

Abstract
Yersinia pestis, the causative agent of plague, uses a type III secretion system (T3SS) to inject cytotoxic Yop proteins directly into the cytosol of mammalian host cells. The T3SS can also be activated in vitro at 37°C in the absence of calcium. The chromosomal gene rfaL (waaL) was recently identified as a virulence factor required for proper function of the T3SS. RfaL functions as a ligase that adds the terminal N-acetylglucosamine to the lipooligosaccharide core of Y. pestis. We previously showed that deletion of rfaL prevents secretion of Yops in vitro. Here we show that the divalent cations calcium, strontium, and magnesium can partially or fully rescue Yop secretion in vitro, indicating that the secretion phenotype of the rfaL mutant may be due to structural changes in the outer membrane and the corresponding feedback inhibition on the T3SS. In support of this, we found that the defect can be overcome by deleting the regulatory gene lcrQ. Consistent with a defective T3SS, the rfaL mutant is less virulent than the wild type. We show here that the virulence defect of the mutant correlates with a decrease in both T3SS gene expression and ability to inject innate immune cells, combined with an increased sensitivity to cationic antimicrobial peptides.
INTRODUCTION
Yersinia pestis is the causative agent of bubonic, pneumonic, and septicemic plague (1). Y. pestis and the other two pathogenic Yersinia species, Y. enterocolitica and Y. pseudotuberculosis, use a type III secretion system (T3SS) as their main virulence factor to cause disease within the mammalian host (2). The T3SS is encoded by a conserved 70-kb virulence plasmid known as pCD1 in Y. pestis (3). This system is composed of approximately 25 coordinately regulated proteins that form a syringe-like complex called the injectisome (2). The base of the apparatus is a macromolecular complex that spans the inner membrane, cell wall, and outer membrane of the bacterium (4–10). The outer portion of the injectisome consists of a needle projecting from the bacterial surface, which is a polymer of a single protein called YscF (11). Y. pestis uses the injectisome to passage cytotoxic proteins, called Yops, from the bacterium to the cytosol of eukaryotic cells (12, 13). Passage into the eukaryotic cell requires a pore that is composed of three proteins: LcrV, which forms a complex on the tip of the YscF needle, and YopB and YopD, which together form a pore in the membrane of the eukaryotic cell (2, 14).
Activation of the T3SS occurs upon entry into a mammalian host. Transition to 37°C activates the expression of an AraC-like transcriptional activator, known as VirF (15, 16). This activation, along with the degradation of a small histone-like protein, YmoA, by the ClpXP/Lon proteases leads to expression of the T3SS genes (17). The presence of certain amino acids (glutamine, glutamate, aspartate, and asparagine) and serum proteins such as albumin leads to formation of the injectisome complex and secretion of early Yops, such as YopBD and LcrV (18). Contact with a host cell triggers directional translocation of late Yops (YopE, -H, -M, -T, -J, and -K; LcrQ; and YpkA) into the host cell (13, 19). Alternatively, the T3SS can be partially activated in vitro by growth at 37°C in calcium-replete medium (20, 21). Growth under these conditions leads to injectisome assembly and secretion of early Yops (22). Upon calcium depletion, growth ceases and secretion of late Yops occurs (20, 21, 23, 24). The growth cessation concomitant with Yop secretion is known as the low-calcium response (LCR) (25) and is a result of a buildup of Na+ and l-glutamic acid and a drop in culture pH (26).
Yop secretion is a highly regulated process that relies on a number of regulatory proteins. The presence of the class I regulators, YscB, SycN, TyeA, and YopN, ensures that only early Yops are secreted prior to cell contact (27–31). Deletion of any class I genes leads to the loss of calcium-dependent regulation and secretion of early and late Yops in the presence and absence of calcium, a phenotype known as “calcium blindness” (32). Another set of regulators, the class II genes (YopD, LcrH, and LcrQ), regulate Yop synthesis, and loss of any of these leads to increased synthesis of Yop proteins (33–36). It has also been shown previously that the needle protein YscF plays a role in regulating secretion of Yops (37, 38). Mutations of specific residues in the YscF protein can result in a calcium-blind phenotype (constitutive secretion) as well as a dominant negative phenotype (constitutive inhibition of secretion) (37–39).
Upon entering the mammalian host, pathogens must find a way to survive attacks made against them by the host's immune system. Y. pestis utilizes the T3SS to evade the immune system, in part by specifically targeting cells of the innate immune system for Yop injection (40). In addition to the T3SS, other virulence attributes include the ability to survive and replicate inside macrophages and to resist the bactericidal action of cationic antimicrobial peptides (CAMPs) and serum (41–44). Many Gram-negative pathogens rely on lipopolysaccharide (LPS) (also called enterotoxin) to resist the effects of serum and CAMPs (45, 46). LPS consists of a variable polysaccharide chain (O antigen) attached to a relatively conserved core oligosaccharide, which is anchored in the outer leaflet of the outer membrane by lipid A. Y. pestis does not make O antigen and therefore synthesizes a rough lipooligosaccharide (LOS) consisting of lipid A and a core oligosaccharide, which is capped by an N-acetylglucosamine (GlcNac) residue (47–50). Y. pestis LOS contributes to CAMP and serum resistance, and defects in LOS biosynthesis correlate with virulence defects (51).
In Y. pestis, RfaL (also referred to as WaaL) attaches the terminal GlcNac residue to the core oligosaccharide (48). We have previously demonstrated that a Y. pestis rfaL mutant isolated from a transposon mutagenesis screen has defective Yop secretion. This mutant displays three distinct but conflicting phenotypes: (i) the mutant is able to secrete early Yops in vitro at 37°C in the presence of calcium but is unable to secrete any Yops in the absence of calcium, (ii) the mutant is able to inject Yops into tissue culture cells, and (iii) the mutant is less virulent in a mouse model of infection (52). In this paper, we further characterize the ΔrfaL mutant to gain insight into the phenomenon underlying these interesting phenotypes.
MATERIALS AND METHODS
Bacterial strains and media.
Table S1 in the supplemental material lists the strains used in this study. Yersinia pestis KIM5, an attenuated (nonpigmented) variant of Y. pestis bv. medievalis KIM lacking the 102-kb pgm locus (53), was used as the parent strain for this work. KIM5 strains and derivatives were propagated on heart infusion agar (HIA) plates at 26°C for 2 days. Overnight cultures were grown in heart infusion broth (HIB) at 26°C. Antibiotics were added as appropriate to a final concentration of 20 μg/ml chloramphenicol, 50 μg/ml ampicillin, or 50 μg/ml kanamycin. Escherichia coli strains were propagated in Luria-Bertani broth or agar, supplemented with 20 μg/ml chloramphenicol, 100 μg/ml ampicillin, or 25 μg/ml kanamycin, at 37°C.
Strain construction.
Tables S1 and S2 in the supplemental material list the plasmids and primers, respectively, used in this work. The ΔrfaL strain, CC003, was constructed using λ-Red recombination (54). The cat cassette from pKD13 was amplified using primers y3762KO-P1 and y3762KO-P2 containing ∼30 nucleotides of homologous DNA upstream and downstream of rfaL, respectively (52). The resulting PCR product was electroporated into Y. pestis KIM5(pKD46) induced with 20 mM arabinose. The transformations were plated onto HIA plates containing chloramphenicol. The resulting colonies were screened for proper recombination of the cat cassette using primers y3762up and y3762down, which amplify a region that spans 500 bp upstream and downstream of the insertion site, respectively. Correct mutants were designated CC001. pKD46 was cured from CC001 by electroporating pMM1 into this strain. pMM1 was cured from CC001 by counterselection on 5% sucrose. pCP20 was then introduced in order to remove the cat cassette, and then pCP20 was cured by growing the cells at 37°C. Loss of the cat cassette was confirmed using y3762up and y3762down. The resulting strain, CC003, contains an unmarked deletion of rfaL.
The ΔyscU mutant, MEL20, was also constructed via λ-Red recombination (54). The cat cassette from pKD13 was amplified using the primers yscU.P1 and yscU.P2. The resulting PCR product was electroporated into Y. pestis KIM5 expressing the Red genes on pMM1. The transformations were plated on HIA plates containing chloramphenicol. Proper insertion of the cat cassette was confirmed using the primers yscU.up and yscU.down. Correct mutants were designated MEL17. pMM1 was cured by counterselection on HIA containing 5% sucrose, and the resulting unmarked ΔyscU mutant was renamed MEL20.
The ΔyopN mutant, MEL18, was constructed via allelic exchange, as previously described, using the plasmid pMM61 to insert a stop codon and a +1 frameshift immediately following the 27th codon (55). The plasmid was constructed using primers yopN.ko.EcoI, yopN.ko.Eco2, yopN.ko.SalI, and yopN.ko.XbaI. The resulting PCR products were digested and ligated directly into pLC28 (55). pMM61 was transformed into Y. pestis KIM5, and mutants were screened for chloramphenicol resistance. The chloramphenicol-resistant merodiploids were resolved by counterselection on HIA plus 5% sucrose. The ΔrfaL ΔyopN mutant, CC004, was constructed via allelic exchange by introducing pMM61 into CC003. The plasmid backbone was cured as described for MEL18. Insertion of the stop codon and the +1 frameshift was confirmed by amplifying yopN using the PCR primers yopN.ko.SalI and yopN.ko.XbaI. The resulting PCR product was digested with EcoRI to confirm the presence of the EcoRI site. Complementation of the ΔrfaL mutant was performed by electroporating pDONR::rfaL into the mutant (52).
The ΔrfaL ΔlcrQ mutant, CC006, was made by introducing a stop codon followed by a +1 frameshift mutation after the start codon of lcrQ. This was done by electroporating pMM54 (56) into CC003 and selecting by growth on HIA with chloramphenicol. The plasmid was cured by growing the mutants on HIA with 5% sucrose. Insertion of the stop codon and a +1 frameshift was confirmed by amplifying lcrQ by PCR using the primers lcrQKO-SalI and lcrQKO-PstI (56). The resulting PCR product was digested with EcoRI to confirm the introduction of the EcoRI site.
