

Abstract
Phospholipase C (PLC) of Cryptococcus neoformans (CnPlc1) is crucial for virulence of this fungal pathogen. To investigate the mechanism of CnPlc1-mediated signaling, we established that phosphatidylinositol 4,5-bisphosphate (PIP2) is a major CnPlc1 substrate, which is hydrolyzed to produce inositol trisphosphate (IP3). In Saccharomyces cerevisiae, Plc1-derived IP3 is a substrate for the inositol polyphosphate kinase Arg82, which converts IP3 to more complex inositol polyphosphates. In this study, we show that in C. neoformans, the enzyme encoded by ARG1 is the major IP3 kinase, and we further demonstrate that catalytic activity of Arg1 is essential for cellular homeostasis and virulence in the Galleria mellonella infection model. IP3 content was reduced in the CnΔplc1 mutant and markedly increased in the CnΔarg1 mutant, while PIP2 was increased in both mutants. The CnΔplc1 and CnΔarg1 mutants shared significant phenotypic similarity, including impaired thermotolerance, compromised cell walls, reduced capsule production and melanization, defective cell separation, and the inability to form mating filaments. In contrast to the S. cerevisiae ARG82 deletion mutant (ScΔarg82) strain, the CnΔarg1 mutant exhibited dramatically enlarged vacuoles indicative of excessive vacuolar fusion. In mammalian cells, PLC-derived IP3 causes Ca2+ release and calcineurin activation. Our data show that, unlike mammalian PLCs, CnPlc1 does not contribute significantly to calcineurin activation. Collectively, our findings provide the first evidence that the inositol polyphosphate anabolic pathway is essential for virulence of C. neoformans and further show that production of IP3 as a precursor for synthesis of more complex inositol polyphosphates is the key biochemical function of CnPlc1.
INTRODUCTION
Cryptococcus neoformans var. grubii is a human fungal pathogen that predominantly infects immunocompromised hosts. After inhalation from the environment, infection spreads from the lung to the brain via the bloodstream, causing life-threatening meningoencephalitis. Several factors contribute to the virulence of C. neoformans, including the ability to grow at physiological temperature (37°C) and to produce melanin and capsule (1–3). Our previous work demonstrated that these virulence determinants are attenuated by deletion of a single gene encoding a key signaling molecule, phosphatidylinositol (PI)-specific phospholipase C (PLC) (Plc1). In addition, the C. neoformans PLC1 deletion mutant (CnΔplc1) had a defective cell wall and compromised virulence in both mouse (37°C) and Caenorhabditis elegans (25°C) models of cryptococcosis (4). A second PLC homolog, Plc2, appears to have no significant role in the biology and virulence of C. neoformans (4).
In mammalian cells, PI-specific phospholipase C enzymes (PLCs) catalyze the hydrolysis of PI-(4,5)-bisphosphate (PIP2) to diacylglycerol (DAG) and d-myo-inositol-(1,4,5)-trisphosphate (IP3). DAG directly activates protein kinase C (PKC), while IP3 triggers release of calcium from intracellular stores via interaction with IP3 receptors in the endoplasmic reticulum (ER). Elevated cytosolic calcium activates calcium-dependent signaling enzymes, such as calcineurin. In the model yeast Saccharomyces cerevisiae, a single PLC isoform has similar catalytic activity to the mammalian PLC-δ isoform (5). However, the role of the PIP2 breakdown products and the mechanism of Plc1-dependent signaling remain to be fully elucidated. Similarly, in C. neoformans, the identity and fate of the CnPlc1 hydrolysis products have not been determined.
In fungi, the role of PIP2-derived IP3 in initiating calcium influx is controversial as no homologs of mammalian IP3 receptors have been identified in fungal genomes. In the filamentous fungus Neurospora crassa, the existence of a functional IP3 receptor equivalent has been inferred on the basis of an IP3-dependent increase in cytoplasmic Ca2+. N. crassa PLC is thought to be activated by membrane stretch, generating IP3, which triggers Ca2+ release from hyphal tip vesicles, thus maintaining a high Ca2+ concentration required for hyphal tip growth (6). In S. cerevisiae, however, transient IP3 increase in nitrogen-depleted cells following addition of ammonium sulfate did not trigger a spike in intracellular calcium (7). In glucose-starved S. cerevisiae, glucose addition caused PLC-dependent influx of extracellular calcium accompanied by a transient increase in IP3. However, it is unclear as to whether calcium influx is triggered by IP3 or via direct interaction of PLC with a plasma membrane calcium channel (8, 9).
In fungi, increased cytosolic calcium triggered by stress or morphological changes activates the protein phosphatase calcineurin via calcium-bound calmodulin. In C. neoformans, calcineurin responds to stress caused by cell-wall-perturbing agents, host physiological temperature, high CO2, alkaline pH, and high cation concentration. Moreover, calcineurin is essential for virulence and hyphal elongation during mating and monokaryotic fruiting (10, 11). In S. cerevisiae, calcineurin conducts most of its transcriptional regulation via activation of the zinc finger transcription factor Crz1. In C. neoformans, the Crz1 ortholog is also activated by calcineurin and, during starvation, by protein kinase C, to regulate cell wall integrity (12, 13).
An alternative fate of PLC-generated IP3 in S. cerevisiae is phosphorylation by inositol polyphosphate kinases (IPKs). The resulting inositol polyphosphates (IPs) influence chromatin remodeling, transcriptional regulation, mRNA nuclear export, telomere length, vacuole morphogenesis, endocytosis, and cell division via an effect on kinetochore activity (14; for review, see reference 15). In S. cerevisiae, the single IPK Arg82 converts Plc1-derived IP3 into IP4 and IP5, which can be further phosphorylated to IP6 by Ipk1. Kcs1 and Vip1 then convert IP6 to different isoforms of the inositol pyrophosphate (PP-IP) PP-IP5. PP-IPs contain two phosphates on the same position of the inositol ring. Arg82 and Kcs1 can also use IP5 as a substrate to generate two isoforms of PP-IP4 (16). Similar to deletion of PLC1 in C. neoformans, deletion of ARG82 in S. cerevisiae results in a pleiotropic phenotype. The S. cerevisiae ARG82 deletion mutant (ScΔarg82) abnormalities include temperature sensitivity, sterility, and defective sporulation. We therefore hypothesized that the key role of Plc1 in C. neoformans is to produce IP3 as a precursor for the synthesis of more complex IPs and that the deletion of the ARG82 ortholog in C. neoformans would produce a pleiotropic phenotype similar to that of the CnΔplc1 mutant due to the lack of IP4–8 species in both mutants.
In this study, we investigated the biochemical mechanism underlying Plc1-mediated signaling in C. neoformans. Our findings suggest that the main function of cryptococcal Plc1 is to supply IP3 as a substrate for an inositol polyphosphate kinase, Arg1 and that the catalytic activity of Arg1 is essential for cellular homeostasis and virulence of C. neoformans.
MATERIALS AND METHODS
Strains and media.
Wild-type (WT) C. neoformans var. grubii strain H99 (serotype A, MATa) was used in this study. The Δcna1 mutant and the KN99 (MATα) strain were a kind gift from Joseph Heitman's laboratory (Duke University, Durham, NC). The Δmpk1 mutant was also a gift from Jenny Lodge's laboratory (Washington University, St. Louis, MO). C. neoformans strains were routinely cultured on YPD medium (1% yeast extract, 2% peptone, 2% dextrose). Mating was induced on 5% V8 juice (pH 5)–2% agar plates. Minimal medium (MM) (15 mM glucose, 10 mM MgSO4, 29.4 mM KH2PO4, 13 mM glycine, 3 μM thiamine) was used for capsule induction. MM agar plates supplemented with 1 mM l-DOPA (l-3,4-dihydroxyphenylalanine) were used to assess melanization.
CnPlc1 expression in S. cerevisiae.
The C. neoformans PLC1 (CNAG_02867) coding sequence (see Table 1 for primer sequences) was amplified from a C. neoformans cDNA library (a gift from Peter Williamson, NIH), cloned into the pYES2/NT vector (Life Technologies), and transformed into S. cerevisiae strain INVSc1. Expression of CnPLC1 was induced by growing the cells in synthetic defined (SD) medium without uracil, replacing glucose with galactose as the sole carbon source. For Plc1 protein purification, yeast cells were processed as previously described (5). In brief, the cells were lysed in buffer A (50 mM Tris [pH 7.9], 400 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], protease inhibitor cocktail) by vigorous vortexing with 0.5-mm glass beads. The protein extracts were clarified by centrifugation and subjected to Ni2+-affinity chromatography. The eluted fractions that were demonstrated to contain Plc1 protein were combined and dialyzed against buffer B (25 mM HEPES-HCl [pH 7.2], 50 mM NaCl, 1 mM EDTA).
Table 1.
