

Abstract
Mineralocorticoid receptor (MR), highly expressed in the hippocampus, binds corticosteroid hormones and coordinately participates, with the glucocorticoid receptor (GR), to the control of stress responses, memorization and behavior. To investigate the impact of MR in neuronal survival, we generated murine embryonic stem (ES) cells that overexpress human MR (P1-hMR) and are induced to differentiate into mature neurons. We showed that recombinant MR expression increased throughout differentiation and is 2-fold higher in P1-hMR ES-derived neurons compared to wild type (WT) controls while GR expression was unaffected. Althought proliferation and early neuronal differentiation were comparable in P1-hMR and WT ES cells, MR overexpression was associated with higher late neuronal marker expression (MAP2, β-tubulin III). This was accompanied by a shift towards neuron survival with an increased ratio of anti- vs pro-apoptotic molecules and 50% decreased caspase 3 activity. Knocking down MR overexpression by small interfering RNAs drastically reversed neuroprotective effects with reduced Bcl2/Bax ratio and decreased MAP2 expression. P1-hMR neurons were protected against oxidative stress-induced apoptosis through reduced caspase 3 activation and drastically increased Bcl2/Bax ratio and β-tubulin III expression. We demonstrated the involvement of MR in neuronal differentiation and survival and identify MR as an important neuroprotective mediator opening potential pharmacological strategies.
Keywords: Animals; Apoptosis; Cell Differentiation; Cell Survival; Embryonic Stem Cells; cytology; Gene Expression Regulation, Developmental; Humans; Mice; Microscopy, Confocal; methods; Microscopy, Fluorescence; methods; Neurons; cytology; metabolism; Oxidative Stress; RNA, Small Interfering; metabolism; Receptors, Mineralocorticoid; biosynthesis
Keywords: neuronal differentiation, mineralocorticoid receptor, apoptosis, embryonic stem cells
Introduction
The mineralocorticoid receptor (MR), a ligand-dependent transcription factor, is highly expressed in the brain, notably in the hippocampus, where it is physiologically occupied and activated by glucocorticoid hormones (GC) (1). MR plays an important role in the neuroendocrine and behavioral responses to stress and in establishing cognitive functions (2). The classical nuclear MR is involved in the stability and integrity of neuronal networks (3). However, recent evidences suggest that rapid effects of GC depend on a membrane-located MR that modulates neuronal excitability (4–5). The central actions exerted by GC are also mediated by the lower affinity glucocorticoid receptor (GR). Thus, the MR/GR balance is of crucial importance to normalize brain activity and to regulate hippocampal plasticity (6).
Several pharmacological studies and analyses of mouse models have shown that MR activation, in contrast to GR activation, is required for neuronal survival in the hippocampus (7–9). While stimulation of anti-apoptotic pathways by MR may partially explain its neuroprotective role (10–11), a rapid increase of MR expression following neuronal injury was reported (12), thus establishing a positive relationship between MR expression and neuroprotection. We have recently demonstrated that MR expression via transcriptional activation of its two promoters increase during neuronal differentiation of murine embryonic stem (ES) cells (13). However, the mechanisms by which MR promotes neuronal differentiation and maintains neuron survival remain unclear.
The hippocampus is a major site of neurogenesis in adulthood. Specific MR activation enhances neonatal neurogenesis (14) thus promoting cognitive processes. Forebrain MR over-expression improves memory processes in mice (9), while MR knockout animals exhibit impaired learning abilities (15). Moreover, hippocampal neurons greatly decrease with age (16) in parallel with the hippocampal MR expression (17), indicating that reduced MR expression is associated with neuronal dysfunction in the hippocampus of older individuals.
To investigate the impact of MR on neuronal survival and/or differentiation and better elucidate the molecular mechanisms involved, we exploited an ES cell model that could be committed to neuronal differentiation (13) and compared wild-type and hMR over-expressing ES cell lines derived from mice overexpressing hMR (18–19). These cell-based systems offer a unique opportunity to examine the functional consequences of MR over-expression on the regulation of the apoptosis signaling pathway during neuronal differentiation and in mature neurons. We showed that MR over-expression increases expression of the late neuronal markers that in turn, is associated with an increase in the ratio between anti- and pro-apoptotic molecules, providing direct evidence for an anti-apoptotic impact of neuronal MR.
Materials and Methods
Cell Culture
A murine hMR-overexpressing ES cell line, in which the P1 promoter drives hMR cDNA expression, was derived as described (19). The wild-type D3 ES cell line (ATCC no. CRL-11693) and the hMR ES cells were grown on 0.1% gelatin-coated plates (Sigma-Aldrich, Lyon, France) and on feeder cells (STO Neomycin LIF, kindly provided by Dr Alan Bradley, The Wellcome Trust Sanger Institute, UK) pretreated with 15 μg/ml mitomycin C (Sigma-Aldrich) for 4 h. Cells were cultured at 37°C in a humidified incubator in presence of 5% CO2.
Reagents
ES medium was composed of DMEM (PAA, Les Mureaux, France) containing 15% fetal calf serum (FCS specifically tested for ES culture (AbCys SA, Paris, France), 1X non-essential amino acids (PAA), 2 mM glutamine (PAA), 100 U/ml penicillin (PAA), 100 μg/ml streptomycin (PAA), 20 mM HEPES (PAA) and 100 μM β-mercaptoethanol (Sigma-Aldrich). Embryoid Bodies (EB) medium had a similar composition but contained 10% FCS without β-mercaptoethanol. Cortisol and aldosterone concentrations in the serum batch used for all experiments were measured at 30.25 nM and 41 pM, respectively. Neuron medium was similar to EB medium but was supplemented with 5 μg/ml insulin (Sigma-Aldrich), 5 μg/ml transferrin (Sigma-Aldrich), and 29 nM sodium selenate (Sigma-Aldrich).
Differentiation of ES cells into Neuronal-like cells
Neuronal differentiation was induced in ES medium containing 15% FCS with retinoic acid (RA), as previously described, via embryoid bodies (EB) formation (13). Of note, we were unable to achieve neuronal differentiation of ES cells when cultivated during two weeks with medium containing Dextran-Charcoal Coated (DCC) serum. Briefly, ES cells formed EB when exposed to 10−6 M Retinoic acid (Sigma-Aldrich) for 5 days in non-adhesive bacterial dishes. At day 7, EB were dissociated and incubated in neuron medium until day 14 in adherence in tissue culture dishes. Cells were washed in PBS and froze before RNA or protein extraction.
Cell Treatment
For hormonal treatment, after 24 h incubation in DCC medium, aldosterone (Acros Organics, Halluin, France), or corticosterone (Sigma-Aldrich), and/or RU486 (Mifepristone) (Sigma-Aldrich) were added to the culture medium at day 13 of the neuronal differentiation. After 6 h, total RNA was extracted with Trizol and gene expression was measured by quantitative real-time PCR. For apoptosis induction, cells were treated at day 14 with 400 μM tert-butylhydroperoxide (t-BHP) (Sigma-Aldrich) for 3 h in neuron medium containing 10% FCS. Successively, proteins were extracted and quantified by Western blot.
Flow cytometry
Cells were fixed and permeabilized using the Foxp3 Staining Buffer Set (eBioscience). Cells were then stained with anti-Ki67 antibody or with isotype control (BD Bioscience) for 30 min on ice. Flow cytometry was performed with a FACSCanto cytometer (BD Biosciences) and data files were analyzed using FlowJo software (Tree Star Inc.).