YscF point mutants were generated as follows. AH020 and AH021 were created by electroporating pCD1 (ΔyopE-sycE::kan ΔyscF) from KIM5-3001.P61 (ΔyscF) (37) into KIM5 and CC003, respectively. Mutants were selected by plating on HIA with kanamycin. Next, pBCSK-YscF (D46C) or pBSCK-YscF (D46A) (37) was electroporated into AH020 and AH021 and selected by growth on HIA with chloramphenicol.
Secretion assays.
Bacteria were grown overnight in HIB and then diluted 1:20 into fresh Dulbecco's modified Eagle's medium (DMEM) (Cellgro catalog no. 15-012-CV) supplemented with 10% fetal bovine serum (FBS) and 2 mM Glutamax (Invitrogen catalog no. 35050). Calcium was chelated with addition of 5 mM EGTA in order to induce secretion. Cultures were incubated for 2 to 2.5 h at 26°C and then switched to 37°C for 3 h. Fractionation, protein precipitation, and immunoblotting have been previously described (40). For the divalent cation rescue assays, overnight cultures were diluted 1:20 into DMEM containing 10% FBS, 2 mM Glutamax, and 25 mM HEPES. Medium was treated with 5 mM EGTA and then supplemented with 5 mM CaCl2, ZnCl2, MgSO4, or SrCl2.
YscF cross-linking assay.
Strains were grown overnight in HIB at 26°C. Bacteria at 8 × 107 CFU were inoculated into 4 ml of TMH (57) with or without 2.5 mM CaCl2. Cultures were incubated for 2.5 h at 26°C and then shifted to 37°C for 3 h to induce secretion. An aliquot of cells from each culture was removed for cross-linking, and the remaining culture was subjected to fractionation for standard secretion assays. For cross-linking, the optical density at 600 nm (OD600) of each sample was measured and 2.0 × 108 CFU was centrifuged at 8,000 × g for 5 min at 4°C. The supernatants were removed, and the pellets were suspended in 25 mM HEPES, pH 8.0, with or without 2.5 mM CaCl2. The suspended pellets were split into two equal-volume fractions. The two fractions were treated with either 1 mM bis(sulfosuccinimidyl)suberate (BS3; Pierce) or water, and then fractions were incubated at 37°C for 30 min. To quench the reaction, 5 mM Tris-HCl (pH 8.0) was added, followed by incubation at room temperature for 15 min. The fractions were centrifuged at 12,200 × g for 5 min. Supernatants were aspirated, and the pellets were vigorously suspended in SDS-sample buffer and then analyzed by SDS-PAGE and immunoblotting.
Pla activation assay.
Strains were cultured in brain heart infusion (BHI) broth overnight at 26°C, back diluted to 8 × 107 CFU/ml, and cultured for 6 h at 26°C or 37°C in the presence or absence of 2.5 mM CaCl2. Plasminogen (Pla) activity was measured as previously described (58). Briefly, bacteria (8 × 106 CFU) were incubated with purified human Glu-plasminogen (Hematologic Technologies) (4 μg) and the chromogenic substrate d-AFK-ANSNH-iC4H9-2HBr (SN5; Hematologic Technologies) (50 μM) in a total volume of 200 μl phosphate-buffered saline (PBS). Reaction mixtures were incubated in triplicate for 3 h at 37°C, and the absorbance at 460 nm was measured every 10 to 11 min in a Molecular Devices SpectraMax M5 fluorescence microplate reader. To evaluate Pla expression levels, the same Y. pestis cultures used in the plasminogen activation assays were incubated with lysozyme (50 μg/ml) for 30 min on ice followed by sonication. Lysate protein concentration was determined by the Bradford assay. Equal amounts of protein (25 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes for immunoblot analysis. Western blotting assays were performed using a rat polyclonal antibody raised against the mature form of Pla.
Pla antibody production.
The gene encoding the mature form of Pla (lacking the N-terminal signal sequence) was amplified by PCR from Y. pestis strain CO92 and cloned into the SphI-HindIII sites of plasmid pQE30 to create pWL223, which contains an N-terminal 6×His tag, as described previously (58). Recombinant His-tagged Pla was purified from the BL21 strain of E. coli also carrying the lac repressor plasmid pREP4 (Invitrogen) under denaturing conditions according to the methods outlined in the QiaExpressionist handbook (Qiagen). Purified Pla protein was sent to Covance Inc. for production of rat polyclonal antibodies.
Mouse survival analysis.
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The work was approved by the Institutional Animal Care and Use Committees at Indiana University. Bacterial strains were grown overnight in HIB at 26°C and then diluted to the appropriate concentration using PBS. Six- to 7-week-old female C57BL/6NHsd inbred mice were obtained from Harlan Laboratories (Indianapolis, IN). For survival analysis, mice were anesthetized with isoflurane and then infected via retro-orbital injection of KIM5, CC003, or CC003(pDONR::rfaL) at a dose of ∼102 CFU, with five mice per dose. The mice were monitored until they either recovered or became moribund, at which point they were euthanized. The criteria for euthanasia of moribund mice are symptomatic of acute and nonrecoverable septicemia: hunched posture, severely matted fur and absence of grooming, emaciation, and immobility. A Kaplan-Meier curve was generated, and survival was analyzed by the log rank test.
Colonization and in vivo injection analysis.
Mice were infected via retro-orbital injection of KIM5 carrying pMM83 (YopM-Bla) at a dose of ∼104 CFU or CC003 carrying pMM83 at a dose of ∼106 CFU. Spleens were harvested from mice at 24-hour intervals postinfection. Following harvest, the spleens were homogenized in 1 ml of RPMI (Cellgro catalog no. 10-040-CV) and passed through a 70-μm filter. A portion of the homogenized spleen was serially diluted and plated in triplicate on HIA plates to determine the average bacterial load per spleen. The remaining homogenate was incubated for 5 min with red blood cell lysis buffer (0.13 mM NH4Cl, 0.017 mM Tris, 500 ml H2O, pH 7.2) at room temperature. Five milliliters of Hanks' buffered salt solution Flow (HBSS Flow) (0.5 mM EDTA, 25 mM HEPES, 2% [wt/vol] bovine serum albumin [BSA], and 1× HBSS, pH 7.4) was added. A 6× dye stock containing 3-[(5′-diacetylfluoroscein)thio]methyl)-7-(7″-butyryloxy-6″-chloro-coumarin-3″-carbonylglycinamido)-3-cephem-4-(4-acetylmethoxy)carboxylate (CCF2-AM) (Invitrogen catalog no. K1032) was prepared according to the manufacturer's instructions and was added to cells at a final concentration of 0.2×, followed by incubation for 1 h at room temperature. Propidium iodine (PI) (0.1 μg/ml) was added to each sample before analysis on the flow cytometer. Flow cytometry was performed at the Indiana University Flow Cytometry Core Facility (IU FCCF) on the BD FACSAria II cytometer. Live cells were identified as PI negative and then analyzed for blue and green fluorescence to distinguish injected from noninjected cells. Triplicate samples from individual mice were analyzed by flow cytometry, and five mice per time point were used to determine means and standard errors of the means (SEM) for injection by YopM-Bla-expressing strains. Data were analyzed using one-way analysis of variance (ANOVA) and the Bonferroni post hoc test.
Ex vivo injection assays.
Spleens harvested from three naïve mice were homogenized in 1 ml of RPMI and passed through a 70-μm filter. The homogenate was incubated for 5 min at room temperature with red blood cell lysis solution. The homogenates were treated with 1 μg/ml cytochalasin D (Sigma catalog no. C8273) for 30 min. Prior to the start of the infection, mid-exponential-phase Y. pestis cultures carrying pMM83 (YopM-Bla) were incubated for 1.5 h at 37°C to preinduce the T3SS. Bacteria were added to splenocytes at a multiplicity of infection (MOI) of 5 to 10, and the infection mixture was centrifuged at 500 × g for 5 min to facilitate cell contact. The infections were incubated at 37°C for up to 4 h. The infection was stopped by adding kanamycin (50 μg/ml). Cells were stained with CCF2-AM at a final concentration of 0.2× for 1 h at room temperature. Cells were pelleted at 1,500 × g for 3 min, and the supernatant was removed. Next, the cells were stained with Fc Block (eBiosciences; clone 93) for 25 min on ice. Cells were pelleted at 1,500 × g for 3 min, and the supernatant was removed. The cells were then stained with either CD19-PeCy7 (BD Pharmingen; clone 1D3), CD4-APC-eFluor 780 (eBiosciences; clone RM4-5), CD8-PerCP-eFluor 710 (eBiosciences; clone 53-6.7), NK1.1-APC (BD Pharmingen; clone PK136), CD11b-PeCy7 (BD Pharmingen; clone 1D3), CD11c-APC (BD Pharmingen; clone HL3), major histocompatibility complex class II (MHC-II)-PerCP-eFluor 710 (eBiosciences; clone M5/114.15.2), or GR-1-APC-Cy7 (BD Pharmingen; clone RB6-8C5) for 35 min on ice. Following the staining, the cells were pelleted and resuspended in HBSS Flow containing PI. Flow cytometry was done in the IU FCCF on the BD FACSAria II or the BD LSRII cytometer. Live cells were first identified as PI negative and then sent to subsequent gates to identify cell types, and then each population was evaluated for the percentage of blue (injected) cells (frequency of injection for each cell type). B cells were CD4− CD8− CD19+ NK1.1−, T-helper cells were CD4+ CD8−, T cytotoxic cells were CD4− CD8+, NK cells were CD4− CD8− CD19− NK1.1+, dendritic cells were CD11c+, macrophages were MHC class II− CD11c− CD11b+ GR-1−, and neutrophils were MHC class II− CD11c− CD11b+ GR-1+.
