

Abstract
Members of the TMEM16 (Anoctamin) family of membrane proteins have been shown to be essential constituents of the Ca2+-activated Cl− channel (CaCC) in many cell types. In this study, we have investigated the electrophysiological properties of mouse TMEM16F. Heterologous expression of TMEM16F in HEK293 cells resulted in plasma membrane localization and an outwardly rectifying ICl,Ca that was activated with a delay of several minutes. Furthermore, a significant Na+ current was activated, and the two permeabilities were correlated according to PNa = 0.3 PCl. The current showed an EC50 of 100 µM intracellular free Ca2+ concentration and an Eisenman type 1 anion selectivity sequence of PSCN > PI > PBr > PCl > PAsp. The mTMEM16F-associated ICl,Ca was abolished in one mutant of the putative pore region (R592E) but retained in two other mutants (K616E and R636E). The mutant K616E had a lower relative permeability to iodide, and the mutant R636E had an altered anion selectivity sequence (PSCN = PI = PBr = PCl > PAsp). Our data provide evidence that TMEM16F constitutes a Ca2+-activated anion channel or a pore-forming subunit of an anion channel with properties distinct from TMEM16A.
INTRODUCTION
Anion channels are involved in several physiological processes, including housekeeping functions such as maintenance of cytoplasmic ion composition, pH regulation, and cell volume regulation (Nilius and Droogmans, 2003; Hoffmann et al., 2009; Verkman and Galietta, 2009) and more specialized physiological functions such as trans-epithelial electrolyte/fluid transport in absorbing and excreting epithelia (Larsen, 2011) and muscle contraction and excitability (Jentsch et al., 2002; Hartzell et al., 2005). Furthermore, intracellular Cl− channels are important for maintaining electroneutrality when protons and Ca2+ ions are transported into intracellular compartments (Jentsch et al., 2002). These functions are maintained by various types of Cl− channels whose mechanisms of activation and molecular identities are often not known. Activation stimuli include ligand-gating, cAMP-dependent phosphorylation, voltage-gating, cell swelling, and increases in intracellular Ca2+ (Jentsch et al., 2002; Nilius and Droogmans, 2003; Eggermont, 2004). Ca2+-activated Cl− channels (CaCCs) were first described in Xenopus laevis oocytes (Miledi et al., 1982; Barish, 1983) and Salamander rods (Bader et al., 1982) and have since been found in many cell types where they have important functions in, e.g., membrane excitability in cardiac muscle and neurons (André et al., 2003; Guo et al., 2008), olfactory transduction (Matthews and Reisert, 2003), epithelial secretion (Kunzelmann et al., 2007; Rock et al., 2009), regulation of vascular tone (Angermann et al., 2006), and photoreception (Lalonde et al., 2008).
At low Ca2+ concentrations, CaCCs are characterized by time-dependent outward rectification and activation in a voltage-dependent manner by raised intracellular concentration of free Ca2+ ([Ca2+]i). At subsaturating [Ca2+]i, CaCCs are slowly activated and deactivated by depolarizing and hyperpolarizing plasma membrane potentials, respectively. A higher Ca2+ sensitivity at depolarizing potentials contributes to the outward rectification (Nilius et al., 1997). At saturating [Ca2+]i, the channels are fully activated at all physiologically relevant membrane potentials, and rectification is decreased with the current-voltage relationship approaching linearity (Kuruma and Hartzell, 2000; Nilius and Droogmans, 2003; Hartzell et al., 2005). The [Ca2+]i required for half maximal activation was reported to be ∼200 nM (dependent on voltage) with a Hill coefficient of Ca2+ binding > 1 (Nilius et al., 1997; Pedersen et al., 1998b; Kuruma and Hartzell, 2000; Klausen et al., 2007). CaCCs show an Eisenman type 1 permeation profile given by PSCN > PI > PBr > PCl > PAsp, a single channel conductance of 0.5–5 pS, and sensitivity to nonselective Cl− channel blockers like DIDS, NPPB, NPA, and niflumic acid (Jentsch et al., 2002; Nilius and Droogmans, 2003). CaCCs in some cells are activated directly by Ca2+, whereas CaCCs in other cells are dependent on the activity of CAMKII (Hartzell et al., 2005).
Recently, TMEM16A, officially named anoctamin 1 (ANO1), was shown, by three independent laboratories to be associated with a Ca2+-activated Cl− current (ICl,Ca; Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008). TMEM16A expression produced robust ICl,Ca with Ca2+ sensitivity, current-voltage relationship, ion selectivity, and pharmacology closely resembling canonical CaCCs. No obvious Ca2+-binding sites have been found in the protein (Hartzell et al., 2009). However, Yu et al. (2012) found that two amino acids, E702 and E705, are important for Ca2+ binding. In the model of TMEM16A structure, these amino acids are placed in the extracellular loop, and Yu et al. (2012) have therefore proposed a new structural model in which there is no reentrant loop and E702 and E705 are thus intracellular. TMEM16A belongs to the TMEM16 family of transmembrane (TM) proteins consisting of 10 members in mammals, TMEM16A–K (ANO 1–10). Hydropathy analysis predicts members of the TMEM16 family to have eight TM domains, with the N and C termini located in the cytosol (Tsutsumi et al., 2005; Yang et al., 2008; Bolduc et al., 2010; Milenkovic et al., 2010) The hydrophobic region between TM5 and TM6 seems to form a reentrant loop, which constitutes a part of the channel pore (Das et al., 2008), and mutations of positively charged amino acids in this region result in a reduction in the anion/cation permeability ratio (Caputo et al., 2008; Yang et al., 2008). It has been suggested that the TMEM16s work as multimeric complexes (Schreiber et al., 2010), and recent data have shown that TMEM16A can form dimers (Sheridan et al., 2011; Fallah et al., 2011), a functional structure it may share with other members of the family.
Some TMEM16 family members show tissue-specific expression in mammals. TMEM16B, TMEM16C, and TMEM16D are primarily expressed in neuronal tissue, and TMEM16E shows strong expression in skeletal muscle and the thyroid gland, whereas TMEM16F, TMEM16H, and TMEM16K have been found expressed in almost all tissue examined (Rock and Harfe, 2008; Stöhr et al., 2009; Gritli-Linde et al., 2009; Schreiber et al., 2010). TMEM16A shows broad expression but is most prominently expressed in electrolyte-transporting tissue, neurons, and interstitial cells (Yang et al., 2008; Ousingsawat et al., 2009; Hwang et al., 2009; Schreiber et al., 2010). TMEM16A and TMEM16B have previously been shown to produce ICl,Ca’s, however, TMEM16B with somewhat different characteristics when compared with those of TMEM16A (Pifferi et al., 2009). TMEM16B is in particular expressed in photoreceptors (Stöhr et al., 2009) and in the cilium of olfactory cells (Rasche et al., 2010), where it appears to play a role in sensory transduction (Pifferi et al., 2012).
It is somewhat uncertain whether the remaining members of the TMEM16 family are CaCCs. Duran et al. (2012) recently published that TMEM16F and several other TMEM16s are completely intracellular. In contrast, Schreiber et al. (2010) found that expression of TMEM16F and TMEM16G in FRT cells induced a small ATP-stimulated I− influx. However, the rate of I− uptake by TMEM16A was ∼18-fold higher than the uptake rates by TMEM16F and TMEM16G, and a short isoform of TMEM16G, with intracellular localization and no predicted TM domains, also took up I− at roughly the same rate as the long isoform of TMEM16G and TMEM16F (Schreiber et al., 2010; Duran and Hartzell, 2011). In whole-cell patch clamp experiments, the authors reported a slowly activating Ca2+-sensitive membrane conductance in TMEM16F- and TMEM16K-expressing cells that was similar to that of mock-transfected FRT cells, within an 8-min observation period (Schreiber et al., 2010). Martins et al. (2011) found that TMEM16F was an essential component of the outwardly rectifying Cl− current that can be stimulated by cAMP in the presence of CFTR. Recently, Shimizu et al. (2013) showed that TMEM16F was a Ca2+-activated anion selective channel. In contrast, adding a great deal of confusion, it was recently reported that TMEM16F is a Ca2+-activated nonselective cation channel (Yang et al., 2012) and that TMEM16F is essential for Ca2+-dependent phospholipid scramblase activity involved in phosphatidylserine exposure to the surface of blood platelets. A patient with Scott syndrome caused by a defect in phospholipid scrambling activity had a mutation in TMEM16F (Suzuki et al., 2010; Castoldi et al., 2011; Nurden and Nurden, 2011; Yang et al., 2012). Finally, a study by Almaça et al. (2009) has suggested TMEM16s to be implicated in cell volume regulation. However, this effect seems predominantly to be secondary to ATP release and purinergic signaling, resulting in CaCC activation (Kunzelmann et al., 2012), and Shimizu et al. (2013) found that neither knockdown nor overexpression of TMEM16F affected the volume-sensitive Cl− current in HEK293T (human embryonic kidney 293 T) cells.
The present investigation aims at the function of TMEM16F in mediating a TM Cl− current. We show that mouse TMEM16F localizes to the surface membrane and generates ICl,Ca when expressed in HEK293 cells. The current is activated after a relatively long delay (minutes) in response to high [Ca2+]i. We determine the Ca2+ dependence, relative anion/cation selectivity, anion selectivity sequence, and the effect of mutations in the putative pore domain.
MATERIALS AND METHODS
Cell culture and transfection
Two different model cell lines were used: the mouse-derived Ehrlich ascites tumor cells (EATCs), and the human-derived HEK293 cells. The cells were kept at 37°C with 5% CO2/95% air. HEK293 cells were kept in DMEM and EATCs in RPMI1640. 10% fetal bovine serum and 1% penicillin/streptomycin were added to each media. HEK293 cells were transfected by Trans-IT-293 (Mirus Bio LLC) and used for electrophysiological recordings 24–48 h after transfection.