Quantitative Real Time PCR
Gene expression was quantified by real time PCR. Total RNA was processed for real time PCR on an ABI 7300 Sequence Detector (Applied Biosystems, Courtaboeuf, France). Briefly, 1 μg of total RNA was treated using the DNase I Amplification Grade procedure (Invitrogen). RNA was then reverse-transcribed with 50 U MultiScribe reverse transcriptase (Applied Biosystems). After 10-fold dilution, 1/20th of the reverse transcription reaction was used for PCR using the Fast SYBR® Green PCR master mix (Applied Biosystems). Final primer concentrations were 300 nM for each primer (see Supplemental Table 1 for sequences). Reaction parameters were 50 °C for 2 min followed by 40 cycles at 95°C for 15 s, and 60 °C for 1 min. For standard preparation amplicons were purified from agarose gel and subcloned into pGEMT-easy plasmid (Promega), then sequenced to confirm the identity of each fragment. Standard curves were generated using serial dilutions of linearized standard plasmids, spanning 6 orders of magnitude and yielding correlation coefficients >0.98 and efficiencies of at least 0.95, in all experiments. Standard and sample values were determined in duplicate from independent experiments. Relative expression within a given sample was calculated as the ratio: attomol of specific gene/femtomol of 18S. Results are mean ± S.E.M and represent the relative expression compared with that obtained with control cells, which was arbitrary set at 1.
Western blot
Total protein extracts were prepared from ES cells and neuron cultures. Cells were lysed, in lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM EDTA, 30 mM Na pyrophosphate, 50 mM Na fluoride, 1% Triton X100, 1X protease inhibitor from Sigma) on ice. Immunoblots were incubated 1 h at room temperature in 5% fat free milk-Tris buffer saline − 0.1% Tween 20 (TBS-T) before overnight incubation at 4°C with one of the following antibodies: rabbit anti-MR 39N (1/1,000), mouse anti-β-tubulin III TU-20 (1/1,000) (Millipore, Molsheim, France), rabbit anti-Bcl2 (1/500) (Ozyme, Saint-Quentin-en-Yvelines, France), mouse anti-PCNA (1/1,000) (Dako, Trappes, France), rabbit anti-caspase 3 (1/1,000) (Ozyme), rabbit anti-Bax (1/15,000) (Ozyme) and mouse anti-GR (clone FIGR, Millipore, 1/500). After extensive washing, membranes were incubated for 30 min at room temperature with peroxydase-conjugated goat anti-rabbit (1/15,000) or anti-mouse (1/15,000) secondary antibodies (Vector Laboratory, Burlingame, CA). After washing, the antigen-antibody complex was visualized by the ECL+ detection kit (GE Healthcare Europe, Orsay, France). For loading normalization, membranes were incubated with rabbit anti-GAPDH (1/5,000) (Sigma-Aldrich) or mouse anti-α-tubulin (1/10,000) (Sigma-Aldrich). Signal intensities were quantified with QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). Alternatively, the Odyssey imaging system (LI-COR Biosciences, Bad Homburg, Germany) was used for quantification with IRDye© 800CW or 680LT near-infrared fluorescent secondary antibodies.
Confocal Immunofluorescence Microscopy
Cells grown on sterile coverslips were fixed with methanol for 10 min, rinsed with PBS-0.1% Tween 20 and incubated with a PBS, 5% BSA, 0.1% casein block for 20 min followed by overnight incubation at 4°C with the anti-MR 39N polyclonal antibody (4 μg/ml) then with Alexa Fluor 555 goat anti-rabbit (1/1,000) (Molecular Probes) for 1 h at room temperature. The cells were next rinsed in PBS, and incubated with the anti-β-tubulin III TU-20 monoclonal antibody (1/100) (AbCys) for 2 h at room temperature followed by washing and incubation with Alexa Fluor 488 goat anti-mouse antibody (1/1,000) (Molecular Probe) for 1 h at room temperature. The coverslips were then mounted with Fluorescence Mounting Medium (Dako), before analysis and imaging by confocal fluorescence microscopy (Zeiss HAL confocal microscope).
MR knockdown by siRNA
Neurons were transiently transfected at day 11 with 100 nM siRNA (Invitrogen; see Supplemental Table 1 for sequences), using Lipofectamine RNAiMAX (Invitrogen) in Opti-MEM Reduced Serum Medium (Invitrogen) according to the manufacturer’s recommendations. Six hours post-transfection, cells were incubated in neuron medium for 48 h. At day 14, total RNA were extracted and gene expression was measured by qPCR.
Statistical Analyses
Results represent mean ± SEM of at least 6 samples for each condition unless stated otherwise. Statistical analyses were performed using a non parametric Mann-Whitney test (Prism4, Graphpad Software, Inc., San Diego, CA).
Results
MR over-expression during neuronal differentiation of ES cells
The hMR over-expressing ES cell line was established from transgenic P1-hMR mouse blastocysts (19). The transgenic mice were generated using 1.2 kb of the human proximal MR promoter, named P1, to drive hMR cDNA expression (18). To investigate the impact of MR over-expression during neuronal differentiation, we first examined the expression of hMR transgene mRNA in the recombinant ES cells by quantitative real-time PCR and showed that hMR transcript levels rose approximately 3.5-fold in mature neurons compared to undifferentiated ES cells (Fig. 1A). We next analyzed MR protein expression during neuronal differentiation in transgenic ES cells (P1-hMR) compared with wild-type (WT) using an anti-MR antibody recognizing both the endogenous murine MR and recombinant human MR (20). Western blot analyses revealed an approximately 1.6-fold increase of MR expression in the P1-hMR ES cells compared to WT ES cells and 1.7-fold increase in the P1-hMR neurons compared to WT neurons (Fig. 1B). In parallel, we showed that while endogenous mMR mRNA expression remains identical in undifferentiated P1-hMR and WT ES cells, differentiated neurons of both genotypes under the same experimental conditions exhibit a 3-fold increase in mMR transcripts without significant difference between transgenic and WT ES cell lines (Fig. 1C). Similarly, the presence of the transgene did not modify the expression of the closely related glucocorticoid receptor (GR) both at the mRNA and protein levels as measured by real-time qPCR during neuronal differentiation and western blot at d14 (Fig. 1D and E). This indicated that hMR overexpression does not affect endogenous corticosteroid receptor abundance in mature neurons. Double-immunolabeling experiments with the anti-MR and anti-β-tubulin III antibodies clearly reveal a colocalization of MR and β-tubulin III (Merge panel Fig. 1F) showing that MR is almost exclusively expressed in mature, β-tubulin III-positive neurons. Altogether, these results demonstrate that ES cell-derived neurons provide an effective cell-based system to investigate the functional consequences of hMR over-expression during neuronal differentiation.
Figure 1. MR over-expression during neuronal differentiation.