Primers used in this study
| Descriptiona | Primerb |
|||
|---|---|---|---|---|
| Sense | Sequence | Antisense | Sequence | |
| PLC1 expression in S. cerevisiae | PLC1-BamHI-s | gccgccggatccATTGCTCTTACCGATGAGATTTATTTCC | PLC1-XbaI-a | gccgcctctagaTTAAATCAAACGTGATTTCTCACG |
| Verification of Δarg1 transformants | Trp-s | CTACAGACAACAATACCATCCTTCC | ActP-a | TGTTGTTACCATCATCCTCTCCTC |
| Verification of Δarg1 transformants | ARG1-ots-s | TGCCTATAATCCATGGTTCG | ARG1-ots-a | TCCTTTTCTTCCCCTCGTCT |
| ARG1 deletion construct and Δarg1::ARG1 rec gDNA fragment | ARG1-5′-s | CAAGGAGGAGCCATGATTTG | ARG1-5′-a-NEO | ctccagctcacatcctcgcaGCGGTGGACTGTTTGAAAGGA |
| ARG1 deletion construct and Δarg1::ARG1 rec gDNA fragment | ARG1-3′-s-NEO | tcctcaggatcttcatggctccGTTGAACGGGGTTGTGTTTGA | ARG1-3′-a | AGCAGGATCAGCAGGAAAGA |
| Neomycin resistance cassette | NEO-s | CTGCGAGGATGTGAGCTGGAG | NEO-a | GGAGCCATGAAGATCCTGAGG |
| Internal ARG1 primers | ARG1-s | CCGACCCCGAAAACTCACCA | ARG1-a | CGCTCGTCTTTCGTCCTTCTTC |
| ARG2 deletion construct | ARG2-5′-s | TACTGCCGACTCAACCCAAC | ARG2-5′-a-NEO | ctccagctcacatcctcgcaGCGAAAGTCTGGCAGGTTCTG |
| ARG2 deletion construct | ARG2-3′-s-NEO | cctcaggatcttcatggctccGCGCCATGATACAGCAGAG | ARG2-3′-a | ATGCCGAGTGACGTGATTTC |
| Verification of Δarg2 transformants | ARG2-ots-s | AAAGCACACGATTGCATGAT | ARG2-ots-a | TGGCTACGTAATGGCTGGAC |
| Internal ARG2 primers | Arg2-s | AAACTAGGGAAACGATGGTGGG | Arg2-a | GTCTCCAATCCATCGTCAACACC |
| CHS6 expression | CHS6-s | TCATCAGAGGAAGCAAGCCACA | CHS6-2 | CTTCAACTTATCCAGCACCTCCTC |
| ACT1 (housekeeping gene) | ACT-s | GCCCAGTCTTCTCAGCTTGAAA | ACT1-a | ACTTTCGGTGGACGATTGAGG |
rec, reconstituted; gDNA, genomic DNA.
The lowercase letters represent sequence with no homology to template DNA, whereas homologous regions are shown by uppercase letters.
PLC activity assay.
The PLC-specific activity of purified CnPlc1 and the crude protein extracts was determined using a radiometric enzyme assay, with PI or PIP2 as the substrate, as described previously (5) with modifications. For the PIP2 hydrolysis assay, the substrate was prepared by combining 10 nmol of cold l-α-phosphatidylinositol 4,5-diphosphate (Sigma; P9763) dissolved in 20 μl chloroform-methanol-water–1 N HCl (20:10:1:1 [vol/vol]) with 1.5 μl (0.015 μCi) of hot phosphatidylinositol-4,5-biphosphate [inositol-2-3H(N)] (Perkin-Elmer; NET895005UC) per reaction. PIP2 substrate mix was dried under nitrogen and resuspended in 25 μl of 2× PIPLC-PIP2 assay buffer (PPAB) (1.6 mM Triton X-100, 100 mM HEPES [pH 7.0], 200 mM NaCl, 2 mM EGTA, 2 mM CaCl2) per reaction. Substrate dissolution was aided by sonication in a water bath sonicator (Soniclean, South Australia, Australia) for 10 min with occasional vortexing. Each assay was initiated by the addition of 25 μl of substrate-PPAB to 5 to 25 μg protein. The final volume of the reaction mixture was adjusted to 50 μl. Incubation was carried out at 30°C for 15 min. The tubes were placed on ice, and 100 μl of 1% bovine serum albumin (BSA) was added to each tube followed by 250 μl of 10% trichloroacetic acid (TCA). The tubes were centrifuged, and the 3H radioactivity in the supernatants (350 μl) was determined by scintillation counting.
To determine the PIPLC activity using PI substrate, the assay was conducted at 37°C in a final volume of 100 μl. For each reaction, 25 nmol of cold l-α-phosphatidylinositol (PI) (Sigma; P8443) dissolved in chloroform and 0.125 μl (0.025 μCi) of phosphatidylinositol, l-α-[myo-inositol-2-3H(N)] (Perkin-Elmer; NET862010UC) were dried under nitrogen and resuspended in 50 μl of 2× PIPLC-PI assay buffer (PIAB) (0.2% sodium deoxycholate, 100 mM HEPES [pH 7.0], 200 mM NaCl, 4 mM EGTA, 0.1% BSA). Substrate dissolution was aided by sonication in a water bath sonicator (Soniclean, South Australia, Australia) for 10 min with occasional vortexing. The assay was initiated by adding 50 μl of substrate-PIAB to 10 to 25 μg protein in a final reaction volume of 100 μl. The reaction mixtures were incubated at 37°C for 10 min and terminated by adding 0.5 ml chloroform-methanol-HCl (100:100:0.6 [vol/vol]) followed by 0.15 ml of 1 M HCl containing 5 mM EGTA and vigorous vortexing. The reaction tubes were centrifuged at 21,000 × g for 1 min to achieve phase separation. 3H radioactivity in the aqueous phase (250 μl) was determined by scintillation counting.
PIP2 hydrolysis was quantified as follows: μmol PIP2 hydrolyzed/min/mg of protein = ([released cpm − blank cpm]/total cpm) × 0.01 μmol PIP2/(mg of protein × 15 min). Similarly, PI hydrolysis was quantified as follows: μmol PI hydrolyzed/min/mg of protein = ([released cpm − blank cpm]/total cpm) × 0.025 μmol PI/(mg of protein × 10 min). “Blank” refers to a reaction mixture without protein, which was included in each assay to determine the level of spontaneous substrate breakdown.
IP3 and PIP2 quantification.
C. neoformans cultures grown overnight in YPD broth were pelleted, resuspended in water to an optical density at 600 nm (OD600) of 10, spotted onto YPD plates, and incubated overnight at 30°C. Forty to 80 mg of cryptococcal cells was scraped from the plates, weighed, quenched by adding 4 ml of ice-cold (<−20°C) 60% methanol, and vortexed vigorously (7, 9). Quenched cells were pelleted by centrifugation and resuspended in ice-cold 4% perchloric acid (1 ml per 75 mg cells). To release IP3, glass beads were added to the tubes, and the cells were homogenized in a bead beater (4× 30-s cycles of beating at 4°C at 1-min intervals). Soluble and insoluble fractions were separated by centrifugation (5 min at 10,000 rpm at 4°C) to quantify IP3 and PIP2, respectively.
The pH of the IP3-containing supernatant was adjusted to 7.5 by titration with ice-cold 1.5 M KOH–60 mM HEPES. pH was monitored during the titration using a universal pH indicator strip. The KClO4 formed during the titration was sedimented by centrifugation at 2,000 × g for 15 min at 4°C. The inositol-1,4,5-trisphosphate [3H] radioreceptor assay kit (PerkinElmer Life Sciences) was then used to measure the IP3 concentration in the supernatant (100 μl/reaction) following the manufacturer's instructions.
To quantify PIP2, the insoluble pellet obtained by centrifugation of the homogenized cells was washed with 1.5 ml of ice-cold water, resuspended in 940 μl chloroform-methanol-HCl (80:40:1 [vol/vol]), and vortexed for 15 min. The lipid and aqueous phases were resolved by adding 310 μl chloroform and 560 μl 0.1 M HCl, vortexing, and centrifugation at 1,000 × g for 15 min at room temperature. Four hundred microliters of the lower phase was dried under a stream of nitrogen. The dried samples were dissolved in 250 μl of 1 M KOH and heated at 100°C for 15 min to hydrolyze PIP2. After cooling on ice, the samples were supplemented with 15 μl HEPES (1 M [pH 7.2]) and further neutralized to pH 7.5 by titration with ice-cold 4% perchloric acid. The samples were centrifuged at 2,000 × g for 15 min at 4°C to remove KClO4 sediment. The IP3 produced as a result of PIP2 hydrolysis (100 μl of supernatant/reaction) was quantified using the inositol-1,4,5-trisphosphate [3H] radioreceptor assay kit (PerkinElmer Life Sciences).
Generation of transgenic strains.
Plasmid pJAF (containing a neomycin resistance [NEOR] cassette) was kindly provided by John R. Perfect, Duke University, Durham, NC. The ARG1 and ARG2 gene deletion constructs were made by overlap PCR, joining the 5′ flank, NEOR from pJAF (ACT1 promoter, neomycin phosphotransferase, TRP1 terminator) and the 3′ flank (see Fig. S1A in the supplemental material for diagram and Table 1 for primer sequences). Transformation was carried out by the biolistic method (17). NEO-resistant transformants were selected on YPD agar plates supplemented with 0.5 M sorbitol and 100 μg/ml G418. Correctly targeted integration of the ARG1 and ARG2 deletion constructs was confirmed by PCR amplification across the junction point of integration of the construct with genomic DNA (gDNA), using a forward primer that anneals outside the region of integration and a reverse primer that anneals within the construct (see Fig. S1 in the supplemental material). The Δarg1:ARG1 reconstituted strain was created by transforming the Δarg1 mutant with a genomic fragment of ARG1 that included 1,169 bp upstream and 1,133 bp downstream of the coding region (primers 5′-s to 3′-a). To select for ARG1-expressing colonies, transformed cells were plated on YPD agar supplemented with 0.5 M sorbitol and 0.005% Congo red and incubated at 37°C. To confirm that the colonies which grew under these conditions were true Δarg1:ARG1 reconstituted strains, the cells were tested for the presence of both ectopically integrated ARG1 and the ARG1 deletion cassette by PCR (see Fig. S1).