Isolation of RNA, reverse transcription (RT), PCR, and cDNA cloning
Total RNA was isolated from EATCs using the RNeasy Mini kit (QIAGEN) according to the manufacturer’s protocol. cDNA was prepared in a total volume of 40 µl by hybridization of 500 ng oligo (dT) primers to 4 µg RNA at 65°C for 5 min, followed by extension at 42°C for 50 min in the presence of 200 U Superscript II RT (Invitrogen), 500 µM dNTP, 10 µM DTT, 50 mM Tris-HCl, 75 mM KCl, and 3 mM MgCl2, pH 8.3. Finally, the RT was inactivated at 70°C for 15 min. PCR was performed in a total volume of 20 µl containing 1 µl of the RT reaction, 0.5 mM dNTPs, 0.5 µM of each primer, 2 mM MgCl2, and 2 U Taq polymerase in PCR buffer (conventional PCR) or 2 U Pfu Ultra II fusion HS polymerase (Agilent Technologies) in Pfu PCR buffer (for cloning). PCR consisted of a denaturizing step of 2 min at 95°C, followed by 35 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 2 min. Primers used for PCR were 16A_for, 5′-GACCTGGGCTATGAGGTTCA-3′; 16A_rev, 5′-GGCTGATGTCTTTGGGGATA-3′; 16B_for, 5′-CCAGGGAAGCAGAGTTCTTG-3′; 16B_rev, 5′-TGTTGTTGGCTCGAGAACAG-3′; 16C_for, 5′-TGACAAGTCAGCGTTTCCAG-3′; 16C_rev, 5′-CAGCCAGGCGAAGTATAAGC-3′; 16D_for, 5′-TTTGAGCACCTCGTGTTTTG-3′; 16D_rev, 5′-TCATGGCCACTCATTGTGAT-3′; 16E_for, 5′-TCTGGATCATGCCTGAACTG-3′; 16E_rev, 5′-ATGGTTAGCTGGGTGGTCAG-3′; 16F_for, 5′-GCAGCCCTTGGATCTTATCA-3′; 16F_rev, 5′-TGCTGTAGCTCAACGGTGTC-3′; 16G_for, 5′-TCTTCCTGGTGGGATGTTTC-3′; 16G_rev, 5′-CCACAGTGCCATGAACAAAC-3′; 16H_for, 5′-ACGCAGGCAGAGCTAGAGAG-3′; 16H_rev, 5′-ACACTGGCCAATGAGGTAGC-3′; 16J_for, 5′-TGAAGGATGGGGTTTTTGAG-3′; 16J_rev, 5′-TGCCATGAAGATGGCAAATA-3′; 16K_for, 5′-TCTGAGTGGACCAGCCTTCT-3′; 16K_rev, 5′-AGAAGAGTGAGGCGAAGCAA-3′; ARP_for, 5′-CGACCTGGAAGTCCAACTAC-3′; and ARP-rev, 5′-ATCTGCATCTGCTTG-3′. Real-time quantitative PCR was performed in triplicates using an MX4000 Real-Time PCR system (Agilent Technologies) and SYBR green PCR master mix (Applied Biosystems) in a total volume of 20 µl containing 1 µl of the RT reaction, 200 nM of primers, and 10 µl of 2× master mix. Primers used are listed above (16F and ARP). Target expression level was normalized to the reference gene level (r18S), and the relative expression ratio was calculated using the equation
where Etarget and Eref are the PCR amplification efficiencies for the target and reference gene (ARP), respectively, and ΔC(t)target and ΔC(t)ref are the change in C(t) values for target and reference genes. Primers used to clone and generate attB-flanked gateway-compatible PCR products were 16F_for, 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGCAGATGATGACTAGGAAGGTC-3′; 16F_rev, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTCATTCGAGTTTTGGCCGCACG-3′; 16F_rev_fusion, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTTCGAGTTTTGGCCGCACGCT-3′; 16A_for, 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGAGGGTCCCCGAGAAGTAC-3′; 16A_rev, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTACAGCGCGTCCCCATGGGA-3′; 16A_rev_fusion, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCAGCGCGTCCCCATGGTACT-3′; 16K_for, 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGAGAGTGACTTTATCAACGCTG-3′; and 16K_rev, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTCAGGTAGCTTCCTTCCCATCT-3′. PCR products were excised from 0.8% agarose gels and DNA extracted using an E.Z.N.A gel extraction kit (Omega Bio-tek). PCR products of mTMEM16F were then recombined into pDONR221 using BP Clonase according to the manufacturer’s instructions (Invitrogen) to generate mTMEM16F entry clones. After transformation into Escherichia coli and selection using 50 µg/ml kanamycin, DNA was purified and inserts in entry clones confirmed by full-length DNA sequencing. Finally, using LR-clonase mix, the inserts from entry clones were recombined into pcDNA6.2/EmGFP-Bsd/V5-DEST and pcDNA3.1-DEST47 to generate vectors either coexpressing mTMEM16F and EmGFP or an mTMEM16F-GFP C-terminal fusion. Expression plasmids were produced in E. coli using 100 µg/ml ampicillin as selection agent.
Site-directed mutagenesis
The mTMEM16F R592E, K616E, R636E, and I342A mutations were constructed using a multisite-directed mutagenesis kit (Agilent Technologies). Primers were R592E, 5′-TTCCTGGGCAAATACGAGAGCGAAGAGTGTG-3′; K616E, 5′-ATCATGGGGGGAGAGGCAATCTGGAACAAC-3′; R636E, 5′-CCCATGGGTTATGAATCTAATTGGAGAGTATAAAAGAGTCTCGGG-3′; and I342A, 5′-GGTCTGTGATCCTGACGCCGGTGGCCAGATCCTG-3′. PCR was performed in a total volume of 25 µl containing 100 ng pcDNA6.2/EmGFP-Bsd/V5-mTMEM16F-DEST or pcDNA-mTMEM16F-DEST47(GFP-fusions), 0.5 mM dNTPs, 0.5 µM of primer, 2.5 µl of 10× reaction buffer, 0.75 µl of quick solution, and 1 µl of multienzyme blend (Agilent Technologies). PCR consisted of a denaturizing step of 1 min at 95°C, followed by 30 cycles of 95°C for 1 min, 55°C for 1 min, and 65°C for 16 min. The PCR reaction was treated with DpnI for 10 min at 37°C before being transformed into E. coli and selected for ampicillin resistance (100 µg/ml). Base pair substitutions were confirmed by DNA sequencing.
Electrophysiological recordings
Cells were plated on poly-l-lysine–coated coverslips. Whole-cell voltage-clamp recordings were performed with the Axopatch 200B amplifier interfaced to a Digidata 1440A using pClamp10 for recording and analysis (Molecular Devices). Analogue signals were acquired at 2.5 kHz and filtered at 1 kHz. All recordings were performed at room temperature (20°C). Patch pipettes were fabricated from borosilicate glass capillaries using a DMZ-Universal Puller (Zeitz Instruments) with a resistance of 2–3 MΩ when filled with the internal solution. For activation of ICl,Ca, the pipette solution contained (in mM): 100 Cs-Aspartate, 40 CsCl, 1 MgCl2, 1.5 Na2-ATP, 5 EGTA, and 10 HEPES, pH 7.2 with CsOH, osmolarity adjusted to 290 mosmol/kg. EGTA stock solutions were prepared by NaOH titration giving a final Na+ concentration in the pipette solution of 15.3 ± 0.6 mM as measured by flame spectrophotometry using a FLM3 flame photometer (Radiometer). CaCl2 was varied between experiments to investigate the role of 0, 0.19, 4.86, 4.98, 5.11, 5.23, 5.41, and 5.70 mM Ca2+. The free [Ca2+]i was measured using a Ca2+-selective electrode (Perfection; Mettler Toledo) in combination with a DrDAQ data logger and Pico Logger software (Pico Technology). Calibration curve was performed by varying [CaCl2] in a calibration solution of (in mM) 100 Cs-Aspartate, 40 CsCl2, and 10 HEPES. Because of the strong buffer capacity of the pipette solutions, we observed super-Nernstian potentials at concentrations <1 µM as the result of Ca2+ depletion of the electrode at standard conditions (Sokalski et al., 1999). Thus, the free [Ca2+] for low-level CaCl2 was measured using the Ca2+-sensitive fluorescent probe, Fura-2P (Molecular Probes), as previously described (Pedersen et al., 1998a). The superfusate was a modified Ringer’s solution and contained (in mM) 140 NMDG-Cl, 10 NaCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4 with NaOH, osmolarity adjusted to 315 mosmole/kg. In the anion selectivity experiment, NMDG-Cl was substituted by equimolar concentrations of NaCl, NaI, NaBr, NaSCN, or Na-aspartate (applied in a randomized order). The reference electrode was an agar bridge composed of 3% agar and 97% of the above mentioned superfusate solution containing NMDG-Cl. Liquid junction potentials (Vlj) were calculated using the Clampex software (Molecular Devices), and Vtest was corrected (Vm = Vtest − Vlj) in all the figures by −19.3 mV, −14.3 mV, −11.9 mV, −14.4 mV, −14.7 mV, and 1.9 mV for NMDG-Cl, NaCl, NaSCN, NaI, NaBr, and NaAsp, respectively. Series resistance was compensated by 60–70%. A ramp protocol ranging from Vtest = −100 to 100 mV over 1 s with a holding potential of Vtest = 0 mV was run continuously with 15-s intervals to follow the current activation. A step protocol with steps ranging from Vtest = −100 to 100 mV with 20-mV increments and a step length of 1 or 5 s was recorded at peak ICl,Ca.