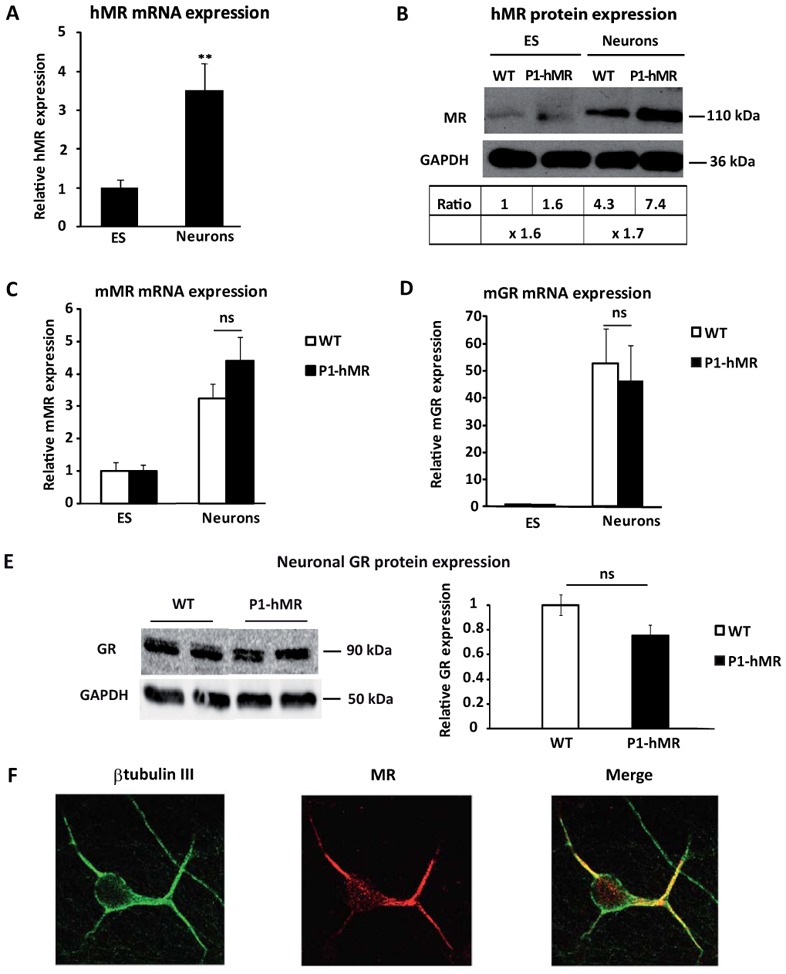
A) Relative hMR mRNA expression levels were determined using qPCR in undifferentiated ES cells and neurons. Results are means ± SEM of two independent experiments of six samples performed in duplicate for each developmental stage indicating the relative expression compared with basal levels of ES (arbitrarily set at 1). ** P<0.01. Mann Whitney test. Relative mRNA expression is normalized to 18S rRNA expression (see Materials and Methods section). B) Western blot analyses of MR protein expression in WT and P1-hMR ES cell lines. Undifferentiated ES and neurons lysates from each ES cell line were processed for immunoblotting with anti-MR antibody. GAPDH was used as loading control. MR was normalized to GAPDH protein levels after digitalization on a gel scanner with QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). Results are presented as MR/GAPDH ratio and as compared with basal levels of WT ES (arbitrarily set at 1). C–D) Relative mMR and mGR mRNA expression levels were determined using qPCR in undifferentiated ES cells and neurons from WT and P1-hMR ES cell lines. Results are means ± SEM of two independent experiments on six samples performed in duplicate for each developmental stage and represent the relative expression compared with basal levels of ES (arbitrarily set at 1). Mann Whitney test. Relative mRNA expression is normalized to 18S rRNA expression (see Materials and Methods section). E) Western blot analysis of GR expression in WT and P1.hMR neurons and signal quantification of the GR/GAPDH ratio (n = 6), ns: non significant. WT mean value arbitrarily set at 1. F) Double-immunolabeling of P1-hMR neurons with antibodies against β-tubulin III (green) (left panel) and MR (red) (middle panel); merged images are shown on the right. Original magnification × 40.
Impact of MR over-expression on neuronal differentiation
In order to examine the impact of MR over-expression, transgenic and WT ES cells were differentiated into neurons, and the variations of the expression levels of several specific neuronal markers were analyzed by quantitative real-time PCR. Under our experimental conditions where the ligand-dependent transcription factor MR was activated by corticosteroid hormones present in the serum containing medium, the expression profile of the neuronal progenitor marker nestin was similar in the ES cell lines of both genotypes during neuronal differentiation (Fig. 2A), suggesting that MR over-expression does not affect early neuronal commitment. Besides, the expression of the mature neuronal marker Microtubule-Associated Protein 2 (MAP2) was very low in undifferentiated ES cells but readily increased, as expected, in mature neurons. We performed neuronal differentiation of another WT ES cell line (19), assessing the expression of two late neuronal markers MAP2 and synaptophysin compared to the WT D3 ES cell line and did not found any significant difference (see supplemental Fig. S1). Interestingly, we showed that the MAP2 mRNA level was 4.5-fold higher in P1-hMR neurons than in WT controls (Fig. 2B). In addition, western blot analysis showed a 1.7-fold increase of another late neuronal marker β-tubulin III in P1-hMR neurons compared to WT neurons (Fig. 2C). Several hypotheses could account for these observations: MR over-expression might facilitate the differentiation of precursors into neuronal lineage and could promote the growth of mature neurons. An alternative and not mutually exclusive hypothesis is that MR-over-expression is associated with an increased survival of newly differentiated neurons.
Figure 2. MR over-expression stimulates late neuronal markers without increasing neuronal proliferation.
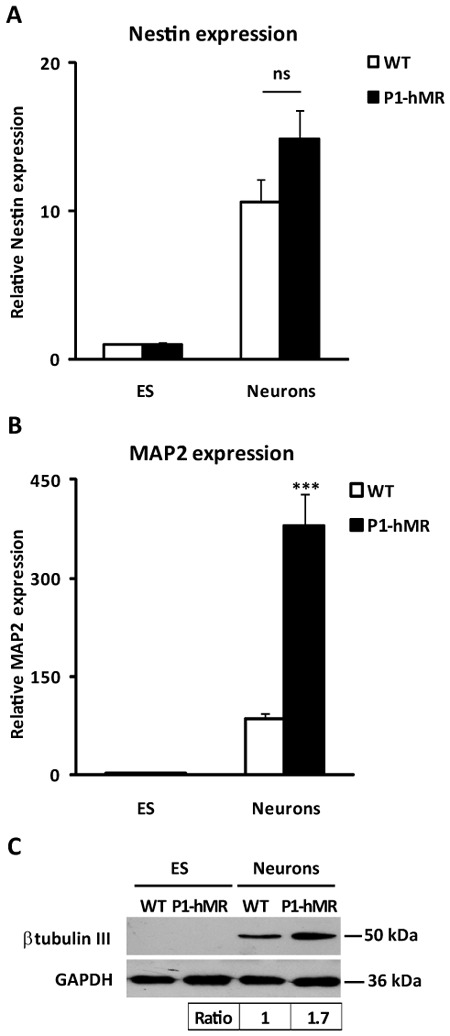
A–B) Relative nestin and MAP2 mRNA expression levels were determined using qPCR in undifferentiated ES cells and neurons. Results are means ± SEM of two independent experiments of six samples performed in duplicate for each developmental stage and represent the relative expression compared with levels of WT. P1-hMR and WT undifferentiated ES cell set arbitrarily at 1. *** P<0.001 Mann Whitney test. Relative mRNA expression is normalized to 18S rRNA expression (see Materials and Methods section). C) Western blot analyses of β-tubulin III protein expression in WT and P1-hMR ES cell lines. Undifferentiated ES and neurons lysates from each ES cell line were processed for immunoblotting with anti-β-tubulin III antibody. GAPDH was used as loading control. b-tubulin III levels were normalized to GAPDH protein levels after digitalization on a gel scanner with QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). Results are presented as β-tubulin III/GAPDH ratio and as compared with basal levels of WT neurons (arbitrarily set at 1).
MR over-expression favors the increased survival of neurons
The increased expression of late neuronal markers reflects an increase of neuronal proliferation or survival. We thus examined by Western blot the expression of the proliferation marker, PCNA (Proliferating Cell Nuclear Antigen) in neurons and did not detect any significant difference between WT or P1-hMR neurons (Fig. 3A). This result was confirmed by Fluorescence Activated Cell Sorting method, using an anti-Ki67 (another proliferation marker) antibody, (56.3% Ki67 positive WT cells vs 57.0% Ki67 positive P1.hMR cells at d13 of differentiation, see supplemental Fig. S2), thus indicating that MR over-expression has no major impact on neuron proliferation. We then examined by Western blot the cleavage of caspase 3, as an index of caspase 3 activation and an indirect marker of apoptosis, and showed a 57% reduction of caspase 3 cleavage in MR over-expressing neurons compared to WT (Fig. 3B), suggesting that MR over-expression may confer resistance to apoptotic cell death thus facilitating neuron survival.
Figure 3. MR over-expression does not affect cell proliferation but reduces Caspase 3 activity in neurons.
