis with resultant increased activity of glucose transporters and increased glucose uptake [97]. FDG-PET is considered to be the preferred functional imaging modality for staging and treatment monitoring of SDHB-related metastatic PGLs.
The catecholamine precursors dihydroxyphenylalanine (DOPA) and dopamine are both transported into chromaffin cells by the human norepinephrine transporters. When labeled with 18F, PCCs and PGLs can be detected with a high sensitivity and specificity. 18F-fluorodopamine (FDA)-PET has been shown to be superior to MIBG for localization of metastatic disease [98, 99]. The sensitivity of 18F-FDA-PET for metastatic PCCs/PGLs ranges from 88%–100% per patient or 70%–97% on a per-lesion basis [86, 98–99]. It is especially useful for the identification of bony metastatic disease, for which it is superior to CT, MRI, 18F-FDG, and 123/131I-MIBG [100].
The sensitivity and specificity of 18F-dihydroxyphenylalanine (FDOPA) for malignant PCCs/PGLs is significantly affected by genotype. Initial reports suggested limited utility in metastatic disease [101], which may have been due to inclusion of SDHB patients. More recent reports indicate a high false-negative rate for 18F-DOPA scans in SDHB patients in contrast to non-SDHB patients, for whom F-DOPA has the highest sensitivity of 93% compared to FDA (76%), FDG (62%), or MIBG (59%) [86].
Somatostatin Receptor Imaging
PCCs/PGLs express somatostatin receptors, predominantly SSTR2, SSTR3, and SSTR5, which represent the molecular basis for the use of somatostatin analogues for both localization of disease and treatment for specific subgroups of these patients. Labeling the somatostatin receptor imaging (SRS) with indium-111 DTPA is widely used for the detection of neuroendocrine tumors [102]. Although it appears to have a lower detection rate for malignant PCCs/PGLs than I123 MIBG [103], a high sensitivity has been reported in head and neck PCCs/PGLs [104]. Recent reports on the use of positron-emitting 68Ga-labeled somatostatin analogues (DOTATOC, DOTATATE, or DOTANOC) show higher sensitivity for PCC/PGL detection than indium-111 due to higher affinity and superior resolution of PET [105, 106]. Reports of small series to date also indicate that Ga-68 DOTATATE imaging may be superior to I123 MIBG in the diagnosis and staging of metastatic PCCs/PGLs [107, 108]. These findings have yet to be validated in larger series with genotype data.
Pathology
The histological diagnosis of PCCs/PGLs is relatively straightforward. Characteristically, tumor cells demonstrate a nested Zellballen pattern surrounded by sustentacular cells, which stain positive for S100 protein on immunohistochemistry. Tumors exhibit immunopositivity for synatophysin and chromogranin A and may express neurofilament [109]. The differentiation of PGLs from other neuroendocrine tumors may be facilitated by immunopositivity for enzymes involved in catecholamine synthesis, such as tyrosine hydroxylase [110, 111]. The difficulty therefore remains to correctly identify malignant disease. At present, there are no absolute histological criteria for the diagnosis of malignancy and no means to identify PCCs/PGLs, which are at risk of recurrence or metastatic spread using standard histopathological techniques. Contributing factors to this difficulty include the rarity of PCCs/PGLs, their variable location in sites where basement membrane penetration may not be assessable, and long latency to metastasis.
Histological Scoring Systems
Attempts have been made to devise a histological algorithm or scoring system to guide pathologists in the diagnosis of malignancy. The Pheochromocytoma of the Adrenal Gland Scaled Score (PASS), devised by Thompson in 2002 [21], is the most widely accepted. PASS uses a range of histological criteria—including tumor necrosis, mitotic rate, tumor cell spindling and the presence of large cell nests—to group adrenal pheochromocytomas into those with potential for biologically aggressive behavior and those likely to behave in a benign manner (Table 2). This system was devised using 100 pheochromocytomas of the adrenal gland—50 histologically benign and 50 histologically malignant—with each histological feature given a weighted score of 1 or 2. The original report cited a threshold score (PASS score) of 4, below which PCC exhibited benign behavior. This scoring system has been evaluated subsequently with varied results. Strong et al. [22] found that a higher threshold of 6 was indicative of malignant behavior but recommended that patients with a PASS score >4 should be closely followed. Wu et al. [112] found significant interobserver and intraobserver variation in assignment of PASS, with variable interpretation of the underlying components. Thus, the reliability of this scoring system has not been unequivocally established, and it is recommended that it be used with caution.
Table 2.
Proposed Pheochromocytoma of the Adrenal Gland Scaled Score (PASS)
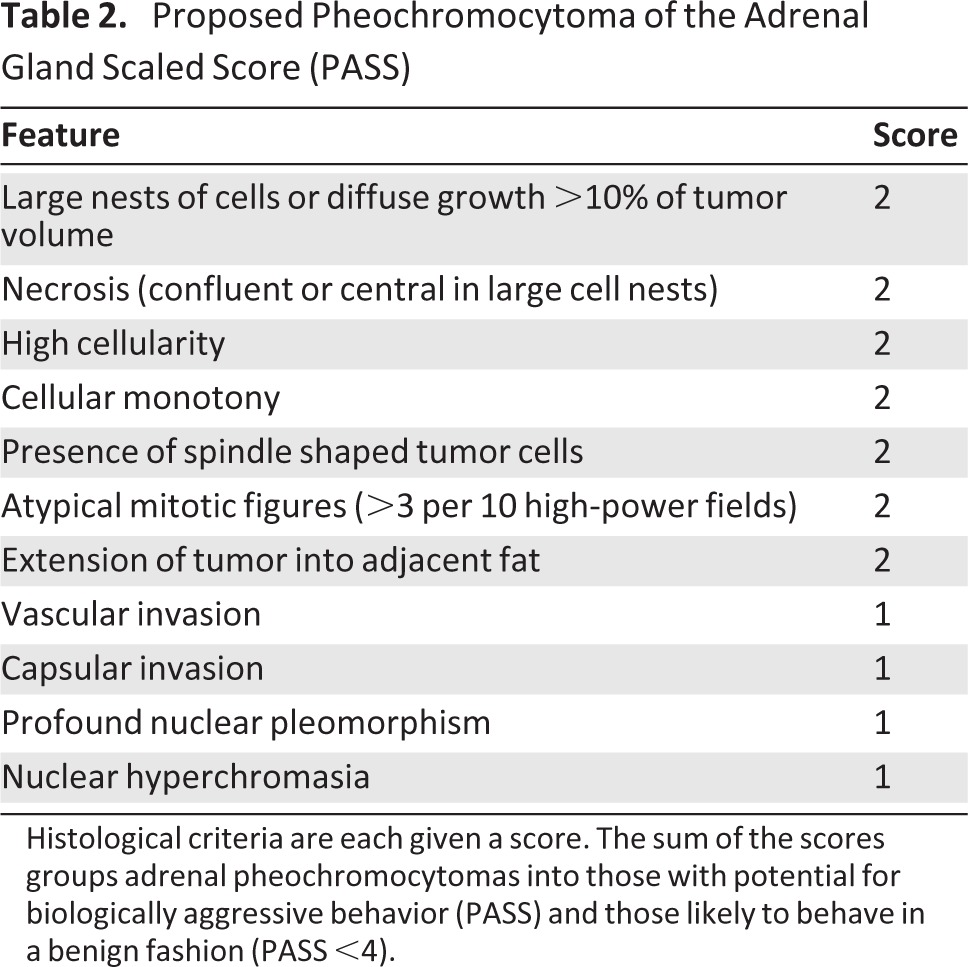
Histological criteria are each given a score. The sum of the scores groups adrenal pheochromocytomas into those with potential for biologically aggressive behavior (PASS) and those likely to behave in a benign fashion (PASS <4).
An alternative scoring system was devised by Kimura et al. [113] using both adrenal PCCs and extra-adrenal sympathetic PGLs (Table 3). This model, developed using 146 tumors—38 of which were metastatic—combined histological criteria with the tumor Ki67 sores and the type of catecholamine produced by the tumor. The higher the score achieved by individual tumors, the greater the correlation with metastatic potential and patient survival, although there was less than 100% discrimination. This scoring system requires further validation to determine its utility and applicability to the clinical setting.
Table 3.
Scoring system devised for both pheochromocytomas and extra-adrenal paragangliomas
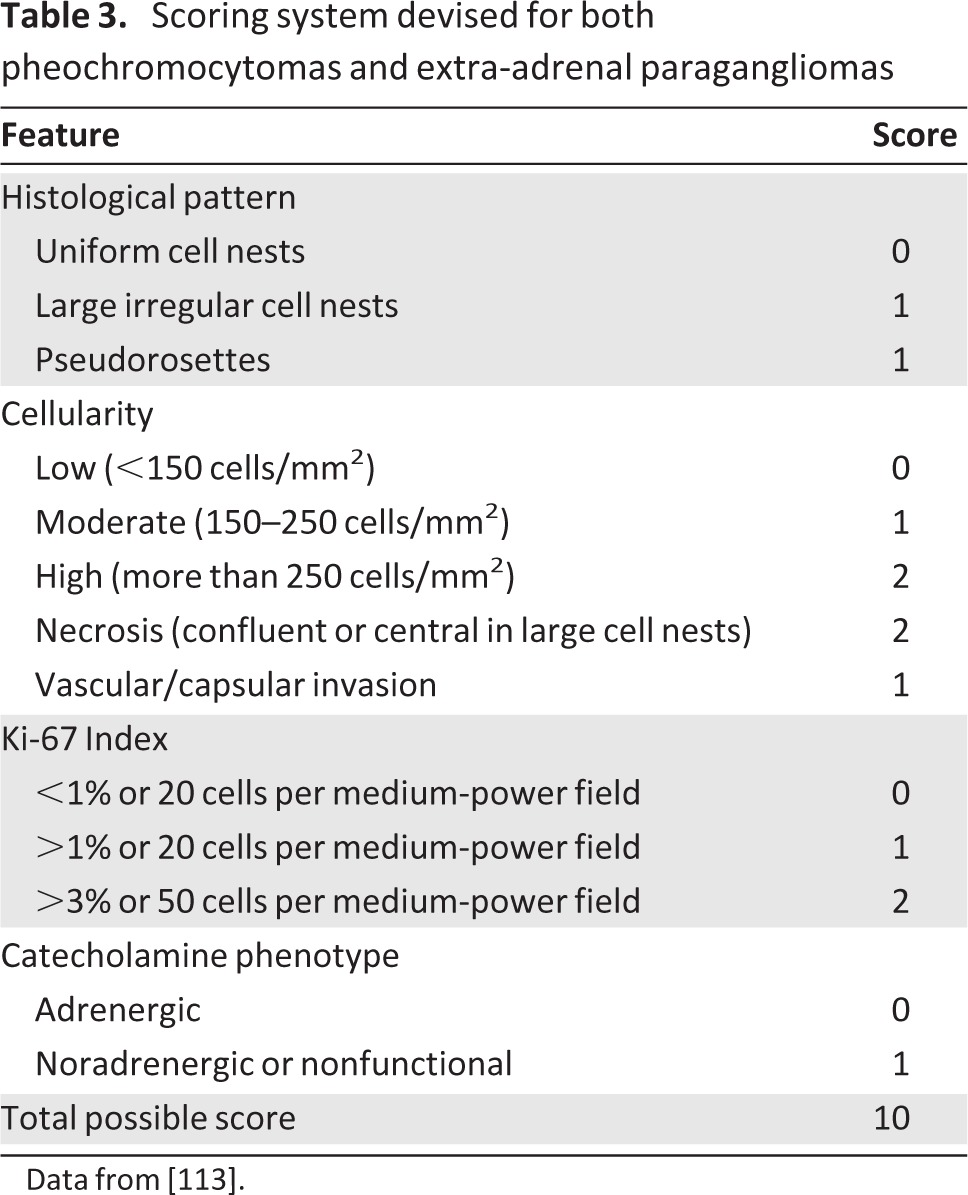
Data from [113].