Immunofluorescence
For images in Fig. 2, HEK293 cells were seeded on poly-l-lysine–coated coverslips in 6-well plates and grown until 50–60% confluence. Cells in each well were transfected with mTMEM16F-GFP fusions using 5 µl PEI transfection reagent and 1 µg DNA. After 24-h incubation, the transfection medium was replaced with fresh medium and the cells were incubated overnight. The cells were then washed in modified Krebs solution and incubated for 20 min at 37°C with modified Krebs solution containing 500 µM free Ca2+ and wheat germ agglutinin (WGA; 1:200). The cells were washed in PBS, fixed with 2% paraformaldehyde in PBS for 15 min, washed twice in PBS, and permeabilized for 10 min with 0.1% Triton X-100 and 1% BSA in PBS. The permeabilized cells were washed and then blocked in 2% BSA in PBS for 40 min and then labeled with primary antibody (1:300 rabbit anti-GFP in PBS with 2% BSA) for 45 min. The cells were then washed three times in PBS with 2% BSA and incubated with Alexa Fluor 488 (1:300 in PBS with 2% BSA) for 45 min. After two washes in PBS with 2% BSA, the cells were incubated with DAPI (1:600 in PBS with 2% BSA) for 5 min and then washed three times in PBS. Finally, the slides were mounted (90% glycerol, 10% of 10× PBS, and 2% wt/vol N-propyl gallate). Images were recorded using a BX-63 epifluorescence microscope (Olympus). Primary antibody was GFP (A6455; 1:500 Life Technologies). Secondary antibody was Alexa Fluor 488 (A11055; 1:1,000; Life Technologies). For images in Fig. 4 E, mTMEM16F-GFP was expressed in HEK293 cells. Immediately before fixation, the cells were exposed to ionomycin for 20 min in a solution containing 500 µM free Ca2+. The images were taken using an epifluorescence microscope. mTMEM16F-GFP was visualized with anti-GFP and Alexa Fluor 488 (green), whereas the plasma membrane was stained using rhodamine-conjugated WGA (red). Nucleus was stained with DAPI (blue). Images in Fig. 6 F were recorded in the EasyRatioPro software (PTI) using an Eclipse TI inverted microscope (Nikon) equipped with CoolSNAP HQ2 digital camera (Photometrics) and Deltaram X monochromator (PTI). HEK293 cells were seeded in 6-well plates and grown until 50–60% confluence. Cells in each well were transfected with mTMEM16F-GFP fusions using 6 µl Mirus293 transfection reagent and 2 µg DNA. After 24-h incubation, cells were trypsinized and 200 µl of cell suspensions transferred to new wells containing poly-l-lysine–coated coverslips. After 1 h, the settled cells were fixed in a final concentration of 2% paraformaldehyde, washed in PBS, and permeabilized for 15 min in 0.1% Triton X-100 in PBS. The permeabilized cells were washed, blocked in 2.5% BSA for 30 min, and labeled with primary antibody (1:300 rabbit anti-GFP in PBS with 1% BSA) overnight. The cells were then washed three times in PBS with 1% BSA and incubated with FITC-conjugated anti–rabbit secondary antibody (1:400) for 2 h. After three washes in PBS, slides were mounted (90% glycerol, 10% of 10× PBS, and 2% wt/vol N-propyl gallate).
Figure 2.
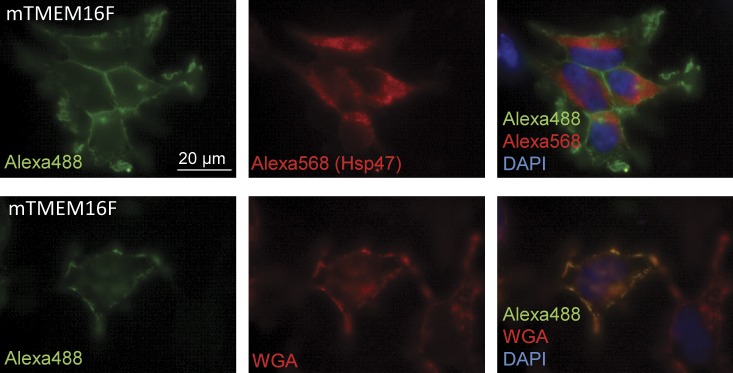
Subcellular localization experiments. mTMEM16F-GFP expressed in HEK293 cells. The images were taken using an epifluorescence microscope. mTMEM16F-GFP was visualized with anti-GFP and Alexa Fluor 488. Anti-Hsp47 and Alexa Fluor 568 were used to label the ER, whereas the plasma membrane was stained using rhodamine-conjugated WGA. The nucleus was stained with DAPI.
Figure 4.

Ca2+ activation kinetics of mTMEM16F. (A) Typical current-voltage relationships of mTMEM16F at max current with 90, 190, or 250 µM free [Ca2+]i. (B) The mean mTMEM16F I-V relation at maximal activation with the indicated free [Ca2+]i, generated from ramp protocol sweeps (from −120 to 80 mV). Data are presented as mean ± SE of three to nine replicates. (C) Hill plot of normalized mTMEM16F ICl,Ca densities at 70 mV. Data are presented as mean ± SE of 3–12 replicates. (D) Ca2+ dependence of mTMEM16F versus time from whole start of whole-cell recording to peak ICa,Cl activation. Linear regression is included to illustrate the decline in the time to peak activation with increasing free [Ca2+]i and is given by f(x) = −0.04x + 14.11. (E) mTMEM16F-GFP expressed in HEK293 cells. Immediately before fixation, the cells were exposed to ionomycin for 20 min in a solution containing 500 µM free Ca2+. The images were taken using an epifluorescence microscope. mTMEM16F-GFP was visualized with anti-GFP and Alexa Fluor 488, whereas the plasma membrane was stained using rhodamine-conjugated WGA. The nucleus was stained with DAPI.
Figure 6.

Effect of single amino acid mutations on whole-cell current and anion selectivity. (A) Mean peak ICa,Cl current density when activated by 250 µM free [Ca2+]i of mock, mTMEM16F, and single amino acid mutants in the putative pore site of mTMEM16F (the control mutation I342A and the pore region mutants R592E, K616E, and R636E) measured at 70 mV and −110 mV, respectively. Data are presented as mean ± SE; you can read the amount of replicates inside the 110-mV bar graphs; “*” and “†” indicate significant (P < 0.05, when tested with one-way ANOVA) difference from mock and mTMEM16F, respectively. (B–D) Anion permeabilities recorded in whole-cell mode after simulation with 250 µM free [Ca2+]i for I342A (B)-, K616E (C)-, and R636E-transfected (D) HEK293 cells. Data are presented as mean ± SE; the number of replicates are indicated in the figures; “*” indicates significant (P < 0.05) difference from PCl, and “†” indicates significant difference (P < 0.05) between the relevant anion permeabilities, when tested with paired t test. Only experiments in which the anion permeabilities were paired were included in the statistical test. (E) Putative topology of the TMEM16F channel with the mutated amino acids’ approximate positions. (F) Immunofluorescence images of localization of TMEM16F mutants expressed in HEK293 cells. The images were taken using an epifluorescence microscope. GFP was enhanced by anti-GFP and Alexa Fluor 488 (green). (G) Presence of mTMEM16F-GFP in the plasma membrane fraction and total cell lysate of HEK293 cells expressing either GFP-tagged WT mTMEM16F or one of the mutants I342A, R592E, K616E, and R636E. Plasma membrane proteins were isolated using a biotinylation assay. β-Actin and NPTII were used as controls for loading and transfection efficiency, respectively.
Surface protein biotinylation
Isolation of plasma membrane proteins used for the measurements in Fig. 6 G was performed using a Pierce Cell Surface Protein Isolation kit (Thermo Fisher Scientific). In brief, cells where washed in ice-cold PBS and incubated for 30 min in PBS with 250 µg/ml Sulpho-NHS-SS-Biotin. The reaction was quenched and cells were collected. After a single wash, cells were dissolved in lysis buffer and disrupted by sonication. 50 µl of the clarified lysate (10,000 g, 2 min, 4°C) was collected for protein concentration measurement (DC protein assay [Bio-Rad Laboratories] using BSA as a standard) and total protein analysis. As protein concentration did not differentiate between samples, 600 µl of clarified lysate was incubated for 1 h with NeutrAvidin agarose beads in a centrifugation column on an end-over-end mixer for affinity purification of biotinylated proteins. Beads were washed once, and protein elution was achieved using NuPAGE LDS sample buffer (Life Technologies) with 50 mM DTT. Expression of specific proteins was analyzed using SDS-PAGE and immunostaining.
SDS-PAGE and immunostaining
SDS-PAGE was performed using the NuPAGE system (Life Technologies). Samples were diluted in LDS Sample Buffer (Life Technologies) with DTT (total lysate = 42 mM; biotinylated membrane protein = 50 mM). Proteins where separated by gel electrophoresis on a precast 10% Bis-Tris polyacrylamide gel (Life Technologies) using MOPS-SDS running buffer. Proteins were electrotransferred to Protran nitrocellulose membranes (Whatman), stained with Ponceau S red (Sigma-Aldrich).
RESULTS
Cloning of TMEM16F and localization experiments in HEK293
TMEM16F was cloned from EATCs. As seen in Fig. 1 A, EATCs express mRNA for TMEM16A, TMEM16B, TMEM16F, TMEM16H, and TMEM16K. In Fig. 1 B, this is also shown on the protein level for TMEM16A, TMEM16F, and TMEM16K by Western blotting with antibodies against A, F, and K. Full-length cDNA of the coding region of mouse TMEM16F was PCR amplified from EATCs. We also cloned the coding regions of mTMEM16A and mTMEM16K. As described in Materials and methods, cDNA was cloned using Invitrogen gateway technology and recombined into pcDNA6.2/EmGFP-Bsd/V5-DEST, which coexpresses EmGFP for easy detection of transfected cells, and into pcDNA3.1-DEST47 to generate a C-terminal GFP fusion for subcellular localization studies.