Complement sensitivity assay.
Pooled defibrinated normal human serum (NHS) was purchased from Innovative Research Inc. Heat inactivation was performed by incubating serum at 56°C for 30 min. KIM5, the rfaL mutant, and the ail mutant [KIM5 (Δy1324) (Δail) (42)] were grown overnight in HIB or HIB plus 5 mM EGTA and then diluted in PBS. Each strain was then added at 2 × 107 CFU to 0.2 ml of the NHS, heat-inactivated NHS (HIS), or PBS. To chelate calcium, 10 mM EGTA was added to the reaction mixtures. After a 1-hour incubation at 37°C, the reaction mixtures were incubated on ice for 15 min and then serially diluted and plated in triplicate onto HIA plates.
PMB sensitivity assay.
To measure sensitivity to polymyxin B (PMB), HIA medium was prepared with polymyxin B sulfate (Calbiochem) concentrations ranging from 0 μg/ml to 50 μg/ml. For the calcium-depleted condition, the same medium was prepared except that 20 mM sodium oxalate and 2.5 mM MgCl2 were added. Bacterial strains were grown overnight in HIB, and then 1 × 108 CFU/ml was serially diluted in PBS. The dilutions were plated in triplicate on the polymyxin B-containing medium. Plates were incubated for 2 days at 26°C. Significance was calculated using one-way ANOVA with the Bonferroni post hoc test.
The assay for growth in the presence of polymyxin B was performed as follows. Strains were grown in HIB to an OD600 between 0.6 and 0.8. The cells were pelleted and then washed once in PBS. Cells were diluted in DMEM plus 10% FBS plus 2 mM Glutamax without phenol red to an OD600 of ∼0.2. Where indicated, 5 mM EGTA, 5 mM MgSO4, or 25 μg/ml polymyxin B was added to the medium. The cultures were incubated at 26°C, and the OD600 of each culture was measured every hour for 4 h. Significance was determined at the 4-h time point using one-way ANOVA with the Tukey post hoc test.
RESULTS
RfaL is required for secretion of Yop effectors.
In a screen designed to identify chromosomal genes contributing to regulation or function of the T3SS in Y. pestis, we identified a mutant, CHI1800, which contains a transposon insertion in rfaL (also called waaL) (52). This mutant is able to secrete the early Yops (such as YopBDR and LcrV) in the presence of calcium in vitro but is unable to secrete any of the early or late Yops (such as YopEHM) in the absence of calcium. To further investigate the role of rfaL, a mutant harboring an unmarked deletion of rfaL was constructed, and in vitro secretion assays were performed to confirm the mutant phenotype. Secretion was monitored by immunoblotting for YopD and YopE, as representative early and late Yops, respectively. Both the wild type (WT) and the ΔrfaL mutant secreted YopD in the presence of calcium, but the mutant did not secrete either YopE or YopD in the absence of calcium (see Fig. S1A in the supplemental material). Ectopic expression of rfaL under the control of its own promoter complemented the mutant phenotype and restored calcium-regulated secretion of early and late Yops (see Fig. S1A). Thus, the ΔrfaL mutant (CC003) displays the same secretion defect as does the transposon mutant (CHI1800) (52).
The ΔrfaL mutant is less virulent than the wild type.
Previous characterization of the rfaL transposon mutant, CHI1800, demonstrated that while it is able to inject Yops into tissue culture cells, it is severely attenuated (∼10,000-fold) in vivo after septicemic infection of BALB/c mice (52). This is in contrast to other work that demonstrated only a slight defect (∼6- to 13.5-fold) for rfaL mutants after subcutaneous infection of BALB/c mice (48, 59). The different observations could be due to differences in strain background: the previous work utilized strains derived from Y. pestis bv. antiqua (strain 231) and Y. pestis bv. orientalis (strain CO92), while our mutant is derived from Y. pestis bv. medievalis strain KIM5. We therefore wanted to confirm the virulence defect of our new ΔrfaL mutant (CC003), which is in the KIM5 strain background. We infected C57BL/6 mice with ∼100 CFU of the wild type, the ΔrfaL mutant, and the complemented ΔrfaL mutant by retro-orbital inoculation. Although a mouse was lost on day 8 for the ΔrfaL mutant, the remaining mice infected with the mutant had all recovered by then. On the other hand, all mice infected with the wild type died by day 8.The median survival times for infections with the wild type and the complemented mutant were 7 and 8 days, respectively, and not statistically different (see Fig. S1B in the supplemental material). In contrast, the virulence defect of the mutant compared to the WT is significant (P < 0.005), and the mutant survival time remained undefined.
The ΔrfaL mutant translocates Yops in vivo but has a colonization defect.
Given that the ΔrfaL mutant is able to inject Yops into tissue culture cells (52), it is somewhat surprising that it is so attenuated in vivo. To investigate the nature of the virulence defect and to determine whether the T3SS is functional in vivo, C57BL/6 mice were infected with wild-type or ΔrfaL strains carrying β-lactamase fused to YopM (YopM-Bla) as a means to directly monitor injection (40). To ensure consistent colonization of the spleen, a dose of 106 CFU was used for the ΔrfaL mutant strains, while a dose of 104 CFU was used for wild-type strains. At these doses, all mice should become colonized without becoming moribund during the timeline of the experiment. Spleens were harvested for analysis at 24-h intervals (days 1 to 2 not shown). Spleen homogenates were stained with the fluorescent β-lactamase substrate CCF2-AM and analyzed by flow cytometry to detect injection of the Bla reporter. The homogenates were also serially diluted and plated to determine the bacterial load in each spleen.
Spleen colonization by the wild type and the ΔrfaL mutant is shown in Fig. 1A, while the percentage of injected splenocytes (positive for the YopM-Bla reporter) is shown in Fig. 1B. Individual mice are distinguished by the presence (closed circles) or absence (open circles) of injected cells within infected spleens. Comparison of the patterns of open and closed circles in Fig. 1A and B reveals a threshold level of colonization, approximately 104 CFU, above which injected splenocytes were routinely detected (Fig. 1 and data not shown). Below that threshold, no injection was detected. From day 2 to day 3, all mice infected with the wild-type strain showed a 10- to 100-fold increase in bacterial loads, and correspondingly, the average level of injection rose from 0.466% on day 2 to 1.28% on day 3 (P < 0.01) (Fig. 1 and data not shown). Unlike the wild type, the mutant did not reach high CFU loads, and by day 3, two mice were starting to recover. Likewise, there were far fewer injected cells from ΔrfaL mutant-infected mice than from wild-type-infected mice (0.51% for the ΔrfaL mutant and 1.28% for WT, P < 0.05). Taken together, these results indicate that the ΔrfaL mutant is capable of Yop delivery in vivo; however, the mutant was defective for persisting in spleens.
Fig 1.
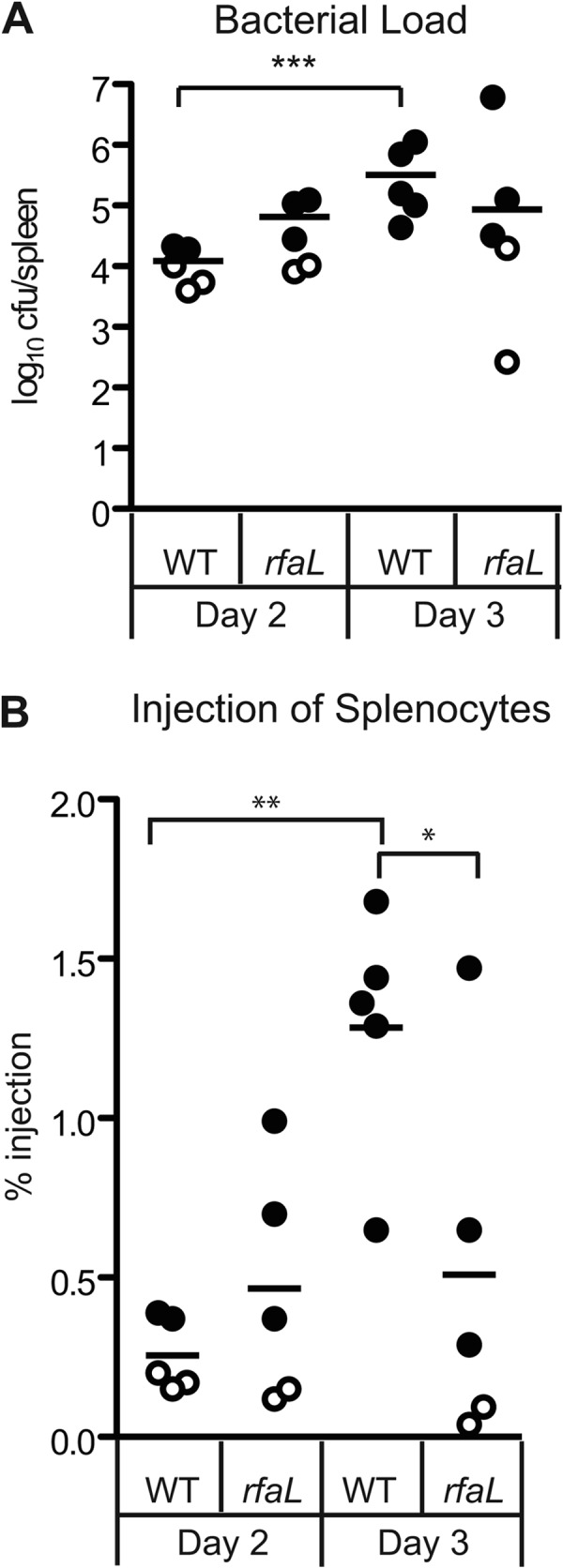
In vivo injection phenotype of the wild type and the ΔrfaL mutant. Six- to 8-week-old C57BL/6 mice were infected with the wild type or the ΔrfaL mutant carrying YopM-Bla. Spleens were harvested 3 days postinfection and homogenized. (A) Colonization of spleens by the wild type and the ΔrfaL mutant. Diluted spleen homogenates were plated in triplicate, and means are shown. The limit of detection was 102 CFU. Data were analyzed by two-tailed Student's t test (***, P < 0.001). (B) Injection of splenocytes by the wild type and the ΔrfaL mutant. The homogenized spleens were stained with CCF2-AM, flow cytometry was performed to determine the percentage of blue cells per spleen, and means are shown. Data were analyzed by one-way ANOVA and the Bonferroni post hoc test. **, P < 0.01; *, P < 0.05. Circles, YopM-Bla. Open circles correspond to samples that had only background levels of blue cells, similar to the glutathione S-transferase–Bla negative control (not shown).