Immunohistochemical Markers of Malignancy
A multitude of immunohistochemical (IHC) markers of malignancy in PCCs/PGLs have been proposed. However, not one has emerged that can be translated into routine clinical practice. Neuroendocrine- and catecholamine-related markers (neuropeptide Y, 3,4-dihydroxyphenylalanine) [114], granin-derived peptides (EM66, secretogranin II) [81, 82, 115], CD-44s [116], angiogenic markers and regulators (vascular endothelial growth factor [VEGF] and VEGFR) [117], heat shock protein 90 [118], and telomerase complex proteins [119] have all been studied with varying degrees of success. The most likely to be of clinical utility is the Ki-67 proliferative index of the tumor, which has been shown to correlate with malignancy in a number of studies [120–122]. A Ki-67 index >3% is considered to be a useful parameter in predicting malignant potential because benign PCCs have never been shown to have scores of >3%. However, despite a high specificity for malignancy, Ki-67 index lacks sensitivity; indices of <3% have been shown in patients with malignant PCCs/PGLs [22, 121, 123–124].
Immunohistochemistry may also allow identification of tumors with loss of SDHB expression. Therefore, these patients can be prioritized for mutation analysis because it has been shown that negative SDHB immunostaining carries a high likelihood for the presence of SDHx mutations (including SDHB, SDHC, and SDHD). Blank et al. [125] demonstrated 100% sensitivity and 84% specificity for the association of SDHB IHC and the presence of an SDHx mutation. Furthermore, patients with IHC-negative SDHB have a significantly poorer prognosis and survival than patients with SDHB IHC-positive tumors. Similarly, SDHA IHC has been used to identify patients with SDHA-mutated PGLs [43]. It has been confirmed that IHC of PCCs/PGLs is a useful strategy for triaging patients to appropriate genetic testing [126].
Pathological Findings in Context of Tumor Genotype
Two physical parameters have been associated with malignancy, tumor size, and location. There are conflicting data regarding tumor size and malignancy, with some authors reporting an association [83, 127, 128] and others finding none [22, 129]. It has been demonstrated that tumors greater than 5 cm in diameter are more likely to be malignant [128] and are associated with a reduction in overall survival [127]. However, using tumor size alone as a predictor of malignancy lacks sensitivity because malignancy has been reported in smaller tumors [22]. Extra-adrenal tumor location has consistently been reported as a risk factor for malignancy [83, 128, 130, 131]. However, many of these reports are limited by lack of tumor genotyping, which is now known to be a significant contributory factor for the risk of malignancy [51, 83, 132]. SDHB-mutated tumors are more likely to be extra-adrenal and large in size at the time of presentation, and 50% of malignancy in extra-adrenal PGL are associated with SDHB mutation [50]. It has recently been reported that the risk of metastatic disease remains elevated in extra-adrenal tumors (3.4-fold) even in the absence of an SDHB mutation [83]. In addition, tumor size and location are independent risk factors for malignancy, which may account largely for the increased risk conferred by SDHB mutations [83].
Molecular Profiling
The sequencing of the human genome and the advent of high-throughput molecular profiling has facilitated comprehensive analysis of transcriptional variation at the genomic level, resulting in an exponential increase in our understanding of the molecular biology of malignancy. Gene expression profiling using microarray technology has been productively applied to many areas of cancer research, resulting in the development of diagnostic and prognostic tools that have been translated to the clinical setting [133]. For PCCs/PGLs, there has been much interest in the application of this technology as a tool to differentiate between benign and malignant tumors.
cDNA-Based Analysis
Comparative genomic hybridization (CGH) studies have identified genetic losses associated with underlying germline mutations; losses of 1p and 3p in sporadic and MEN-2 mutation PCCs/PGLs [134, 135] have been reported, along with ch11 losses in VHL-associated PCCs/PGLs [136] and 11q deletions in head and neck PGLs [137]. Sandgren et al. [138] applied high-resolution whole-genome array CGH in a series of 53 PCCs/PGLs and reported that DNA gain was more frequent in malignancy. DNA gain at 1q was identified in malignant PGL, whereas gain at 19q, trisomy 12, and loss of 11q were associated with malignant PCCs. Validation of these findings in a larger cohort is required to rule out any confounding variables from underlying genetic mutations.
Gene Expression Profiling
Gene expression studies in PCCs/PGLs have demonstrated that specific gene expression profiles correlate with both the underlying mutation (germline or somatic) and the catecholamine biochemical phenotype [139]. PCCs/PGLs can be split into two clusters based on their gene expression profiles [140]. Cluster 1 tumors are predominantly those with mutations in VHL and SDHx; they exhibit a pseudohypoxic phenotype, with activity of hypoxia-inducible transcription factors (Fig. 1) [141]. Cluster 2 tumors include MEN-2 and NF-1 mutations; they are characterized by a RAS/RAF/ERK signaling phenotype, with increased expression of genes involved in adrenergic metabolism (Fig. 2) [142]. The clustering of PCCs/PGLs indicates differing routes to tumorigenesis, which has implications for the development of novel targeted therapeutics.
Figure 1.

Cell signaling pathways for pheochromocytoma and paraganglioma susceptibility genes, showing cluster 1 genes and related signaling pathways. Cluster 1 tumors predominantly have mutations in Von Hippel Lindau syndrome and SDHx and exhibit a pseudohypoxic phenotype with activity of hypoxia-inducible transcription factors.
Abbreviations: HIF, hypoxia-inducible factor; SDH, succinate dehydrogenase; VEGF, vascular endothelial growth factor; VHL, Von Hippel Lindau syndrome.
Figure 2.

Cell signaling pathways for pheochromocytoma and paraganglioma susceptibility genes, showing cluster 2-related kinase signaling pathways. Cluster 2 tumors include RET and NF-1 mutations and are characterized by a RAS/RAF/ERK signaling phenotype with increased expression of genes involved in adrenergic metabolism.
Several gene expression array studies have been undertaken to identify gene signatures that may discriminate between benign and malignant disease [143–146]; indeed, numerous differentially expressed genes and proteins have been reported in this manner. Brouwers et al. [143] analyzed 90 tumors, of which 20 were malignant, and identified a large numbers of dysregulated genes. The majority of genes associated with malignant potential appear to be downregulated, indicating that malignant PCCs/PGLs are less differentiated. Thouennon et al. [145] also reported downregulation of gene expression in association with malignancy, with many of the downregulated genes encoding for neuroendocrine factors.
Suh et al. [146] performed genomewide expression analysis of 58 PCCs/PGLs. They identified a set of six differentially expressed genes (CFC1, FAM62B, HOMER1, LRRN3, TBX3, ADAMTS), which in combination had an area under the receiver operating characteristic curve of 0.96 for distinguishing benign versus malignant disease. Similarly, Waldman et al. [144] reported 132 differentially expressed genes between benign and malignant tumors, six of which were validated using real-time quantitative polymerase chain reaction (RQ-PCR): calsequestrin, NNAT, neurogranin, secreted protein acidic and rich in cysteine [SPARC], EGR2, and MAOB).
Despite the identification of differentially expressed genes, there is little overlap between the studies, and they are limited by small numbers, lack of control for confounding factors, use of differing transcriptomic platforms, and lack of validation. To date, there is no individual molecular marker and no gene signature to distinguish benign from malignant pheochromocytoma that has been translated to use in the clinical setting.
MicroRNA Expression Profiling
The recognition that microRNA (miRNA) represent a crucial link in the cancer biology picture has prompted the investigation of these molecules as potential diagnostic and prognostic biomarkers of malignancy. Meyer-Rochow et al. [147] investigated miRNA expression in 12 malignant and 12 benign PCC using microarray expression profiling and identified 18 miRNAs that were differentially expressed between benign and malignant samples. Three of these (miR-15a, miR-16, and miR-483–5p) were validated using RQ-PCR. Interestingly, miR-15a and miR-16 are well described as oncomirs [148], and miR-483–5p has recently been identified as a marker of malignancy in adrenocortical carcinoma [149]. Tömböl et al. [150] reported on miRNA expression profiling in benign and recurrent PCCs/PGLs; taking mutational status into account, they identified significantly higher expression of miR-885–5p and miR-1225–3p in MEN-2 and sporadic recurring pheochromocytomas, respectively. More recently, Patterson et al. [151] used microarray analysis to identify eight miRNAs that are differentially expressed between benign and malignant pheochromocytomas; of these, miR-483–5p, miR-183, and miR-101 were validated using RT-PCR.
One characteristic of miRNAs that makes them particularly attractive as biomarkers is the ability to measure circulating serum miRNA. Indeed, Patterson et al. [151] confirmed that the subset of miRNAs identified on microarray were also detected in the serum of patients with pheochromocytomas. At present, diagnostic, prognostic, and predictive miRNA signatures and markers remain hypothesis generating; they require validation in larger, independent clinical cohorts prior to any consideration for clinical applications. MiRNAs possess the additional attraction of potential for development as therapeutic targets because of their ability to regulate gene expression. It is likely that future microarray studies will adopt an integrated approach of miRNA and mRNA expression analysis in an attempt to decipher regulatory pathways and therapeutic targets in addition to expression patterns.
Therapeutic Advances
The treatment of malignant PCCs and PGLs includes surgical resection and debulking, pharmacological control of catecholamine-mediated symptoms, radiotherapy, and systemic therapy [28]. Because these are relatively rare tumors, the evidence for each of these treatment modalities is limited.
Surgery
Surgical intervention for malignant PCCs/PGLs should be undertaken in specialist centers with the benefit of a multidisciplinary approach. Prior to initiation of treatment, it is imperative that a comprehensive diagnostic workup is undertaken to identify the presence of metastatic disease, as well as the presence of underlying genetic mutations that may predispose patients to bilateral disease, because this may influence the surgical approach [152]. Preoperatively, hypertension needs to be tightly controlled with the potential need for both α- and β-blockade. Appropriate fluid resuscitation in conjunction with this is critical to achieve a successful perioperative surgical outcome [153].
For patients with no preoperative evidence of metastatic disease, complete excision is undertaken due to the long-term consequences of catecholamine excess. Local excision via a minimally invasive approach has become the criterion standard for surgical management [19]. The two most common approaches are the lateral transperitoneal approach and the posterior retroperitoneal approach. Laparoscopic adrenalectomy was first described in 1992 by Gagner et al. using the transperitoneal approach [154]. This procedure has become widely accepted with benefits including decreased analgesic requirement, improved patient satisfaction, shorter hospital stay, and shorter recovery time when compared to open surgery [155]. A lateral transperitoneal approach offers excellent exposure due to the effects of gravity; it can be used for large tumors up to a maximum diameter of 8–12 cm [156, 157]. For such tumors, surgical technique must be meticulous to avoid capsular rupture, which increases the risk of tumor seeding and locoregional recurrence [158].
Retroperitoneoscopic adrenalectomy was developed in 1993 [159] and brought into clinical practice by Martin Walz [160]. This approach has gained popularity because it offers advantages, including direct access to the gland and the option of bilateral adrenalectomy without a requirement for repositioning if a prone approach is used. This approach is relatively contraindicated in superobese patients (BMI >40 kg/m2). A prospective study by Rubinstein et al. compared the transperitoneal and retroperitoneal approaches and found no difference in operative time, blood loss, analgesic requirement, hospital stay, or complication rates [161]. Retrospective series have had varying reports of no difference between the two approaches or only differences in operative time, but these results must be interpreted in the context of the operating surgeons' experience [162–164]. Both laparoscopic approaches compare favorably with open adrenalectomy. There remains no clear consensus among surgeons with regard to the two laparoscopic approaches; the choice currently depends predominantly on the experience and preference of surgeon.