Virulence in Galleria mellonella.
C. neoformans WT and mutant strains were grown overnight, pelleted by centrifugation, and resuspended in phosphate-buffered saline (PBS) at a concentration of 108 cells/ml. G. mellonella larvae (10 per strain) were inoculated with 10 μl of cell suspension (106 yeast cells) by injection into the hemocoel via the lower pro-legs. The viability of each inoculum was assessed by performing serial 10-fold dilutions, plating the lowest dilutions on Sabouraud agar (SAB) plates, and counting the CFU after 3 days of incubation at 30°C. Inoculated larvae were monitored daily for 10 days. The Kaplan-Meier method in the SPSS version 21 statistical software was used to estimate the differences in survival (log rank test) and to plot the survival curves. In all cases, a P value of <0.05 was considered statistically significant.
The number of viable fungal cells in larvae was determined by homogenizing each larva in 1 ml PBS and preparing and plating serial dilutions of each homogenate as described for the inoculum.
Gene expression.
To compare levels of chitin synthase CHS6 (CNAG_00546) expression, the cells were cultured in YPD overnight at 30°C and diluted to an OD of 0.5. After 4 h of incubation, the cells were treated with calcofluor white (CFW) (2.2 mg/ml) with or without FK506 (10 μg/ml) for 1 h. The cells were collected and snap-frozen. RNA extraction and cDNA synthesis were performed as described previously (12). Real-time PCR was performed using the actin-encoding gene (ACT1) for normalization.
Microscopy.
For staining of C. neoformans vacuoles, the cells were grown in YPD overnight, diluted 1:10 in fresh medium, and incubated for 4 h. The cells were pelleted and resuspended in 50 mM sodium citrate buffer (pH 5), supplemented with 1 μM carboxy-DCFDA[carboxy-5(and-6)-chloromethyl-2′,7-dichlorodihydrofluorescein diacetate] (Life Technologies), and incubated for 20 min. Measurements of vacuolar size were performed using ImageJ software (NIH). For the FM-4-64 pulse-chase experiment, the YPD-grown cells were loaded with 10 μM FM-4-64 in YPD for 5 min (time point 0), pelleted, and resuspended in fresh YPD medium. The tubes were incubated at 30°C with shaking. Aliquots were withdrawn at the indicated times, and the cells were pelleted and kept on ice until observed by microscopy.
For F-actin staining, the cells were grown overnight in YPD, pelleted and fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. The cells were washed twice with PBS and permeabilized with 1% Triton X-100 in PBS for 8 min. Following permeabilization, the cells were washed twice with PBS and incubated with 1% BSA in PBS for 20 min, followed by staining with Alexa Fluor 488-conjugated phalloidin in PBS (Life Technologies; 25 U/100 μl cells) for 90 min. DAPI (4′,6-diamidino-2-phenylindole; 1 μg/ml) was added for the final 3 min of the 90-min incubation with Alexa Fluor 488-conjugated phalloidin.
RESULTS
CnPlc1 preferentially hydrolyzes PIP2.
To establish that Plc1 is enzymatically active and to determine its substrate preference, we expressed recombinant His-tagged CnPLC1 in S. cerevisiae under the regulation of a galactose-inducible promoter (Fig. 1A). The enzyme was then purified by nickel affinity chromatography. Purified enzyme and crude cell lysates were assayed for PI-PLC hydrolytic activity using PI or PIP2 as a substrate in the presence or absence of calcium (Fig. 1B). No activity was obtained when calcium was omitted from the buffer (not shown). However, in the presence of calcium, PIP2, but not PI, was hydrolyzed by both pure and crude CnPlc1, confirming that PIP2 is a preferred substrate of CnPlc1. These data support our previous finding where the PIP2 hydrolytic activity associated with Δplc1 lysates was significantly lower than the PI hydrolytic activity compared to the respective WT levels (4). Preference for PIP2 indicates that CnPlc1 can produce both IP3 and DAG, rather than DAG alone, as would occur if PI was the preferred substrate.
Fig 1.

The recombinant CnPlc1 strain preferentially hydrolyzes PIP2. (A) Expression of HIS-tagged CnPLC1 in S. cerevisiae. CnPLC1-expressing and control strains were grown under noninducing (glucose) and inducing (galactose) conditions. Lysates were subjected to SDS-PAGE and Western blotting with antibodies against the histidine tag. Sacch-EV, empty pYES2/NT vector; Sacch-LacZ, β-galactosidase-expressing positive control; Sacch-CnPLC1(1) and Sacch-CnPLC1(2), two CnPLC1-expressing recombinants. (B) Purified CnPlc1 and crude lysates were assayed for hydrolytic activity against PI or PIP2, and release of 3H-inositol from each substrate was measured by β-counting. Results represent means ± standard errors (SE) (n ≥ 3). *, statistically significant difference relative to Sacch-CnPlc1 lysate and nickel affinity (His)-purified Plc1 (P < 0.001, using a two-tailed t test). #, statistically significant difference relative to nickel affinity (His)-purified Plc1 (P = 0.01).
Identification of the major inositol polyphosphate kinase (Arg1) in C. neoformans.
In S. cerevisiae, PLC-derived IP3 is converted to IP4 and IP5 by the inositol polyphosphate multikinase Arg82 (16, 18). Using the S. cerevisiae protein in a similarity search, we identified two Arg82 homologs in the C. neoformans genome: CnArg1 (CNAG_06500; NCBI accession no. AFR98730) and CnArg2 (CNAG_02802; NCBI accession AFR93890), which share 22% and 15% identity, respectively, with ScArg82, and 17% identity with each other (see Fig. S2 in the supplemental material for alignment). Similar to ScArg82, CnArg1 and CnArg2 contain a conserved PDKG motif essential for the catalytic activity of IP3 kinases (19, 20).
To determine whether CnArg1 and CnArg2 are functional IP3 kinases, we created ARG1 and ARG2 deletion mutants (CnΔarg1 and CnΔarg2 strains, respectively. We then compared the IP3 contents of CnΔarg1 and CnΔarg2 cells with those of the WT and CnΔplc1 mutant. Due to the absence of functional CnPlc1, we expected the IP3 content of CnΔplc1 to be reduced. Due to the absence of functional IP3 kinase, we expected to see an accumulation of Plc1-derived IP3 in CnΔarg1 and/or CnΔarg2 cells. Consistent with our predictions, IP3 content was reduced 3-fold in CnΔplc1 cells and markedly increased (>5-fold) in CnΔarg1 cells (Fig. 2A). In contrast to the CnΔarg1 strain, the IP3 content in the CnΔarg2 cells was similar to that in the WT (see Fig. S3A in the supplemental material). To ascertain the authenticity of the CnΔarg1 phenotype, we created the CnΔarg1:ARG1 reconstitution strain by transforming CnΔarg1 with a genomic ARG1 fragment (see Fig. S1 in the supplemental material). The IP3 content in the CnΔarg1:ARG1 cells was intermediate between those of the WT and CnΔarg1 mutant, consistent with partial restoration of the Δarg1 phenotype by the introduction of ARG1. Accumulation of IP3 in the CnΔarg1 mutant establishes CnArg1 as the major IP3 kinase in C. neoformans.
Fig 2.
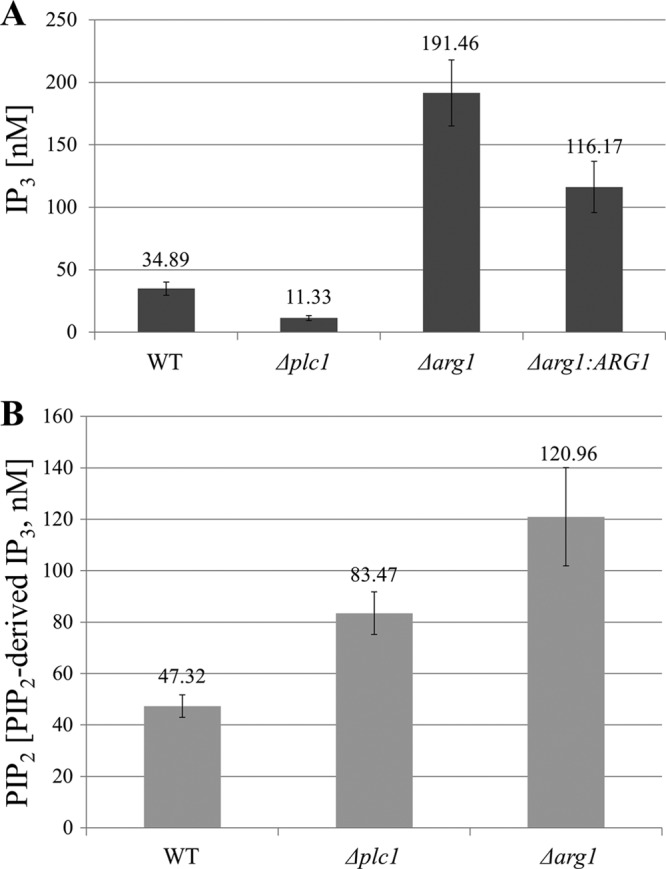
Quantification of IP3 (A) and PIP2 (B) in the Δplc1, Δarg1, and WT strains. Results represent the means and standard deviations of three IP3 measurements. The differences in IP3 and PIP2 contents between any two strains are statistically significant (P < 0.01 using the two-tailed t test). Each experiment was performed 3 times with similar results. In panels A and B, IP3 was measured using the IP3 assay kit. In panel B, membrane-associated PIP2 was quantified indirectly by measuring IP3 released by alkaline hydrolysis.