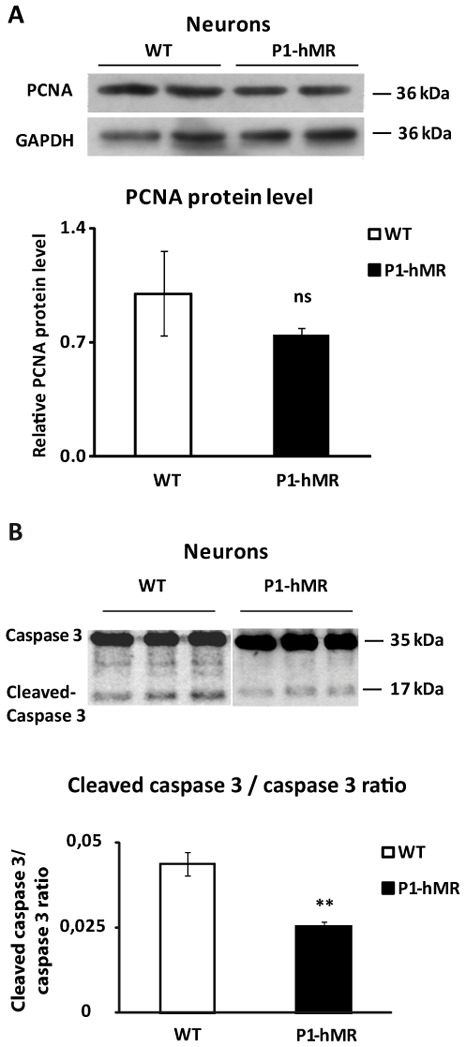
A) Western blot analyses of PCNA protein expression in WT and P1-hMR neurons. Neurons lysates from each ES cell line were processed for immunoblotting with anti-PCNA antibody. GAPDH was used as loading control. PCNA was normalized to GAPDH protein levels after digitalization on a gel scanner with QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). Results are presented as ratio PCNA/GAPDH and as compared with basal levels of WT neurons (arbitrarily set at 1). B) Caspase 3 activity was analyzed by western blot expression in WT and P1-hMR neurons. Lysates were processed for immunoblotting with an antibody recognizing both caspase 3 and cleaved-caspase 3. Protein levels were quantifued after digitalization on a gel scanner using QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). Results are presented as ratio cleaved-caspase 3/caspase 3 of 6 samples and as compared with basal levels of WT neurons (arbitrarily set at 1). ** P<0.01. Mann Whitney test.
Functional consequences of MR over-expression on neuron survival
To determine the impact of MR over-expression on neuron survival, we studied the expression of transcripts and proteins encoded by the Bcl2 gene family during neuronal differentiation of ES cell lines of both genotypes. The Bcl2 gene family is a major regulatory component of the apoptotic pathway comprising death inducers and death repressors. These proteins are activated by different stimuli and represent upstream events leading to the conclusive phase of the apoptotic process involving caspase 3 activation. The ratio between death inducers and repressors is a key element determining cell survival or death (21–22). Specifically we examined the expression of two anti-apoptotic markers: Bcl2 and BclxL, and two pro-apoptotic markers: Bax and Bak. Quantitative real-time PCR analysis indicated that the expression of Bcl2 transcripts increases 6.5- and 23.7-fold during neuronal differentiation of WT and P1-hMR ES cell lines, respectively (Fig. 4A), Bcl2 mRNA expression being significantly and reproducibly higher in P1-hMR than in WT neurons. Moreover, P1-hMR neurons exhibited a 2.5-fold rise of BclxL transcript levels compared to undifferentiated P1-hMR ES cells, whereas no statistical difference in BclxL expression was detected during neuronal differentiation of the WT ES cell line (Fig. 4B). Taken together, these findings strongly support a positive relationship between MR over-expression and an increase in the expression of anti-apoptotic markers. In contrast, steady state levels of Bax and Bak mRNA decreased by approximately 50% in WT ES cell-derived neurons but remained constant in P1-hMR neurons during neuronal differentiation (Figure 4C and 4D). Finally, of major interest, the ratios of both mRNA (Fig. 4E) and protein (Fig. 4F) between anti-apoptotic and pro-apoptotic markers were always higher in P1-hMR that in WT neurons, providing strong evidence that MR over-expression promotes anti-apoptotic factors expression, thus facilitating neuronal survival.
Figure 4. Anti-apoptotic factors expression is increased in P1-hMR neurons.
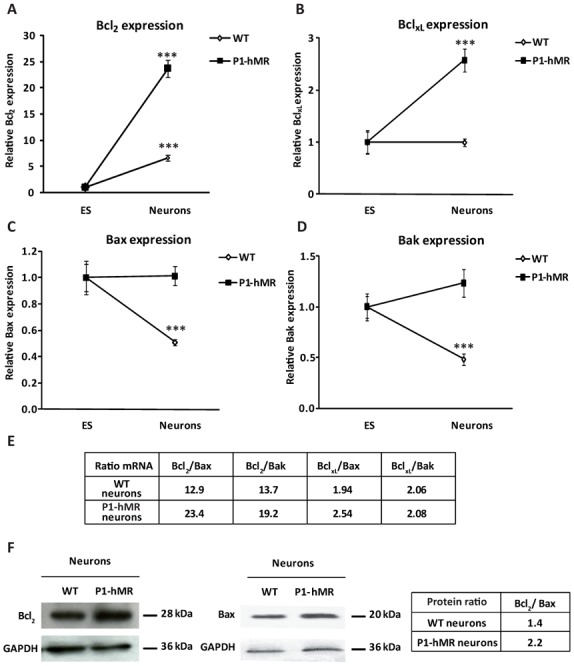
A–B–C–D) Relative Bcl2, BclxL, Bax, and Bak mRNA expression levels were determined using qPCR in undifferentiated ES cells and neurons. Results are means ± SEM of two independent experiments of six samples performed in duplicate for each developmental stage and represent the relative expression compared with levels of undifferentiated ES cell set arbitrarily at 1. *** P<0.001 Mann Whitney test. Relative mRNA expression is normalized to 18S rRNA expression (see Materials and Methods section). E) The table represents the ratio between the mean of each anti-apoptotic marker expression to the mean of each pro-apoptotic marker expression in neuronal state. F) Western blot analyses of Bcl2 and Bax protein expression in WT and P1-hMR neurons. Lysates were processed for immunoblotting with anti-Bcl2 or Bax antibody. GAPDH was used as loading control. Bcl2 and Bax were normalized to GAPDH protein levels after digitalization on a gel scanner by using QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). The table represents the ratio between Bcl2 to Bax expression in neuronal state.
MR knockdown inhibits neuronal-specific increase in anti-apoptotic markers
To confirm that MR over-expression enhances neuronal differentiation and stimulates neuronal survival, a small interfering RNA (siRNA) strategy was exploited using two unrelated MR specific siRNAs in P1-hMR ES-derived neurons. In Fig. 5 is illustrated the decrease of mMR and hMR mRNA expression (approximately 67 % and 57 %, respectively), obtained 48h post-transfection with the respective siRNA compared with scrambled siRNA (Fig. 5A–B). This reduction was accompanied not only by a significant and concomitant diminution of the mRNA levels of the late neuronal marker MAP2 (98% and 86 %) but also by a decrease of the anti-apoptotic marker Bcl2 (Fig. 5C–D). In parallel, the two MR siRNAs induced about a 50% increase of the relative expression of the pro-apoptotic marker Bax (Fig. 5E). Finally, of major interest, the two MR siRNAs caused a marked reduction of the anti-apoptotic to pro-apoptotic ratio (66%) (Fig. 5F). Collectively, these findings bring additional support for MR involvement in the increased expression of late neuronal markers. We also provide evidence that MR knock-down blunts the increase of anti-apoptotic markers associated with MR over-expression while facilitating the decrease of pro-apoptotic markers expression, validating the anti-apoptotic role of this receptor.
Figure 5. MR down-regulation inhibits MR-mediated neuroprotective effects.