The ΔrfaL mutant targets the same cell populations as does the wild type.
Y. pestis displays a preference in targeting innate immune cells for Yop injection during septicemic infection (40). Given that the virulence of the ΔrfaL mutant is dramatically decreased despite its ability to inject Yops into splenocytes in vivo, we wondered whether the mutant's virulence defect is due to defective target cell selection. To investigate that possibility, ex vivo infections were performed on spleens from naïve mice. Spleen homogenates were infected in triplicate with either the wild type or the ΔrfaL mutant carrying the YopM-Bla reporter. Following the infection, cells in the homogenate were stained with CCF2-AM and with antibodies for immunophenotyping. Flow cytometry was performed to determine the distribution of cell types within the total cell population (Fig. 2A) and the frequency of injection for each cell type (Fig. 2B). As expected, B cells, T-helper cells, and the T-cytotoxic cells make up the majority of the total splenocyte population, with innate immune cells being present in minor amounts (Fig. 2A). However, both the wild type and the ΔrfaL mutant showed a clear preference for neutrophils, since this cell population had the highest percentage of injected cells (Fig. 2B). B cells and dendritic cells were also injected at slightly higher frequencies than were other cell types. Compared to the wild type, there was less injection for each cell type by the ΔrfaL mutant. This was particularly evident for the neutrophil population, which showed 83.5% injection by the wild type versus 52.5% injection by the mutant (P < 0.001). These results indicate that although both the wild type and the ΔrfaL mutant have a preference for innate immune cells, particularly neutrophils, the mutant appears to be less effective at delivering Yops into those cells. Thus, it appears that the ΔrfaL mutant is still capable of target cell selection and injection of Yops, which suggests that the attenuation in vivo is not due to a simple absence of Yop translocation into the appropriate immune cells.
Fig 2.

Ex vivo target cell selection of the wild type and the ΔrfaL mutant. Spleens from three healthy 6- to 8-week-old C57BL/6 mice were harvested and homogenized. The homogenates were infected with the wild type carrying YopM-Bla or the ΔrfaL mutant carrying YopM-Bla at 37°C. Cells were stained with antibodies and CCF2-AM and then analyzed by flow cytometry. The experiment was done in triplicate, and data are pooled from the three replicates. Means and SEM are shown. (A) Overall distribution of spleen populations. The inset is a magnified view of the indicated populations. (B) Target cell injection. Individual cell types were evaluated to determine the percentage of injected cells within that population. Differences in injection between the WT and the rfaL mutant for individual cell types were determined by two-tailed Student's t tests. ***, P < 0.001; **, P = 0.001 to 0.01; *, P = 0.01 to 0.05. TH, T-helper cells; TC, T-cytotoxic cells; NK, natural killer cells; DC, dendritic cells; Mac, macrophages; Neut, neutrophils.
The ΔrfaL mutant assembles YscF needles on its surface in the absence of calcium.
To investigate the cause of the Yop injection defect, we addressed the possibility that the phenotype stemmed from structural defects or instability of the injectisome. LPS is a major component of the outer membrane and therefore plays an important role in the folding and activity of some outer membrane proteins (60–64). It is possible that alteration of the LOS in Y. pestis, due to lack of RfaL, leads to structural changes in proteins or macromolecular complexes embedded within the outer membrane. To determine if the needle subunit, YscF, was polymerizing on the bacterial surface, we performed an in vitro cross-linking assay, using the nonpermeable cross-linking agent BS3 in order to capture needles (37). Bacteria were grown in medium with or without calcium at 37°C, and then BS3 was added to cross-link the assembled needles. To visualize the injectisome needles, whole-cell samples were subjected to SDS-PAGE followed by immunoblotting for the needle component, YscF. The wild-type strain produced polymers of YscF in the presence and absence of calcium, as demonstrated by the ladder-like pattern seen with an anti-YscF antibody (Fig. 3). This pattern was not observed for the ΔyscF and ΔrfaL ΔyscF mutants. Interestingly, the ΔrfaL mutant also produced a pattern of YscF polymers similar to that of the wild type (Fig. 3). However, the amount of polymer made under both conditions appears to be smaller than that produced by the wild type, and the ΔrfaL mutant also produced less YscF monomer than did the wild type (Fig. 3). Thus, even though the mutant cannot secrete Yops in vitro in the absence of calcium, it does harbor YscF polymers with a pattern similar to that of the wild type, suggesting that needles may be present, though in fewer numbers. However, we do not know from these data whether these needles are functional or if they are structurally similar to those in the wild type.
Fig 3.

In vitro YscF cross-linking assay. Wild-type, ΔrfaL, ΔyscF, or ΔrfaL ΔyscF strains were incubated in TMH with or without CaCl2 (+Ca or −Ca) at 37°C. Cultures were centrifuged, and the cell pellets were gently resuspended in HEPES with or without CaCl2 and then incubated with BS3 (+) or with water (−). Proteins were trichloroacetic acid precipitated and immunoblotted using anti-YscF and anti-RpoA antibodies.
The secretion defect of an ΔrfaL mutant cannot be rescued by constitutive YscF point mutations.
Since the cross-linking experiment suggests that the rfaL mutant makes YscF needles in vitro in the absence of calcium (Fig. 3), we wondered if the lack of secretion resulted from an inability of needles to respond to external stimuli. It has been shown previously in Y. pestis that the YscF needle has a role in regulating Yop secretion in response to calcium or cell contact (37). We therefore asked whether we could rescue the secretion defect of the mutant by introducing mutations in YscF that induce constitutive Yop secretion in the presence and absence of calcium. Mutating the aspartic acid residue at position 46 to either an alanine or a cysteine is an example of this type of mutation (37). We introduced plasmids carrying either YscF D46A or D46C mutations into wild-type and ΔrfaL strains lacking native yscF and performed our standard in vitro secretion assay. Though these point mutations caused secretion of early and late Yops in the presence of calcium, as expected, no secretion was observed for the ΔrfaL mutant in the absence of calcium (see Fig. S2 in the supplemental material).
A mutation in yopN cannot rescue the ΔrfaL secretion phenotype.
Since YscF point mutants that should lock needles into an active state could not rescue the ΔrfaL mutant (see Fig. S2 in the supplemental material), we reasoned that the blockade on secretion might result from an internal regulatory phenomenon. YopN is an intracellular regulator that prevents late Yop secretion in the presence of calcium, and yopN mutants have a calcium-blind phenotype whereby all Yops are secreted in both the presence and the absence of calcium (65, 66). We hypothesized that if the secretion defect in the ΔrfaL mutant was due to an internal calcium-dependent regulatory block, then introducing a yopN mutation should restore secretion. We constructed a double mutant containing deletions in both rfaL and yopN and performed in vitro secretion assays (Fig. 4A). As before, the ΔrfaL mutant secreted YopD as the wild type did in the presence of calcium and, unlike the wild type, did not secrete any Yops in the absence of calcium (Fig. 4A). The ΔyopN mutant secreted all Yops in both the presence and the absence of calcium; however, the ΔrfaL ΔyopN mutant secreted Yops only in the presence of calcium (Fig. 4A). Thus, the deletion of yopN cannot restore secretion to the rfaL mutant.
Fig 4.

Deletion of lcrQ but not yopN can partially rescue the secretion defect of the ΔrfaL mutant. (A) Secretion assays performed with wild-type, ΔrfaL, ΔyopN, and ΔrfaL ΔyopN strains. (B) Secretion assays performed with wild type, ΔrfaL, ΔlcrQ, and ΔrfaL ΔlcrQ strains. Assays were done by growing bacteria in DMEM at 37°C with or without EGTA (−Ca or +Ca, respectively). Following incubation, cultures were centrifuged to separate secreted proteins (S) from cell pellet (P). The proteins in both fractions were trichloroacetic acid precipitated, and immunoblotting was performed using the indicated antibodies. (C) Needle cross-linking by the wild type or ΔlcrQ, ΔrfaL, ΔrfaL ΔlcrQ, ΔyscF, or ΔrfaL ΔyscF mutants. Bacteria were incubated in TMH with or without CaCl2 (+Ca or −Ca, respectively) at 37°C. Cultures were centrifuged, and the cell pellets were gently resuspended in HEPES with or without CaCl2 and then incubated with BS3 (+) or with water (−). Proteins were trichloroacetic acid precipitated and immunoblotted using anti-YscF and anti-RpoA antibodies. mon., monomer.
A mutation in lcrQ can partially rescue the ΔrfaL mutant phenotypes.