In addition to reduced IP3 content, deletion of PLC1 potentially leads to accumulation of its PIP2 substrate, with a resultant deleterious effect on yeast cells (21). We therefore measured the PIP2 membrane content in Δplc1 and Δarg1 cells indirectly, by measuring IP3 produced by alkaline hydrolysis of PIP2 (Fig. 2B). Surprisingly, PIP2 accumulation was even more pronounced in Δarg1 cells than in Δplc1 cells. This finding implies product inhibition of Plc1 activity in Δarg1 mutants by the high concentration of IP3.
Arg1 is essential for virulence, cell wall integrity, and mating in C. neoformans.
Changes in IP3 content in Δplc1 and Δarg1 cells (Fig. 2A) demonstrate that Plc1 and Arg1 function within the same signaling pathway, with Plc1 providing a substrate (IP3) for Arg1. Arg1 catalytic products and their derivatives are expected to be absent in both mutants, as is the case in S. cerevisiae (22, 23). To assess the role of the Arg1 protein and its products in virulence and homeostasis, we characterized the Δarg1 phenotype, as compared to the Δplc1 mutant and the wild-type strain.
Similar to the Δplc1 mutant, growth of the Δarg1 mutant was delayed under normal growth conditions (30°C, YPD). However, growth of both strains was markedly inhibited at human physiological temperature (37°C) and in the presence of the cell-wall-perturbing agent Congo red (Fig. 3A). Melanization on minimal medium containing l-DOPA (Fig. 3B) and the formation of mating filaments in a unilateral cross with KN99 (MATa) (Fig. 3C) were also reduced. In the case of temperature stress and mating, the phenotype of the Δarg1:ARG1 strain was completely restored to that of the WT. However, in the case of Congo red sensitivity and melanization, the phenotype of the Δarg1:ARG1 strain was intermediate between those of the WT and Δarg1 strains, consistent with partial restoration of the Δarg1 phenotype by the ectopically integrated ARG1. Both the Δplc1 and Δarg1 mutants formed smaller capsules than the WT cells under capsule-inducing conditions, with capsule size restored to that of WT in the Δarg1:ARG1 strain (Fig. 3D). Unlike the Δarg1 mutant, the Δarg2 strain exhibited WT-like tolerance to elevated temperature and cell wall stress (see Fig. S3B in the supplemental material). The Δarg2 strain also produced melanin and capsules similar to the WT strain (see Fig. S3C and D). The lack of phenotypic defects in the Δarg2 strain is consistent with unaltered IP3 content in this mutant (see Fig. S3A).
Fig 3.
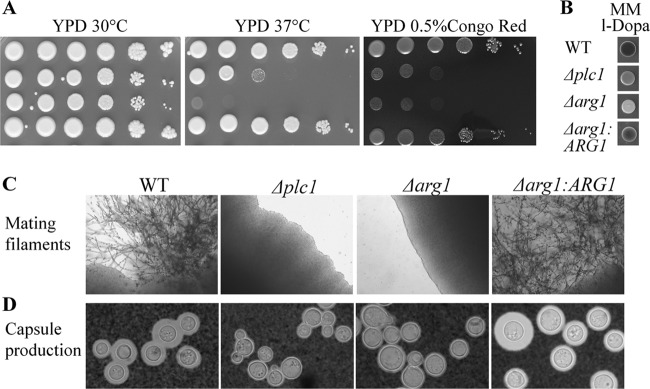
Phenotypic characterization of the Δarg1 mutant compared to the Δplc1, WT, and Δarg1:ARG1 strains. (A) WT H99 and the Δarg1, Δarg1:ARG1, and Δplc1 strains (serially diluted 10-fold from 106 to 10 cells/spot from left to right) were spotted on YPD plates and incubated at 30°C or 37°C and onto a Congo red-supplemented plate to assess cell wall integrity at 30°C. (B) Melanization of WT strain H99 and the mutant strains was compared on minimal medium supplemented with the laccase substrate l-DOPA. (C) WT H99 and the Δarg1, Δarg1:ARG1, and Δplc1 (MATα) strains were crossed with KN99 (MATa) on V8 agar plates and incubated for 10 days to assess formation of mating filaments. (D) Wild-type and mutant strains were grown in minimal medium (broth) to induce capsule production.
The virulence of the Δarg1 and Δplc1 mutants was compared with that of the WT in larvae of the greater wax moth, Galleria mellonella. The G. mellonella infection model was chosen over the mouse inhalational model as larvae can be maintained at the permissive growth temperature of the mutant strains (30°C). At the mouse body temperature (∼37°C), the growth of both mutants was severely compromised (Fig. 3A). Larvae were inoculated with 1 × 106 cryptococci. The inoculum was plated out to quantify the inoculum as CFU. These were determined to be 0.8 × 106, 0.5 × 106, and 1.1 × 106 CFU for the WT, Δplc1 mutant, and Δarg1 mutant, respectively. While all of the larvae inoculated with WT C. neoformans died within 3 days, Δarg1 strain-inoculated insects died more slowly (median survival of ∼5 days) (Fig. 4). Unlike the Δarg1 mutant, infection with the Δplc1 strain caused the death of only 1 larva out of 10 over the entire infection period. Survival of the larvae was analyzed using the Kaplan-Meier method (log rank test). Overall, the difference in survival rates was statistically significant (P < 0.001). For the pairwise comparisons, the survival rates of PBS-treated and Δplc1 strain-infected larvae were comparable (P = 0.317). Survival of PBS-treated and Δplc1 strain-infected larvae was significantly greater than that of Δarg1 strain-infected larvae (P = 0.001), which in turn was significantly greater than that of the WT-infected larvae (P < 0.001).
Fig 4.
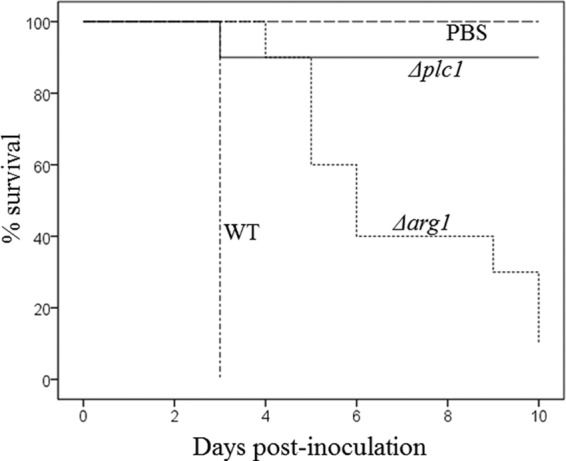
Comparison of the virulence of Δplc1, Δarg1, and WT strains using a Galleria mellonella infection model. G. mellonella larvae were inoculated with the WT and mutant strains as described in Materials and Methods. The health of the larvae was monitored for 10 days, and their deaths were recorded.
Fungal cells were retrieved from infected insects and quantified. WT cells had propagated dramatically within larvae that had succumbed to infection on day 3 postinoculation, with ∼128-fold more CFU recovered compared with the number inoculated. The Δarg1 cells also propagated within larvae, but not to the same extent as the WT, with ∼25-fold more CFU recovered from larvae at death on day 4 postinoculation. Interestingly, the Δplc1 cells did not propagate in larvae, as similar CFU to those inoculated were recovered from healthy larvae on day 6 postinoculation.
Altered vacuolar morphology, cell separation, and endocytosis in Δplc1 and Δarg1 mutants.
We previously demonstrated that Δplc1 mutant cells had irregular morphology and contained enlarged vacuoles, compared with the WT strain (4). Similar to the Δplc1 cells, Δarg1 cells appeared to have enlarged vacuoles (Fig. 3D). To further investigate vacuolar morphology, vacuolar lumens were stained with carboxy-DCFDA and vacuole size and number per cell were documented (Fig. 5A and B). Cells of all strains contained numerous small vacuoles. However, Δarg1 cells often contained one markedly enlarged vacuole, implying excessive vacuolar fusion in this mutant (Fig. 5A, arrowheads). The average vacuole size in Δarg1 cells (2.2 μm) is 3-fold larger than that in WT cells (0.63 μm), while the vacuoles of Δplc1 cells (average size, 0.97 μm) are only slightly larger than the vacuoles of the WT (Fig. 5B). In all strains, the number of vacuoles per cell inversely correlated with vacuole size. On average, WT cells contained 3.5 vacuoles, compared to 1.3 and 2 vacuoles in Δarg1 and Δplc1 cells, respectively (Fig. 5B).
Fig 5.
