

Abstract
The bacterial pathogen Listeria monocytogenes causes serious food-borne illnesses in pregnant women and the immunocompromised. L. monocytogenes promotes its internalization into host epithelial cells and then uses an F-actin-dependent motility process to spread from infected cells to surrounding healthy cells. In cultured enterocytes, efficient spread of L. monocytogenes requires the secreted bacterial protein InlC. InlC promotes dissemination by physically interacting with and antagonizing the function of the human adaptor protein Tuba. Here we examine the role of InlC and its interaction with host Tuba during infection in mice. The study took advantage of a single-amino-acid substitution (K173A) in InlC that impairs binding to human Tuba but does not affect InlC-mediated inhibition of the NF-κB pathway. Mice were inoculated intravenously with the wild-type L. monocytogenes strain EGD, an isogenic strain deleted for the inlC gene (ΔinlC), or a strain expressing K173A mutant InlC (inlC.K173A). The 50% lethal doses (LD50) for the ΔinlC or inlC.K173A mutant strain were approximately 4- or 6-fold greater than that for the wild-type strain, indicating a role for inlC in virulence. Compared to the wild-type strain, the inlC.K173A mutant strain exhibited lower bacterial loads in the liver. Histological analysis of livers indicated that the two inlC mutant strains produced smaller foci of infection than did the wild-type strain. These smaller foci are consistent with a role for InlC in cell-to-cell spread in vivo. Taken together, these results provide evidence that interaction of InlC with host Tuba is important for full virulence.
INTRODUCTION
Listeria monocytogenes is a food-borne, intracellular pathogen that causes gastroenteritis, abortions, or meningitis with a high mortality rate (1, 2). L. monocytogenes replicates within mammalian cells and spreads from one cell to another through a motility process that is dependent on the host F-actin cytoskeleton (3). Spreading is initiated by the bacterial surface protein ActA, which stimulates the formation of F-actin “comet tails” that propel L. monocytogenes through the cytoplasm. Motile bacteria contact the inner surface of the host cell plasma membrane and induce the formation of pathogen-containing protrusions that are engulfed by neighboring cells. Cell-to-cell spread is critical for virulence, since an L. monocytogenes strain containing a null mutation in the actA gene is severely compromised for disease in a mouse model (4). The 50% lethal dose (LD50) for this actA mutant is approximately 3 logs higher than that of the isogenic wild-type (wt) strain (4).
ActA is thought to be required for spreading in all cell types. In cells that do not form tight barriers, such as macrophages or fibroblasts, actin-dependent motility may be sufficient for formation of bacterial protrusions (5). In these cell types, the force provided by motility may be the sole factor controlling protrusion generation. However, many tissues infected by L. monocytogenes contain polarized cells connected by cell-cell junctions. Such tissues include enterocytes lining the intestinal lumen, hepatocytes, and the brain endothelium (2). In polarized enterocytes, the secreted bacterial protein InlC acts after ActA-mediated comet tail formation to enhance the production of L. monocytogenes protrusions (6). InlC stimulates protrusion formation by antagonizing the function of a human signaling protein called Tuba. Tuba is a large scaffolding protein that contains several functional domains, including a carboxyl-terminal Src homology 3 (SH3) domain that interacts with the human actin regulatory protein N-WASP (7). Genetic and biochemical data indicate that InlC inhibits host Tuba by binding to this SH3 domain, thereby disrupting Tuba/N-WASP complexes (6). How InlC-mediated inhibition of host Tuba and N-WASP leads to enhanced protrusion formation is not fully understood. Based on the effects of L. monocytogenes infection on the morphology of apical junctions in enterocytes, it was hypothesized that InlC might dissipate cortical tension, thereby reducing the force required to deform the plasma membrane into protrusions (6).
To date, InlC is one of only two bacterial proteins known to bind to mammalian SH3 domains. The other bacterial ligand of an SH3 domain is the enteropathogenic Escherichia coli (EPEC) protein EspF, which interacts with the mammalian trafficking protein SNX9 (8). SH3 domains are common and extensively studied protein-protein interaction motifs in eukaryotes (9). Binding of eukaryotic SH3 domain ligands is mediated by short peptide sequences of the consensus PxxP (where x is any amino acid) or, less commonly, other sequences, such as PxxDY or RxxK (9). Interestingly, interaction of InlC with the Tuba SH3 domain involves an RxxK sequence in InlC (6). Replacement of the lysine residue in this sequence with an alanine results in an InlC protein (InlC.K173A) that folds normally but is partly defective in binding to the Tuba SH3 domain (6). The InlC.K173A protein binds to the SH3 domain of human Tuba at about 1/6 the affinity of wild-type InlC. Importantly, an L. monocytogenes inlC.K173A strain expressing the mutant InlC protein is compromised for protrusion formation and cell-to-cell spread in the polarized human enterocyte cell line Caco-2 BBE1 (6). These results indicate that efficient spreading of L. monocytogenes in cultured enterocytes requires the ability of InlC to interact with the carboxyl-terminal SH3 domain of Tuba.
In addition to interacting with Tuba, InlC affects host gene expression mediated by the transcription factor NF-κB (10). NF-κB stimulates expression of genes involved in cell growth, cell survival, neuronal development, and the immune response (11). InlC associates with host IKKα, a positive regulator of NF-κB. InlC-IKKα interaction prevents NF-κB activation, thereby suppressing expression of proinflammatory cytokines that would otherwise restrict Listeria infection (10).