In our secretion assays, we noticed that in addition to a lack of secretion, the ΔrfaL mutant exhibited decreased production of T3SS components compared to the wild type when calcium was absent (Fig. 3 and 4A) (see also Fig. S1 and S2 in the supplemental material). In cases of a nonfunctional T3SS, the lack of secretion leads to a buildup of LcrQ, which inhibits the synthesis of the T3SS genes in a phenomenon known as feedback inhibition (67, 68). It is possible that insufficient production of injectisome components prevents detectable levels of Yop secretion or that Yops are not synthesized in sufficient amounts to be detected if secreted. To address these possibilities and to determine whether feedback inhibition interferes with the production of T3SS components and secretion of Yops in the ΔrfaL mutant, an ΔrfaL ΔlcrQ mutant was constructed and secretion assays were performed. As in previous secretion assays, the ΔrfaL mutant secreted YopD in the presence of calcium but did not secrete either YopD or YopE in the absence of calcium. It also produced less YscD in the absence of calcium than did the wild type (Fig. 4B). In contrast, the ΔrfaL ΔlcrQ mutant secreted Yops in the presence and absence of calcium, similar to the wild type; however, the amount of Yop secretion in the absence of calcium was reduced (Fig. 4B). The ΔrfaL ΔlcrQ mutant also produced YscD at levels similar to those of the wild type in the absence of calcium (Fig. 4B). Furthermore, when needle cross-linking assays are performed, the ΔrfaL ΔlcrQ mutant produces normal amounts of YscF monomer and polymers (Fig. 4C). This is consistent with previous research showing that buildup of LcrQ inside the cell represses synthesis of T3SS components (67, 68). These data suggest that the lack of secretion by the ΔrfaL mutant is partially due to a decrease in overall synthesis of the T3SS proteins as a result of feedback inhibition. That inhibition can be relieved to some extent by deleting lcrQ; however, the ΔrfaL ΔlcrQ double mutant still secreted smaller amounts of Yops in the absence of calcium than did the wild type.
The ΔrfaL mutant secretion defect can be partially rescued by divalent cations.
Chelating agents are known to destabilize the outer membrane of Gram-negative bacteria, and the secretion defect of the ΔrfaL mutant manifests primarily in the absence of calcium. Since RfaL is responsible for adding the terminal GlcNac to the LOS, we hypothesized that the altered LOS of the rfaL mutant might compromise cell envelope integrity and that the defect is exacerbated by chelating calcium. We investigated the possibility that other divalent cations may suffice to restore the cation cross-bridging between lipid A molecules and thereby restore calcium-regulated secretion to the ΔrfaL mutant. Secretion assays were performed in which EGTA was added to chelate calcium from the culture medium, followed by the addition of strontium, zinc, magnesium, and/or calcium. We chose to test zinc and strontium because they have been shown to overcome the growth cessation phenotype associated with the LCR (69). Magnesium was tested as well because it, along with calcium, plays a role in stabilizing the outer membrane of Gram-negative bacteria (46).
When both calcium and strontium were present, both the wild type and the ΔrfaL mutant secreted YopD (Fig. 5). Similar results were obtained when both calcium and magnesium were present. However, when strontium alone was added back to chelated medium, both strains secreted only YopD, similar to the effect of adding calcium alone (Fig. 5). This demonstrates that strontium can function in place of calcium in terms of allowing secretion of early Yops while preventing secretion of late Yops. Surprisingly, when magnesium alone was added back to the medium, both strains were able to secrete both YopE and YopD (Fig. 5). However, secretion by the ΔrfaL mutant under this condition was reduced compared to that by the wild type (Fig. 5). This suggests that magnesium can partially rescue the ΔrfaL mutant phenotype by restoring calcium-regulated secretion. Notably, addition of either strontium or magnesium increased production of T3SS proteins (Fig. 5, YscD). Therefore, in cultures that contained both calcium and strontium, a lack of secretion cannot be attributed to insufficient synthesis of injectisome components. The addition of zinc in any combination was unable to restore secretion to the ΔrfaL mutant (data not shown). As a control, we performed this assay with a mutant lacking YscU, a component of the injectisome, which is essential for T3SS function. As expected, the ΔyscU mutant was unable to secrete YopE or YopD under any condition (Fig. 5). Because magnesium was able to restore secretion of late Yops in the absence of calcium, these data support the possibility of structural instability in the outer membrane that affects injectisome structure and/or function in the rfaL mutant.
Fig 5.

Mg2+ and Sr2+ can partially rescue the secretion defect of the ΔrfaL mutant. Wild-type, ΔrfaL, and ΔyscU strains were grown at 37°C in buffered DMEM with or without EGTA, CaCl2, SrCl2, or MgSO4 added to the medium. Following incubation, cultures were centrifuged to separate secreted proteins (S) from cells (P). Proteins in both fractions were trichloroacetic acid precipitated and immunoblotted using antibodies against YscD, YopE, YopD, and RpoA.
Rescue of the ΔrfaL mutant phenotype in vitro.
Since neither the loss of lcrQ nor the addition of Mg2+ individually was able to fully restore Yop synthesis and secretion by the ΔrfaL mutant, we asked whether a combination of the two would fully rescue the ΔrfaL mutant's phenotypes. As previously demonstrated, when magnesium was added to chelated cultures, the ΔrfaL mutant secreted YopD and YopE, albeit at a decreased level compared to the wild type (Fig. 6). In contrast, addition of magnesium to the ΔrfaL ΔlcrQ mutant restored the synthesis and secretion of both YopD and YopE in the absence of calcium (Fig. 6). We also tested whether the addition of magnesium could restore Yop secretion to the ΔrfaL ΔyopN mutant, and indeed, the ΔrfaL ΔyopN mutant secreted both YopD and YopE like the wild type did (Fig. 6). These data demonstrate that by mitigating potential membrane instability and relieving feedback inhibition through a combination of magnesium supplementation and the loss of either lcrQ or yopN, we can fully rescue the in vitro expression and secretion defects of the ΔrfaL mutant.
Fig 6.

The combination of Mg2+ and deletion of lcrQ can fully rescue the ΔrfaL mutant. Wild-type, ΔrfaL, ΔlcrQ, ΔrfaL ΔyopN, and ΔrfaL ΔlcrQ strains were grown at 37°C in buffered DMEM with or without EGTA, CaCl2, or MgSO4 added to the medium. Following incubation, cultures were centrifuged to separate secreted proteins (S) from cells (P). Proteins in both fractions were trichloroacetic acid precipitated and immunoblotted using antibodies against YscD, YopE, YopD, and RpoA.
Rescue of the ΔrfaL mutant phenotype during infection.
We next turned to ex vivo infections to correlate our in vitro findings with events that happen during infection. Spleen homogenates were infected with the wild type, the ΔlcrQ mutant, the ΔrfaL mutant, or the ΔrfaL ΔlcrQ mutant, each carrying YopM-Bla. To monitor injection of the Bla reporter, cells were stained with CCF2-AM and analyzed by flow cytometry (Fig. 7). As expected, injection levels for the ΔlcrQ mutant were dramatically higher than those for all other strains under all conditions (P < 0.001). The ΔrfaL mutant injected fewer cells than did the wild type under all conditions (P < 0.001). In contrast, higher injection levels were observed for the ΔrfaL ΔlcrQ mutant than for the wild type (P < 0.001), though this was true only in the presence of calcium or magnesium. When divalent cations were chelated from the culture medium during infection, injection levels for all strains were dramatically decreased. Though deletion of lcrQ increased Yop injection in the absence of calcium, the mutation alone was not sufficient to completely rescue the rfaL mutant phenotype (P < 0.01 for the ΔrfaL ΔlcrQ mutant compared to the wild type). As with the in vitro secretion assays (Fig. 6), the combination of magnesium supplementation and lcrQ deletion was required to counteract the effect of calcium depletion. Together, the in vitro and ex vivo data support the hypothesis that RfaL is important for membrane stability and, by extension, proper function of the T3SS.
Fig 7.

Addition of Mg2+ and deletion of lcrQ restore Yop translocation by the ΔrfaL mutant. Spleens from three healthy 6- to 8-week-old C57BL/6 mice were harvested and homogenized. Splenocytes were infected with wild-type (WT), ΔlcrQ, ΔrfaL, and ΔrfaL ΔlcrQ strains carrying YopM-Bla in RPMI (untreated) with or without EGTA or MgSO4 at 37°C. Cells were stained with CCF2-AM and then analyzed by flow cytometry. The experiment was done in triplicate, and data are pooled from the three replicates, in order to determine the percentage of total splenocytes injected. Means and SEM are shown. Differences between strains were determined by one-way ANOVA and the Tukey post hoc test. ***, P < 0.001; **, P < 0.01.
CAMP sensitivity of the rfaL mutant.
To further characterize the nature of the mutant's attenuation, we looked at other factors that may be contributing to the virulence and colonization defects. CAMPs are a vital part of the mammalian innate immune system (44). Outer membrane polysaccharides such as O antigen can act as a barrier to CAMPs, providing many Gram-negative bacteria with protection against these compounds (46). We hypothesized that since RfaL modifies the LOS core of Y. pestis, and since magnesium supplementation is required to rescue the T3SS defect of the mutant, then the ΔrfaL mutant may be more susceptible to CAMPs (48, 70–72). We therefore tested the ΔrfaL mutant's sensitivity to the CAMP polymyxin B. The ΔrfaL mutant and the wild type were grown on HIA (with or without a calcium chelator) containing increasing concentrations of polymyxin B. In calcium-replete medium, the wild type displayed approximately 10-fold less viability in the presence of polymyxin B (Fig. 8A). In contrast, the viability of the ΔrfaL mutant was decreased by several orders of magnitude. Chelating calcium from the medium increased the effect of polymyxin B on both the wild type and the ΔrfaL mutant (Fig. 8A). This effect was greatest on the ΔrfaL mutant, which displayed a decrease in viability of approximately 8 orders of magnitude at the highest concentration of polymyxin B. When this assay was performed at 37°C in the presence of calcium, the mutant also exhibited an increased sensitivity to polymyxin B compared to the wild type, but the defect was even more substantial (data not shown). The results from this assay clearly demonstrate that the ΔrfaL mutant has an increased sensitivity to polymyxin B and that this sensitivity is exaggerated when calcium is absent from the growth medium. The collective increased sensitivity to CAMPs may help to explain the virulence defect of the mutant.
Fig 8.