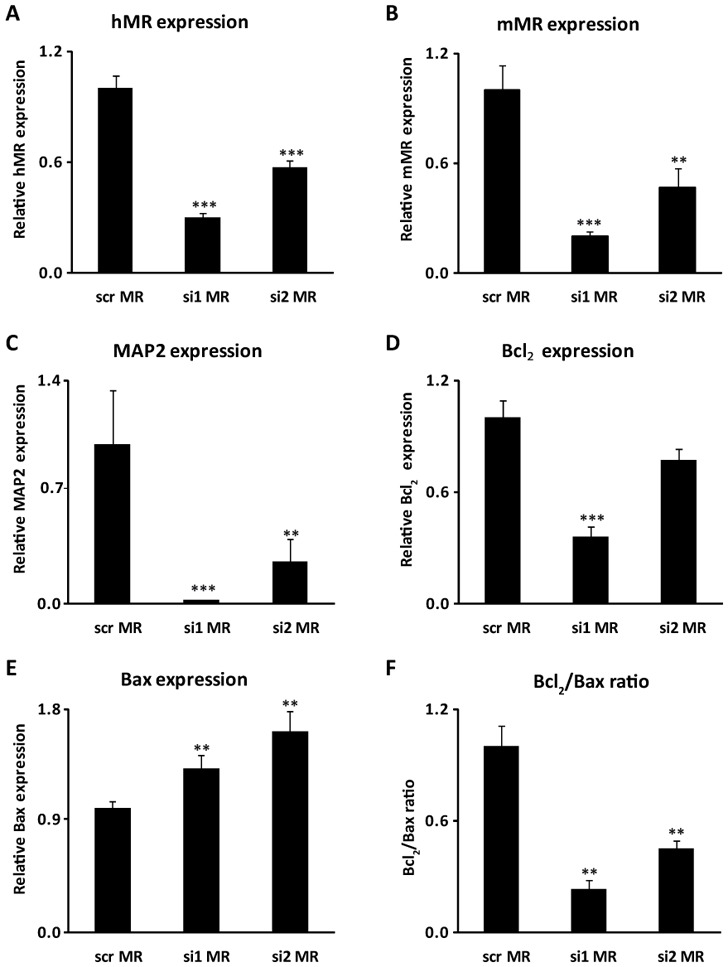
P1-hMR neurons were transfected with either the control scrambled siRNA (scr MR) or by two unrelated MR siRNA (si1 MR, si2 MR). A–E) Relative mMR, hMR, MAP2, Bcl2, Bax mRNA expression levels were determined using qPCR. Results are means ± SEM of six samples performed in duplicate and represent the relative expression compared with basal levels of control scrambled siRNA transfected neurons (scr MR). **P<0.01, ***P<0.001. Mann Whitney test. Relative mRNA expression is normalized to 18S rRNA expression (see Materials and Methods section). F) The graph represents the ratio between the mean of anti-apoptotic marker Bcl2 expression to the mean of pro-apoptotic marker Bax expression in each experimental condition.
The relative level of MR is crucial for the anti-apoptotic effect
We decided then to investigate the impact of steroid hormones on the ratio of anti-apoptotic to pro-apoptotic markers, in ES-cell derived neurons. Cells were incubated with aldosterone or corticosterone at d13 of differentiation. Steroid-induced modification of Bcl2 and Bax mRNA expression was measured after 6 h treatment using quantitative real-time PCR. As shown in Fig. 6, 100 nM aldosterone had no consequence on the ratio of anti-apoptotic to pro-apoptotic marker on both genotypes. In sharp contrast, corticosterone had a differential effect on P1-hMR neurons compared to WT neurons. A 35% decrease of Bcl2 to Bax ratio was observed in WT neurons, whereas a 2.3-fold increase was observed in P1-hMR neurons. Corticosterone-induced effects were not affected by RU486, a GR antagonist, unambiguously demonstrating MR involvement in controlling the anti-apoptotic/pro-apoptotic signal balance. Collectively, these findings show that MR not only controls gene expression of death repressors and inducers but more importantly that neuronal MR abundance also dictates the extent and the direction towards pro- or anti-apoptotic phenotype.
Figure 6. Opposite effects of corticosterone-activated MR signaling on Bcl2/Bax ratio in WT and P1-hMR neurons.
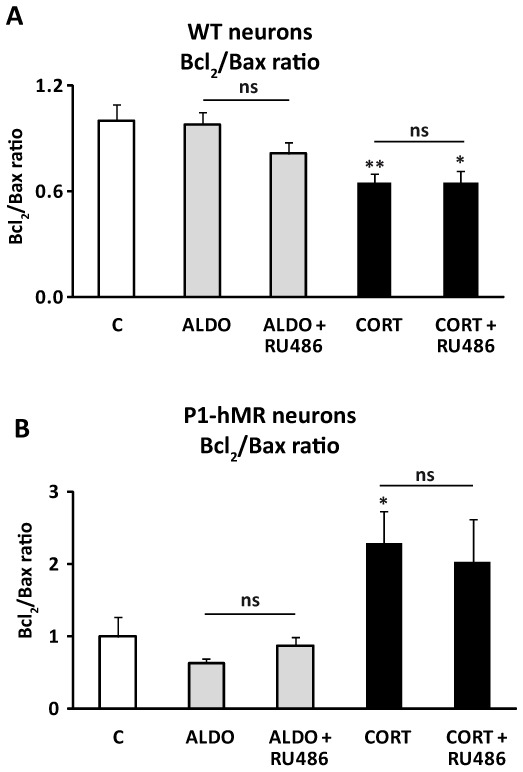
WT and P1-hMR neurons were exposed to 100 nM aldosterone (ALDO) and corticosterone (CORT) in the absence or presence of 1 μM RU486. A–B) Results are means ± SEM (six samples performed in duplicate), of ratio between the mean of anti-apoptotic marker Bcl2 expression to the mean of pro-apoptotic marker Bax expression in neurons. ** P<0.01, * P<0.05. Mann Whitney test.
Neuronal MR over-expression reduces t-BHP-induced cell death
To examine the effect of MR over-expression on oxidative stress-induced apoptosis, we compared the survival of WT and of MR over-expressing neurons after 3 h exposure to 400 μM tert-butylhydroperoxide (t-BHP). t-BHP treatment led to characteristic WT cell morphological changes including round shape of neurons, beading followed by extensive degeneration of the neurites and lost of neuronal integrity, many cells detaching from the culture plate. In contrast, under similar experimental conditions, P1-hMR neurons appeared almost normal with only few floating cells. Western blot analyses show that the t-BHP-induced caspase 3 cleavage is 3-fold higher in WT than in P1-hMR neurons (Fig. 7A). Likewise, the ratio between anti-apoptotic and pro-apoptotic markers is 5-fold higher in P1-hMR than in WT neurons (Fig. 7B). Additionally, exposure of cultures to t-BHP induces to a drastic reduction of β-tubulin III protein expression in WT compared to P1-hMR neurons (Fig. 7C), supporting the morphological changes. Altogether, these data demonstrate that MR over-expression is associated with a significant protection against t-BHP-induced neuronal death.
Figure 7. Neuronal MR over-expression confers resistance to oxidative stress-induced cell death.