In patients with bilateral adrenal pheochromocytoma or for patients with MEN-2 and VHL who have a high incidence of synchronous and metachronous disease, bilateral cortical-sparing adrenalectomy has been advocated [165]. This approach has been advocated as a means of steroid independence, avoiding the need for lifelong corticosteroid replacement; it also reduces the risk of Addisonian crises following bilateral adrenalectomy. To retain sufficient adrenal function, at least one third of the gland must be preserved [166, 167]; this has been shown to be feasible using minimally invasive techniques with a prone retroperitoneoscopic approach with or without radiological guidance with intraoperative ultrasound [19, 165, 168].
Walz et al. recently published a series of 66 patients treated for bilateral pheochromocytoma; 89 cortical-sparing resections were performed, resulting in a corticosteroid-free postoperative course in 91% of cases (n = 60); no recurrent disease was noted during 48 months of follow-up [165]. However, earlier series with longer follow-up times reported recurrence rates in the range of 10%–60% [169–171]. In addition, partial adrenalectomy does not always ensure cortisol independency postoperatively [171], so the benefits and risks of total versus partial (cortical sparing) adrenalectomy should be discussed with the patient in each individual case.
Lifelong clinical and biochemical surveillance is warranted for any patient undergoing subtotal adrenalectomy to detect recurrent disease. At present, it is not clear whether partial adrenalectomy may harbor an increased risk of recurrence in malignant pheochromocytomas. Advocates of this approach argue that if the pheochromocytoma is resected completely with no capsular rupture, an increased risk of recurrence is unlikely. A central consideration in the selection of patients for partial adrenalectomy is genotype; subtotal/cortical sparing adrenalectomy is recommended for patients with MEN-2A or VHL mutations in whom there is frequently bilateral disease but a low risk of malignancy. For patients with SDHB mutations, in whom the risk of malignancy is increased, there is no evidence to support subtotal/cortical sparing adrenalectomy for PCCs [172]. Further advances in minimally invasive adrenalectomy include single-site access [173, 174], natural orifice transluminal endoscopic surgery [175], and robot-assisted surgery [176], which have been described and developed but are not yet in widespread clinical use.
For cases in which locoregional infiltration is diagnosed preoperatively or there is a high suspicion of malignancy, the goal of surgery remains complete surgical excision, which may be best facilitated with an open surgical approach [177]. This approach may require extensive resection of adjacent tissues and involved organs (pancreas, spleen, liver, kidney, vena cava). It is reasonable to initially explore these patients laparoscopically/retroperitoneoscopically; however, local invasion usually mandates conversion to open surgery [177]. Intraoperative use of 123I-MIBG and a γ-probe may be helpful in localizing metastatic deposits or identifying residual disease when attempting complete excision.
For patients with confirmed metastatic disease in whom disease eradication is impossible, surgery is palliative, aiming to reduce tumor burden and minimize the effects of excess catecholamine secretion. Intended curative surgery is only feasible in a minority (10%) of patients who present with metastatic disease [178, 179]. Despite the fact that complete eradication of disease may often be unachievable, debulking/cytoreductive surgery is still advocated because it improves symptoms caused by local invasion and catecholamine secretion. Consensus guidelines recommend surgery for liver metastases in well-differentiated neuroendocrine tumors (NETs) if complete resection or debulking of approximately 90% of the tumor load is feasible [180]. In patients with liver metastases from NETs, an aggressive surgical approach to resection is also supported by improved survival [178, 181, 182]. A recent systematic review by Saxena et al. [183] of 1,469 patients in 29 studies evaluating outcome in patients undergoing hepatic resection for NET metastases reports symptomatic relief in a median of 95% of patients undergoing cytoreductive surgery. Furthermore, hepatic resection of NET metastases was associated with favorable 5- and 10-year survival rates of 70.5% (31%–100%) and 42% (0%–100%), respectively. Importantly, despite the fact that many patients in these series had extensive metastatic disease, the median mortality was <3% (range: 0%–9%); the median morbidity of 23% (range: 3%–45%) is comparable to that seen in liver resection for other metastatic disease [183, 184].
In patients with very advanced disease for whom surgical resection is not immediately feasible, locoregional therapies including embolization, radiofrequency ablation, or selective internal radiation therapy may be used to downstage disease and achieve symptomatic relief. Radiofrequency ablation (RA) is advocated predominantly as an adjunct to cytoreductive surgery. Retrospective series provide promising data for RA in the management of hepatic neuroendocrine metastases; however, randomized trials comparing cytoreductive surgery with RA are lacking.
In patients with very advanced disease for whom surgical resection is not immediately feasible, locoregional therapies including embolization, radiofrequency ablation, or selective internal radiation therapy may be used to downstage disease and achieve symptomatic relief [185–187]. Radiofrequency ablation (RA) is advocated predominantly as an adjunct to cytoreductive surgery. Retrospective series provide promising data for RA in the management of hepatic neuroendocrine metastases; however, randomized trials comparing cytoreductive surgery with RA are lacking [188]. An aggressive cytoreductive approach (surgery alone or in combination with RA) has been compared with other treatment modalities. Osborne et al. [182] reported improved symptom control in patients with hepatic neurodendocrine metastases treated with cytoreduction (69%) compared to transarterial embolization (69%). A survival benefit was demonstrated by Chamberlain et al., who compared patients treated with surgical resection (1-, 3-, and 5-year survival rates: 94%, 83%, and 76%, respectively) to those treated medically (1-, 3-, and 5-year survival rates: 76%, 39%, and 0%, respectively). For this reason, a surgical approach is advocated when feasible.
The goal of treatment for thoracoabdominal PGLs is the same as that for adrenal PCCs: to achieve complete resection where possible. Surgery in these cases is complex and technically challenging. It should be centralized to specialist centers [19].
Radionuclide Treatment
Radionuclide treatment may be indicated for patients with malignant/metastatic disease for whom surgical resection is not feasible. Radionuclide treatment is performed using β-emitting isotopes coupled to either MIBG or somatostatin analogues. The structural similarity between MIBG and noradrenaline allows uptake and concentration of MIBG in chromaffin cells, which forms the basis of its utility in diagnostic imaging for sympathetic PCCs and PGLs.
I131-labeled MIBG therapy has been used for the adjuvant treatment of malignant PCCs and PGLs since the 1980s when I131, which was already in use for the treatment of thyroid malignancy, was attached to the MIBG molecule; when concentrated in the chromaffin cells, I131 produced MIBG-I131 at high enough doses to be used as a selective radiation therapy agent [189–191]. Approximately 60% of metastatic PCCs/PGLs are 131I-MIBG avid. Of these, 30% exhibit a complete/partial respond to I131-labeled MIBG; disease stabilization can be achieved in a further 43% [25].
Tumor response to 131I-MIBG can be assessed radiologically and biochemically [192]. A phase II clinical trial of high dose 131I-MIBG therapy in 50 patients with metastatic PCCs/PGLs showed that 65% of tumors exhibited at least a partial response, with a 64% 5-year overall survival rate [193]. Safford et al. [194] reported on 33 patients with metastatic PCCs/PGLs who were treated with I131-labeled MIBG and demonstrated a median survival of 4.7 years. However, treatment is associated with significant side effects, including nausea, vomiting, hypertension, and hypothyroidism. High dose 131I-MIBG is also myeloablative and, therefore, should be considered only in highly selected patients.
The presence of SSTR in PCCs/PGLs has similarly facilitated treatment with radiolabeled somatostatin analogues, the most commonly used of which are yttrium-90-DOTATOC (90Y-DOTATOC) and lutetium-177-DOTA0-Tyr3-octreotate (177Lu-DOTATATE) [195, 196]. The likelihood of tumor response is predicted by high tumor uptake of these agents on pretreatment scintigraphy. Symptomatic relief and tumor stabilization have been reported, and the results reported for 177Lu-DOTATATE in metastatic NETs are encouraging in terms of tumor regression [197]. Kwekkeboom et al. [196] analyzed the response to 177Lu-DOTATATE in 301 patients with NETs at 3 months following treatment. They reported an overall objective tumor response rate, including complete remission, partial remission, and minor response of 46%, with high uptake on diagnostic Octreoscan imaging strongly predictive of tumor remission. Therapy was well tolerated with low toxicity. Although this therapeutic modality is in the early stages of investigation, it may become an alternative strategy when surgical intervention is not possible.
Radiation Therapy
Malignant PCCs/PGLs are not radiosensitive and external beam radiotherapy is not a primary treatment modality. Recent reports suggest that it may be effective in the management of locally advanced malignant paraganglioma when used in conjunction with radionuclide therapy (131I-MIBG). In a small series of patients with malignant PCCs/PGLs of the thorax, abdomen, and pelvis, Fishbein et al. [198] reported a durable response rate and excellent performance status at up to 24 months in patients with widespread systemic metastases treated with sequential 131I-MIBG and external beam radiotherapy, supporting a role for this treatment strategy in local control [198]. External beam radiotherapy may also have a role in conjunction with surgical excision for the treatment of isolated bony metastases. There have been several case reports and series of patients with isolated spinal metastases for whom en-bloc resection of the metastasis followed by external beam radiotherapy has resulted in disease-free intervals of more than 5 years [199–201], with the longest reported survival of 26 years using this treatment strategy [202].
Systemic Chemotherapy
Systemic chemotherapy is only indicated in patients who are either not amenable to surgical resection or are not responsive to radionuclide therapy. The most effective chemotherapeutic regimen is a combination of cyclophosphamide, vincristine, and dacarbazine (CVD), which has shown response rates of 50%–55%. Partial response is also associated with palliation of symptoms [203–205]. Huang et al. [203] reported a 22-year follow-up of patients treated with CVD who exhibited tumor shrinkage and symptomatic improvement, but no survival benefit was shown in patients whose tumors responded. Based on the 55% of tumors exhibiting a significant reduction in size, CVD may play a role as neoadjuvant therapy to render large tumors amenable to surgical intervention, although as yet this has not been demonstrated in clinical practice.
Targeted Therapy
The lack of survival improvement with standard adjuvant treatments highlights the need for novel targeted therapies. The insights gained from the identification of genetic mutations in PCCs/PGLs and the molecular alterations associated with malignancy may provide a springboard for their development. The division of PCCs/PGLs via molecular profiling into two major groups or clusters (pseudohypoxic phenotype and RAS/RAF/ERK signaling phenotype) [141, 142] has focused the attention of researchers on these discrete routes to tumorigenesis in an attempt to identify appropriate therapeutic targets that may modulate the activity of key enzymes involved in these signaling pathways, thus modulating the malignant transformation of PCCs/PGLs.
Heat Shock Protein 90
Heat shock protein 90 (Hsp90) plays an important role in molecular stability, maintaining the folding and conformation of multiple proteins that may be involved in oncogenic pathways [206]. Hsp90 is overexpressed in malignant PCCs [119], indicating that it may represent a potential therapeutic target [207]; because of the multiple oncogenic signaling pathways regulated by Hsp90, inhibitors may target several oncogenic proteins simultaneously [208]. Hsp90 inhibitors 17-allylamino and 17-demethyoxygeldamycin have been developed and show promise in other malignancies, specifically in early studies of Her2+, trastuzumab-refractory breast cancer, for which objective responses were seen on a weekly schedule of tanespimycin [209, 210]. These agents have yet to undergo a clinical trial for malignant PCCs/PGLs. However, they are well tolerated and side effects consist predominantly of hepatic, gastrointestintal, and constitutional symptoms that are readily managed with supportive medications [211].