Polymyxin B sensitivity assays. (A) The wild type and the ΔrfaL mutant were serially diluted and plated in triplicate onto HIA plates with or without added sodium oxalate (−Ca or +Ca) containing the indicated concentrations of polymyxin B (PMB). Plates were incubated for 2 days at 26°C. Colonies were counted, and the average log10 CFU/ml was determined. The data shown are representative of three independent trials. Significance was determined using one-way ANOVA and the Bonferroni post hoc test. ***, P < 0.001. (B and C) The wild type and the ΔrfaL mutant were diluted to an OD600 of ∼0.2 into DMEM with or without EGTA (−Ca or +Ca). Where noted, MgSO4 and/or polymyxin B was added. The cultures were incubated at 26°C, and the OD600 of each culture was taken every hour for 4 h. The experiment was performed in triplicate, and the data were pooled. Shown are the means and SEM of the combined trials.
CAMPs like polymyxin B work by displacing divalent cations like calcium and magnesium from the outer membrane (44, 46). Since the addition of magnesium is able to restore T3SS function to the mutant in the absence of calcium (Fig. 5 to 7), we tested whether the addition of magnesium could also restore polymyxin B resistance. We followed the growth of the wild type and the ΔrfaL mutant in DMEM supplemented with combinations of the EGTA chelator, magnesium, and polymyxin B. Growth of the ΔrfaL mutant under all conditions lacking polymyxin B was normal (Fig. 8B). However, after 4 h, the addition of polymyxin B to the medium in all cases dramatically reduced growth of the ΔrfaL mutant (P < 0.001 for PMB+ cultures versus PMB− cultures at 4 h). Unlike its effect on T3SS function, the addition of extra magnesium was unable to overcome the ΔrfaL mutant's sensitivity to polymyxin B when calcium was absent from the medium. A similar pattern was seen for the wild type, except that the growth of the wild type was only mildly affected by polymyxin B (Fig. 8C).
Lack of RfaL is not severely pleiotropic.
Our data indicate that the outer membrane of the ΔrfaL mutant is compromised, the consequences of which dramatically affect T3SS function and CAMP resistance. However, we were interested to see if other outer membrane properties were also affected in the ΔrfaL mutant, which could reflect a widespread pleiotropic phenomenon. We therefore investigated whether the outer membrane proteins Pla and Ail were functioning properly in the ΔrfaL mutant. Ail, along with Pla and PsaA, functions as an adhesin, which helps mediate Yop translocation into host cells (73, 74). If an outer membrane defect is preventing the adhesins from localizing and functioning properly, this might contribute to the decreased splenocyte injection displayed by the mutant. To determine whether Pla localization to the outer membrane and its enzymatic activity were affected by lack of rfaL, we measured Pla activity by conducting a plasminogen activation assay. The ΔrfaL mutant exhibited only a slight decrease in activity compared to the wild type under all of the conditions tested (see Fig. S3A to C in the supplemental material). Though this small decrease may not be biologically relevant, Student's t tests comparing the WT and the rfaL mutant at the 40-min time point indicate that this difference is significant (P values of 0.015 [26°C], 0.007 [37°C without Ca], and 0.005 [37°C with Ca]). Pla activity in the mutant is also evident by the degradation of YopE or YopH in our secretion assays (Fig. 4 and 6; see also Fig. S2). Similar to YscD and YscF, less Pla is produced by the mutant than by the wild type (see Fig. S3D). The ΔrfaL mutant also exhibited less autoprocessing of Pla than did the wild type, which is consistent with the slight decrease in Pla activity displayed by the mutant (see Fig. S3D). Thus, while Pla activity is reduced, it is clearly still functioning in the ΔrfaL mutant and the defect is mild.
Outer membrane polysaccharides play an important role in the resistance of many Gram-negative bacteria to serum (45). Y. pestis does not make O antigen, but it is still resistant to serum (75). This resistance is attributed to Ail, an outer membrane protein and adhesin (41, 42). Therefore, to test Ail function, we investigated whether the rfaL mutant retained serum resistance. To that end, we incubated wild-type, ΔrfaL, and Δail strains in normal human serum or heat-inactivated serum for 1 h. As expected, the wild type was resistant to the serum while the Δail mutant showed a 10,000-fold decrease in survival, as shown previously (42) (see Fig. S4 in the supplemental material). Surprisingly, the ΔrfaL mutant survived exposure to human serum, similar to the wild type. This was also true when the calcium was chelated from the reaction mixture, as well as when the calcium was chelated from both the medium used to grow the bacteria and the reaction mixture (see Fig. S4). These data indicate that the observed virulence defect of the ΔrfaL mutant is not due to an increased sensitivity to serum. The fact that the mutant is still resistant to human complement indicates that Ail is still localizing and functioning properly. These data bolster the idea that any effect on the outer membrane caused by lack of RfaL is not severely pleiotropic.
DISCUSSION
In our previous study, a screen for mutations that led to a loss of Yop secretion uncovered a mutant containing a transposon insertion in rfaL (52). RfaL (also called WaaL) catalyzes the ligation of GlcNAc to the lipid A core of the Y. pestis LOS (70). Though a role for RfaL in T3SS function had not been described previously, we demonstrated that the mutant cannot secrete effector Yops in vitro and is severely attenuated in vivo (52). In this study, using a clean ΔrfaL background, we confirm the secretion and virulence defects and also investigate the mechanism leading to those phenotypes. Our data demonstrate that the virulence defect of the ΔrfaL mutant is most likely due to an increased sensitivity to CAMPs and a decrease in immune cell targeting as a result of reduced T3SS capabilities. As to the foundation for these phenotypes, our results suggest that they are due to a structural defect in the mutant's outer membrane. This is supported by several observations. (i) Expression of T3SS genes was reduced and there were fewer YscF needles, which is indicative of feedback inhibition invoked by a nonfunctional T3SS. (ii) Deleting lcrQ relieved the feedback inhibition and partially rescued the T3SS expression and secretion defects. (iii) Magnesium partially rescued these defects as well, and together, addition of magnesium and deletion of lcrQ fully complemented the ΔrfaL mutant. (iv) The mutant was very sensitive to polymyxin B, and calcium depletion exacerbated the problem.
In the absence of RfaL, calcium appears to be necessary for certain processes relying on outer membrane stability (type III secretion, CAMP resistance, and to a lesser extent Pla activity). The need for calcium can be bypassed through the addition of MgSO4 or SrCl2, which restored T3SS function to the ΔrfaL mutant. Interestingly, strontium mimicked calcium in its regulation of the T3SS. The addition of strontium in the absence of calcium allowed the wild type and the ΔrfaL mutant to secrete YopD (early Yop), but like calcium, it prevented both strains from secreting YopE (late Yop) (Fig. 5). In contrast, magnesium did not function as a “calcium signal.” Instead, it restored T3SS function and the ability to respond to the low-calcium signal and to perform calcium-regulated Yop secretion (Fig. 5). It is unclear whether the divalent cations rescue the rfaL mutant by enhancing the cross-bridging between lipid A molecules or provide some other function such as stabilizing protein folding.
The ΔrfaL mutant injected splenocytes less efficiently than did the wild type, and this defect was exacerbated by calcium depletion (Fig. 7). Deletion of lcrQ only partially rescued the mutant phenotype in the absence of calcium. However, when magnesium was added, injection was restored in the ΔrfaL ΔlcrQ mutant. These data correlate with the in vitro secretion assays (Fig. 6) and suggest that magnesium only partially mends the outer membrane defect that is triggering feedback inhibition of the T3SS in the rfaL mutant.
Previous work demonstrated that the O-antigen ligase of Y. pestis functions to attach a GlcNac residue to the terminal sugar in the outer core of the lipooligosaccharide (48, 70). However, it was also demonstrated that the rfaL mutants of Y. pestis strains 231 and CO92 had much milder phenotypes (48, 59) than those in our findings. In both cases, the mutants were resistant to human serum and had slightly reduced Pla activity. However, the 231 and CO92 rfaL mutants displayed polymyxin B resistance near that of the wild-type strain, in contrast to our KIM5 rfaL mutant, whose polymyxin B sensitivity was increased by several orders of magnitude. The reason for the different observations is unknown; however, the different strain backgrounds could give rise to distinct expression patterns for genes involved in LOS biosynthesis and modification.
Though these data support the hypothesis that the T3SS defect of the rfaL mutant stems from membrane instability due to altered LOS, the problems caused by a lack of RfaL are distinct rather than pervasive. For example, magnesium partially rescues the T3SS defect (Fig. 5), but it has no effect on the CAMP sensitivity (Fig. 8B). Moreover, in the rfaL mutant Pla activity is only slightly reduced (see Fig. S3 in the supplemental material), and Ail activity is normal (see Fig. S4). In addition, we did not see any difference between the wild type and the rfaL mutant during macrophage survival assays (data not shown).
Defects in the outer membrane and compounds like CAMPs that disrupt the outer membrane of Gram-negative bacteria often trigger stress response pathways. Two of these systems, the CpxRA and RcsBD systems, have been previously shown to impair T3SS and flagellar biosynthesis in other Gram-negative bacteria when activated (76–80). We investigated whether an activated CpxRA or RcsBD pathway could be involved in the secretion defect of the mutant. However, we deleted the response regulators and the histidine kinases for these systems in our ΔrfaL background, and none of these mutations were able to rescue the secretion defect of the ΔrfaL mutant (data not shown). We also investigated whether the RpoE system was upregulated in the ΔrfaL mutant, but it appears to be expressed at a level equivalent to that in the wild type at both 26°C and 37°C with or without calcium in the medium (data not shown). These three systems are not the only ones activated by outer membrane stress (81, 82), and more work is required to determine if the secretion defect is due to an activation of a stress response pathway. Additionally, it remains unclear how the signal stemming from outer membrane defects is relayed to ultimately trigger the LcrQ-mediated feedback inhibition. Understanding how this and other information regarding the physiological state of the bacterium contributes to proper T3SS function could provide useful targets for pharmaceutical intervention.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Marketon lab for their helpful discussions.