Figure 1.

TMEM16 expression in EATCs. (A) PCR for all known mouse TMEM16 genes in EATCs. (B) Western blots showing expression of TMEM16F, TMEM16FA, and TMEM16FK protein in EATCs. Presented blots are representative of three blots made.
Previous localization studies of TMEM16 proteins have reported plasma membrane expression of TMEM16A, TMEM16B, TMEM16E, TMEM16F, and TMEM16G (Bera et al., 2004; West et al., 2004; Rock et al., 2008; Schroeder et al., 2008; Yang et al., 2008). In agreement with this, expression of GFP-tagged mTMEM16F in HEK293 cells showed significant staining at the plasma membrane (Fig. 2). This is seen from an almost complete overlap between the GFP-tagged TMEM16F and the plasma membrane marker WGA and no overlap with the ER marker Hsp47 (Fig. 2, right, merged images).
Whole-cell experiments of the ICl,Ca in mTMEM16F-expressing HEK293 cells
To study the putative ICl,Ca’s of mTMEM16F, we heterologously expressed mTMEM16F in HEK293 cells and measured whole-cell currents of cells showing expression of GFP. To suppress endogenous K+ currents, the whole-cell voltage-clamp experiments were performed under conditions in which K+ was replaced by Cs+ in the pipette solutions. We did not observe activation of ICl,Ca at submicromolar free [Ca2+]i, but with [Ca2+]i in the range of 55–250 µM, a delayed and transiently activated current was identified. In the first set of experiments, mTMEM16F-transfected HEK293 cells were dialyzed with intracellular solutions containing a high free [Ca2+]i (250 µM). Fig. 3 A (left) shows a representative current trace of the time course of the Ca2+-activated current determined by measuring the current at the beginning and at the end of the 1-s voltage ramps from −110 to 70 mV elicited at 15-s intervals (Fig. 3 A, left, inset). The outward current increased from 8.9 ± 1.2 to 86.1 ± 19.8 pA/pF (Vm = 70 mV, n = 6, P < 0.05) within 7.9 ± 1.4 min. The voltage- and time-dependent current activated by 250 µM free [Ca2+]i was measured at the peak of the current using 5-s voltage steps at potentials ranging from −120 to 80 mV as shown in the right panel of Fig. 3 A. Depolarizing voltage steps (from 40 to 80 mV) resulted in outward currents with two components: an initial ohmic current followed by a delayed activation. Hyperpolarizing voltage steps (from −40 to −120 mV) also resulted in currents of two components: a small instant inward current followed by inactivation (not visible in Fig. 3 A because of scaling). Because of these time-dependent current changes, the steady-state currents become strongly outwardly rectifying with relatively small components at hyperpolarizing potentials. Mock-transfected HEK293 cells showed a similar but significantly smaller current response to whole-cell patch clamp with high [Ca2+]i, but retained the same time-dependent activation/deactivation at the peak of the response resulting in strong outward current rectification (Fig. 3 B). These currents are supposedly carried by the native human TMEM16F, which is expressed in HEK293 cells (Kunzelmann et al., 2009; Shimizu et al., 2013). In our study, the variation in the response among the mock-transfected cells was fairly large, so the mean ICl,Ca of a sample of five cells turned out to be nonsignificant with a change from 14.7 ± 5.9 to 26.6 ± 5.0 pA/pF at 70 mV (Vm = 70 mV, n = 5, P < 0.17) with the peak current at 6.3 ± 2.1 min.
Figure 3.

ICl,Ca densities in mock-, mTMEM16F-, and mTMEM16A-transfected HEK293 cells. (A) Typical current development recorded by a ramp protocol run every 15 s (left; see inset) and representative current-voltage relationships (right) of mTMEM16F (250 µM free [Ca2+]i). Inward currents were initially inactivated similar to those in C; however, this inactivation is not visible at the given scale. (B and C) Mock (250 µM free [Ca2+]i; B) and mTMEM16A (0.07 µM free [Ca2+]i; C). “a” indicates where the step protocols were recorded. (D) Mean peak ICa,Cl current density when activated by 250 µM free [Ca2+]i of mock and mTMEM16F, measured at 70 mV and −110 mV, respectively. Data are presented as mean ± SE of 14 mock- and 8 mTMEM16F-transfected cells and tested by one-way ANOVA: *, P < 0.05.
Importantly, we believe that the ICl,Ca from mock-transfected cells shown here is not carried by an endogenously expressed TMEM16A, as expression of mTMEM16A generated instantly activated, submicromolar Ca2+-sensitive currents similar to those reported in the original studies (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008). In fact, at an output gain of 1 mV/pA and a [Ca2+]i of 1 µM, we lost control of TMEM16A-generated currents at potentials positive to 40 mV. The currents shown in Fig. 3 C were recorded at 70 nM [Ca2+]i.
Ca2+ activation kinetics of mTMEM16F
The Ca2+ dependency of the mTMEM16F-associated ICl,Ca was determined by measuring the above mentioned peak current as a function of [Ca2+]i in the range of 55–415 µM. As shown in Fig. 4 A, with the typical time course of the current obtained by application of the step protocol, we observed a larger instantaneous current, a subsequent faster activation, and an increase in outward rectification of the Cl− current with increasing [Ca2+]i. The resulting current-voltage relationships (Fig. 4 B) were obtained as described in Fig. 3 A. The mean ICl,Ca densities at 70 mV were depicted as a function of [Ca2+]i and fitted by the Hill equation giving an EC50 of 105.6 ± 1.2 µM [Ca2+]i and a Hill coefficient of 2.00 ± 0.56 (Fig. 4 C). The Cl− current was strongly outwardly rectifying both at low (90 µM) and high (190–250 µM) [Ca2+]i and did not approach linearity at increasing [Ca2+]i as seen for endogenous CaCC (Kuruma and Hartzell, 2000), TMEM16A, and TMEM16B currents (Yang et al., 2008; Pifferi et al., 2009; Scudieri et al., 2012). Rather, there was a tendency for the outward rectification to be more pronounced at increasing free Ca2+ concentration (Fig. 4 D), which was also observed for the endogenous ICl,Ca. The currents showed strong time- and voltage-dependent activation at both low and high Ca2+ concentrations (Fig. 4 A). In spite of a fairly large random variation among the cells, it was indicated that the time from whole-cell breakthrough to activation of the ICl,Ca (compare with Fig. 3 A) depends on [Ca2+]i, i.e.; short delays with little variation were observed at the highest [Ca2+]i (Fig. 4 D). To investigate the mechanism of activation, we treated HEK293 cells expressing mTMEM16F-GFP with 10 µM ionomycin in an extracellular solution containing 500 µM Ca2+ and made immunofluorescence preparations after 20-min treatment (Fig. 4 E). We did not observe any change in localization of mTMEM16F-GFP between nontreated and ionomycin-treated cells (compare with Fig. 2).
Anion selectivity of mTMEM16F-induced currents
The anion selectivity of mTMEM16F-induced ICl,Ca was examined by measuring the relative permeability to various anions by replacing Cl− with equimolar concentrations of SCN−, Br−, I−, or Asp− in the bath solution. The above bath solutions were prepared from Na salts of the respective anions. In a recent preliminary study, it was suggested that TMEM16F displays a cation conductance (Yang et al., 2011). In our study, the reversal potential of [Ca2+]i-activated membrane currents was significantly displaced toward positive values when NMDG-Cl of the bath solution was replaced by NaCl, which indicates a significant conductive Na+ permeability (see below). In a more detailed quantitative analysis, presented below, the ratio of the plasma membrane’s Na+ and Cl− permeability will be shown, PNa/PCl ≈ 0.3. Applying the Goldman-Hodgkin-Katz voltage equation for PAsp/PCl < 1, see below, and a PNa/PCl ratio different from 0, the expressions for the reversal potentials before and after partial substitution of Cl− in bath with a monovalent anion, X−, are:
R is the universal gas constant (8.31 J/mol/K), T is the absolute temperature (293 K), F is the Faraday (96,485 C/mol), P denotes permeability, subscript p refers to pipette solution, and o and s refer to bath solution before and after substitution, respectively. With the measured change in reversal potential given the symbol one proceeds as follows:
| (1) |
As [Na+]p = 15.3 mM and PNa/PCl ≈ 0.34 (see below), in Eq. 1, (PNa/PCl) × [Na+]p ≈ 5.2 mM, which amounts to <5% of [Cl−]o. Thus, as a first approximation, we neglect the term and obtain
| (2) |
Fig. 5 A shows representative current-voltage relationships constructed from the linear voltage ramps of mTMEM16F-transfected HEK293 cells. Using Eq. 2, the shifts in reversal potential resulting from the change of anions in the bath solution were used to calculate the relative permeability (Px/PCl; Table 1). The anion permeability sequence of the native ICl,Ca in mock-expressing cells (putatively because of native hTMEM16F) was PSCN > PI > PBr = PCl > PAsp (Fig. 5, A and C), closely resembling an Eisenman type 1 selectivity sequence. The anion permeability sequence of the ICl,Ca in mTMEM16F-expressing cells was PSCN > PI > PBr > PCl > PAsp (Fig. 5, B and D), an Eisenman type 1 selectivity sequence. Not only was the permeability sequence different, we also found that the ICl,Ca of mTMEM16F-expressing cells had a significantly higher PBr/PCl than the ICl,Ca in mock-expressing cells.
Figure 5.

Anion selectivity of the mTMEM16F-induced current. (A and B) Representative anion selectivity current traces recorded in whole-cell configuration after stimulation with 250 µM [Ca2+]i of mock (A)- and mTMEM16F-transfected (B) HEK293 cells. (C and D) Mean relative anion permeabilities (PX/PCl) for mock (C)- and mTMEM16F-transfected (D) HEK293 cells. PX/PCl are presented as a mean of seven to nine cells and are tested by one way ANOVA; “*” indicates significant (P < 0.05) difference from PCl, and “†” indicates significant difference (P < 0.05) between the relevant anion permeabilities when tested with paired t test. Only experiments in which the anion permeabilities were paired were included in the statistical test.