Mammalian Target of Rapamycin
In vitro studies have implicated the PI3/Akt/mTOR pathway in the pathogenesis of malignant neuroendocrine tumors, including pheochromocytoma [212]. This pathway is responsible for the regulation of cell growth and survival. Therefore, dysfunction of this pathway, with mTOR upregulation, leads to increased cell proliferation, angiogenesis, and decreased apoptosis, thus potentiating malignant transformation [213]. Everolimus (a compound that inhibits mTOR signaling) has been evaluated in a small number of patients with malignant PCCs/PGLs with disappointing results [212]. It has been postulated that this lack of efficacy may be due to compensatory P13K/AKT and ERK activation in response to mTOR inhibition, which has been previously described in neuroendocrine tumor cell lines [214]. Preclinical data suggest that the concomitant inhibition of more than one pathway may reverse drug resistance and more successfully affect tumor growth than targeting a single pathway. There are currently phase I and phase II studies underway evaluating the efficacy of everolimus combined with multiple other specific molecular drugs, including ertolinib (NCT00843531, phase II), VEGFR inhibitor PTK787/ZK 222584 (NCT00655655, phase I), octreotide, and cixutumumab (NCT01204476, phase I). Initial results of these trials are still awaited.
Receptor Tyrosine Kinase Inhibitors
Sunitinib is a receptor tyrosine kinase inhibitor that inhibits VEGF-R, PDGF, and c-KIT and exhibits both antiangiogenic and antitumor activity [215]. Patients with malignant PCCs/PGLs who were treated with sunitinib have achieved partial tumor regression, improved performance status, and a reduction in tumor marker levels. However, it appears that a pathway of drug resistance develops quickly after an initial tumor stabilization period [215–217].
The efficacy of sunitinib is currently being evaluated in an international, multicenter, randomized, double-blinded phase II study initiated jointly by members of the Pheochromocytoma and paraganglioma RESearch Support ORganization (PRESSOR) consortium and the European Network for the Study of Adrenal Tumors. The First International Randomized Study in Malignant Progressive Pheochromocytoma and Paraganglioma (FIRSTMAPP; NCT01371201) is currently recruiting patients and aims to determine the efficacy of sunitinib on progression-free survival at 12 months in patients with progressive malignant PCCs and PGLs.
The results with other tyrosine kinase inhibitors, including imatinib mesylate, have not shown efficacy in the treatment of PCCs and PGLs [218]. However, preclinical results for the insulin-like growth factor-R inhibitor NVP-AEW541 indicate potential antitumor activity in neuroendocrine tumor cells and animal models. Grossmann et al. [219] tested the antitumor potential of NVP-AEW541 in mouse PCC cell lines; they demonstrated a decrease in cell viability, which was both dose- and time-dependent. It has been postulated that this is due to significant inhibition of PI3K/AKT and compensatory activation of ERK and mTORC1/p70S6K signaling. These findings require further investigation and validation.
Antiangiogenic Therapy
Malignant PCCs/PGLs are highly vascularized tumors that are likely to be dependent on angiogenesis-mediated growth [117, 220, 221]. Targeting the VEGF pathway has become a commonly used strategy in malignancy [222] and may hold promise for malignant PCCs and PGLs. Kulke et al. [223] evaluated the efficacy of thalidomide, an antiangiogenic agent, in combination with temozolamide in patients with metastatic carcinoid tumors, PCCs, and pancreatic NETs. This regimen resulted in an objective biochemical response rate in 40% of patients and a radiological response rate in 25% of patients overall (33% for PCCs). However, this study included only 3 patients with PCCs, one of whom exhibited a partial response [223].
It is likely that genetic testing may play a role in the selection of patients who may benefit from antiangiogenic therapy. Patients with mutations in VHL and SDHx develop PCCs/PGLs with a pseudohypoxic phenotype (cluster 1), exhibiting activation of HIF and upregulation of VEGF and PDGF, which may be targeted with therapy involving the VEGF signaling pathway. Antiangiogenic agents also may represent an attractive therapeutic option in combination with other agents, such as the VEGF receptor tyrosine kinase inhibitor PTK787/ZK 222584, which is currently being trialed in combination with the mTOR inhibitor everolimus for patients with solid tumors, including PCCs/PGLs (NCT00655655). The rationale is that the combination of an mTOR inhibitor and tyrosine kinase inhibitor may have a synergistic effect on both tumor growth and angiogenesis. This is a phase I trial that will predominantly evaluate the side effects and optimal dosing for these agents in combination.
Future Directions
The optimal management of patients with malignant PCCs/PGLs remains a clinical challenge. Insights gained from the identification of genetic mutations and molecular alterations have been central to the recent developments in diagnostic approaches and targeted therapeutics. Due to the rarity of this tumor, large-scale clinical studies that would progress clinical practice are rare. Therefore, it remains important to use all available resources, including cell line and animal models, for ongoing research into appropriate therapeutics [224]. Even more crucial is the requirement for international collaboration [225], which will not only facilitate optimal management but also allow accurate data collection and biobanking of specimens. Large-scale international multicenter studies will be required to effectively exploit and clinically translate the molecular and genetic knowledge gained in the last decade into effective targeted molecular therapies.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Author Contributions
Conception/Design: Aoife J. Lowery, Enda W. McDermott, Ruth S. Prichard
Data analysis and interpretation: Aoife J. Lowery
Manuscript writing: Aoife J. Lowery, Siun Walsh, Enda W. McDermott, Ruth S. Prichard
Final approval of manuscript: Aoife J. Lowery, Siun Walsh, Enda W. McDermott, Ruth S. Prichard
Disclosures
The authors indicated no financial relationships.
Section editors: Herbert Chen: None; Stan Sidhu: None
Reviewer “A”: None
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.DeLellis RA, Lloyd RV, Heitz PU, et al. Tumours of endocrine organs. Lyon, France: IARC Press; 2004. [Google Scholar]
- 2.Stenstrom G, Svardsudd K. Pheochromocytoma in Sweden, 1958–1981: An analysis of the National Cancer Registry Data. Acta Med Scand. 1986;220:225–232. [PubMed] [Google Scholar]
- 3.Andersen GS, Toftdahl DB, Lund JO, et al. The incidence rate of phaeochromocytoma and Conn's syndrome in Denmark, 1977–1981. J Hum Hypertens. 1988;2:187–189. [PubMed] [Google Scholar]
- 4.Beard CM, Sheps SG, Kurland LT, et al. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58:802–804. [PubMed] [Google Scholar]
- 5.Hartley L, Perry-Keene D. Phaeochromocytoma in Queensland, 1970–83. Aust N Z J Surg. 1985;55:471–475. [PubMed] [Google Scholar]
- 6.Baysal BE. Hereditary paraganglioma targets diverse paraganglia. J Med Genet. 2002;39:617–622. doi: 10.1136/jmg.39.9.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erickson D, Kudva YC, Ebersold MJ, et al. Benign paragangliomas: Clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. 2001;86:5210–5216. doi: 10.1210/jcem.86.11.8034. [DOI] [PubMed] [Google Scholar]
- 8.Sinclair AM, Isles CG, Brown I, et al. Secondary hypertension in a blood pressure clinic. Arch Intern Med. 1987;147:1289–1293. [PubMed] [Google Scholar]
- 9.Omura M, Saito J, Yamaguchi K, et al. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27:193–202. doi: 10.1291/hypres.27.193. [DOI] [PubMed] [Google Scholar]
- 10.Young WF., Jr Management approaches to adrenal incidentalomas. A view from Rochester, Minnesota. Endocrinol Metab Clin North Am. 2000;29:159–185. doi: 10.1016/s0889-8529(05)70122-5. [DOI] [PubMed] [Google Scholar]
- 11.Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 12.Mannelli M, Ianni L, Cilotti A, et al. Pheochromocytoma in Italy: A multicentric retrospective study. Eur J Endocrinol. 1999;141:619–624. doi: 10.1530/eje.0.1410619. [DOI] [PubMed] [Google Scholar]
- 13.Cohen DL, Fraker D, Townsend RR. Lack of symptoms in patients with histologic evidence of pheochromocytoma: A diagnostic challenge. Ann NY Acad Sci. 2006;1073:47–51. doi: 10.1196/annals.1353.005. [DOI] [PubMed] [Google Scholar]
- 14.Zelinka T, Widimsky J, Weisserova J. Diminished circadian blood pressure rhythm in patients with asymptomatic normotensive pheochromocytoma. Physiol Res. 2001;50:631–634. [PubMed] [Google Scholar]
- 15.Lai EW, Perera SM, Havekes B, et al. Gender-related differences in the clinical presentation of malignant and benign pheochromocytoma. Endocrine. 2008;34:96–100. doi: 10.1007/s12020-008-9108-4. [DOI] [PubMed] [Google Scholar]
- 16.Zarnegar R, Kebebew E, Duh QY, et al. Malignant pheochromocytoma. Surg Oncol Clin N Am. 2006;15:555–571. doi: 10.1016/j.soc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Offergeld C, Brase C, Yaremchuk S, et al. Head and neck paragangliomas: Clinical and molecular genetic classification. Clinics (Sao Paulo) 2012;67(suppl 1):19–28. doi: 10.6061/clinics/2012(Sup01)05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proye C, Vix M, Goropoulos A, et al. High incidence of malignant pheochromocytoma in a surgical unit. 26 cases out of 100 patients operated from 1971 to 1991. J Endocrinol Invest. 1992;15:651–663. doi: 10.1007/BF03345810. [DOI] [PubMed] [Google Scholar]
- 19.Walz MK, Alesina PF, Wenger FA, et al. Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: Results of 161 tumors in 126 patients. World J Surg. 2006;30:899–908. doi: 10.1007/s00268-005-0373-6. [DOI] [PubMed] [Google Scholar]
- 20.Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63:5615–5621. [PubMed] [Google Scholar]
- 21.Thompson LD. Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551–566. doi: 10.1097/00000478-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Strong VE, Kennedy T, Al-Ahmadie H, et al. Prognostic indicators of malignancy in adrenal pheochromocytomas: Clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery. 2008;143:759–768. doi: 10.1016/j.surg.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Tischler AS. Pheochromocytoma: time to stamp out “malignancy”? Endocr Pathol. 2008;19:207–208. doi: 10.1007/s12022-008-9047-x. [DOI] [PubMed] [Google Scholar]
- 24.Timmers HJ, Brouwers FM, Hermus AR, et al. Metastases but not cardiovascular mortality reduces life expectancy following surgical resection of apparently benign pheochromocytoma. Endocr Relat Cancer. 2008;15:1127–1133. doi: 10.1677/ERC-08-0049. [DOI] [PubMed] [Google Scholar]