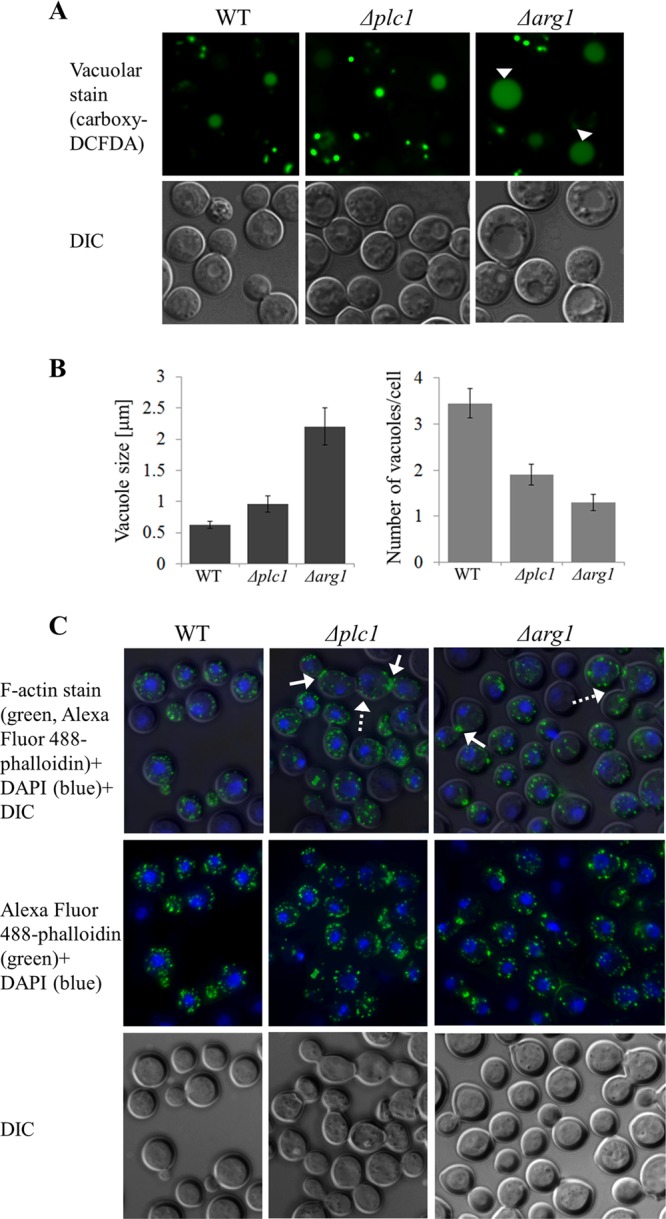
Vacuolar morphology and cytoskeletal organization in Δplc1, Δarg1, and WT cells. (A) C. neoformans cells grown in YPD were stained with the vacuolar lumen marker carboxy-DCFDA. Large vacuoles in Δarg1 cells are indicated by arrowheads. (B) Vacuole size and number per cell were quantified in the WT and mutant strains, as indicated. Bars represent the mean and standard error (n = 20 cells for each strain). The differences between the WT and Δarg1 strains and between the WT and Δplc1 strains are statistically significant (P ≤ 0.05 using a two-tailed Mann-Whitney test) (C) YPD-grown cells were fixed and stained with Alexa Fluor 488-phalloidin (green) to visualize F-actin and DAPI (blue) to visualize nuclei. Broken arrows indicate multicellular aggregates in Δarg1 and Δplc1 cells. Solid arrows indicate actomyosin rings separating mother and daughter cells.
Δplc1 and Δarg1 cells often formed aggregates of three or four conjoined cells (Fig. 5C, broken arrows), while WT cells were only joined in pairs. Since defects in morphology and cell separation might reflect abnormal polarized growth, WT, Δplc1, and Δarg1 cells were stained with Alexa Fluor 488-conjugated phalloidin to visualize F-actin distribution. In all strains, actin patches were distributed throughout the cytosol of mature cells and accumulated in the emerging buds, as described previously (24). In the WT, most of the conjoined cells were premitotic, with nuclei visible only in the mother cell. However, in the Δplc1 and Δarg1 mutants, cells often failed to separate after mitosis, and nuclei were visible in both mother and daughter cells. Furthermore, brightly stained actomyosin rings were often observed at the junctions between these conjoined mutant cells (Fig. 5C, solid arrows). These actomyosin rings eventually disappeared, and the actin patches became redistributed throughout the conjoined cells. Despite being joined, these cells sometimes formed new buds. Actomyosin rings were rarely observed in WT cells. A similar cell separation defect accompanied by increased localization of F-actin in the medial ring was observed in Schizosaccharomyces pombe deficient in the IP5 2-kinase, Ipk1 (25).
In S. cerevisiae, inositol pyrophosphates regulate endocytic trafficking (23). As we expected inositol pyrophosphates to be absent from the CnΔplc1 and CnΔarg1 mutants, we investigated the dynamics of endocytosis in these mutants by tracking internalization of the lipophilic dye FM4-64 (Fig. 6). The cells were labeled with the dye for 5 min and then chased with fresh medium for different time periods to follow the progress of endocytosis. After a 10-min chase, FM4-64 staining in WT cells was visible in small vesicles and endosomal and, possibly, vacuolar membranes, while in Δarg1 cells, FM4-64 had reached the membranes of large vacuoles, a hallmark of this mutant (Fig. 5). In most Δplc1 cells, FM4-64 was still associated with the plasma membrane after a 10-min chase. Between 10 and 60 min, the FM4-64 staining patterns of WT and Δarg1 cells remained largely unchanged. In the Δplc1 mutant, the dye became visible inside the cells after a 40-min chase, and by 60 min had stained the endosomal and vacuolar membranes (Fig. 6).
Fig 6.
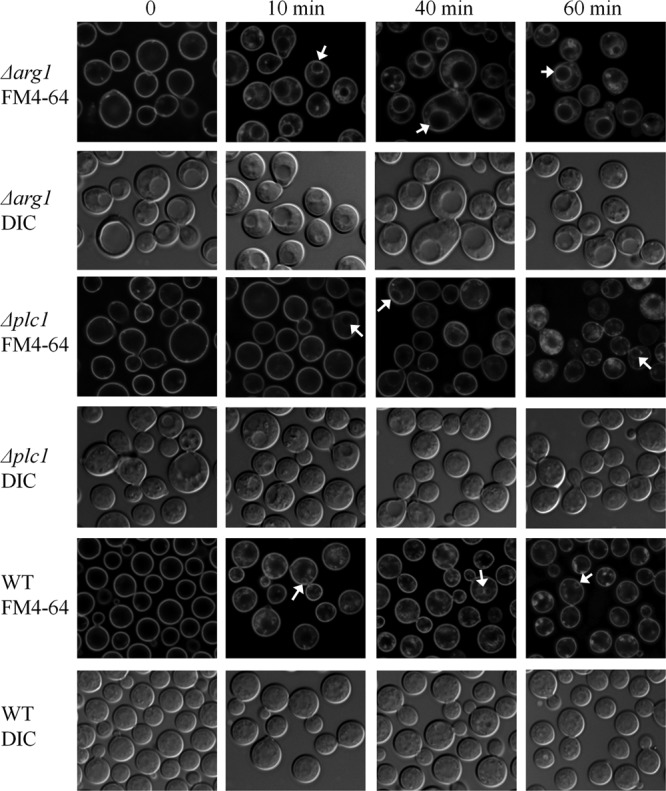
Tracking endocytosis in Δplc1, Δarg1, and WT cells with FM4-64. Internalization of the lipophilic dye FM4-64 was observed immediately after staining (time point 0) and at 10, 40, and 60 min of incubation in fresh medium. Arrows indicate vacuoles labeled with FM4-64. DIC, differential interference contrast.
CnPlc1 does not contribute significantly to calcineurin activation.
In mammalian cells, PLC1 is a well-established regulator of IP3-mediated calcium release into the cytosol and cytosolic calcium activates calcineurin. In C. neoformans, the Δplc1 mutant shares several phenotypic characteristics with the Δcna1 calcineurin deletion mutant: enlarged cells at 30°C, sensitivity to Congo red, growth retardation at 37°C, and a mating defect (Fig. 3) (4, 10, 12). However, there are also significant phenotypic differences between these mutants: only the Δcna1 mutant is sensitive to calcofluor white (CFW) (see below) and has enlarged capsules (our unpublished observation).
To determine whether Plc1 is essential for calcineurin activation, we investigated whether the Δplc1 mutant defects, which are common to the Δcna1 strain, could be rescued by supplying exogenous calcium to the Δplc1 growth medium. Our previous findings established that, similar to other fungal species, C. neoformans calcineurin is activated by the addition of exogenous calcium to the growth medium (12). Furthermore, in filamentous fungi, addition of calcium restored some of the defects observed in the Δplc1 mutant, such as growth rate in Cryphonectria parasitica, and formation of infection structures (appressoria) in Magnaporthe oryzae (26, 27). In S. cerevisiae, addition of calcium to the medium allowed the Δplc1 mutant to grow at 38°C (28). However, addition of calcium to the C. neoformans Δplc1 mutant (50 mM or 5 mM CaCl2) did not restore mating ability, growth at 37°C, or resistance to cell wall stress, indicating that impaired calcineurin activation in the absence of Plc1 is not the cause of the defects observed in this mutant (Fig. 7).
Fig 7.