Table 1.
Anion permeability ratios relative to PCl of mock-, mTMEM16F-, I342A-, K616E-, and R636E-transfected HEK293 cells
| Px/PCl | Mock | TMEM16F | I342A | K616E | R636E |
| PSCN/PCl | 4.20 ± 1.03 (NS) | 3.46 ± 0.80 | 2.62 ± 0.38 (NS) | 2.19 ± 0.23 (NS) | 1.64 ± 0.37a |
| PI/PCl | 1.47 ± 0.16 (NS) | 1.82 ± 0.20 | 1.85 ± 0.22 (NS) | 1.35 ± 0.11a | 0.86 ± 0.09a |
| PBr/PCl | 0.96 ± 0.11a | 1.32 ± 0.06 | 1.32 ± 0.21 (NS) | 1.09 ± 0.08 (NS) | 0.86 ± 0.11a |
| PAsp/PCl | 0.57 ± 0.08 (NS) | 0.51 ± 0.06 | 0.53 ± 0.11 (NS) | 0.70 ± 0.06 (NS) | 0.50 ± 0.06 (NS) |
The ratios were calculated by Eq. 2.
Significant differences between relative anion permeabilities of mTMEM16F-WT and mock or mutant mTMEM16F-expressing cells are indicated (P < 0.05), when tested with one-way ANOVA and using a Student-Newman-Keuls post hoc test.
Anion selectivity of mTMEM16F mutants
A previous study on TMEM16A mutants (Yang et al., 2008) indicated that the pore region lies within the highly conserved extracellular domain between TM5 and TM6. Mutation of positively charged amino acids R621, K645, and K668 to the negatively charged glutamic acid within this region resulted in changes in the kinetics of anion currents and a reduction in the anion/cation permeability ratio. To investigate the function of this region in TMEM16F, we introduced mutations in R592, K616, and R636 (Fig. 6 E) by replacing them with glutamic acid (E) and constructed a control mutation I342A, which we anticipated would not affect the conductive properties of the putative channel pore. We introduced the R592, K616, and R636 mutations into both GFP coexpression vectors and the C-terminal GFP fusions. Fig. 6 A shows the peak ICl,Ca activation of HEK293 cells transfected with mock, mTMEM16F, or the single amino acid mutants of mTMEM16F. Compared with the maximal whole-cell Ca2+-activated (250 µM) Cl− current in mTMEM16F-WT, the mutation R592E reduced the current strongly and was no longer significantly different from currents in mock transfectants. In contrast, the maximal current was not significantly affected in the I342A, K616E, and R636E mutants (Fig. 6 A). Fig. 6 (B–D) and Table 1 show the relative anion permeabilities recorded in whole-cell mode after stimulation with 250 µM [Ca2+]i for I342A-, K616E-, and R636E-expressing HEK293 cells, respectively. The PBr/PCl of the control mutation I342A was not significantly different from mTMEM16F-WT (Table 1). However, the observation that in mTMEM16F-WT PBr was significantly higher than PCl was not reproduced with the I342A control mutation, perhaps as a result of the variability in the data. Although the permeability sequence was not changed in K616E, the relative permeability of iodide was lower when compared with mTMEM16F-WT (Table 1). In R636E, a marked reduction was found in the relative permeability to thiocyanate, iodide, and bromide (Fig. 6, C and D; and Table 1) when compared with those found in mTMEM16F-WT (Table 1).
Subcellular localization of mTMEM16F mutants
The subcellular localization of the R636E, K616E, and R592E single amino acid mutants are shown in Fig. 6 F. Prominent staining is seen in the plasma membrane as well as in intracellular compartments of the mTMEM16F-WT as well as mutant-expressing cells (Figs. 2 and 6 F), suggesting that the loss of current in the mutants is not caused by trafficking problems. To investigate the findings from the immunofluorescence imaging further, we performed a biotinylation assay in which plasma membrane proteins were first isolated and TMEM16F then visualized by Western blotting. The membrane proteins were isolated from HEK293 cells transfected with plasmids containing the respective TMEM16F-GFP constructs (WT and the four mutants I342A, R592E, K616E, and R636E). As seen in Fig. 6 G, mTMEM16F-WT along with all of the mutants localizes to the plasma membrane, consistent with the results from the immunofluorescence imaging.
The Na+ current component of membrane currents in TMEM16F-expressing cells
The protocols applied above allowed for a quantitative analysis of anion selectivity ratios that were independent of the plasma membrane’s Na+ permeability, however large it might have been. Here, we analyze absolute Na+ and Cl− permeabilities of HEK293 cells expressing WT TMEM16F and the various mutants of the channel. Fig. 7 A shows instantaneous and (near) steady-state current-voltage relationships of a TMEM16F-WT–expressing HEK293. With NMDG+ in bath, the instantaneous current, IInst, could be fitted by the GHK Cl− current equation
| (3) |
In Eq. 3, [Cl−]i = 44.7 mM and [Cl−]o = 155 mM; i.e., the Cl− equilibrium potential is ECl = −31 mV. With the Cl− permeability as free parameter, we obtained PCl = 12.9 (±0.6) × 10−14 cm3/s/pF. Replacement of NMDG+ with Na+ resulted in rightward displacement of the IInst/Vm relationship as expected if the membrane current also contains an Na+ component. The full line of Fig. 7 A (left) indicates the nonlinear fit of Eq. 4, which expresses the instantaneous currents as the sum of two GHK-current components carried by Cl− and Na+, respectively:
| (4) |
With [Na+]i = 15.3 mM, [Na+]o = 150 mM, and the two ion permeabilities being free parameters, we obtained PCl = 31.7 (±1.0) × 10−14 cm3/s/pF and PNa = 9.7 (±0.5) × 10−14 cm3/s/pF. The Fig. 7 A (right) shows the corresponding outwardly rectifying (near) steady-state current-voltage relationships. ISt-St with NMDG-Cl in bath intersected the x axis at Vrev ≈ ECl. However, with NaCl in bath and ENa ≈ 57 mV, the reversal potential was displaced to the right of ECl, in casu Vrev ≈ −12 mV, confirming that the conductance of the plasma membrane is governed also by a significant Na+ permeability (Fig. 7 A, inset).
Figure 7.

Instantaneous (IInst) and near steady-state (ISt-St) current-voltage relationships of HEK293 cells in whole-cell patch-clamp configuration. Green symbols show I-V relationships obtained with NMDG-Cl in bath. Blue symbols show I-V relationships obtained with NaCl in bath. IInst was recorded immediately after the voltage step from the holding potential, Vhold = 0 mV, to the new Vm. ISt-St was recorded toward the end of the 1-s voltage pulse. Thus, rather than estimating the PNa/PCl of each cell from a reversal potential obtained by interpolation, we could take advantage of IInst-Vm data points covering a broader range about Vrev. Current-voltage relationships are shown for the following (all permeabilities in units of 10−14 cm3/s/pF). (A) HEK293 cells expressing TMEM16F-WT (with NMDG-Cl in bath, PCl = 12.9 ± 0.6; with NaCl in bath, PCl = 31.7 ± 1.0 and PNa = 9.7 ± 0.5). (B) HEK293 cells expressing the neutral mutant TMEM16F-I342A (with NMDG-Cl in bath, PCl = 19.8 ± 0.8; with NaCl in bath, PCl = 93.8 ± 4.2 and PNa = 13.3 ± 3.6). (C) HEK293 cells expressing the anion sequence modifying TMEM16F-R636E mutant (with NMDG-Cl in bath, PCl = 16.9 ± 1.5; with NaCl in bath, PCl = 22.9 ± 1.7 and PNa = 8.0 ± 1.4). It is noted that for all three ISt-St/Vm relationships obtained with NMDG-Cl in bath that Vrev ≈ ECl, whereas the reversal potentials with NaCl in bath are significantly displaced to the right of ECl, which is emphasized by the insets of expanded axis scales.
Similar acceptable nonlinear fits of Eqs. 3 and 4 were obtained with all other mutants. This is exemplified in Fig. 7 B with HEK293 cells expressing the TMEM16F-I342A, mutated at a site outside the pore region, and in Fig. 7 C with TMEM16F-R636E, mutated in the pore region which significantly altered the anion selectivity ratios. Table 2 collects the results of the analysis of all IInst/Vm relationships generated by the above protocols. There was no systematic variation of PNa/PCl between the groups, indicating that the ratio is not affected by the mutations introduced here. As shown Fig. 8, cells with large PCl also exhibited large PNa. The cross correlation coefficient obtained by a least squares fitting to all data was R2 = 0.903, with a linear relationship between the two permeabilities given by PNa = 0.334 × PCl (±SD = 0.028; n = 15).
Table 2.