We acknowledge membership within and support from the Region V “Great Lakes” Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium (GLRCE; National Institute of Allergy and Infectious Diseases award 1-U54-AI-057153). Partial support for this work was provided by the Office of the Vice Provost of Research at Indiana University—Bloomington through a Faculty Research Support Program Award to M.M.M. W.W.L. acknowledges NIH/NIAID grant R01 AI093727, and A.J.C. was supported by NIH/NIAID grant T32 AI007476.
Footnotes
Published ahead of print 28 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01417-12.
REFERENCES
- 1. Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 10:35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cornelis GR. 2006. The type III secretion injectisome. Nat. Rev. Microbiol. 4:811–825 [DOI] [PubMed] [Google Scholar]
- 3. Hu P, Elliott J, McCready P, Skowronski E, Garnes J, Kobayashi A, Brubaker RR, Garcia E. 1998. Structural organization of virulence-associated plasmids of Yersinia pestis. J. Bacteriol. 180:5192–5202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Spreter T, Yip CK, Sanowar S, Andre I, Kimbrough TG, Vuckovic M, Pfuetzner RA, Deng W, Yu AC, Finlay BB, Baker D, Miller SI, Strynadka NC. 2009. A conserved structural motif mediates formation of the periplasmic rings in the type III secretion system. Nat. Struct. Mol. Biol. 16:468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Plano GV, Barve SS, Straley SC. 1991. LcrD, a membrane-bound regulator of the Yersinia pestis low-calcium response. J. Bacteriol. 173:7293–7303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Allaoui A, Woestyn S, Sluiters C, Cornelis GR. 1994. YscU, a Yersinia enterocolitica inner membrane protein involved in Yop secretion. J. Bacteriol. 176:4534–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fields KA, Plano GV, Straley SC. 1994. A low-Ca2+ response (LCR) secretion (ysc) locus lies within the lcrB region of the LCR plasmid in Yersinia pestis. J. Bacteriol. 176:569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diepold A, Wiesand U, Cornelis GR. 2011. The assembly of the export apparatus (YscR,S,T,U,V) of the Yersinia type III secretion apparatus occurs independently of other structural components and involves the formation of an YscV oligomer. Mol. Microbiol. 82:502–514 [DOI] [PubMed] [Google Scholar]
- 9. Diepold A, Wiesand U, Amstutz M, Cornelis GR. 2012. Assembly of the Yersinia injectisome: the missing pieces. Mol. Microbiol. 85:878–892 [DOI] [PubMed] [Google Scholar]
- 10. Diepold A, Amstutz M, Abel S, Sorg I, Jenal U, Cornelis GR. 2010. Deciphering the assembly of the Yersinia type III secretion injectisome. EMBO J. 29:1928–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoiczyk E, Blobel G. 2001. Polymerization of a single protein of the pathogen Yersinia enterocolitica into needles punctures eukaryotic cells. Proc. Natl. Acad. Sci. U. S. A. 98:4669–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cornelis GR. 2002. Yersinia type III secretion: send in the effectors. J. Cell Biol. 158:401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosqvist R, Magnusson KE, Wolf-Watz H. 1994. Target cell contact triggers expression and polarized transfer of Yersinia YopE cytotoxin into mammalian cells. EMBO J. 13:964–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mueller CA, Broz P, Muller SA, Ringler P, Erne-Brand F, Sorg I, Kuhn M, Engel A, Cornelis GR. 2005. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 310:674–676 [DOI] [PubMed] [Google Scholar]
- 15. Cornelis G, Sluiters C, de Rouvroit CL, Michiels T. 1989. Homology between virF, the transcriptional activator of the Yersinia virulence regulon, and AraC, the Escherichia coli arabinose operon regulator. J. Bacteriol. 171:254–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lambert de Rouvroit C, Sluiters C, Cornelis GR. 1992. Role of the transcriptional activator, VirF, and temperature in the expression of the pYV plasmid genes of Yersinia enterocolitica. Mol. Microbiol. 6:395–409 [PubMed] [Google Scholar]
- 17. Jackson MW, Silva-Herzog E, Plano GV. 2004. The ATP-dependent ClpXP and Lon proteases regulate expression of the Yersinia pestis type III secretion system via regulated proteolysis of YmoA, a small histone-like protein. Mol. Microbiol. 54:1364–1378 [DOI] [PubMed] [Google Scholar]
- 18. Lee VT, Mazmanian SK, Schneewind O. 2001. A program of Yersinia enterocolitica type III secretion reactions is activated by specific signals. J. Bacteriol. 183:4970–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee VT, Anderson DM, Schneewind O. 1998. Targeting of Yersinia Yop proteins into the cytosol of HeLa cells: one-step translocation of YopE across bacterial and eukaryotic membranes is dependent on SycE chaperone. Mol. Microbiol. 28:593–601 [DOI] [PubMed] [Google Scholar]
- 20. Michiels T, Wattiau P, Brasseur R, Ruysschaert JM, Cornelis G. 1990. Secretion of Yop proteins by yersiniae. Infect. Immun. 58:2840–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Straley SC, Plano GV, Skrzypek E, Haddix PL, Fields KA. 1993. Regulation by Ca2+ in the Yersinia low-Ca2+ response. Mol. Microbiol. 8:1005–1010 [DOI] [PubMed] [Google Scholar]
- 22. Lee VT, Schneewind O. 1999. Type III machines of pathogenic yersiniae secrete virulence factors into the extracellular milieu. Mol. Microbiol. 31:1619–1629 [DOI] [PubMed] [Google Scholar]
- 23. Higuchi K, Carlin CE. 1958. Studies on the nutrition and physiology of Pasteurella pestis. II. A defined medium for the growth of Pasteurella pestis. J. Bacteriol. 75:409–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brubaker RR, Surgalla MJ. 1964. The effect of Ca++ and Mg++ on lysis, growth, and production of virulence antigens by Pasteurella Pestis. J. Infect. Dis. 114:13–25 [DOI] [PubMed] [Google Scholar]
- 25. Goguen JD, Yother J, Straley SC. 1984. Genetic analysis of the low calcium response in Yersinia pestis mu d1(Ap lac) insertion mutants. J. Bacteriol. 160:842–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brubaker RR. 2005. Influence of Na(+), dicarboxylic amino acids, and pH in modulating the low-calcium response of Yersinia pestis. Infect. Immun. 73:4743–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Skryzpek E, Straley SC. 1993. LcrG, a secreted protein involved in negative regulation of the low-calcium response in Yersinia pestis. J. Bacteriol. 175:3520–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boland A, Sory MP, Iriarte M, Kerbourch C, Wattiau P, Cornelis GR. 1996. Status of YopM and YopN in the Yersinia Yop virulon: YopM of Y. enterocolitica is internalized inside the cytosol of PU5-1.8 macrophages by the YopB, D, N delivery apparatus. EMBO J. 15:5191–5201 [PMC free article] [PubMed] [Google Scholar]
- 29. Nilles ML, Williams AW, Skrzypek E, Straley SC. 1997. Yersinia pestis LcrV forms a stable complex with LcrG and may have a secretion-related regulatory role in the low-Ca2+ response. J. Bacteriol. 179:1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jackson MW, Day JB, Plano GV. 1998. YscB of Yersinia pestis functions as a specific chaperone for YopN. J. Bacteriol. 180:4912–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng LW, Schneewind O. 2000. Yersinia enterocolitica TyeA, an intracellular regulator of the type III machinery, is required for specific targeting of YopE, YopH, YopM, and YopN into the cytosol of eukaryotic cells. J. Bacteriol. 182:3183–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng LW, Kay O, Schneewind O. 2001. Regulated secretion of YopN by the type III machinery of Yersinia enterocolitica. J. Bacteriol. 183:5293–5301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rimpilainen M, Forsberg A, Wolf-Watz H. 1992. A novel protein, LcrQ, involved in the low-calcium response of Yersinia pseudotuberculosis shows extensive homology to YopH. J. Bacteriol. 174:3355–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Price SB, Straley SC. 1989. lcrH, a gene necessary for virulence of Yersinia pestis and for the normal response of Y. pestis to ATP and calcium. Infect. Immun. 57:1491–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bergman T, Hakansson S, Forsberg A, Norlander L, Macellaro A, Backman A, Bolin I, Wolf-Watz H. 1991. Analysis of the V antigen lcrGVH-yopBD operon of Yersinia pseudotuberculosis: evidence for a regulatory role of LcrH and LcrV. J. Bacteriol. 173:1607–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Williams AW, Straley SC. 1998. YopD of Yersinia pestis plays a role in negative regulation of the low-calcium response in addition to its role in translocation of Yops. J. Bacteriol. 180:350–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Torruellas J, Jackson MW, Pennock JW, Plano GV. 2005. The Yersinia pestis type III secretion needle plays a role in the regulation of Yop secretion. Mol. Microbiol. 57:1719–1733 [DOI] [PubMed] [Google Scholar]