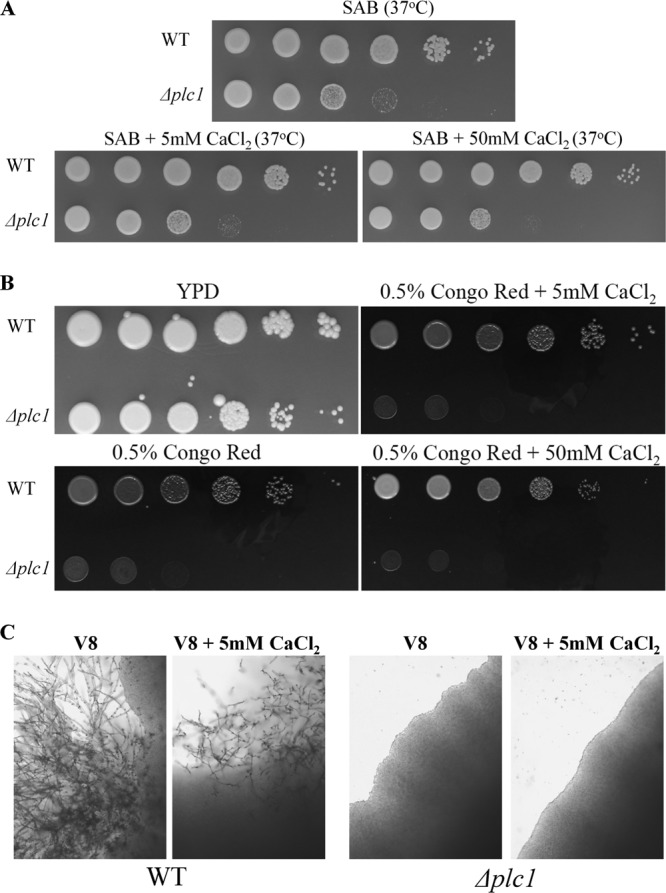
Addition of exogenous calcium does not rescue the high-temperature growth, cell wall, and mating defects of the Δplc1 mutant. (A) Sabouraud agar (SAB) was supplemented with 5 or 50 mM CaCl2. The plates were spotted with WT and Δplc1 cells (serially diluted 10-fold from 106 to 10 cells/spot from left to right) and incubated at 37°C. (B) YPD plates were supplemented with Congo red and CaCl2 as indicated. Fungal cells were spotted onto the plates as described in panel A, and the plates were incubated at 30°C. (C) WT and Δplc1 (MATα) strains were crossed with the KN99 (MATa) strain on V8 agar plates with or without 5 mM CaCl2 to assess formation of mating filaments.
We previously demonstrated that the expression of the chitin synthase-encoding gene, CHS6, is upregulated in response to cell wall perturbation with CFW and that CHS6 expression requires functional calcineurin (12). To assess the activity of calcineurin in the absence of Plc1, we tested CHS6 expression in the Δplc1 mutant, compared to that in the WT and Δcna1 strains. CHS6 expression was reduced ∼2-fold in the Δplc1 mutant, compared to the WT. However, upon addition of the calcineurin inhibitor FK506, CHS6 expression dropped by ∼12- and 25-fold in the WT and Δplc1 strains, respectively. The reduction of CHS6 expression in the Δplc1 strain indicates that calcineurin is active in this mutant (Fig. 8A).
Fig 8.
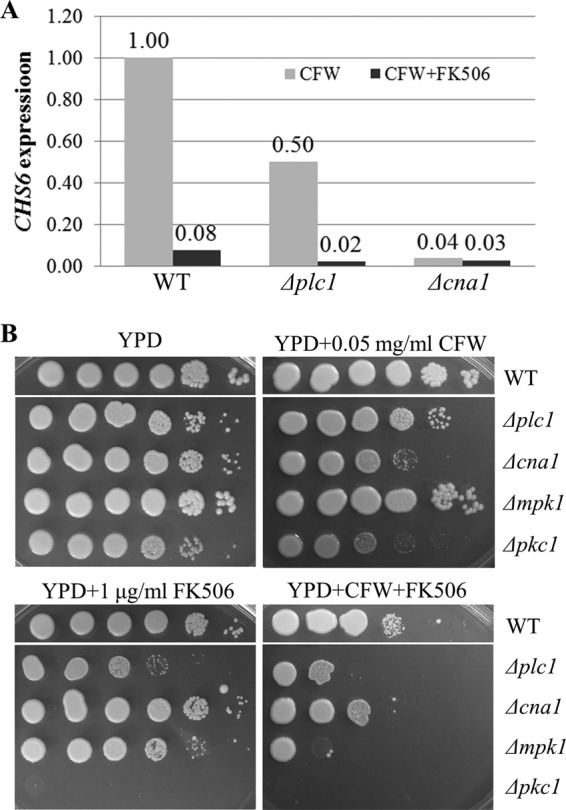
Inhibition of calcineurin in the Δplc1 mutant causes reduced expression of the calcineurin-dependent chitin synthase gene, CHS6, and growth retardation. (A) WT, Δplc1, and Δcna1 strains were grown in YPD broth and incubated for 1 h with calcofluor white (CFW) (2.2 mg/ml) in the presence or absence of the calcineurin inhibitor FK506 (10 μg/ml). CHS6 expression was quantified by quantitative reverse transcription-PCR (qRT-PCR), and the results are expressed relative to the WT plus CFW, which has been normalized to 1.00. (B) Cell suspensions of WT strain H99, the Δplc1 mutant, and the Δmpk1 and Δpkc1 cell wall integrity mutants were serially diluted 10-fold (from 106 to 10 cells/spot from left to right) and spotted onto YPD plates supplemented with 0.5 M sorbitol (essential for Δpkc1 cell growth). CFW and FK506 were added as indicated.
Furthermore, growth of the Δplc1 mutant was inhibited in the presence of FK506 at 30°C, while the Δcna1 mutant grew normally, indicating that calcineurin is essential at this growth temperature in the absence of Plc1 (Fig. 8B). Moreover, calcineurin inhibition caused the Δplc1 mutant to become more sensitive to CFW than the Δcna1 mutant. In C. neoformans, protein kinase C regulates cell wall integrity via activation of the mitogen-activated protein (MAP) kinase cascade, in which Mpk1 is a terminal kinase (29, 30). This pathway functions independently of calcineurin, although cross talk between the two pathways has been observed (30, 31). Similar to the Δplc1 mutant, growth of the cell wall integrity pathway mutants (Δpkc1 and Δmpk1) was inhibited by FK506 in the presence and absence of CFW. These findings suggest that Plc1, calcineurin, and the Pkc1/Mpk1 cell wall integrity pathway function in parallel to maintain cell wall integrity. Although we cannot rule out the possibility of cross talk between the Plc1 and calcineurin pathways, collectively our results suggest it is unlikely that Plc1 contributes significantly to calcineurin activation in C. neoformans.
DISCUSSION
In this report, we present for the first time evidence that metabolism of inositol polyphosphates is essential for the phenotypic expression of virulence determinants in human fungal pathogens. We have studied this pathway in the basidiomycete C. neoformans and show that the main biochemical and cellular role of Plc1 is to produce IP3 as a substrate for the inositol polyphosphate kinase Arg1. We have shown that Plc1 and Arg1, and therefore the inositol polyphosphate anabolic pathway, are key regulators of several important virulence attributes of C. neoformans, including cell wall integrity, growth at elevated temperature, melanin production, and capsule biosynthesis, suggesting a broad role for inositol polyphosphates in cellular homeostasis, growth, and pathogenicity of C. neoformans. Two further important findings from our study are that, unlike in mammalian cells, CnPlc1-derived IP3 does not contribute significantly to calcineurin activation and that IP3 homeostasis in C. neoformans regulates cellular function differently from that in the nonpathogenic model yeast, S. cerevisiae.
We have demonstrated that CnPlc1 has similar catalytic activity to the mammalian PLC-δ isoform since it preferentially hydrolyzes PIP2 over PI in a Ca2+-dependent manner, to produce IP3 and DAG. CnPlc1 activity is consistent with the activity and substrate preference of other eukaryotic PLC enzymes and the fact that hallmark domains of eukaryotic PLC proteins, the X and Y catalytic domains, and a C2 calcium binding motif are present in CnPlc1 (32). In mammalian cells, the C2 motif is involved in calcium-dependent binding of PLC to membrane phospholipids and orientation of the catalytic domain toward the membrane surface (33). The ability of CnPlc1to bind to the plasma membrane via its C2 domain would enable Plc1 to sense and respond to cell surface perturbations arising due to changes in the external environment. C. neoformans also has a second PLC, Plc2, which has a catalytic domain similar to bacterial PLCs, suggesting the functional similarity to prokaryotic enzymes. However, deletion of PLC2 produced no changes in cellular function or virulence (4).
We identified two putative inositol polyphosphate multikinases in the C. neoformans genome, designated Arg1 and Arg2, with Arg1 being the closest homolog of ScArg82. Accumulation of IP3 in the Δarg1 mutant, but not in the Δarg2 mutant, established Arg1 as the major IP3 kinase in C. neoformans. This observation coupled with the fact that IP3 content is reduced in Δplc1 cells, confirms that there is a block within the IP-processing pathway in both mutants. These data also demonstrate that Plc1 and Arg1 function within the same signaling pathway, with Plc1 providing a substrate (IP3) for Arg1.
While accumulation of the Plc1 substrate PIP2 was expected in Δplc1 cells, even higher PIP2 levels were detected in Δarg1 cells. A similar phenomenon was observed in the ScΔarg82 mutant (23). It is possible that the excessive IP3 in CnΔarg1 competes with PIP2 for binding to the C2 domain of Plc1 and thus inhibits Plc1 enzyme activity. This hypothesis is supported by the finding that the C2 domain-containing PLC of a squid binds IP3 (34). A functionally similar phenomenon of feedback inhibition was described for mammalian PLC-δ1, which binds PIP2-containing membranes via its pleckstrin homology domain. The IP3 produced following receptor-stimulated activation of PLC-δ1 antagonizes its binding to PIP2, causing translocation of PLC-δ1 from the plasma membrane to the cytosol and rendering it inactive (35, 36).