Cl− and Na+ permeabilities obtained from GHK fits to instantaneous current-voltage relationships of 15 HEK293 cells
| Cell type | NMDG-Cl in bath | NaCl in bath | PNa/PCl | PCl−NMDG-Cl/PCl−NaCl | ||||
| PCl | ±SD | PCl | ±SD | PNa | ±SD | |||
| 10−14 cm3/s/pF | 10−14 cm3/s/pF | 10−14 cm3/s/pF | 10−14 cm3/s/pF | 10−14 cm3/s/pF | 10−14 cm3/s/pF | |||
| WT | 30.4 | 1.9 | 29.6 | 1.2 | 7.1 | 0.6 | 0.24 | 1.03 |
| WT | 12.9 | 0.6 | 31.7 | 1.0 | 9.7 | 0.5 | 0.31 | 0.41 |
| I342A | 26.8 | 0.8 | 55.6 | 2.0 | 14.7 | 1.7 | 0.26 | 0.48 |
| I342A | 19.8 | 0.8 | 93.8 | 4.2 | 13.3 | 3.6 | 0.14 | 0.21 |
| I342A | 138.5 | 15.0 | 230.2 | 1.6 | 77.9 | 1.4 | 0.34 | 0.60 |
| I342A | 87.2 | 6.9 | 127.6 | 2.4 | 67.7 | 1.9 | 0.53 | 0.68 |
| K616E | 204.1 | 16.0 | 176.8 | 12.7 | 45.8 | 10.6 | 0.26 | 1.15 |
| K616E | 45.7 | 4.7 | 137.7 | 7.7 | 30.6 | 6.4 | 0.22 | 0.33 |
| K616E | 12.8 | 1.0 | 23.4 | 2.5 | 12.7 | 2.1 | 0.54 | 0.55 |
| R592E | 10.6 | 3.4 | 12.7 | 2.3 | 9.7 | 2.0 | 0.76 | 0.83 |
| R592E | 42.4 | 1.3 | 72.7 | 3.5 | 11.2 | 3.0 | 0.15 | 0.58 |
| R636E | 16.2 | 1.7 | 27.3 | 1.8 | 3.1 | 1.5 | 0.11 | 0.59 |
| R636E | 90.0 | 2.4 | 130.4 | 1.2 | 69.5 | 1.0 | 0.53 | 0.69 |
| R636E | 16.9 | 1.5 | 22.9 | 1.7 | 8.0 | 1.4 | 0.35 | 0.74 |
| R636E | 150.0 | 8.5 | 232.0 | 4.6 | 79.9 | 3.8 | 0.34 | 0.65 |
As explained in Eq. 4 (compare with Fig. 7, A–C). The error of each estimate is indicated by the SD of the fit given by the Origin fitting routine (OriginLab).
Figure 8.

Relationship between PCl and PNa of HEK293 cells expressing WT and engineered TMEM16F constructs. The permeabilities were obtained as described in the legend of Fig. 7. For expressing permeability in the conventional units of cm/s, the values given in Table 2 have been recalculated using the empirical approximation that 106 pF corresponds to a membrane area of 1 cm2. The linear fit to all data is given by PNa = 0.334 × PCl (±SD = 0.028), R2 = 0.903, and n = 15.
Comparing whole-cell Cl− permeabilities obtained with NMDG+ and Na+ in bath showed that in 13 of the 15 cells studied, PCl increased significantly by replacing NMDG+ with Na+. This result was independent of the expressed channel construct. The mean ratio (±SEM) of PCl with NMDG+ outside and PCl with Na+ outside was 0.64 (±0.07; Table 2).
DISCUSSION
The current carried by TMEM16F
Mutations in genes of the subfamily among the human TMEM16 proteins (TMEM16E and TMEM16F) have been associated with gnathodiaphyseal dystrophy (Katoh and Katoh, 2004; Tsutsumi et al., 2005) and Scott syndrome (Suzuki et al., 2010; Castoldi et al., 2011), respectively, and TMEM16F has been shown to be necessary for the Ca2+-dependent phospholipid scramblase activity (Suzuki et al., 2010; Yang et al., 2012). The literature about the putative function of TMEM16F as an ion channel is contradictive. Caputo et al. (2008) found that siRNA knockdown of TMEM16F did not affect the anion current in their halide-sensitive YFP assay, and Duran et al. (2012) recently published that TMEM16F is an intracellular protein. In contrast, although not characterized in detail, other studies have suggested that TMEM16F produces Ca2+-sensitive currents in FRT and HeLa cells (Schreiber et al., 2010; Kunzelmann et al., 2012). To complete the confusion, it has been published that TMEM16F forms a Ca2+-activated nonselective cation channel (Yang et al., 2012). In the present study, we characterized the electrophysiological properties of the TMEM16F-associated anion currents in WT TMEM16F and in constructs mutated in the putative pore region. Moreover, we investigated the localization of TMEM16F and the relation between PNa and PCl after activation of TMEM16F.
TMEM16F localizes to the surface membrane
Although we noted relatively strong mTMEM16F staining of intracellular organelles, we also observed distinct mTMEM16F localization to the plasma membrane in HEK293 cells (Figs. 2 and 4). This is consistent with previous localization studies of TMEM16 family members including TMEM16A, TMEM16B, TMEM16E, and TMEM16G (Bera et al., 2004; West et al., 2004; Rock et al., 2008; Schroeder et al., 2008; Yang et al., 2008).
Ca2+ simultaneously activates with delay voltage- and time-dependent anion and cation currents in mTMEM16F-expressing cells
We have shown that overexpression of mTMEM16F in HEK293 cells results in [Ca2+]i-regulated Cl− currents and a significant cation conductance. Furthermore, the observed anion activity shows significantly different characteristics to what has been reported for both TMEM16A and TMEM16B as well as native CaCCs in various tissues (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008; Pifferi et al., 2009; Stöhr et al., 2009; Schreiber et al., 2010). The current was not activated immediately after establishment of whole-cell configuration with high [Ca2+] in the pipette solution. However, after a delay of several minutes, a transiently activated and strongly outwardly rectifying current that peaked after a mean of ∼9 min was observed. In the study by Schreiber et al. (2010), a slowly activating current was reported for TMEM16F-expressing FRT cells, which has little resemblance with our observations, perhaps because their observation period was no more than 8 min. However, in a recent study by Shimizu et al. (2013), treatment with ionomycin induced a delayed and slowly activating current, which is similar to our findings. In our study, mTMEM16F was characterized by a strong outwardly rectifying current in asymmetrical solutions, a time-dependent activation at depolarizing potentials (positive to 40 mV), and a time-dependent deactivation at hyperpolarizing potentials (Vhold = −20 mV), which resulted in large steady-state outward currents and relatively very small steady-state inward currents, similar to what was found for TMEM16B (Pifferi et al., 2009; Stöhr et al., 2009). The very slow activation of Ca2+ is not yet explained. We considered that it was caused by delayed trafficking, for example if recruitment of TMEM16F to the plasma membrane depends on a slow Ca2+ activation of a second messenger. This hypothesis was tested; however, under the conditions of our experiments, we did not find support for this hypothesis (Fig. 4 E). Shimizu et al. (2013) investigated more specifically whether CAMKII has a role in this delay, but they failed to see any effect using a CamKII inhibitor (KN93), and the very slow activation is yet to be explained.
Ca2+ activation kinetics of mTMEM16F
We found that the delayed ICl,Ca activations in mTMEM16F-expressing cells depend on [Ca2+]i in the micromolar range (Fig. 4, A–D). The EC50 for Ca2+ is ∼76 µM, and when measured at depolarizing potentials, the Hill coefficient was calculated to 2, in line with Yang et al. (2012), suggesting cooperative Ca2+ activation (Fig. 4 C). In comparison, an EC50 of 0.4 µM and 3.3 µM with an nH of 2 and >2 has been reported for TMEM16A and TMEM16B, respectively (Yang et al., 2008; Pifferi et al., 2009; Scudieri et al., 2012). For TMEM16F, it was recently reported that the EC50 value was 3.4 µM (Yang et al., 2012) and 9.6 µM (Shimizu et al., 2013). Both groups have used EGTA-buffered solutions and calculated the free Ca2+ concentration using Cabuf software. Using the same software, we find an EC50 value of 12 µM Ca2+. Because the Ca2+ concentrations of use are well outside the dynamic range for the EGTA buffers (Dweck et al., 2005), we have measured the actual Ca2+ concentration of the pipette solution (see Materials and methods), finding a significantly higher free Ca2+ concentration, shifting the dose–response curve rightwards. Thus, the difference in Ca2+ dependence between the three groups most likely reflects methodical difference in [Ca2+]free estimation rather than significantly different affinities. However, the Ca2+ dependence is dramatically different from the results for TMEM16A (compare also with Fig. 3 C).
The slope of the I/V relationship of the strongly outwardly rectifying TMEM16F Cl− current did not approach linearity at increasing intracellular Ca2+ concentrations (Fig. 4 A), as seen for endogenous CaCC (Kuruma and Hartzell, 2000), TMEM16A (Schroeder et al., 2008), and TMEM16B (Pifferi et al., 2009) currents. Contrary to what has been reported for TMEM16A (Schroeder et al., 2008; Yang et al., 2008) and TMEM16B (Pifferi et al., 2009), the TMEM16F-generated current shows time-dependent activation at depolarizing potentials even at a saturating free [Ca2+]i, (Fig. 4, A and D). This would indicate that full activation of the mTMEM16F-generated current is not governed solely by [Ca2+]i.
The Na+ ion permeability of TMEM16F-expressing cells
Surprisingly, replacing extracellular NMDG+ with Na+ stimulated the Cl− permeability in a large majority of studied TMEM16F constructs (Table 2). The mechanism of the interaction between extracellular Na+ and the Cl− current is not known. Our study also showed that TMEM16F-expressing HEK293 cells conduct a significant Na+ current and that cells expressing the mutated TMEM16F exhibited a significant Na+ permeability as well. Moreover, within the range of PCl from ∼1 × 10−7 to 25 × 10−7 cm/s, the two permeabilities were correlated according to PNa = 0.334 (±0.028) × PCl (Fig. 8). In the face of the anion selectivity series obtained, it is unlikely that Na+ shares the TM pore with Cl−. Alternative interpretations would be that trafficking of Na+ and Cl− channels to the surface membrane is coupled or, perhaps, that the TMEM16F molecule expresses parallel Cl− and Na+ selective pores. In connection to this, it is interesting that TMEM16F has been described both as a Ca2+-activated nonselective cation channel with PNa/PCl = 6.8 (Yang et al., 2012) and as a Ca2+-activated selective anion channel (Shimizu et al., 2013). Although Yang et al. (2012) was unable to observe any shift in reversal potential upon shifting from Cl− to the larger anion Mes, Shimizu et al. (2013) did not find any change when shifting Na+ cations to the bigger NMDG ions. In contrast, we observed a significant shift in Erev, both when substituting Na+ with NMDG and when substituting Cl− with aspartate. The background of this difference is not clear, but an important point is that mutations in TMEM16F have been observed to change the permeability profile for anions (K616E and R636E; this study) and the measured cation current (Q559K; Yang et al., 2012), indicating that the observed currents are carried by TMEM16F. However, it is unclear whether the decrease in PNa/PCl observed for the Q559K mutation (Yang et al., 2012) actually is a decrease in PNa or potentially could relate to an increased Cl− permeability. Thus, it is so far unclear whether the selectivity filter for cations is to be found in the TM5–6 region as seen for the anion selectivity filter (Fig. 6; Yang et al., 2008). This will have to be further tested in the future, preferably by single channel analysis.