- 25.Chrisoulidou A, Kaltsas G, Ilias I, et al. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2007;14:569–585. doi: 10.1677/ERC-07-0074. [DOI] [PubMed] [Google Scholar]
- 26.Nomura K, Kimura H, Shimizu S, et al. Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab. 2009;94:2850–2856. doi: 10.1210/jc.2008-2697. [DOI] [PubMed] [Google Scholar]
- 27.Sisson JC, Shulkin BL, Esfandiari NH. Courses of malignant pheochromocytoma: Implications for therapy. Ann N Y Acad Sci. 2006;1073:505–511. doi: 10.1196/annals.1353.053. [DOI] [PubMed] [Google Scholar]
- 28.Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: Tecommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab. 2007;3:92–102. doi: 10.1038/ncpendmet0396. [DOI] [PubMed] [Google Scholar]
- 29.Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: Updates. Arch Pathol Lab Med. 2008;132:1272–1284. doi: 10.5858/2008-132-1272-PAEPU. [DOI] [PubMed] [Google Scholar]
- 30.Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–333. doi: 10.1055/s-0031-1301302. [DOI] [PubMed] [Google Scholar]
- 31.Machens A, Brauckhoff M, Gimm O, et al. Risk-oriented approach to hereditary adrenal pheochromocytoma. Ann N Y Acad Sci. 2006;1073:417–428. doi: 10.1196/annals.1353.045. [DOI] [PubMed] [Google Scholar]
- 32.Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet. 2011;19:617–623. doi: 10.1038/ejhg.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 34.Astuti D, Douglas F, Lennard TW, et al. Germline SDHD mutation in familial phaeochromocytoma. Lancet. 2001;357:1181–1182. doi: 10.1016/S0140-6736(00)04378-6. [DOI] [PubMed] [Google Scholar]
- 35.Peczkowska M, Cascon A, Prejbisz A, et al. Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab. 2008;4:111–115. doi: 10.1038/ncpendmet0726. [DOI] [PubMed] [Google Scholar]
- 36.Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–233. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlisio S, Kenchappa RS, Vredeveld LC, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008;22:884–893. doi: 10.1101/gad.1648608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685–2692. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- 39.Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–667. doi: 10.1038/ng.861. [DOI] [PubMed] [Google Scholar]
- 40.Zhuang Z, Yang C, Lorenzo F, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:922–930. doi: 10.1056/NEJMoa1205119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bourgeron T, Rustin P, Chretien D, et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11:144–149. doi: 10.1038/ng1095-144. [DOI] [PubMed] [Google Scholar]
- 42.Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korpershoek E, Favier J, Gaal J, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96:E1472–E1476. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- 44.Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kunst HP, Rutten MH, de Monnink JP, et al. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res. 2011;17:247–254. doi: 10.1158/1078-0432.CCR-10-0420. [DOI] [PubMed] [Google Scholar]
- 46.Bayley JP, Kunst HP, Cascon A, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–372. doi: 10.1016/S1470-2045(10)70007-3. [DOI] [PubMed] [Google Scholar]
- 47.Schiavi F, Milne RL, Anda E, et al. Are we overestimating the penetrance of mutations in SDHB? Hum Mutat. 2010;31:761–762. doi: 10.1002/humu.21269. [DOI] [PubMed] [Google Scholar]
- 48.Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 49.Ricketts CJ, Forman JR, Rattenberry E, et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41–51. doi: 10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- 50.Brouwers FM, Eisenhofer G, Tao JJ, et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: Implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505–4509. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- 51.Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92:3822–3828. doi: 10.1210/jc.2007-0709. [DOI] [PubMed] [Google Scholar]
- 52.Gill AJ, Pachter NS, Clarkson A, et al. Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome type 4. N Engl J Med. 2011;364:885–886. doi: 10.1056/NEJMc1012357. [DOI] [PubMed] [Google Scholar]
- 53.Ricketts CJ, Shuch B, Vocke CD, et al. Succinate dehydrogenase kidney cancer: An aggressive example of the warburg effect in cancer. J Urol. 2012;188:2063–2071. doi: 10.1016/j.juro.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Timmers HJ, Gimenez-Roqueplo AP, Mannelli M, et al. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer. 2009;16:391–400. doi: 10.1677/ERC-08-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burnichon N, Rohmer V, Amar L, et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009;94:2817–2827. doi: 10.1210/jc.2008-2504. [DOI] [PubMed] [Google Scholar]
- 56.Carney JA. The triad of gastric epithelioid leiomyosarcoma, pulmonary chondroma, and functioning extra-adrenal paraganglioma: A five-year review. Medicine (Baltimore) 1983;62:159–169. doi: 10.1097/00005792-198305000-00003. [DOI] [PubMed] [Google Scholar]
- 57.Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 58.Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neumann HP, Sullivan M, Winter A, et al. Germline mutations of the TMEM127 gene in patients with paraganglioma of head and neck and extraadrenal abdominal sites. J Clin Endocrinol Metab. 2011;96:E1279–E1282. doi: 10.1210/jc.2011-0114. [DOI] [PubMed] [Google Scholar]
- 60.Yao L, Schiavi F, Cascon A, et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA. 2010;304:2611–2619. doi: 10.1001/jama.2010.1830. [DOI] [PubMed] [Google Scholar]
- 61.Yeh IT, Lenci RE, Qin Y, et al. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet. 2008;124:279–285. doi: 10.1007/s00439-008-0553-1. [DOI] [PubMed] [Google Scholar]
- 62.Astuti D, Ricketts CJ, Chowdhury R, et al. Mutation analysis of HIF prolyl hydroxylases (PHD/EGLN) in individuals with features of phaeochromocytoma and renal cell carcinoma susceptibility. Endocr Relat Cancer. 2011;18:73–83. doi: 10.1677/ERC-10-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lorenzo FR, Yang C, Ng Tang Fui M, et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl) 2012 Oct 23; doi: 10.1007/s00109-012-0967-z. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burnichon N, Cascon A, Schiavi F, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. 2012;18:2828–2837. doi: 10.1158/1078-0432.CCR-12-0160. [DOI] [PubMed] [Google Scholar]
- 65.Eisenhofer G, Goldstein DS, Kopin IJ, et al. Pheochromocytoma: Rediscovery as a catecholamine-metabolizing tumor. Endocr Pathol. 2003;14:193–212. doi: 10.1007/s12022-003-0012-4. [DOI] [PubMed] [Google Scholar]
- 66.Eisenhofer G, Lenders JW, Pacak K. Choice of biochemical test for diagnosis of pheochromocytoma: Validation of plasma metanephrines. Curr Hypertens Rep. 2002;4:250–255. doi: 10.1007/s11906-002-0015-4. [DOI] [PubMed] [Google Scholar]
- 67.Lenders JW, Pacak K, Eisenhofer G. New advances in the biochemical diagnosis of pheochromocytoma: Moving beyond catecholamines. Ann NY Acad Sci. 2002;970:29–40. doi: 10.1111/j.1749-6632.2002.tb04410.x. [DOI] [PubMed] [Google Scholar]
- 68.Hickman PE, Leong M, Chang J, et al. Plasma free metanephrines are superior to urine and plasma catecholamines and urine catecholamine metabolites for the investigation of phaeochromocytoma. Pathology. 2009;41:173–177. doi: 10.1080/00313020802579284. [DOI] [PubMed] [Google Scholar]
- 69.Unger N, Pitt C, Schmidt IL, et al. Diagnostic value of various biochemical parameters for the diagnosis of pheochromocytoma in patients with adrenal mass. Eur J Endocrinol. 2006;154:409–417. doi: 10.1530/eje.1.02097. [DOI] [PubMed] [Google Scholar]
- 70.Sawka AM, Prebtani AP, Thabane L, et al. A systematic review of the literature examining the diagnostic efficacy of measurement of fractionated plasma free metanephrines in the biochemical diagnosis of pheochromocytoma. BMC Endocr Disord. 2004;4:2. doi: 10.1186/1472-6823-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eisenhofer G, Pacak K, Huynh TT, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer. 2011;18:97–111. doi: 10.1677/ERC-10-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eisenhofer G, Lenders JW, Timmers H, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57:411–420. doi: 10.1373/clinchem.2010.153320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schlumberger M, Gicquel C, Lumbroso J, et al. Malignant pheochromocytoma: Clinical, biological, histologic and therapeutic data in a series of 20 patients with distant metastases. J Endocrinol Invest. 1992;15:631–642. doi: 10.1007/BF03345807. [DOI] [PubMed] [Google Scholar]
- 74.Proye C, Fossati P, Fontaine P, et al. Dopamine-secreting pheochromocytoma: An unrecognized entity? Classification of pheochromocytomas according to their type of secretion. Surgery. 1986;100:1154–1162. [PubMed] [Google Scholar]
- 75.Foo SH, Chan SP, Ananda V, et al. Dopamine-secreting phaeochromocytomas and paragangliomas: Clinical features and management. Singapore Med J. 2010;51:e89–e93. [PubMed] [Google Scholar]
- 76.Zelinka T, Musil Z, Duskova J, et al. Metastatic pheochromocytoma: Does the size and age matter? Eur J Clin Invest. 2011;41:1121–1128. doi: 10.1111/j.1365-2362.2011.02518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.John H, Ziegler WH, Hauri D, et al. Pheochromocytomas: Can malignant potential be predicted? Urology. 1999;53:679–683. doi: 10.1016/s0090-4295(98)00612-8. [DOI] [PubMed] [Google Scholar]
- 78.Januszewicz W, Wocial B, Januszewicz A, et al. Dopamine and dopa urinary excretion in patients with pheochromocytoma–diagnostic implications. Blood Press. 2001;10:212–216. doi: 10.1080/08037050152669729. [DOI] [PubMed] [Google Scholar]
- 79.Rao F, Keiser HR, O'Connor DT. Malignant pheochromocytoma Chromaffin granule transmitters and response to treatment. Hypertension. 2000;36:1045–1052. doi: 10.1161/01.hyp.36.6.1045. [DOI] [PubMed] [Google Scholar]
- 80.Guerin M, Guillemot J, Thouennon E, et al. Granins and their derived peptides in normal and tumoral chromaffin tissue: Implications for the diagnosis and prognosis of pheochromocytoma. Regul Pept. 2010;165:21–29. doi: 10.1016/j.regpep.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 81.Yon L, Guillemot J, Montero-Hadjadje M, et al. Identification of the secretogranin II-derived peptide EM66 in pheochromocytomas as a potential marker for discriminating benign versus malignant tumors. J Clin Endocrinol Metab. 2003;88:2579–2585. doi: 10.1210/jc.2002-021748. [DOI] [PubMed] [Google Scholar]
- 82.Guillemot J, Thouennon E, Guerin M, et al. Differential expression and processing of secretogranin II in relation to the status of pheochromocytoma: Implications for the production of the tumoral marker EM66. J Mol Endocrinol. 2012;48:115–127. doi: 10.1530/JME-11-0077. [DOI] [PubMed] [Google Scholar]