- 38. Davis AJ, Mecsas J. 2007. Mutations in the Yersinia pseudotuberculosis type III secretion system needle protein, YscF, that specifically abrogate effector translocation into host cells. J. Bacteriol. 189:83–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davis AJ, Diaz DA, Mecsas J. 2010. A dominant-negative needle mutant blocks type III secretion of early but not late substrates in Yersinia. Mol. Microbiol. 76:236–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. 2005. Plague bacteria target immune cells during infection. Science 309:1739–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bliska JB, Falkow S. 1992. Bacterial resistance to complement killing mediated by the Ail protein of Yersinia enterocolitica. Proc. Natl. Acad. Sci. U. S. A. 89:3561–3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bartra SS, Styer KL, O'Bryant DM, Nilles ML, Hinnebusch BJ, Aballay A, Plano GV. 2008. Resistance of Yersinia pestis to complement-dependent killing is mediated by the Ail outer membrane protein. Infect. Immun. 76:612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daha MR. 2010. Role of complement in innate immunity and infections. Crit. Rev. Immunol. 30:47–52 [DOI] [PubMed] [Google Scholar]
- 44. Guani-Guerra E, Santos-Mendoza T, Lugo-Reyes SO, Teran LM. 2010. Antimicrobial peptides: general overview and clinical implications in human health and disease. Clin. Immunol. 135:1–11 [DOI] [PubMed] [Google Scholar]
- 45. Goebel EM, Wolfe DN, Elder K, Stibitz S, Harvill ET. 2008. O antigen protects Bordetella parapertussis from complement. Infect. Immun. 76:1774–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nikaido H, Vaara M. 1985. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 49:1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vinogradov EV, Lindner B, Kocharova NA, Senchenkova SN, Shashkov AS, Knirel YA, Holst O, Gremyakova TA, Shaikhutdinova RZ, Anisimov AP. 2002. The core structure of the lipopolysaccharide from the causative agent of plague, Yersinia pestis. Carbohydr. Res. 337:775–777 [DOI] [PubMed] [Google Scholar]
- 48. Dentovskaya SV, Anisimov AP, Kondakova AN, Lindner B, Bystrova OV, Svetoch TE, Shaikhutdinova RZ, Ivanov SA, Bakhteeva IV, Titareva GM, Knirel AY. 2011. Functional characterization and biological significance of Yersinia pestis lipopolysaccharide biosynthesis genes. Biochemistry (Mosc.) 76:808–822 [DOI] [PubMed] [Google Scholar]
- 49. Knirel YA, Dentovskaya SV, Senchenkova SN, Shaikhutdinova RZ, Kocharova NA, Anisimov AP. 2006. Structural features and structural variability of the lipopolysaccharide of Yersinia pestis, the cause of plague. J. Endotoxin Res. 12:3–9 [DOI] [PubMed] [Google Scholar]
- 50. Knirel YA, Lindner B, Vinogradov EV, Kocharova NA, Senchenkova SN, Shaikhutdinova RZ, Dentovskaya SV, Fursova NK, Bakhteeva IV, Titareva GM, Balakhonov SV, Holst O, Gremyakova TA, Pier GB, Anisimov AP. 2005. Temperature-dependent variations and intraspecies diversity of the structure of the lipopolysaccharide of Yersinia pestis. Biochemistry 44:1731–1743 [DOI] [PubMed] [Google Scholar]
- 51. Knirel YA, Anisimov AP. 2012. Lipopolysaccharide of Yersinia pestis, the cause of plague: structure, genetics, biological properties. Acta Naturae 4:46–58 [PMC free article] [PubMed] [Google Scholar]
- 52. Houppert AS, Kwiatkowski E, Glass EM, Debord KL, Merritt PM, Schneewind O, Marketon MM. 2012. Identification of chromosomal genes in Yersinia pestis that influence type III secretion and delivery of Yops into target cells. PLoS One 7:e34039 doi:10.1371/journal.pone.0034039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brubaker RR. 1969. Mutation rate to non-pigmentation in Pasteurella pestis. J. Bacteriol. 98:1404–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cheng LW, Anderson DM, Schneewind O. 1997. Two independent type III secretion mechanisms for YopE in Yersinia enterocolitica. Mol. Microbiol. 24:757–765 [DOI] [PubMed] [Google Scholar]
- 56. Sorg JA, Miller NC, Marketon MM, Schneewind O. 2005. Rejection of impassable substrates by Yersinia type III secretion machines. J. Bacteriol. 187:7090–7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Straley SC, Bowmer WS. 1986. Virulence genes regulated at the transcriptional level by Ca2+ in Yersinia pestis include structural genes for outer membrane proteins. Infect. Immun. 51:445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513 [DOI] [PubMed] [Google Scholar]
- 59. Filippov AA, Sergueev KV, He Y, Huang XZ, Gnade BT, Mueller AJ, Fernandez-Prada CM, Nikolich MP. 2011. Bacteriophage-resistant mutants in Yersinia pestis: identification of phage receptors and attenuation for mice. PLoS One 6:e25486 doi:10.1371/journal.pone.0025486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eren E, van den Berg B. 2012. Structural basis for activation of an integral membrane protease by lipopolysaccharide. J. Biol. Chem. 287:23971–23976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kukkonen M, Suomalainen M, Kyllonen P, Lahteenmaki K, Lang H, Virkola R, Helander IM, Holst O, Korhonen TK. 2004. Lack of O-antigen is essential for plasminogen activation by Yersinia pestis and Salmonella enterica. Mol. Microbiol. 51:215–225 [DOI] [PubMed] [Google Scholar]
- 62. Kukkonen M, Lahteenmaki K, Suomalainen M, Kalkkinen N, Emody L, Lang H, Korhonen TK. 2001. Protein regions important for plasminogen activation and inactivation of alpha2-antiplasmin in the surface protease Pla of Yersinia pestis. Mol. Microbiol. 40:1097–1111 [DOI] [PubMed] [Google Scholar]
- 63. Kramer RA, Vandeputte-Rutten L, de Roon GJ, Gros P, Dekker N, Egmond MR. 2001. Identification of essential acidic residues of outer membrane protease OmpT supports a novel active site. FEBS Lett. 505:426–430 [DOI] [PubMed] [Google Scholar]
- 64. Kramer RA, Brandenburg K, Vandeputte-Rutten L, Werkhoven M, Gros P, Dekker N, Egmond MR. 2002. Lipopolysaccharide regions involved in the activation of Escherichia coli outer membrane protease OmpT. Eur. J. Biochem. 269:1746–1752 [DOI] [PubMed] [Google Scholar]
- 65. Yother J, Chamness TW, Goguen JD. 1986. Temperature-controlled plasmid regulon associated with low calcium response in Yersinia pestis. J. Bacteriol. 165:443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Forsberg A, Viitanen AM, Skurnik M, Wolf-Watz H. 1991. The surface-located YopN protein is involved in calcium signal transduction in Yersinia pseudotuberculosis. Mol. Microbiol. 5:977–986 [DOI] [PubMed] [Google Scholar]
- 67. Stainier I, Iriarte M, Cornelis GR. 1997. YscM1 and YscM2, two Yersinia enterocolitica proteins causing downregulation of yop transcription. Mol. Microbiol. 26:833–843 [DOI] [PubMed] [Google Scholar]
- 68. Pettersson J, Nordfelth R, Dubinina E, Bergman T, Gustafsson M, Magnusson KE, Wolf-Watz H. 1996. Modulation of virulence factor expression by pathogen target cell contact. Science 273:1231–1233 [DOI] [PubMed] [Google Scholar]
- 69. Higuchi K, Kupferberg LL, Smith JL. 1959. Studies on the nutrition and physiology of Pasteurella pestis. III. Effects of calcium ions on the growth of virulent and avirulent strains of Pasteurella pestis. J. Bacteriol. 77:317–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Knirel YA, Dentovskaya SV, Bystrova OV, Kocharova NA, Senchenkova SN, Shaikhutdinova RZ, Titareva GM, Bakhteeva IV, Lindner B, Pier GB, Anisimov AP. 2007. Relationship of the lipopolysaccharide structure of Yersinia pestis to resistance to antimicrobial factors. Adv. Exp. Med. Biol. 603:88–96 [DOI] [PubMed] [Google Scholar]
- 71. Kiljunen S, Datta N, Dentovskaya SV, Anisimov AP, Knirel YA, Bengoechea JA, Holst O, Skurnik M. 2011. Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage phiA1122. J. Bacteriol. 193:4963–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kiljunen S, Hakala K, Pinta E, Huttunen S, Pluta P, Gador A, Lonnberg H, Skurnik M. 2005. Yersiniophage phiR1-37 is a tailed bacteriophage having a 270 kb DNA genome with thymidine replaced by deoxyuridine. Microbiology 151:4093–4102 [DOI] [PubMed] [Google Scholar]
- 73. Felek S, Tsang TM, Krukonis ES. 2010. Three Yersinia pestis adhesins facilitate Yop delivery to eukaryotic cells and contribute to plague virulence. Infect. Immun. 78:4134–4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Felek S, Krukonis ES. 2009. The Yersinia pestis Ail protein mediates binding and Yop delivery to host cells required for plague virulence. Infect. Immun. 77:825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Skurnik M, Bengoechea JA. 2003. The biosynthesis and biological role of lipopolysaccharide O-antigens of pathogenic yersiniae. Carbohydr. Res. 338:2521–2529 [DOI] [PubMed] [Google Scholar]
- 76. Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59:379–405 [DOI] [PubMed] [Google Scholar]
- 77. Lin D, Rao CV, Slauch JM. 2008. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J. Bacteriol. 190:87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Carlsson KE, Liu J, Edqvist PJ, Francis MS. 2007. Extracytoplasmic-stress-responsive pathways modulate type III secretion in Yersinia pseudotuberculosis. Infect. Immun. 75:3913–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Carlsson KE, Liu J, Edqvist PJ, Francis MS. 2007. Influence of the Cpx extracytoplasmic-stress-responsive pathway on Yersinia sp.-eukaryotic cell contact. Infect. Immun. 75:4386–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Raivio TL, Silhavy TJ. 1999. The sigmaE and Cpx regulatory pathways: overlapping but distinct envelope stress responses. Curr. Opin. Microbiol. 2:159–165 [DOI] [PubMed] [Google Scholar]
- 81. MacRitchie DM, Buelow DR, Price NL, Raivio TL. 2008. Two-component signaling and gram negative envelope stress response systems. Adv. Exp. Med. Biol. 631:80–110 [DOI] [PubMed] [Google Scholar]
- 82. Groisman EA. 2001. The pleiotropic two-component regulatory system PhoP-PhoQ. J. Bacteriol. 183:1835–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.