Anion selectivity of mTMEM16F-WT and mTMEM16F mutants: TMEM16F is a functional anion channel
The anion substitution experiments showed that the ICl,Ca of mTMEM16F follows the Eisenman type 1 selectivity sequence, PSCN > PI > PBr > PCl > PAsp, indicating weak interactions of the permeating ion with the channel pore, which has been reported also for CaCCs and volume-regulated anion channels (Nilius and Droogmans, 2003), TMEM16A (Yang et al., 2008), and TMEM16B (Pifferi et al., 2009).
To investigate whether the TMEM16F protein constitutes a channel for halide ion currents, we performed mutations of amino acids in the putative pore region between TM5 and TM6. Within this region, mutations in TMEM16A of the positively charged amino acids R621, K645, and K668 to the negatively charged glutamic acid resulted in diminished selectivity ratios relative to Cl− for several anions (Yang et al., 2008). We introduced the following single amino acid mutations, R592E, K616E, and R636E, replacing the positively charged arginine-592, lysine-616, and arginine-636 with the negatively charged glutamic acid. Although these mutants all trafficked to the membrane, the mutant R592E reduced the current strongly, whereas the maximal current was not affected in the K616E and R636 mutants (Fig. 6 A). Notably, we found a reduced PI/PCl in K616E when compared with mTMEM16F-WT (Table 1) and an anion selectivity sequence changed to PSCN = PI = PBr = PCl > PAsp in the R636E mutant (Fig. 6 C). The above results suggest that these residues are involved in the anion selectivity of the pore. In contrast, the current density and anion selectivity sequence was unaffected in the control mutant I342A, in which the amino acid substitution was located outside the putative pore region (Fig. 6, B and E). Collectively, these findings provide the evidence that TMEM16F is a pore-forming subunit of an anion channel.
Conclusion
Mouse TMEM16F heterologously expressed in HEK293 cells localizes to the plasma membrane and induces an ICl,Ca with properties different from canonical ICl,Ca and from Ca2+-activated anion currents associated with TMEM16A and TMEM16B. Furthermore, a significant Na+ current was activated, and the two permeabilities were correlated according to PNa = 0.3 PCl.
Acknowledgments
Søren L. Johansen, Arne Nielsen and Birthe J. Hansen are acknowledged for excellent technical assistance.
The present work was supported by the Danish Council for Independent Research/Natural Sciences (grants 09-064182 and 10-085373), Lundbeck Foundation (J Nr R32-A3102), and Carlsberg Foundation (grant 2008-01-0639).
Edward N. Pugh Jr. served as editor.
Footnotes
Abbreviations used in this paper:
- CaCC
- Ca2+-activated Cl− channel
- EATC
- Ehrlich ascites tumor cell
- RT
- reverse transcription
- TM
- transmembrane
- WGA
- wheat germ agglutinin
References
- Almaça J., Tian Y., Aldehni F., Ousingsawat J., Kongsuphol P., Rock J.R., Harfe B.D., Schreiber R., Kunzelmann K. 2009. TMEM16 proteins produce volume-regulated chloride currents that are reduced in mice lacking TMEM16A. J. Biol. Chem. 284:28571–28578 10.1074/jbc.M109.010074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- André S., Boukhaddaoui H., Campo B., Al-Jumaily M., Mayeux V., Greuet D., Valmier J., Scamps F. 2003. Axotomy-induced expression of calcium-activated chloride current in subpopulations of mouse dorsal root ganglion neurons. J. Neurophysiol. 90:3764–3773 10.1152/jn.00449.2003 [DOI] [PubMed] [Google Scholar]
- Angermann J.E., Sanguinetti A.R., Kenyon J.L., Leblanc N., Greenwood I.A. 2006. Mechanism of the inhibition of Ca2+-activated Cl− currents by phosphorylation in pulmonary arterial smooth muscle cells. J. Gen. Physiol. 128:73–87 10.1085/jgp.200609507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader C.R., Bertrand D., Schwartz E.A. 1982. Voltage-activated and calcium-activated currents studied in solitary rod inner segments from the salamander retina. J. Physiol. 331:253–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barish M.E. 1983. A transient calcium-dependent chloride current in the immature Xenopus oocyte. J. Physiol. 342:309–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera T.K., Das S., Maeda H., Beers R., Wolfgang C.D., Kumar V., Hahn Y., Lee B., Pastan I. 2004. NGEP, a gene encoding a membrane protein detected only in prostate cancer and normal prostate. Proc. Natl. Acad. Sci. USA. 101:3059–3064 10.1073/pnas.0308746101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc V., Marlow G., Boycott K.M., Saleki K., Inoue H., Kroon J., Itakura M., Robitaille Y., Parent L., Baas F., et al. 2010. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am. J. Hum. Genet. 86:213–221 10.1016/j.ajhg.2009.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., Galietta L.J. 2008. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 322:590–594 10.1126/science.1163518 [DOI] [PubMed] [Google Scholar]
- Castoldi E., Collins P.W., Williamson P.L., Bevers E.M. 2011. Compound heterozygosity for 2 novel TMEM16F mutations in a patient with Scott syndrome. Blood. 117:4399–4400 10.1182/blood-2011-01-332502 [DOI] [PubMed] [Google Scholar]
- Das S., Hahn Y., Walker D.A., Nagata S., Willingham M.C., Peehl D.M., Bera T.K., Lee B., Pastan I. 2008. Topology of NGEP, a prostate-specific cell:cell junction protein widely expressed in many cancers of different grade level. Cancer Res. 68:6306–6312 10.1158/0008-5472.CAN-08-0870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran C., Hartzell H.C. 2011. Physiological roles and diseases of Tmem16/Anoctamin proteins: are they all chloride channels? Acta Pharmacol. Sin. 32:685–692 10.1038/aps.2011.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran C., Qu Z., Osunkoya A.O., Cui Y., Hartzell H.C. 2012. ANOs 3-7 in the anoctamin/Tmem16 Cl- channel family are intracellular proteins. Am. J. Physiol. Cell Physiol. 302:C482–C493 10.1152/ajpcell.00140.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweck D., Reyes-Alfonso A., Jr, Potter J.D. 2005. Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347:303–315 10.1016/j.ab.2005.09.025 [DOI] [PubMed] [Google Scholar]
- Eggermont J. 2004. Calcium-activated chloride channels: (un)known, (un)loved? Proc. Am. Thorac. Soc. 1:22–27 10.1513/pats.2306010 [DOI] [PubMed] [Google Scholar]
- Fallah G., Römer T., Detro-Dassen S., Braam U., Markwardt F., Schmalzing G. 2011. TMEM16A(a)/anoctamin-1 shares a homodimeric architecture with CLC chloride channels. Mol. Cell. Proteomics. 10:M110.004697 10.1074/mcp.M110.004697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A., Vaziri Sani F., Rock J.R., Hallberg K., Iribarne D., Harfe B.D., Linde A. 2009. Expression patterns of the Tmem16 gene family during cephalic development in the mouse. Gene Expr. Patterns. 9:178–191 10.1016/j.gep.2008.11.002 [DOI] [PubMed] [Google Scholar]
- Guo D., Young L., Patel C., Jiao Z., Wu Y., Liu T., Kowey P.R., Yan G.X. 2008. Calcium-activated chloride current contributes to action potential alternations in left ventricular hypertrophy rabbit. Am. J. Physiol. Heart Circ. Physiol. 295:H97–H104 10.1152/ajpheart.01032.2007 [DOI] [PubMed] [Google Scholar]
- Hartzell C., Putzier I., Arreola J. 2005. Calcium-activated chloride channels. Annu. Rev. Physiol. 67:719–758 10.1146/annurev.physiol.67.032003.154341 [DOI] [PubMed] [Google Scholar]
- Hartzell H.C., Yu K., Xiao Q., Chien L.T., Qu Z. 2009. Anoctamin/TMEM16 family members are Ca2+-activated Cl- channels. J. Physiol. 587:2127–2139 10.1113/jphysiol.2008.163709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E.K., Lambert I.H., Pedersen S.F. 2009. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89:193–277 10.1152/physrev.00037.2007 [DOI] [PubMed] [Google Scholar]
- Hwang S.J., Blair P.J., Britton F.C., O’Driscoll K.E., Hennig G., Bayguinov Y.R., Rock J.R., Harfe B.D., Sanders K.M., Ward S.M. 2009. Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J. Physiol. 587:4887–4904 10.1113/jphysiol.2009.176198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch T.J., Stein V., Weinreich F., Zdebik A.A. 2002. Molecular structure and physiological function of chloride channels. Physiol. Rev. 82:503–568 [DOI] [PubMed] [Google Scholar]