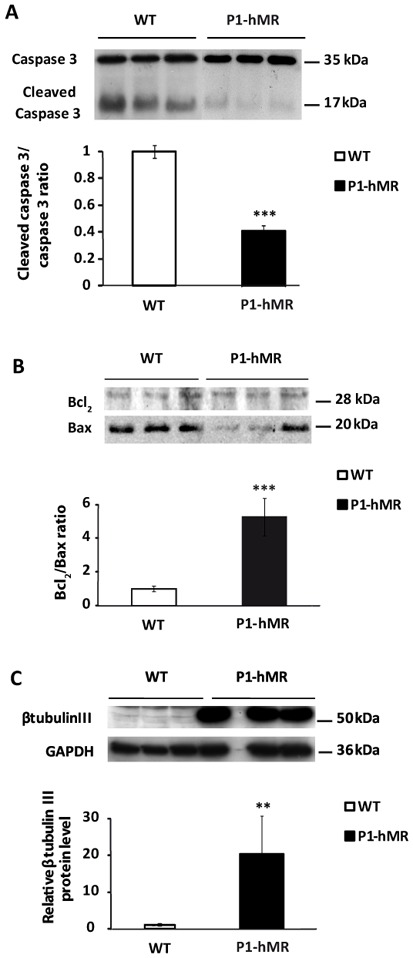
WT and P1-hMR neurons were exposed to 400 μM t-BHP for 4 hours. A) Caspase 3 activity was analyzed by western blot expression in WT and P1-hMR neurons. Lysates were processed for immunoblotting with antibody recognizing both caspase 3 and cleaved-caspase 3. Protein levels after digitalization on a gel scanner by use QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). Results are presented as cleaved-caspase 3/caspase 3 ratio (n = 9) and compared to ratio detected in WT neurons (arbitrarily set at 1). * P<0.05. Mann Whitney test. B) Western blot analyses of Bcl2 and Bax protein expression in WT and P1-hMR neurons. Neuron lysates from each ES cell line were processed for immunoblotting with anti-Bcl2 or anti-Bax antibody. Results are presented as ratio Bcl2/Bax (n=9) and compared with basal levels of WT neurons (arbitrarily set at 1). *** P<0.001. Mann Whitney test.
C) Western blot analyses of β-tubulin III protein expression in WT and P1-hMR neurons. Neurons lysates from each ES cell line were processed for immunoblotting with anti- β-tubulin III antibody. GAPDH was used as loading control. β-tubulin III was normalized to GAPDH protein levels after digitalization on a gel scanner by use QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). Results are presented as ratio β-tubulin III/GAPDH (n = 6, **P<0.01. Mann Whitney test) and as compared with basal levels of WT neurons (arbitrarily set at 1).
Discussion
In this present work, we investigated whether and how MR controls neuronal differentiation and/or survival using a model of MR over-expression in ES cell-derived neurons obtained from P1-hMR transgenic mice (18–19). In this cell-based system, P1-hMR neurons exhibit a 2-fold increase in MR protein expression compared to differentiated WT neurons while GR expression level remains unchanged, leading to a moderately enhanced MR/GR ratio. To our knowledge, this is the first report that directly quantified the extent of neuronal MR overexpression at the protein level. This parameter is lacking in other brain-specific MR overexpression transgenic models (9, 23).
Given that the relative receptor density and their occupancy by corticosteroid hormones are known to greatly affect neuronal maintenance, transmission and damage (2), our ES-derived neurons in which expression of one component of the corticosteroid signaling is specifically modified, constitute an appropriate experimental system. Even though one limitation of our model is that we could not directly control the concentration of corticosteroid hormones provided by the serum during the initial steps of neuronal differentiation, our cell based model remains suitable to clarify neuronal MR influence on cell differentiation, proliferation and susceptibility to cell apoptosis. Herein, we show that MR over-expression from early neuronal developmental stages and onwards is associated with increased expression of late neuronal differentiation markers. We unambiguously establish the pivotal role of MR in controlling the balance between anti- and pro-apoptotic signals as confirmed by knocking down MR expression with small interfering RNA strategy. We finally demonstrate the importance of MR abundance in conferring relative resistance to oxidative stress-induced cell apoptosis thus facilitating neuronal survival.
MR and GR are abundantly expressed in neurons of the limbic areas where they mediate quite opposite effects. MR and GR exhibit distinct functional properties notwithstanding their similarities of structure, and mechanisms of action. Most notably both receptors bind glucocorticoid hormones (cortisol in humans, corticosterone in rodents) but GR presents a low affinity while MR has a 10 fold higher affinity for glucocorticoids (24). In addition, as ligand-dependent transcription factors, MR and GR recruit similar but also distinct coregulators which may partially account for the diversity of neuronal responses to glucocorticoids (25–27). Recent accumulating evidences show that acutely or chronically unbalanced glucocorticoid concentrations differentially affect neuronal function. Under rest conditions, basal levels of glucocorticoids which predominantly activated MR are essential for neuronal development, integrity and function. On the other hand, under stress exposure, high levels of glucocorticoids, which fully occupied and activated GR, are detrimental and induce neuronal death (8) though cell cycle arrest and activation of apoptosis signaling pathways (11, 28–29). Repeated stressful events trigger the damaging effects of GR on neurons and brain functions (6, 30). Thus, the coordinated activation of MR/GR pathways appears to be a major and critical regulator of neuronal function. Yet, the extent of MR signaling activation in the central nervous system seems to depend on the MR abundance beside corticosteroid hormone levels. In this respect, our model of MR over-expressing ES derived neurons conveys important new informations concerning MR influence in neuronal determination and survival.
It is well established that the ratio between death repressors or anti-apoptotic molecules (e.g. Bcl2, BclxL) and death inducers (e.g. Bax, Bak) or pro-apoptotic markers is determinant for cell fate (21–22). Under basal conditions, Bcl2 and BclxL sequester by dimerization Bax and Bak in the cytosol, thus preventing their migration to mitochondria and apoptosis. However, when the amount of repressors is insufficient to neutralize all the pro-apoptotic molecules, apoptotic signals prevail leading to caspase activation and apoptosis (31–32).
We demonstrated that the anti-apoptotic/pro-apoptotic molecule ratio is much higher in MR over-expressing neurons than in WT neurons, supporting a neuroprotective role of MR. As previously reported on other models (10–11), the involvement of neuronal MR in regulating apoptosis signaling pathways is corroborated by several lines of evidence.
First, we show that corticosterone, but not aldosterone exposure of WT neurons significantly increases the pro-apoptotic potential while corticosterone exerts an anti-apoptotic effect on P1-hMR neurons. These opposite actions of corticosterone persist in presence of the GR antagonist RU486, identifying MR as a pro-survival factor and underlying the role of MR over-expression in conferring neuronal resistance to apoptotic signals. These findings are in agreement with previous in vitro and in vivo studies that reported a rapid upregulation of MR (mRNA and protein) associated with an increased survival of rat primary cortical neurons in response to mild injury and in rat hippocampus following hypothermic transient global ischemia (12). The in vivo neuroprotective effect of MR was further demonstrated by transgenic mice presenting specific forebrain MR over-expression. These animals exhibited a decreased sensitivity to stress, anxiety-like behavior and enhanced memory (9, 23). More importantly, these transgenic mice presented with attenuated hippocampal neuron loss after cerebral ischemia, consistent with the increased survival of MR over-expressing ES-derived neurons we described.
Second, to validate the assumption the MR over-expression confers apoptosis resistance, we performed MR knockdown in P1-hMR neurons by a siRNA strategy. Along with the marked reduction of MR expression, a significant decrease of MAP2 expression was observed consistent with a massive loss of mature neurons associated with a reduced anti/pro-apoptotic Bcl2/Bax ratio. Taken together, there is a clear positive relationship between MR abundance, anti-/pro-apoptotic factor expression ratio and neuronal marker level. This observation is in agreement with the forebrain specific genetic disruption of MR in mice, which associates altered learning processes and dentate granule cell degeneration (7, 15). Taken together, these findings provide strong evidence that increased in vitro and in vivo MR expression is directly and causally linked to the promotion of neuronal survival.
Third, additional results corroborate the prominent role of activated MR signaling in preventing neuronal cell death-signaling cascade. We explored cell viability after acute exposure of neurons to t-BHP, a strong inducer of oxidative stress. We show that MR overexpressing neurons were resistant to oxidative injury as revealed by the reduction in caspase 3 cleavage and the sharp increase in b-tubulin III protein expression monitoring neuronal survival. Interestingly, several physiological or pathophysiological conditions are clearly associated with an increased expression of brain MR such as during aging (17), after antidepressant imipramine treatment (33), in depressed patients (34) or after cerebral ischemia (35).