- 83.Eisenhofer G, Lenders JW, Siegert G, et al. Plasma methoxytyramine: A novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012;48:1739–1749. doi: 10.1016/j.ejca.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lumachi F, Tregnaghi A, Zucchetta P, et al. Sensitivity and positive predictive value of CT, MRI and 123I-MIBG scintigraphy in localizing pheochromocytomas: A prospective study. Nucl Med Commun. 2006;27:583–587. doi: 10.1097/00006231-200607000-00006. [DOI] [PubMed] [Google Scholar]
- 85.Maurea S, Cuocolo A, Reynolds JC, et al. Iodine-131-metaiodobenzylguanidine scintigraphy in preoperative and postoperative evaluation of paragangliomas: Comparison with CT and MRI. J Nucl Med. 1993;34:173–179. [PubMed] [Google Scholar]
- 86.Timmers HJ, Chen CC, Carrasquillo JA, et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2009;94:4757–4767. doi: 10.1210/jc.2009-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taieb D, Sebag F, Barlier A, et al. 18F-FDG avidity of pheochromocytomas and paragangliomas: A new molecular imaging signature? J Nucl Med. 2009;50:711–717. doi: 10.2967/jnumed.108.060731. [DOI] [PubMed] [Google Scholar]
- 88.Vaidyanathan G. Meta-iodobenzylguanidine and analogues: Chemistry and biology. Q J Nucl Med Mol Imaging. 2008;52:351–368. [PubMed] [Google Scholar]
- 89.Bombardieri E, Giammarile F, Aktolun C, et al. 131I/123I-metaiodobenzylguanidine (mIBG) scintigraphy: Procedure guidelines for tumour imaging. Eur J Nucl Med Mol Imaging. 2010;37:2436–2446. doi: 10.1007/s00259-010-1545-7. [DOI] [PubMed] [Google Scholar]
- 90.Jacobson AF, Deng H, Lombard J, et al. 123I-meta-iodobenzylguanidine scintigraphy for the detection of neuroblastoma and pheochromocytoma: Results of a meta-analysis. J Clin Endocrinol Metab. 2010;95:2596–2606. doi: 10.1210/jc.2009-2604. [DOI] [PubMed] [Google Scholar]
- 91.Van Der Horst-Schrivers AN, Jager PL, Boezen HM, et al. Iodine-123 metaiodobenzylguanidine scintigraphy in localising phaeochromocytomas: Experience and meta-analysis. Anticancer Res. 2006;26:1599–1604. [PubMed] [Google Scholar]
- 92.Kaji P, Carrasquillo JA, Linehan WM, et al. The role of 6-[18F]fluorodopamine positron emission tomography in the localization of adrenal pheochromocytoma associated with von Hippel-Lindau syndrome. Eur J Endocrinol. 2007;156:483–487. doi: 10.1530/EJE-06-0712. [DOI] [PubMed] [Google Scholar]
- 93.Fonte JS, Robles JF, Chen CC, et al. False-negative (1)(2)(3)I-MIBG SPECT is most commonly found in SDHB-related pheochromocytoma or paraganglioma with high frequency to develop metastatic disease. Endocr Relat Cancer. 2012;19:83–93. doi: 10.1530/ERC-11-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fottner C, Helisch A, Anlauf M, et al. 6–18F-fluoro-L-dihydroxyphenylalanine positron emission tomography is superior to 123I-metaiodobenzyl-guanidine scintigraphy in the detection of extraadrenal and hereditary pheochromocytomas and paragangliomas: Correlation with vesicular monoamine transporter expression. J Clin Endocrinol Metab. 2010;95:2800–2810. doi: 10.1210/jc.2009-2352. [DOI] [PubMed] [Google Scholar]
- 95.Shulkin BL, Koeppe RA, Francis IR, et al. Pheochromocytomas that do not accumulate metaiodobenzylguanidine: Localization with PET and administration of FDG. Radiology. 1993;186:711–715. doi: 10.1148/radiology.186.3.8430179. [DOI] [PubMed] [Google Scholar]
- 96.Timmers HJ, Kozupa A, Chen CC, et al. Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol. 2007;25:2262–2269. doi: 10.1200/JCO.2006.09.6297. [DOI] [PubMed] [Google Scholar]
- 97.Blanchet EM, Martucci V, Pacak K. Pheochromocytoma and paraganglioma: Current functional and future molecular imaging. Front Oncol. 2011;1:58. doi: 10.3389/fonc.2011.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ilias I, Yu J, Carrasquillo JA, et al. Superiority of 6-[18F]-fluorodopamine positron emission tomography versus [131I]-metaiodobenzylguanidine scintigraphy in the localization of metastatic pheochromocytoma. J Clin Endocrinol Metab. 2003;88:4083–4087. doi: 10.1210/jc.2003-030235. [DOI] [PubMed] [Google Scholar]
- 99.Timmers HJ, Eisenhofer G, Carrasquillo JA, et al. Use of 6-[18F]-fluorodopamine positron emission tomography (PET) as first-line investigation for the diagnosis and localization of non-metastatic and metastatic phaeochromocytoma (PHEO) Clin Endocrinol (Oxf) 2009;71:11–17. doi: 10.1111/j.1365-2265.2008.03496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zelinka T, Timmers HJ, Kozupa A, et al. Role of positron emission tomography and bone scintigraphy in the evaluation of bone involvement in metastatic pheochromocytoma and paraganglioma: specific implications for succinate dehydrogenase enzyme subunit B gene mutations. Endocr Relat Cancer. 2008;15:311–323. doi: 10.1677/ERC-07-0217. [DOI] [PubMed] [Google Scholar]
- 101.Timmers HJ, Hadi M, Carrasquillo JA, et al. The effects of carbidopa on uptake of 6–18F-Fluoro-L-DOPA in PET of pheochromocytoma and extraadrenal abdominal paraganglioma. J Nucl Med. 2007;48:1599–1606. doi: 10.2967/jnumed.107.042721. [DOI] [PubMed] [Google Scholar]
- 102.Krenning EP, Bakker WH, Breeman WA, et al. Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet. 1989;1:242–244. doi: 10.1016/s0140-6736(89)91258-0. [DOI] [PubMed] [Google Scholar]
- 103.Limouris GS, Giannakopoulos V, Stavraka A, et al. Comparison of In-111 pentetreotide, Tc-99m (V)DMSA and I-123 mlBG scintimaging in neural crest tumors. Anticancer Res. 1997;17:1589–1592. [PubMed] [Google Scholar]
- 104.Duet M, Sauvaget E, Petelle B, et al. Clinical impact of somatostatin receptor scintigraphy in the management of paragangliomas of the head and neck. J Nucl Med. 2003;44:1767–1774. [PubMed] [Google Scholar]
- 105.Khan MU, Khan S, El-Refaie S, et al. Clinical indications for Gallium-68 positron emission tomography imaging. Eur J Surg Oncol. 2009;35:561–567. doi: 10.1016/j.ejso.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 106.Krausz Y, Freedman N, Rubinstein R, et al. 68Ga-DOTA-NOC PET/CT imaging of neuroendocrine tumors: Comparison with (1)(1)(1)In-DTPA-octreotide (OctreoScan(R)) Mol Imaging Biol. 2011;13:583–593. doi: 10.1007/s11307-010-0374-1. [DOI] [PubMed] [Google Scholar]
- 107.Kroiss A, Putzer D, Uprimny C, et al. Functional imaging in phaeochromocytoma and neuroblastoma with 68Ga-DOTA-Tyr 3-octreotide positron emission tomography and 123I-metaiodobenzylguanidine. Eur J Nucl Med Mol Imaging. 2011;38:865–873. doi: 10.1007/s00259-010-1720-x. [DOI] [PubMed] [Google Scholar]
- 108.Naji M, Zhao C, Welsh SJ, et al. 68Ga-DOTA-TATE PET vs. 123I-MIBG in identifying malignant neural crest tumours. Mol Imaging Biol. 2011;13:769–775. doi: 10.1007/s11307-010-0396-8. [DOI] [PubMed] [Google Scholar]
- 109.Kimura N, Nakazato Y, Nagura H, et al. Expression of intermediate filaments in neuroendocrine tumors. Arch Pathol Lab Med. 1990;114:506–510. [PubMed] [Google Scholar]
- 110.Kimura N, Miura Y, Nagatsu I, et al. Catecholamine synthesizing enzymes in 70 cases of functioning and non-functioning phaeochromocytoma and extra-adrenal paraganglioma. Virchows Arch A Pathol Anat Histopathol. 1992;421:25–32. doi: 10.1007/BF01607135. [DOI] [PubMed] [Google Scholar]
- 111.Meijer WG, Copray SC, Hollema H, et al. Catecholamine-synthesizing enzymes in carcinoid tumors and pheochromocytomas. Clin Chem. 2003;49:586–593. doi: 10.1373/49.4.586. [DOI] [PubMed] [Google Scholar]
- 112.Wu D, Tischler AS, Lloyd RV, et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol. 2009;33:599–608. doi: 10.1097/PAS.0b013e318190d12e. [DOI] [PubMed] [Google Scholar]
- 113.Kimura N, Watanabe T, Noshiro T, et al. Histological grading of adrenal and extra-adrenal pheochromocytomas and relationship to prognosis: A clinicopathological analysis of 116 adrenal pheochromocytomas and 30 extra-adrenal sympathetic paragangliomas including 38 malignant tumors. Endocr Pathol. 2005;16:23–32. doi: 10.1385/ep:16:1:023. [DOI] [PubMed] [Google Scholar]
- 114.Helman LJ, Cohen PS, Averbuch SD, et al. Neuropeptide Y expression distinguishes malignant from benign pheochromocytoma. J Clin Oncol. 1989;7:1720–1725. doi: 10.1200/JCO.1989.7.11.1720. [DOI] [PubMed] [Google Scholar]
- 115.Portela-Gomes GM, Stridsberg M, Grimelius L, et al. Expression of chromogranins A, B, and C (secretogranin II) in human adrenal medulla and in benign and malignant pheochromocytomas: An immunohistochemical study with region-specific antibodies. APMIS. 2004;112:663–673. doi: 10.1111/j.1600-0463.2004.t01-1-apm1121003.x. [DOI] [PubMed] [Google Scholar]
- 116.August C, August K, Schroeder S, et al. CGH and CD 44/MIB-1 immunohistochemistry are helpful to distinguish metastasized from nonmetastasized sporadic pheochromocytomas. Mod Pathol. 2004;17:1119–1128. doi: 10.1038/modpathol.3800160. [DOI] [PubMed] [Google Scholar]
- 117.Favier J, Plouin PF, Corvol P, et al. Angiogenesis and vascular architecture in pheochromocytomas: Distinctive traits in malignant tumors. Am J Pathol. 2002;161:1235–1246. doi: 10.1016/S0002-9440(10)64400-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Scholz T, Schulz C, Klose S, et al. Diagnostic management of benign and malignant pheochromocytoma. Exp Clin Endocrinol Diabetes. 2007;115:155–159. doi: 10.1055/s-2007-970410. [DOI] [PubMed] [Google Scholar]
- 119.Boltze C, Mundschenk J, Unger N, et al. Expression profile of the telomeric complex discriminates between benign and malignant pheochromocytoma. J Clin Endocrinol Metab. 2003;88:4280–4286. doi: 10.1210/jc.2002-021299. [DOI] [PubMed] [Google Scholar]
- 120.de Wailly P, Oragano L, Rade F, et al. Malignant pheochromocytoma: New malignancy criteria. Langenbecks Arch Surg. 2012;397:239–246. doi: 10.1007/s00423-011-0850-3. [DOI] [PubMed] [Google Scholar]
- 121.Tavangar SM, Shojaee A, Moradi Tabriz H, et al. Immunohistochemical expression of Ki67, c-erbB-2, and c-kit antigens in benign and malignant pheochromocytoma. Pathol Res Pract. 2010;206:305–309. doi: 10.1016/j.prp.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 122.Feng C, Li HZ, Yan WG, et al. The significance of Ki-67 antigen expression in the distinction between benign and malignant pheochromocytomas. Zhonghua Wai Ke Za Zhi. 2007;45:1697–1700. [PubMed] [Google Scholar]
- 123.van der Harst E, Bruining HA, Jaap Bonjer H, et al. Proliferative index in phaeochromocytomas: Does it predict the occurrence of metastases? J Pathol. 2000;191:175–180. doi: 10.1002/(SICI)1096-9896(200006)191:2<175::AID-PATH615>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 124.Brown HM, Komorowski RA, Wilson SD, et al. Predicting metastasis of pheochromocytomas using DNA flow cytometry and immunohistochemical markers of cell proliferation: A positive correlation between MIB-1 staining and malignant tumor behavior. Cancer. 1999;86:1583–1589. [PubMed] [Google Scholar]
- 125.Blank A, Schmitt AM, Korpershoek E, et al. SDHB loss predicts malignancy in pheochromocytomas/sympathethic paragangliomas, but not through hypoxia signalling. Endocr Relat Cancer. 2010;17:919–928. doi: 10.1677/ERC-09-0316. [DOI] [PubMed] [Google Scholar]