- Katoh M., Katoh M. 2004. GDD1 is identical to TMEM16E, a member of the TMEM16 family. Am. J. Hum. Genet. 75:927–928 10.1086/425341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausen T.K., Bergdahl A., Hougaard C., Christophersen P., Pedersen S.F., Hoffmann E.K. 2007. Cell cycle-dependent activity of the volume- and Ca2+-activated anion currents in Ehrlich lettre ascites cells. J. Cell. Physiol. 210:831–842 10.1002/jcp.20918 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Milenkovic V.M., Spitzner M., Soria R.B., Schreiber R. 2007. Calcium-dependent chloride conductance in epithelia: is there a contribution by Bestrophin? Pflugers Arch. 454:879–889 10.1007/s00424-007-0245-z [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Kongsuphol P., Aldehni F., Tian Y., Ousingsawat J., Warth R., Schreiber R. 2009. Bestrophin and TMEM16-Ca(2+) activated Cl(-) channels with different functions. Cell Calcium. 46:233–241 10.1016/j.ceca.2009.09.003 [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Schreiber R., Kmit A., Jantarajit W., Martins J.R., Faria D., Kongsuphol P., Ousingsawat J., Tian Y. 2012. Expression and function of epithelial anoctamins. Exp. Physiol. 97:184–192 [DOI] [PubMed] [Google Scholar]
- Kuruma A., Hartzell H.C. 2000. Bimodal control of a Ca2+-activated Cl− channel by different Ca2+ signals. J. Gen. Physiol. 115:59–80 10.1085/jgp.115.1.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde M.R., Kelly M.E., Barnes S. 2008. Calcium-activated chloride channels in the retina. Channels (Austin). 2:252–260 10.4161/chan.2.4.6704 [DOI] [PubMed] [Google Scholar]
- Larsen E.H. 2011. Reconciling the Krogh and Ussing interpretations of epithelial chloride transport - presenting a novel hypothesis for the physiological significance of the passive cellular chloride uptake. Acta Physiol. (Oxf.). 202:435–464 10.1111/j.1748-1716.2010.02239.x [DOI] [PubMed] [Google Scholar]
- Martins J.R., Faria D., Kongsuphol P., Reisch B., Schreiber R., Kunzelmann K. 2011. Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc. Natl. Acad. Sci. USA. 108:18168–18172 10.1073/pnas.1108094108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews H.R., Reisert J. 2003. Calcium, the two-faced messenger of olfactory transduction and adaptation. Curr. Opin. Neurobiol. 13:469–475 10.1016/S0959-4388(03)00097-7 [DOI] [PubMed] [Google Scholar]
- Miledi R., Parker I., Sumikawa K. 1982. Properties of acetylcholine receptors translated by cat muscle mRNA in Xenopus oocytes. EMBO J. 1:1307–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic V.M., Brockmann M., Stöhr H., Weber B.H., Strauss O. 2010. Evolution and functional divergence of the anoctamin family of membrane proteins. BMC Evol. Biol. 10:319 10.1186/1471-2148-10-319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B., Droogmans G. 2003. Amazing chloride channels: an overview. Acta Physiol. Scand. 177:119–147 10.1046/j.1365-201X.2003.01060.x [DOI] [PubMed] [Google Scholar]
- Nilius B., Prenen J., Voets T., Van den Bremt K., Eggermont J., Droogmans G. 1997. Kinetic and pharmacological properties of the calcium-activated chloride-current in macrovascular endothelial cells. Cell Calcium. 22:53–63 10.1016/S0143-4160(97)90089-0 [DOI] [PubMed] [Google Scholar]
- Nurden A., Nurden P. 2011. Advances in our understanding of the molecular basis of disorders of platelet function. J. Thromb. Haemost. 9:76–91 10.1111/j.1538-7836.2011.04274.x [DOI] [PubMed] [Google Scholar]
- Ousingsawat J., Martins J.R., Schreiber R., Rock J.R., Harfe B.D., Kunzelmann K. 2009. Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J. Biol. Chem. 284:28698–28703 10.1074/jbc.M109.012120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen S.F., Pedersen S., Lambert I.H., Hoffmann E.K. 1998a. P2 receptor-mediated signal transduction in Ehrlich ascites tumor cells. Biochim. Biophys. Acta. 1374:94–106 10.1016/S0005-2736(98)00123-0 [DOI] [PubMed] [Google Scholar]
- Pedersen S.F., Prenen J., Droogmans G., Hoffmann E.K., Nilius B. 1998b. Separate swelling- and Ca2+-activated anion currents in Ehrlich ascites tumor cells. J. Membr. Biol. 163:97–110 10.1007/s002329900374 [DOI] [PubMed] [Google Scholar]
- Pifferi S., Dibattista M., Menini A. 2009. TMEM16B induces chloride currents activated by calcium in mammalian cells. Pflugers Arch. 458:1023–1038 10.1007/s00424-009-0684-9 [DOI] [PubMed] [Google Scholar]
- Pifferi S., Cenedese V., Menini A. 2012. Anoctamin 2/TMEM16B: a calcium-activated chloride channel in olfactory transduction. Exp. Physiol. 97:193–199 [DOI] [PubMed] [Google Scholar]
- Rasche S., Toetter B., Adler J., Tschapek A., Doerner J.F., Kurtenbach S., Hatt H., Meyer H., Warscheid B., Neuhaus E.M. 2010. Tmem16b is specifically expressed in the cilia of olfactory sensory neurons. Chem. Senses. 35:239–245 10.1093/chemse/bjq007 [DOI] [PubMed] [Google Scholar]
- Rock J.R., Harfe B.D. 2008. Expression of TMEM16 paralogs during murine embryogenesis. Dev. Dyn. 237:2566–2574 10.1002/dvdy.21676 [DOI] [PubMed] [Google Scholar]
- Rock J.R., Futtner C.R., Harfe B.D. 2008. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev. Biol. 321:141–149 10.1016/j.ydbio.2008.06.009 [DOI] [PubMed] [Google Scholar]
- Rock J.R., O’Neal W.K., Gabriel S.E., Randell S.H., Harfe B.D., Boucher R.C., Grubb B.R. 2009. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl- secretory channel in mouse airways. J. Biol. Chem. 284:14875–14880 10.1074/jbc.C109.000869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R., Uliyakina I., Kongsuphol P., Warth R., Mirza M., Martins J.R., Kunzelmann K. 2010. Expression and function of epithelial anoctamins. J. Biol. Chem. 285:7838–7845 10.1074/jbc.M109.065367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder B.C., Cheng T., Jan Y.N., Jan L.Y. 2008. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 134:1019–1029 10.1016/j.cell.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudieri P., Sondo E., Ferrera L., Galietta L.J. 2012. The anoctamin family: TMEM16A and TMEM16B as calcium-activated chloride channels. Exp. Physiol. 97:177–183 10.1113/expphysiol.2011.058198 [DOI] [PubMed] [Google Scholar]
- Sheridan J.T., Worthington E.N., Yu K., Gabriel S.E., Hartzell H.C., Tarran R. 2011. Characterization of the oligomeric structure of the Ca(2+)-activated Cl- channel Ano1/TMEM16A. J. Biol. Chem. 286:1381–1388 10.1074/jbc.M110.174847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T., Iehara T., Sato K., Fujii T., Sakai H., Okada Y. 2013. TMEM16F is a component of a Ca2+-activated Cl- channel but not a volume-sensitive outwardly rectifying Cl- channel. Am. J. Physiol. Cell Physiol. 2013:111–121 [DOI] [PubMed] [Google Scholar]
- Sokalski T., Zwickl T., Bakker E., Pretsch E. 1999. Lowering the detection limit of solvent polymeric ion-selective electrodes. 1. Modeling the influence of steady-state ion fluxes. Anal. Chem. 71:1204–1209 10.1021/ac980944v [DOI] [Google Scholar]
- Stöhr H., Heisig J.B., Benz P.M., Schöberl S., Milenkovic V.M., Strauss O., Aartsen W.M., Wijnholds J., Weber B.H., Schulz H.L. 2009. TMEM16B, a novel protein with calcium-dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J. Neurosci. 29:6809–6818 10.1523/JNEUROSCI.5546-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J., Umeda M., Sims P.J., Nagata S. 2010. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 468:834–838 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- Tsutsumi S., Inoue H., Sakamoto Y., Mizuta K., Kamata N., Itakura M. 2005. Molecular cloning and characterization of the murine gnathodiaphyseal dysplasia gene GDD1. Biochem. Biophys. Res. Commun. 331:1099–1106 10.1016/j.bbrc.2005.03.226 [DOI] [PubMed] [Google Scholar]
- Verkman A.S., Galietta L.J. 2009. Chloride channels as drug targets. Nat. Rev. Drug Discov. 8:153–171 10.1038/nrd2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West R.B., Corless C.L., Chen X., Rubin B.P., Subramanian S., Montgomery K., Zhu S., Ball C.A., Nielsen T.O., Patel R., et al. 2004. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am. J. Pathol. 165:107–113 10.1016/S0002-9440(10)63279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Jin T., Cheng T., Jan Y.N., Jan L.Y. 2011. Scan: A novel small-conductance Ca2+-activated non-selective cation channel encoded by TMEM16F. Biophys. J. 100:259a 10.1016/j.bpj.2010.12.1631 [DOI] [Google Scholar]
- Yang H., Kim A., David T., Palmer D., Jin T., Tien J., Huang F., Cheng T., Coughlin S.R., Jan Y.N., Jan L.Y. 2012. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 151:111–122 10.1016/j.cell.2012.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.D., Cho H., Koo J.Y., Tak M.H., Cho Y., Shim W.S., Park S.P., Lee J., Lee B., Kim B.M., et al. 2008. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 455:1210–1215 10.1038/nature07313 [DOI] [PubMed] [Google Scholar]
- Yu K., Duran C., Qu Z., Cui Y.Y., Hartzell H.C. 2012. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ. Res. 110:990–999 10.1161/CIRCRESAHA.112.264440 [DOI] [PMC free article] [PubMed] [Google Scholar]