The molecular mechanisms by which MR may regulate gene expression of the Bcl2 family members remain to be established. As a transcription factor, MR may directly or indirectly interact with the regulatory sequences of anti-apoptotic genes to modulate their transcription. Several groups have identified potential hormone responsive elements in BclxL and Bcl2 gene promoters which specifically bind PR and GR in vitro and in vivo (36–38). Given that MR binds to the same consensus HRE sequence, it is tempting to speculate that MR may regulate Bcl2 and BclxL gene expression by acting directly on the HRE sequences located at their promoters, in the context of neuronal survival in rodents (39). This does not exclude that MR activates or represses other specific sets of target genes essential for neuronal survival program.
We also surmise that the shift of the pro-apoptotic/anti-apoptotic balance towards neuron survival may account for the higher expression of late neuronal markers in P1-hMR neurons. Indeed, it has been previously proposed that anti-apoptotic factors facilitate neuronal differentiation, whereas a reduction of pro-apoptotic factors expression was observed by several groups (40–44). Therefore, besides their role in cell death, the proteins of Bcl2 family are largely implicated in neurogenesis.
An additional layer of complexity is given by the putative membrane-located MR which exerts rapid non-genomic actions resulting in the stimulation of the frequency of excitatory postsynaptic glutamate currents in the mouse hippocampus. This effect was blocked by MR specific antagonist spironolactone, and did not occur in brain specific MR knockout mice (5). Surprisingly, this membrane-located MR seems to have a 10 to 20 fold lower affinity for GC than the intracellular MR. Interestingly, MR has been recently detected in the membranes of rat amygdala glutamatergic and GABAergic neurons, with a presynaptic and postsynaptic localization (45). Whether this membrane MR is involved in neuronal survival remains to be elucidated.
In conclusion, we have successfully established a novel model of MR over-expression using the neuronal differentiation of ES cells that was proven to be a suitable cell-based system to investigate many functions of neuronal MR. This alternative approach fully complementing previous cellular and animals models should facilitate the development of therapeutic strategies designed to improve neuronal MR signaling efficiency and thereby opening new means to prevent or attenuate neuronal cell apoptosis in neurodegenerative diseases.
Acknowledgments
This work was supported by fundings from Institut National de la Santé et de la Recherche Médicale (Inserm) and the Université Paris-Sud 11. MM was recipient of fellowships from the Ministère de l’Enseignement Superieur et de la Recherche and the Societe Francaise d’Endocrinologie (SFE).
We thank Federico Simonetta (UMR_S 1012, Le Kremlin Bicêtre, France) for his help with FACS experiments.
Abbreviations
- EB
embryoid bodies
- ES
embryonic stem (cell)
- hMR
human mineralocorticoid receptor
- GC
glucocorticoid
- GR
glucocorticoid receptor
- MAP2
microtubule associated protein 2
- MR
mineralocorticoid receptor
- PCNA
proliferating cell nuclear antigen
- t-BHP
tert-butylhydroperoxide
- WT
wild-type
Footnotes
Disclose summary: The authors have nothing to disclose.
References
- 1.Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- 2.de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neuroscience. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- 3.de Kloet ER, Karst H, Joels M. Corticosteroid hormones in the central stress response: quick-and-slow. Front Neuroendocrinol. 2008;29:268–272. doi: 10.1016/j.yfrne.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Olijslagers JE, de Kloet ER, Elgersma Y, van Woerden GM, Joels M, Karst H. Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur J Neurosci. 2008;27:2542–2550. doi: 10.1111/j.1460-9568.2008.06220.x. [DOI] [PubMed] [Google Scholar]
- 5.Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102:19204–19207. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joels M, Karst H, DeRijk R, de Kloet ER. The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 2008;31:1–7. doi: 10.1016/j.tins.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Gass P, Kretz O, Wolfer DP, Berger S, Tronche F, Reichardt HM, Kellendonk C, Lipp HP, Schmid W, Schutz G. Genetic disruption of mineralocorticoid receptor leads to impaired neurogenesis and granule cell degeneration in the hippocampus of adult mice. EMBO Rep. 2000;1:447–451. doi: 10.1093/embo-reports/kvd088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crochemore C, Lu J, Wu Y, Liposits Z, Sousa N, Holsboer F, Almeida OF. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol Psychiatry. 2005;10:790–798. doi: 10.1038/sj.mp.4001679. [DOI] [PubMed] [Google Scholar]
- 9.Lai M, Horsburgh K, Bae SE, Carter RN, Stenvers DJ, Fowler JH, Yau JL, Gomez-Sanchez CE, Holmes MC, Kenyon CJ, Seckl JR, Macleod MR. Forebrain mineralocorticoid receptor overexpression enhances memory, reduces anxiety and attenuates neuronal loss in cerebral ischaemia. Eur J Neurosci. 2007;25:1832–1842. doi: 10.1111/j.1460-9568.2007.05427.x. [DOI] [PubMed] [Google Scholar]
- 10.McCullers DL, Herman JP. Mineralocorticoid receptors regulate bcl-2 and p53 mRNA expression in hippocampus. Neuroreport. 1998;9:3085–3089. doi: 10.1097/00001756-199809140-00031. [DOI] [PubMed] [Google Scholar]
- 11.Almeida OF, Conde GL, Crochemore C, Demeneix BA, Fischer D, Hassan AH, Meyer M, Holsboer F, Michaelidis TM. Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. Faseb J. 2000;14:779–790. doi: 10.1096/fasebj.14.5.779. [DOI] [PubMed] [Google Scholar]
- 12.Macleod MR, Johansson IM, Soderstrom I, Lai M, Gido G, Wieloch T, Seckl JR, Olsson T. Mineralocorticoid receptor expression and increased survival following neuronal injury. Eur J Neurosci. 2003;17:1549–1555. doi: 10.1046/j.1460-9568.2003.02587.x. [DOI] [PubMed] [Google Scholar]
- 13.Munier M, Meduri G, Viengchareun S, Leclerc P, Le Menuet D, Lombes M. Regulation of mineralocorticoid receptor expression during neuronal differentiation of murine embryonic stem cells. Endocrinology. 2010;151:2244–2254. doi: 10.1210/en.2009-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujioka A, Fujioka T, Ishida Y, Maekawa T, Nakamura S. Differential effects of prenatal stress on the morphological maturation of hippocampal neurons. Neuroscience. 2006;141:907–915. doi: 10.1016/j.neuroscience.2006.04.046. [DOI] [PubMed] [Google Scholar]
- 15.Berger S, Wolfer DP, Selbach O, Alter H, Erdmann G, Reichardt HM, Chepkova AN, Welzl H, Haas HL, Lipp HP, Schutz G. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc Natl Acad Sci U S A. 2006;103:195–200. doi: 10.1073/pnas.0503878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Nicola AF, Pietranera L, Beauquis J, Ferrini MG, Saravia FE. Steroid protection in aging and age-associated diseases. Exp Gerontol. 2009;44:34–40. doi: 10.1016/j.exger.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Choi JH, Hwang IK, Lee CH, Chung DW, Yoo KY, Li H, Won MH, Seong JK, Yoon YS, Lee IS. Immunoreactivities and levels of mineralocorticoid and glucocorticoid receptors in the hippocampal CA1 region and dentate gyrus of adult and aged dogs. Neurochem Res. 2008;33:562–568. doi: 10.1007/s11064-007-9479-6. [DOI] [PubMed] [Google Scholar]