Common phenotypic defects of the Δarg1 and Δplc1 mutants, including sterility, compromised cell wall, thermosensitivity, reduced capsule size, and melanization, may be caused by accumulation of PIP2 or absence of complex IPs. In S. cerevisiae, the lack of detectable PP-IPs in Δarg82 and Δkcs1 mutants is thought to be responsible for vacuolar fragmentation, salt stress sensitivity, and a defective cell wall (22). However, excessive accumulation of PIP2 is also detrimental, as deletion of PIP2-dephosphorylating synaptojanins in S. cerevisiae caused defects in actin organization, endocytosis, and clathrin-mediated sorting between the Golgi apparatus and endosomes (21). It should be noted that despite the overall phenotypic similarity between the C. neoformans Δplc1 and Δarg1 mutants, the extent of each defect was not the same. The Δarg1 mutant was more sensitive than the Δplc1 mutant to a growth temperature of 37°C and did not produce any detectable melanin, while the Δplc1 mutant was slightly pigmented. The virulence of the Δplc1 mutant and the propagation in moth larvae were markedly reduced, while the Δarg1 mutant propagated and caused insect death, albeit more slowly than the WT. These differences may be attributed to the opposing trend in IP3 content in the two mutants and/or the more significant accumulation of PIP2 in the Δarg1 mutant.
Consistent with defects in pathogenicity-related phenotypes, virulence of both Δplc1 and Δarg1 strains was attenuated in G. mellonella, which is a proven invertebrate model of cryptococcosis (37). Larvae infected with the Δplc1 mutant survived longer than larvae infected with the Δarg1 mutant, despite the Δplc1 strain having a higher growth rate than the Δarg1 strain at 30°C in vitro on a standard growth medium. This difference in survival correlated with the extent of propagation of each strain within the host: while both the WT and Δarg1 strains underwent significant propagation, the Δplc1 strain did not. Differences in the ability of the Δplc1 and Δarg1 strains to assimilate nutrients and/or maintain viability within the host may be responsible for the difference in the virulence between these mutants, as the nutritional environment in larval hemocoel differs significantly from that of standard liquid media used to propagate yeast in vitro. In support of this hypothesis, the ScΔplc1 strain exhibits compromised ability to assimilate galactose, raffinose, and glycerol (5) and rapidly loses viability in the absence of a nitrogen source (5).
As Plc1-mediated hydrolysis of PIP2 also generates DAG, it is possible that some of the phenotypic abnormalities of the Δplc1 mutant are caused by the reduction in DAG content. Moreover, if Plc1 is inhibited in the Δarg1 mutant by the excess of IP3, the same defect might be expected in both mutants. However, we propose that it is unlikely that a reduction in DAG content in both mutants is responsible for their phenotypic similarity: first, as the S. cerevisiae Δplc1, Δarg1, and Δkcs1 strains share a significant degree of similarity, the absence of PP-IPs is more likely to be the major cause of common phenotypic defects in these mutants (38, 39). Second, PLCs is not the only source of DAG in C. neoformans: DAG is also produced by Ipc1 (inositol-phosphorylceramide synthase-1), an enzyme of the sphingolipid biosynthetic pathway. Ipc1-derived DAG was shown to facilitate C. neoformans protein kinase C activation and subsequent melanization (40, 41).
In S. cerevisiae, the absence of Plc1, Arg82, and Kcs1 caused an increase in cell size and vacuolar fragmentation. Although the cells of the C. neoformans Δarg1 mutant were also enlarged, we observed the opposite defect in vacuolar morphology: one large vacuole dominated the mutant cells, in contrast to multiple small vacuoles observed in the WT. This defect was significantly more pronounced in the Δarg1 cells than in the Δplc1 cells. As PIP2 plays an essential role in vacuole fusion in S. cerevisiae (42), elevated PIP2 in the C. neoformans Δarg1 strain and, to a lesser extent, in the Δplc1 strain may be responsible for the enlarged vacuoles in these mutants.
We observed a dramatic cell separation defect in both Δplc1 and Δarg1 cells. Postmitotic mother and daughter cells remained fused, often forming new buds. Interestingly, a similar defect was observed in an S. pombe Δipk1 mutant, but not in S. cerevisiae (25). S. pombe Δipk1 cells were defective in dissolution of the septum and the cell wall surrounding the septum, resulting in accumulation of binuclear cells with a medial division septum. This defect was attributed to the absence of IP6 and IP7 in the S. pombe Δipk1 strain, a feature likely to be shared by the C. neoformans Δplc1 and Δarg1 mutants. As phosphoinositides and IPs are involved in multiple aspects of intracellular trafficking, we tracked the endocytic pathway in C. neoformans using the lipophilic styryl dye FM4-64. Endocytosis is initiated at the plasma membrane, which invaginates to form an endocytic vesicle. The endocytic vesicle is then transferred to an endosomal compartment that subsequently fuses with vacuoles. However, in the S. cerevisiae Δarg1 and Δkcs1 mutants, FM4-64 accumulated in aberrant endosomal intermediates juxtaposed to vacuoles (23). In contrast, the C. neoformans Δarg1 mutant accumulated FM4-64 in large vacuoles, which were typical for this mutant, but rarely appeared in wild-type cells. Interestingly, the internalization of FM4-64 was considerably slower in Δplc1 cells than in wild-type and Δarg1 cells. The striking differences in vacuolar morphology and endocytosis between the S. cerevisiae Δarg82 and C. neoformans Δarg1 mutants, despite the similarity of their IP3/PIP2 profiles, imply differences in the regulation of vacuole fusion and/or intracellular trafficking in pathogenic basidiomycetes such as C. neoformans and the nonpathogenic ascomycete S. cerevisiae.
In mammalian cells, Plc1-derived IP3 serves as a precursor for complex IPs, but it also triggers calcium release and subsequent calcineurin activation. Yeast, however, does not possess orthologs of mammalian IP3 receptors, and the ability of PLC to activate calcineurin in yeast is unclear. The limited phenotypic similarity between the CnΔplc1 mutant and the calcineurin (Δcna1) mutant clearly indicates that calcineurin does not depend solely on Plc1 for its activation. We therefore tested the possibility that Plc1 partially contributes to calcineurin activation. Inhibition of calcineurin in the Δplc1 strain at 25°C caused growth retardation, indicating that under conditions that do not normally require calcineurin function, calcineurin is essential to compensate for the absence of Plc1. We previously demonstrated that the chitin synthase-encoding gene CHS6 is regulated by calcineurin and highly expressed in the presence of calcofluor white (12). Although expression of CHS6 was lower in the Δplc1 strain than in the wild type, inhibition of calcineurin in the Δplc1 strain caused a dramatic reduction in CHS6 expression, indicating that calcineurin is active in the Δplc1 strain. To test whether some of the Δplc1 abnormalities, which are shared by the Δcna1 mutant, are due to reduced calcineurin activation, we attempted to rescue these defects by providing extracellular calcium, which activates calcineurin in C. neoformans (12). However, none of the Δplc1 defects tested was rectified by extracellular calcium. Taken together, these findings suggest that Plc1 does not contribute significantly to calcineurin activity. Similar to Plc1 and Cna1, Arg1 is essential for growth at 37°C, formation of mating filaments, and cell wall integrity. It is therefore likely that the Plc1/IPK pathway functions in parallel to calcineurin to regulate these essential functions, although there may potentially be cross talk between the two pathways. This redundancy most likely contributes to the robustness of C. neoformans and its success as a pathogen.
In summary, our study shows that C. neoformans Plc1 produces IP3, which serves as a precursor for the synthesis of more complex IPs by the inositol polyphosphate multikinase Arg1. Arg1-generated IPs are likely to be further phosphorylated, as the C. neoformans genome encodes inositol hexa- and heptakisphosphate kinases. Plc1, Arg1, and their products play multiple roles in cellular homeostasis and have a dramatic impact on virulence of C. neoformans. The role of IPKs and their products has never been addressed in pathogenic fungi, and our findings lay the foundation for investigation of the mechanisms by which IPKs and IP metabolism promote fungal virulence.
Supplementary Material
ACKNOWLEDGMENTS
We thank Wieland Meyer and Carolina Firacative for assistance with propagation and inoculation of G. mellonella larvae and Karen Byth for help with statistical analysis of the survival study.
This work was supported by a University of Sydney Bridging grant to J.T.D. T.C.S. is a Sydney Medical School Foundation Fellow.
Footnotes
Published ahead of print 4 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01421-12.