- 126.Gill AJ, Benn DE, Chou A, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol. 2010;41:805–814. doi: 10.1016/j.humpath.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 127.Ayala-Ramirez M, Feng L, Johnson MM, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: Primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96:717–725. doi: 10.1210/jc.2010-1946. [DOI] [PubMed] [Google Scholar]
- 128.Feng F, Zhu Y, Wang X, et al. Predictive factors for malignant pheochromocytoma: Analysis of 136 patients. J Urol. 2011;185:1583–1590. doi: 10.1016/j.juro.2010.12.050. [DOI] [PubMed] [Google Scholar]
- 129.Agarwal A, Mehrotra PK, Jain M, et al. Size of the tumor and pheochromocytoma of the adrenal gland scaled score (PASS): Can they predict malignancy? World J Surg. 2010;34:3022–3028. doi: 10.1007/s00268-010-0744-5. [DOI] [PubMed] [Google Scholar]
- 130.Amar L, Servais A, Gimenez-Roqueplo AP, et al. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab. 2005;90:2110–2116. doi: 10.1210/jc.2004-1398. [DOI] [PubMed] [Google Scholar]
- 131.Park J, Song C, Park M, et al. Predictive characteristics of malignant pheochromocytoma. Korean J Urol. 2011;52:241–246. doi: 10.4111/kju.2011.52.4.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 133.Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 134.Edstrom E, Mahlamaki E, Nord B, et al. Comparative genomic hybridization reveals frequent losses of chromosomes 1p and 3q in pheochromocytomas and abdominal paragangliomas, suggesting a common genetic etiology. Am J Pathol. 2000;156:651–659. doi: 10.1016/S0002-9440(10)64769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dannenberg H, Speel EJ, Zhao J, et al. Losses of chromosomes 1p and 3q are early genetic events in the development of sporadic pheochromocytomas. Am J Pathol. 2000;157:353–359. doi: 10.1016/S0002-9440(10)64547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu TH, Chen YJ, Wu SF, et al. Distinction between benign and malignant pheochromocytomas. Zhonghua Bing Li Xue Za Zhi. 2004;33:198–202. [PubMed] [Google Scholar]
- 137.Dannenberg H, de Krijger RR, Zhao J, et al. Differential loss of chromosome 11q in familial and sporadic parasympathetic paragangliomas detected by comparative genomic hybridization. Am J Pathol. 2001;158:1937–1942. doi: 10.1016/S0002-9440(10)64662-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sandgren J, Diaz de Stahl T, Andersson R, et al. Recurrent genomic alterations in benign and malignant pheochromocytomas and paragangliomas revealed by whole-genome array comparative genomic hybridization analysis. Endocr Relat Cancer. 2010;17:561–579. doi: 10.1677/ERC-09-0310. [DOI] [PubMed] [Google Scholar]
- 139.Eisenhofer G, Huynh TT, Pacak K, et al. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: Activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer. 2004;11:897–911. doi: 10.1677/erc.1.00838. [DOI] [PubMed] [Google Scholar]
- 140.Dahia PL, Hao K, Rogus J, et al. Novel pheochromocytoma susceptibility loci identified by integrative genomics. Cancer Res. 2005;65:9651–9658. doi: 10.1158/0008-5472.CAN-05-1427. [DOI] [PubMed] [Google Scholar]
- 141.Dahia PL, Ross KN, Wright ME, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005;1:72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Huynh TT, Pacak K, Wong DL, et al. Transcriptional regulation of phenylethanolamine N-methyltransferase in pheochromocytomas from patients with von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2. Ann N Y Acad Sci. 2006;1073:241–252. doi: 10.1196/annals.1353.026. [DOI] [PubMed] [Google Scholar]
- 143.Brouwers FM, Elkahloun AG, Munson PJ, et al. Gene expression profiling of benign and malignant pheochromocytoma. Ann NY Acad Sci. 2006;1073:541–556. doi: 10.1196/annals.1353.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Waldmann J, Fendrich V, Holler J, et al. Microarray analysis reveals differential expression of benign and malignant pheochromocytoma. Endocr Relat Cancer. 2010;17:743–756. doi: 10.1677/ERC-09-0118. [DOI] [PubMed] [Google Scholar]
- 145.Thouennon E, Elkahloun AG, Guillemot J, et al. Identification of potential gene markers and insights into the pathophysiology of pheochromocytoma malignancy. J Clin Endocrinol Metab. 2007;92:4865–4872. doi: 10.1210/jc.2007-1253. [DOI] [PubMed] [Google Scholar]
- 146.Suh I, Shibru D, Eisenhofer G, et al. Candidate genes associated with malignant pheochromocytomas by genome-wide expression profiling. Ann Surg. 2009;250:983–990. doi: 10.1097/SLA.0b013e3181b248bb. [DOI] [PubMed] [Google Scholar]
- 147.Meyer-Rochow GY, Jackson NE, Conaglen JV, et al. MicroRNA profiling of benign and malignant pheochromocytomas identifies novel diagnostic and therapeutic targets. Endocr Relat Cancer. 2010;17:835–846. doi: 10.1677/ERC-10-0142. [DOI] [PubMed] [Google Scholar]
- 148.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Patterson EE, Holloway AK, Weng J, et al. MicroRNA profiling of adrenocortical tumors reveals miR-483 as a marker of malignancy. Cancer. 2011;117:1630–1639. doi: 10.1002/cncr.25724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tombol Z, Eder K, Kovacs A, et al. MicroRNA expression profiling in benign (sporadic and hereditary) and recurring adrenal pheochromocytomas. Mod Pathol. 2010;23:1583–1595. doi: 10.1038/modpathol.2010.164. [DOI] [PubMed] [Google Scholar]
- 151.Patterson E, Webb R, Weisbrod A, et al. The microRNA expression changes associated with malignancy and SDHB mutation in pheochromocytoma. Endocr Relat Cancer. 2012;19:157–166. doi: 10.1530/ERC-11-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Benhammou JN, Boris RS, Pacak K, et al. Functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma in patients with von Hippel-Lindau syndrome after at least 5 years of followup. J Urol. 2010;184:1855–1859. doi: 10.1016/j.juro.2010.06.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Adler JT, Meyer-Rochow GY, Chen H, et al. Pheochromocytoma: Current approaches and future directions. The Oncologist. 2008;13:779–793. doi: 10.1634/theoncologist.2008-0043. [DOI] [PubMed] [Google Scholar]
- 154.Gagner M, Lacroix A, Bolte E. Laparoscopic adrenalectomy in Cushing's syndrome and pheochromocytoma. N Engl J Med. 1992;327:1033. doi: 10.1056/NEJM199210013271417. [DOI] [PubMed] [Google Scholar]
- 155.Gumbs AA, Gagner M. Laparoscopic adrenalectomy. Best Pract Res Clin Endocrinol Metab. 2006;20:483–499. doi: 10.1016/j.beem.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 156.Germain A, Klein M, Brunaud L. Surgical management of adrenal tumors. J Visc Surg. 2011;148:e250–e261. doi: 10.1016/j.jviscsurg.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 157.Ippolito G, Palazzo FF, Sebag F, et al. Safety of laparoscopic adrenalectomy in patients with large pheochromocytomas: A single institution review. World J Surg. 2008;32:840–844. doi: 10.1007/s00268-007-9327-5. [DOI] [PubMed] [Google Scholar]
- 158.Li ML, Fitzgerald PA, Price DC, et al. Iatrogenic pheochromocytomatosis: A previously unreported result of laparoscopic adrenalectomy. Surgery. 2001;130:1072–1077. doi: 10.1067/msy.2001.118373. [DOI] [PubMed] [Google Scholar]
- 159.Brunt LM, Molmenti EP, Kerbl K, et al. Retroperitoneal endoscopic adrenalectomy: An experimental study. Surg Laparosc Endosc. 1993;3:300–306. [PubMed] [Google Scholar]
- 160.Walz MK, Peitgen K, Hoermann R, et al. Posterior retroperitoneoscopy as a new minimally invasive approach for adrenalectomy: Results of 30 adrenalectomies in 27 patients. World J Surg. 1996;20:769–774. doi: 10.1007/s002689900117. [DOI] [PubMed] [Google Scholar]
- 161.Rubinstein M, Gill IS, Aron M, et al. Prospective, randomized comparison of transperitoneal versus retroperitoneal laparoscopic adrenalectomy. J Urol. 2005;174:442–445. doi: 10.1097/01.ju.0000165336.44836.2d. [DOI] [PubMed] [Google Scholar]
- 162.Suzuki K, Kageyama S, Hirano Y, et al. Comparison of 3 surgical approaches to laparoscopic adrenalectomy: A nonrandomized, background matched analysis. J Urol. 2001;166:437–443. [PubMed] [Google Scholar]
- 163.Li QY, Li F. Laparoscopic adrenalectomy in pheochromocytoma: Retroperitoneal approach versus transperitoneal approach. J Endourol. 2010;24:1441–1445. doi: 10.1089/end.2010.0065. [DOI] [PubMed] [Google Scholar]
- 164.Fernandez-Cruz L, Saenz A, Taura P, et al. Retroperitoneal approach in laparoscopic adrenalectomy: Is it advantageous? Surg Endosc. 1999;13:86–90. doi: 10.1007/s004649900907. [DOI] [PubMed] [Google Scholar]
- 165.Alesina PF, Hinrichs J, Meier B, et al. Minimally invasive cortical-sparing surgery for bilateral pheochromocytomas. Langenbecks Arch Surg. 2012;397:233–238. doi: 10.1007/s00423-011-0851-2. [DOI] [PubMed] [Google Scholar]
- 166.Brauckhoff M, Thanh PN, Gimm O, et al. Functional results after endoscopic subtotal cortical-sparing adrenalectomy. Surg Today. 2003;33:342–348. doi: 10.1007/s005950300078. [DOI] [PubMed] [Google Scholar]
- 167.Parnaby CN, Galbraith N, O'Dwyer PJ. Importance of the adrenal gland blood supply during laparoscopic subtotal adrenalectomy. J Laparoendosc Adv Surg Tech A. 2010;20:311–315. doi: 10.1089/lap.2009.0361. [DOI] [PubMed] [Google Scholar]
- 168.Walz MK, Peitgen K, Saller B, et al. Subtotal adrenalectomy by the posterior retroperitoneoscopic approach. World J Surg. 1998;22:621–626. doi: 10.1007/s002689900444. [DOI] [PubMed] [Google Scholar]
- 169.Lee JE, Curley SA, Gagel RF, et al. Cortical-sparing adrenalectomy for patients with bilateral pheochromocytoma. Surgery. 1996;120:1064–1070. doi: 10.1016/s0039-6060(96)80056-0. [DOI] [PubMed] [Google Scholar]
- 170.Inabnet WB, Caragliano P, Pertsemlidis D. Pheochromocytoma: Inherited associations, bilaterality, and cortex preservation. Surgery. 2000;128:1007–1011. doi: 10.1067/msy.2000.110846. [DOI] [PubMed] [Google Scholar]
- 171.Yip L, Lee JE, Shapiro SE, et al. Surgical management of hereditary pheochromocytoma. J Am Coll Surg. 2004;198:525–534. doi: 10.1016/j.jamcollsurg.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 172.Brauckhoff M, Dralle H. Function-preserving adrenalectomy for adrenal tumors. Chirurg. 2012;83:519–527. doi: 10.1007/s00104-011-2192-7. [DOI] [PubMed] [Google Scholar]
- 173.Miyajima A, Hattori S, Maeda T, et al. Transumbilical approach for laparo-endoscopic single-site adrenalectomy: Initial experience and short-term outcome. Int J Urol. 2012;19:331–335. doi: 10.1111/j.1442-2042.2011.02932.x. [DOI] [PubMed] [Google Scholar]
- 174.Walz MK, Groeben H, Alesina PF. Single-access retroperitoneoscopic adrenalectomy (SARA) versus conventional retroperitoneoscopic adrenalectomy (CORA): A case-control study. World J Surg. 2010;34:1386–1390. doi: 10.1007/s00268-010-0494-4. [DOI] [PubMed] [Google Scholar]
- 175.Zou X, Zhang G, Xiao R, et al. Transvaginal natural orifice transluminal endoscopic surgery (NOTES)-assisted laparoscopic adrenalectomy: first clinical experience. Surg Endosc. 2011;25:3767–3772. doi: 10.1007/s00464-011-1786-y. [DOI] [PubMed] [Google Scholar]