- 18.Le Menuet D, Isnard R, Bichara M, Viengchareun S, Muffat-Joly M, Walker F, Zennaro MC, Lombes M. Alteration of cardiac and renal functions in transgenic mice overexpressing human mineralocorticoid receptor. J Biol Chem. 2001;276:38911–38920. doi: 10.1074/jbc.M103984200. [DOI] [PubMed] [Google Scholar]
- 19.Le Menuet D, Munier M, Meduri G, Viengchareun S, Lombes M. Mineralocorticoid receptor overexpression in embryonic stem cell-derived cardiomyocytes increases their beating frequency. Cardiovasc Res. 2010;87:467–475. doi: 10.1093/cvr/cvq087. [DOI] [PubMed] [Google Scholar]
- 20.Viengchareun S, Kamenicky P, Teixeira M, Butlen D, Meduri G, Blanchard-Gutton N, Kurschat C, Lanel A, Martinerie L, Sztal-Mazer S, Blot-Chabaud M, Ferrary E, Cherradi N, Lombes M. Osmotic stress regulates mineralocorticoid receptor expression in a novel aldosterone-sensitive cortical collecting duct cell line. Mol Endocrinol. 2009;23:1948–1962. doi: 10.1210/me.2009-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 22.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rozeboom AM, Akil H, Seasholtz AF. Mineralocorticoid receptor overexpression in forebrain decreases anxiety-like behavior and alters the stress response in mice. Proc Natl Acad Sci U S A. 2007;104:4688–4693. doi: 10.1073/pnas.0606067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reagan LP, McEwen BS. Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J Chem Neuroanat. 1997;13:149–167. doi: 10.1016/s0891-0618(97)00031-8. [DOI] [PubMed] [Google Scholar]
- 25.Obradovic D, Tirard M, Nemethy Z, Hirsch O, Gronemeyer H, Almeida OF. DAXX, FLASH, and FAF-1 modulate mineralocorticoid and glucocorticoid receptor-mediated transcription in hippocampal cells--toward a basis for the opposite actions elicited by two nuclear receptors? Mol Pharmacol. 2004;65:761–769. doi: 10.1124/mol.65.3.761. [DOI] [PubMed] [Google Scholar]
- 26.Tirard M, Jasbinsek J, Almeida OF, Michaelidis TM. The manifold actions of the protein inhibitor of activated STAT proteins on the transcriptional activity of mineralocorticoid and glucocorticoid receptors in neural cells. J Mol Endocrinol. 2004;32:825–841. doi: 10.1677/jme.0.0320825. [DOI] [PubMed] [Google Scholar]
- 27.Pascual-Le Tallec L, Lombes M. The mineralocorticoid receptor: a journey exploring its diversity and specificity of action. Mol Endocrinol. 2005;19:2211–2221. doi: 10.1210/me.2005-0089. [DOI] [PubMed] [Google Scholar]
- 28.Crochemore C, Michaelidis TM, Fischer D, Loeffler JP, Almeida OF. Enhancement of p53 activity and inhibition of neural cell proliferation by glucocorticoid receptor activation. FASEB J. 2002;16:761–770. doi: 10.1096/fj.01-0577com. [DOI] [PubMed] [Google Scholar]
- 29.Sundberg M, Savola S, Hienola A, Korhonen L, Lindholm D. Glucocorticoid hormones decrease proliferation of embryonic neural stem cells through ubiquitin-mediated degradation of cyclin D1. J Neurosci. 2006;26:5402–5410. doi: 10.1523/JNEUROSCI.4906-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joels M. Functional actions of corticosteroids in the hippocampus. Eur J Pharmacol. 2008;583:312–321. doi: 10.1016/j.ejphar.2007.11.064. [DOI] [PubMed] [Google Scholar]
- 31.Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. doi: 10.1146/annurev.neuro.20.1.245. [DOI] [PubMed] [Google Scholar]
- 32.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 33.Brady LS, Whitfield HJ, Jr, Fox RJ, Gold PW, Herkenham M. Long-term antidepressant administration alters corticotropin-releasing hormone, tyrosine hydroxylase, and mineralocorticoid receptor gene expression in rat brain. Therapeutic implications. J Clin Invest. 1991;87:831–837. doi: 10.1172/JCI115086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang SS, Kamphuis W, Huitinga I, Zhou JN, Swaab DF. Gene expression analysis in the human hypothalamus in depression by laser microdissection and real-time PCR: the presence of multiple receptor imbalances. Mol Psychiatry. 2008;13:786–799. 741. doi: 10.1038/mp.2008.38. [DOI] [PubMed] [Google Scholar]
- 35.Lai M, Bae SE, Bell JE, Seckl JR, Macleod MR. Mineralocorticoid receptor mRNA expression is increased in human hippocampus following brief cerebral ischaemia. Neuropathol Appl Neurobiol. 2009;35:156–164. doi: 10.1111/j.1365-2990.2008.00980.x. [DOI] [PubMed] [Google Scholar]
- 36.Viegas LR, Vicent GP, Baranao JL, Beato M, Pecci A. Steroid hormones induce bcl-X gene expression through direct activation of distal promoter P4. J Biol Chem. 2004;279:9831–9839. doi: 10.1074/jbc.M312402200. [DOI] [PubMed] [Google Scholar]
- 37.Gascoyne DM, Kypta RM, Vivanco MM. Glucocorticoids inhibit apoptosis during fibrosarcoma development by transcriptionally activating Bcl-xL. J Biol Chem. 2003;278:18022–18029. doi: 10.1074/jbc.M301812200. [DOI] [PubMed] [Google Scholar]
- 38.Yin P, Lin Z, Cheng YH, Marsh EE, Utsunomiya H, Ishikawa H, Xue Q, Reierstad S, Innes J, Thung S, Kim JJ, Xu E, Bulun SE. Progesterone receptor regulates Bcl-2 gene expression through direct binding to its promoter region in uterine leiomyoma cells. J Clin Endocrinol Metab. 2007;92:4459–4466. doi: 10.1210/jc.2007-0725. [DOI] [PubMed] [Google Scholar]
- 39.Balsamo A, Cicognani A, Gennari M, Sippell WG, Menabo S, Baronio F, Riepe FG. Functional characterization of naturally occurring NR3C2 gene mutations in Italian patients suffering from pseudohypoaldosteronism type 1. Eur J Endocrinol. 2007;156:249–256. doi: 10.1530/eje.1.02330. [DOI] [PubMed] [Google Scholar]
- 40.Trouillas M, Saucourt C, Duval D, Gauthereau X, Thibault C, Dembele D, Feraud O, Menager J, Rallu M, Pradier L, Boeuf H. Bcl2, a transcriptional target of p38alpha, is critical for neuronal commitment of mouse embryonic stem cells. Cell Death Differ. 2008;15:1450–1459. doi: 10.1038/cdd.2008.63. [DOI] [PubMed] [Google Scholar]
- 41.Liste I, Garcia-Garcia E, Martinez-Serrano A. The generation of dopaminergic neurons by human neural stem cells is enhanced by Bcl-XL, both in vitro and in vivo. J Neurosci. 2004;24:10786–10795. doi: 10.1523/JNEUROSCI.3208-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liste I, Garcia-Garcia E, Bueno C, Martinez-Serrano A. Bcl-XL modulates the differentiation of immortalized human neural stem cells. Cell Death Differ. 2007;14:1880–1892. doi: 10.1038/sj.cdd.4402205. [DOI] [PubMed] [Google Scholar]
- 43.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 44.Courtois ET, Castillo CG, Seiz EG, Ramos M, Bueno C, Liste I, Martinez-Serrano A. In vitro and in vivo enhanced generation of human A9 dopamine neurons from neural stem cells by Bcl-XL. J Biol Chem. 2010;285:9881–9897. doi: 10.1074/jbc.M109.054312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prager EM, Brielmaier J, Bergstrom HC, McGuire J, Johnson LR. Localization of mineralocorticoid receptors at mammalian synapses. PLoS One. 2010;5:e14344. doi: 10.1371/journal.pone.0014344. [DOI] [PMC free article] [PubMed] [Google Scholar]