REFERENCES
- 1. Kozubowski L, Heitman J. 2012. Profiling a killer, the development of Cryptococcus neoformans. FEMS Microbiol. Rev. 36:78–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kronstad JW, Attarian R, Cadieux B, Choi J, D'Souza CA, Griffiths EJ, Geddes JM, Hu G, Jung WH, Kretschmer M, Saikia S, Wang J. 2011. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat. Rev. Microbiol. 9:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kozubowski L, Lee SC, Heitman J. 2009. Signalling pathways in the pathogenesis of Cryptococcus. Cell. Microbiol. 11:370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chayakulkeeree M, Sorrell TC, Siafakas AR, Wilson CF, Pantarat N, Gerik KJ, Boadle R, Djordjevic JT. 2008. Role and mechanism of phosphatidylinositol-specific phospholipase C in survival and virulence of Cryptococcus neoformans. Mol. Microbiol. 69:809–826 [DOI] [PubMed] [Google Scholar]
- 5. Flick JS, Thorner J. 1993. Genetic and biochemical characterization of a phosphatidylinositol-specific phospholipase C in Saccharomyces cerevisiae. Mol. Cell. Biol. 13:5861–5876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silverman-Gavrila LB, Lew RR. 2002. An IP3-activated Ca2+ channel regulates fungal tip growth. J. Cell Sci. 115:5013–5025 [DOI] [PubMed] [Google Scholar]
- 7. Bergsma JC, Kasri NN, Donaton MC, De Wever V, Tisi R, de Winde JH, Martegani E, Thevelein JM, Wera S. 2001. PtdIns(4,5)P(2) and phospholipase C-independent Ins(1,4,5)P(3) signals induced by a nitrogen source in nitrogen-starved yeast cells. Biochem. J. 359:517–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tisi R, Baldassa S, Belotti F, Martegani E. 2002. Phospholipase C is required for glucose-induced calcium influx in budding yeast. FEBS Lett. 520:133–138 [DOI] [PubMed] [Google Scholar]
- 9. Tisi R, Belotti F, Wera S, Winderickx J, Thevelein JM, Martegani E. 2004. Evidence for inositol triphosphate as a second messenger for glucose-induced calcium signalling in budding yeast. Curr. Genet. 45:83–89 [DOI] [PubMed] [Google Scholar]
- 10. Odom A, Muir S, Lim E, Toffaletti DL, Perfect J, Heitman J. 1997. Calcineurin is required for virulence of Cryptococcus neoformans. EMBO J. 16:2576–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cruz MC, Fox DS, Heitman J. 2001. Calcineurin is required for hyphal elongation during mating and haploid fruiting in Cryptococcus neoformans. EMBO J. 20:1020–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lev S, Desmarini D, Chayakulkeeree M, Sorrell TC, Djordjevic JT. 2012. The Crz1/Sp1 transcription factor of Cryptococcus neoformans is activated by calcineurin and regulates cell wall integrity. PLoS One 7:e51403 doi:10.1371/journal.pone.0051403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adler A, Park YD, Larsen P, Nagarajan V, Wollenberg K, Qiu J, Myers TG, Williamson PR. 2011. A novel specificity protein 1 (SP1)-like gene regulating protein kinase C-1 (Pkc1)-dependent cell wall integrity and virulence factors in Cryptococcus neoformans. J. Biol. Chem. 286:20977–20990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perera NM, Michell RH, Dove SK. 2004. Hypo-osmotic stress activates Plc1p-dependent phosphatidylinositol 4,5-bisphosphate hydrolysis and inositol hexakisphosphate accumulation in yeast. J. Biol. Chem. 279:5216–5226 [DOI] [PubMed] [Google Scholar]
- 15. Tsui MM, York JD. 2010. Roles of inositol phosphates and inositol pyrophosphates in development, cell signaling and nuclear processes. Adv. Enzyme Regul. 50:324–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang T, Caffrey JJ, Shears SB. 2001. The transcriptional regulator, Arg82, is a hybrid kinase with both monophosphoinositol and diphosphoinositol polyphosphate synthase activity. FEBS Lett. 494:208–212 [DOI] [PubMed] [Google Scholar]
- 17. Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. 1993. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J. Bacteriol. 175:1405–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. El Alami M, Messenguy F, Scherens B, Dubois E. 2003. Arg82p is a bifunctional protein whose inositol polyphosphate kinase activity is essential for nitrogen and PHO gene expression but not for Mcm1p chaperoning in yeast. Mol. Microbiol. 49:457–468 [DOI] [PubMed] [Google Scholar]
- 19. Bertsch U, Deschermeier C, Fanick W, Girkontaite I, Hillemeier K, Johnen H, Weglohner W, Emmrich F, Mayr GW. 2000. The second messenger binding site of inositol 1,4,5-trisphosphate 3-kinase is centered in the catalytic domain and related to the inositol trisphosphate receptor site. J. Biol. Chem. 275:1557–1564 [DOI] [PubMed] [Google Scholar]
- 20. Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. 1999. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr. Biol. 9:1323–1326 [DOI] [PubMed] [Google Scholar]
- 21. Stefan CJ, Audhya A, Emr SD. 2002. The yeast synaptojanin-like proteins control the cellular distribution of phosphatidylinositol (4,5)-bisphosphate. Mol. Biol. Cell 13:542–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dubois E, Scherens B, Vierendeels F, Ho MM, Messenguy F, Shears SB. 2002. In Saccharomyces cerevisiae, the inositol polyphosphate kinase activity of Kcs1p is required for resistance to salt stress, cell wall integrity, and vacuolar morphogenesis. J. Biol. Chem. 277:23755–23763 [DOI] [PubMed] [Google Scholar]
- 23. Saiardi A, Sciambi C, McCaffery JM, Wendland B, Snyder SH. 2002. Inositol pyrophosphates regulate endocytic trafficking. Proc. Natl. Acad. Sci. U. S. A. 99:14206–14211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kopecka M, Gabriel M, Takeo K, Yamaguchi M, Svoboda A, Ohkusu M, Hata K, Yoshida S. 2001. Microtubules and actin cytoskeleton in Cryptococcus neoformans compared with ascomycetous budding and fission yeasts. Eur. J. Cell Biol. 80:303–311 [DOI] [PubMed] [Google Scholar]
- 25. Sarmah B, Wente SR. 2009. Dual functions for the Schizosaccharomyces pombe inositol kinase Ipk1 in nuclear mRNA export and polarized cell growth. Eukaryot. Cell 8:134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rho HS, Jeon J, Lee YH. 2009. Phospholipase C-mediated calcium signalling is required for fungal development and pathogenicity in Magnaporthe oryzae. Mol. Plant Pathol. 10:337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chung HJ, Kim MJ, Lim JY, Park SM, Cha BJ, Kim YH, Yang MS, Kim DH. 2006. A gene encoding phosphatidyl inositol-specific phospholipase C from Cryphonectria parasitica modulates the lac1 expression. Fungal Genet. Biol. 43:326–336 [DOI] [PubMed] [Google Scholar]
- 28. Payne WE, Fitzgerald-Hayes M. 1993. A mutation in PLC1, a candidate phosphoinositide-specific phospholipase C gene from Saccharomyces cerevisiae, causes aberrant mitotic chromosome segregation. Mol. Cell. Biol. 13:4351–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gerik KJ, Bhimireddy SR, Ryerse JS, Specht CA, Lodge JK. 2008. PKC1 is essential for protection against both oxidative and nitrosative stresses, cell integrity, and normal manifestation of virulence factors in the pathogenic fungus Cryptococcus neoformans. Eukaryot. Cell 7:1685–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kraus PR, Fox DS, Cox GM, Heitman J. 2003. The Cryptococcus neoformans MAP kinase Mpk1 regulates cell integrity in response to antifungal drugs and loss of calcineurin function. Mol. Microbiol. 48:1377–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kojima K, Bahn YS, Heitman J. 2006. Calcineurin, Mpk1 and Hog1 MAPK pathways independently control fludioxonil antifungal sensitivity in Cryptococcus neoformans. Microbiology 152:591–604 [DOI] [PubMed] [Google Scholar]
- 32. Bruzik KS, Tsai MD. 1994. Toward the mechanism of phosphoinositide-specific phospholipases C. Bioorg. Med. Chem. 2:49–72 [DOI] [PubMed] [Google Scholar]
- 33. Ochocka AM, Pawelczyk T. 2003. Isozymes delta of phosphoinositide-specific phospholipase C and their role in signal transduction in the cell. Acta Biochim. Pol. 50:1097–1110 [PubMed] [Google Scholar]
- 34. Kishigami A, Ogasawara T, Watanabe Y, Hirata M, Maeda T, Hayashi F, Tsukahara Y. 2001. Inositol-1,4,5-trisphosphate-binding proteins controlling the phototransduction cascade of invertebrate visual cells. J. Exp. Biol. 204:487–493 [DOI] [PubMed] [Google Scholar]
- 35. Downes CP, Gray A, Fairservice A, Safrany ST, Batty IH, Fleming I. 2005. The regulation of membrane to cytosol partitioning of signalling proteins by phosphoinositides and their soluble headgroups. Biochem. Soc. Trans. 33:1303–1307 [DOI] [PubMed] [Google Scholar]
- 36. Hirose K, Kadowaki S, Tanabe M, Takeshima H, Iino M. 1999. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science 284:1527–1530 [DOI] [PubMed] [Google Scholar]
- 37. Mylonakis E, Moreno R, El Khoury JB, Idnurm A, Heitman J, Calderwood SB, Ausubel FM, Diener A. 2005. Galleria mellonella as a model system to study Cryptococcus neoformans pathogenesis. Infect. Immun. 73:3842–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Auesukaree C, Tochio H, Shirakawa M, Kaneko Y, Harashima S. 2005. Plc1p, Arg82p, and Kcs1p, enzymes involved in inositol pyrophosphate synthesis, are essential for phosphate regulation and polyphosphate accumulation in Saccharomyces cerevisiae. J. Biol. Chem. 280:25127–25133 [DOI] [PubMed] [Google Scholar]
- 39. Seeley ES, Kato M, Margolis N, Wickner W, Eitzen G. 2002. Genomic analysis of homotypic vacuole fusion. Mol. Biol. Cell 13:782–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heung LJ, Kaiser AE, Luberto C, Del Poeta M. 2005. The role and mechanism of diacylglycerol-protein kinase C1 signaling in melanogenesis by Cryptococcus neoformans. J. Biol. Chem. 280:28547–28555 [DOI] [PubMed] [Google Scholar]
- 41. Heung LJ, Luberto C, Plowden A, Hannun YA, Del Poeta M. 2004. The sphingolipid pathway regulates Pkc1 through the formation of diacylglycerol in Cryptococcus neoformans. J. Biol. Chem. 279:21144–21153 [DOI] [PubMed] [Google Scholar]
- 42. Mayer A, Scheglmann D, Dove S, Glatz A, Wickner W, Haas A. 2000. Phosphatidylinositol 4,5-bisphosphate regulates two steps of homotypic vacuole fusion. Mol. Biol. Cell 11:807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.