- 176.Morris LF, Perrier ND. Advances in robotic adrenalectomy. Curr Opin Oncol. 2012;24:1–6. doi: 10.1097/CCO.0b013e32834da8e1. [DOI] [PubMed] [Google Scholar]
- 177.Henry JF, Sebag F, Iacobone M, et al. Results of laparoscopic adrenalectomy for large and potentially malignant tumors. World J Surg. 2002;26:1043–1047. doi: 10.1007/s00268-002-6666-0. [DOI] [PubMed] [Google Scholar]
- 178.Sarmiento JM, Heywood G, Rubin J, et al. Surgical treatment of neuroendocrine metastases to the liver: A plea for resection to increase survival. J Am Coll Surg. 2003;197:29–37. doi: 10.1016/S1072-7515(03)00230-8. [DOI] [PubMed] [Google Scholar]
- 179.Basuroy R, Srirajaskanthan R, Ramage JK. A multimodal approach to the management of neuroendocrine tumour liver metastases. Int J Hepatol. 2012;2012:819193. doi: 10.1155/2012/819193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Steinmuller T, Kianmanesh R, Falconi M, et al. Consensus guidelines for the management of patients with liver metastases from digestive (neuro)endocrine tumors: Foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2008;87:47–62. doi: 10.1159/000111037. [DOI] [PubMed] [Google Scholar]
- 181.Touzios JG, Kiely JM, Pitt SC, et al. Neuroendocrine hepatic metastases: Does aggressive management improve survival? Ann Surg. 2005;241:776–783. doi: 10.1097/01.sla.0000161981.58631.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Osborne DA, Zervos EE, Strosberg J, et al. Improved outcome with cytoreduction versus embolization for symptomatic hepatic metastases of carcinoid and neuroendocrine tumors. Ann Surg Oncol. 2006;13:572–581. doi: 10.1245/ASO.2006.03.071. [DOI] [PubMed] [Google Scholar]
- 183.Saxena A, Chua TC, Perera M, et al. Surgical resection of hepatic metastases from neuroendocrine neoplasms: A systematic review. Surg Oncol. 2012;21:e131–e141. doi: 10.1016/j.suronc.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 184.Simmonds PC, Primrose JN, Colquitt JL, et al. Surgical resection of hepatic metastases from colorectal cancer: A systematic review of published studies. Br J Cancer. 2006;94:982–999. doi: 10.1038/sj.bjc.6603033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nazario J, Gupta S. Transarterial liver-directed therapies of neuroendocrine hepatic metastases. Semin Oncol. 2010;37:118–126. doi: 10.1053/j.seminoncol.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 186.Vogl TJ, Naguib NN, Zangos S, et al. Liver metastases of neuroendocrine carcinomas: Interventional treatment via transarterial embolization, chemoembolization and thermal ablation. Eur J Radiol. 2009;72:517–528. doi: 10.1016/j.ejrad.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 187.Cao CQ, Yan TD, Bester L, et al. Radioembolization with yttrium microspheres for neuroendocrine tumour liver metastases. Br J Surg. 2010;97:537–543. doi: 10.1002/bjs.6931. [DOI] [PubMed] [Google Scholar]
- 188.Gamblin TC, Christians K, Pappas SG. Radiofrequency ablation of neuroendocrine hepatic metastasis. Surg Oncol Clin N Am. 2011;20:273–279. doi: 10.1016/j.soc.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 189.Shapiro B, Gross MD, Shulkin B. Radioisotope diagnosis and therapy of malignant pheochromocytoma. Trends Endocrinol Metab. 2001;12:469–475. doi: 10.1016/s1043-2760(01)00492-1. [DOI] [PubMed] [Google Scholar]
- 190.Sisson JC, Shapiro B, Beierwaltes WH, et al. Radiopharmaceutical treatment of malignant pheochromocytoma. J Nucl Med. 1984;25:197–206. [PubMed] [Google Scholar]
- 191.Vetter H, Fischer M, Muller-Rensing R, et al. 131-I-meta-iodobenzylguanidine in treatment of malignant phaeochromocytomas. Lancet. 1983;2:107. doi: 10.1016/s0140-6736(83)90091-0. [DOI] [PubMed] [Google Scholar]
- 192.Fitzgerald PA, Goldsby RE, Huberty JP, et al. Malignant pheochromocytomas and paragangliomas: A phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG) Ann N Y Acad Sci. 2006;1073:465–490. doi: 10.1196/annals.1353.050. [DOI] [PubMed] [Google Scholar]
- 193.Gonias S, Goldsby R, Matthay KK, et al. Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol. 2009;27:4162–4168. doi: 10.1200/JCO.2008.21.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Safford SD, Coleman RE, Gockerman JP, et al. Iodine -131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery. 2003;134:956–962. doi: 10.1016/s0039-6060(03)00426-4. [DOI] [PubMed] [Google Scholar]
- 195.Kaltsas GA, Papadogias D, Makras P, et al. Treatment of advanced neuroendocrine tumours with radiolabelled somatostatin analogues. Endocr Relat Cancer. 2005;12:683–699. doi: 10.1677/erc.1.01116. [DOI] [PubMed] [Google Scholar]
- 196.Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124–2130. doi: 10.1200/JCO.2007.15.2553. [DOI] [PubMed] [Google Scholar]
- 197.Kam BL, Teunissen JJ, Krenning EP, et al. Lutetium-labelled peptides for therapy of neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2012;39:S103–S112. doi: 10.1007/s00259-011-2039-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fishbein L, Bonner L, Torigian DA, et al. External beam radiation therapy (EBRT) for patients with malignant pheochromocytoma and non-head and -neck paraganglioma: Combination with 131I-MIBG. Horm Metab Res. 2012;44:405–410. doi: 10.1055/s-0032-1308992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kasliwal MK, Sharma MS, Vaishya S, et al. Metachronous pheochromocytoma metastasis to the upper dorsal spine-6-year survival. Spine J. 2008;8:845–848. doi: 10.1016/j.spinee.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 200.Richter A, Halm HF, Lerner T, et al. Long-term follow-up after en bloc resection and reconstruction of a solitary paraganglioma metastasis in the first lumbar vertebral body: A case report. J Med Case Rep. 2011;5:45. doi: 10.1186/1752-1947-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lazaro B, Klemz M, Flores MS, et al. Malignant paraganglioma with vertebral metastasis: Case report. Arq Neuropsiquiatr. 2003;61(2B):463–467. doi: 10.1590/s0004-282x2003000300026. [DOI] [PubMed] [Google Scholar]
- 202.Yoshida S, Hatori M, Noshiro T, et al. Twenty-six-years' survival with multiple bone metastasis of malignant pheochromocytoma. Arch Orthop Trauma Surg. 2001;121:598–600. doi: 10.1007/s004020100305. [DOI] [PubMed] [Google Scholar]
- 203.Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: Recommendation from a 22-year follow-up of 18 patients. Cancer. 2008;113:2020–2028. doi: 10.1002/cncr.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: Effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109:267–273. doi: 10.7326/0003-4819-109-4-267. [DOI] [PubMed] [Google Scholar]
- 205.Keiser HR, Goldstein DS, Wade JL, et al. Treatment of malignant pheochromocytoma with combination chemotherapy. Hypertension. 1985;7:I18–I24. doi: 10.1161/01.hyp.7.3_pt_2.i18. [DOI] [PubMed] [Google Scholar]
- 206.Banerji U. Heat shock protein 90 as a drug target: Some like it hot. Clin Cancer Res. 2009;15:9–14. doi: 10.1158/1078-0432.CCR-08-0132. [DOI] [PubMed] [Google Scholar]
- 207.Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: The story unfolds. Expert Opin Biol Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 208.Powers MV, Workman P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer. 2006;13:S125–S135. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- 209.Modi S, Stopeck AT, Gordon MS, et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol. 2007;25:5410–5417. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- 210.Modi S, Stopeck A, Linden H, et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17:5132–5139. doi: 10.1158/1078-0432.CCR-11-0072. [DOI] [PubMed] [Google Scholar]
- 211.Sausville EA, Tomaszewski JE, Ivy P. Clinical development of 17-allylamino, 17-demethoxygeldanamycin. Curr Cancer Drug Targets. 2003;3:377–383. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- 212.Druce MR, Kaltsas GA, Fraenkel M, et al. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: Experience with the mTOR inhibitor everolimus (RAD001) Horm Metab Res. 2009;41:697–702. doi: 10.1055/s-0029-1220687. [DOI] [PubMed] [Google Scholar]
- 213.Yuan R, Kay A, Berg WJ, et al. Targeting tumorigenesis: Development and use of mTOR inhibitors in cancer therapy. J Hematol Oncol. 2009;2:45. doi: 10.1186/1756-8722-2-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zitzmann K, Ruden J, Brand S, et al. Compensatory activation of Akt in response to mTOR and Raf inhibitors: A rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010;295:100–109. doi: 10.1016/j.canlet.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 215.Joshua AM, Ezzat S, Asa SL, et al. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J Clin Endocrinol Metab. 2009;94:5–9. doi: 10.1210/jc.2008-1836. [DOI] [PubMed] [Google Scholar]
- 216.Park KS, Lee JL, Ahn H, et al. Sunitinib, a novel therapy for anthracycline- and cisplatin-refractory malignant pheochromocytoma. Jpn J Clin Oncol. 2009;39:327–331. doi: 10.1093/jjco/hyp005. [DOI] [PubMed] [Google Scholar]
- 217.Jimenez C, Cabanillas ME, Santarpia L, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: Targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab. 2009;94:386–391. doi: 10.1210/jc.2008-1972. [DOI] [PubMed] [Google Scholar]
- 218.Gross DJ, Munter G, Bitan M, et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer. 2006;13:535–540. doi: 10.1677/erc.1.01124. [DOI] [PubMed] [Google Scholar]
- 219.Nolting S, Grossman AB. Signaling pathways in pheochromocytomas and paragangliomas: Prospects for future therapies. Endocr Pathol. 2012;23:21–33. doi: 10.1007/s12022-012-9199-6. [DOI] [PubMed] [Google Scholar]
- 220.Rooijens PP, de Krijger RR, Bonjer HJ, et al. The significance of angiogenesis in malignant pheochromocytomas. Endocr Pathol. 2004;15:39–45. doi: 10.1385/ep:15:1:39. [DOI] [PubMed] [Google Scholar]
- 221.Favier J, Igaz P, Burnichon N, et al. Rationale for anti-angiogenic therapy in pheochromocytoma and paraganglioma. Endocr Pathol. 2012;23:34–42. doi: 10.1007/s12022-011-9189-0. [DOI] [PubMed] [Google Scholar]
- 222.Zhang C, Tan C, Ding H, et al. Selective VEGFR inhibitors for anticancer therapeutics in clinical use and clinical trials. Curr Pharm Des. 2012;18:2921–2935. doi: 10.2174/138161212800672732. [DOI] [PubMed] [Google Scholar]
- 223.Kulke MH, Stuart K, Enzinger PC, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24:401–406. doi: 10.1200/JCO.2005.03.6046. [DOI] [PubMed] [Google Scholar]
- 224.Korpershoek E, Pacak K, Martiniova L. Murine models and cell lines for the investigation of pheochromocytoma: Applications for future therapies? Endocr Pathol. 2012;23:43–54. doi: 10.1007/s12022-012-9194-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Andersen KF, Altaf R, Krarup-Hansen A, et al. Malignant pheochromocytomas and paragangliomas: The importance of a multidisciplinary approach. Cancer Treat Rev. 2011;37:111–119. doi: 10.1016/j.ctrv.2010.07.002. [DOI] [PubMed] [Google Scholar]