The primary goal of this study was to investigate the effect of the inlC.K173A mutation on L. monocytogenes virulence in a mouse model. If this mutation were found to reduce virulence, then such a result would provide evidence for the idea that the interaction of InlC with host Tuba is important for pathogenesis. InlC binds the carboxy-terminal SH3 domain of mouse Tuba, as well as that of human Tuba (6), indicating that mice are an appropriate model for assessing the role of InlC-Tuba interaction in virulence. Prior to carrying out the mouse studies, we performed experiments demonstrating that the InlC.K173A mutation in InlC does not affect activation of NF-κB. These studies indicated that the inlC.K173A mutant is an appropriate tool for investigating the role of InlC/Tuba interaction in virulence. By performing intravenous inoculation of mice, we found that the 50% lethal dose (LD50) for the inlC.K173A mutant strain was approximately 6-fold higher than that for the isogenic wild-type strain. These results indicate that the inlC.K173A mutation reduces virulence. In addition, compared to the wild-type bacterial strain, the inlC.K173A mutant strain was partly defective in replication in the liver. Histological analysis of the liver revealed that the inlC.K173A mutant produced foci of infection that were smaller than those made by the wild-type strain. The reduced size of these foci is consistent with a role for InlC-Tuba interaction in cell-to-cell spread in vivo. Taken together, our findings indicate that interaction of L. monocytogenes InlC with host Tuba is important for full virulence in mice.
MATERIALS AND METHODS
Bacterial strains, mammalian cell lines, and media.
The wild-type Listeria monocytogenes strain EGD and the isogenic ΔinlC and inlC.K173A mutant strains were previously described (6, 12). The ΔinlC strain has an in-frame deletion in the inlC gene (12), and the inlC.K173A strain expresses a mutant InlC protein with a lysine-to-alanine substitution at amino acid 273 (6). These strains were grown in brain heart infusion (BHI) broth and prepared for infection of cultured human cells as described previously (13). For preparation of bacterial cultures for mouse infection, L. monocytogenes strains were grown in BHI overnight and washed in phosphate-buffered saline (PBS), and frozen stocks were prepared in BHI and 40% glycerol. These frozen stocks were used to determine the numbers of CFU per milliliter. Mice were infected with bacteria from frozen stocks after thawing, washing, and resuspension in saline solution at the appropriate dilution.
Cells of the human epithelial cell line HeLa (ATTC CCL-2) were grown in Dulbecco's modified Eagle's medium (DMEM) with 4.5 g of glucose per liter and 2 mM glutamine (11996-065; Life Technologies) supplemented with 10% fetal bovine serum (FBS). Cell growth, stimulation with the cytokine tumor necrosis factor alpha (TNF-α), and bacterial infections were performed at 37°C with 5% CO2.
Antibodies, plasmids, and other reagents.
Rabbit polyclonal antibodies against recombinant InlC or the last SH3 domain in Tuba (SH36) were previously described (6). The commercially available primary antibodies used were rabbit anti-Listeria monocytogenes (223021; Becton Dickenson) and mouse anti-NF-κB (sc-8008; Santa Cruz Biotechnology). Secondary antibodies coupled to Alexa Fluor 488 or Alexa Fluor 647 and phalloidin conjugated to Alexa Fluor 555 were purchased from Life Technologies. 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was from Sigma-Aldrich. A plasmid allowing expression of enhanced green fluorescent protein (EGFP)-tagged wild-type InlC in mammalian cells (EGFP-InlCwt) was constructed by PCR amplification using chromosomal DNA from L. monocytogenes strain EGD as a template and the primers 5′-CCCAAGCTTCTGAGAGTATTCAACGACCAACG-3′ and 5′-GGGGTACCCTAATTCTTGATAGGTTGTGTAAC-3′. (The underlined sequences indicate HindIII and KpnI sites, respectively.) The resulting PCR product was subcloned into the HindIII/KpnI sites of pEGFPC1 (Clontech). A plasmid expressing EGFP-tagged InlC.K173A was constructed through site-directed mutagenesis using a Quikchange kit (Stratagene), the primers 5′-TCTTATCTATTCGTAATAATGCGTTAAAAAGTATTGTGATGC-3′ and 5′-GCATCACAATACTTTTTAACGCATTATTACGAATAGATAAGA-3′, and the EGFP-InlCwt plasmid as a template. The inlC inserts in the EGFP-InlCwt and EGFP-InlC.K173A plasmids were verified by DNA sequencing.
Analysis of NF-κB localization in cultured human cells.
For experiments assessing the effects of L. monocytogenes infection on NF-κB localization, 2 × 105 HeLa cells were seeded on 22- by 22-mm glass coverslips in 6-well plates and grown for about 2 days. Cells were then washed twice in 2 ml of PBS and either left uninfected or infected with the wild-type bacterial strain EGD, the ΔinlC strain, or the inlC.K173A strain in DMEM at a multiplicity of infection (MOI) of ∼10:1. Plates were centrifuged for 2 min at 2,000 rpm at room temperature to enhance contact between bacteria and HeLa cells. The infection was allowed to proceed for 1.5 h at 37°C in 5% CO2 in the absence of antibiotic. Cells were then washed twice in 2 ml of warm DMEM and incubated for another 3.5 h in DMEM containing 20 μg/ml gentamicin. After infection, HeLa cells were treated or not for 30 min with 25 ng/ml TNF-α. Cells were fixed, permeabilized, and labeled with primary antibodies and fluorescent secondary antibody conjugates as described previously (14, 15). The labeling reagents were mouse anti-NF-κB, anti-mouse-Alexa Fluor 488, phalloidin-Alexa Fluor 555 (F-actin staining), and DAPI (DNA staining).
For experiments analyzing the effect of ectopically expressed wild-type (wt) or mutant InlC proteins on NF-κB localization, HeLa cells seeded on 22- by 22-mm glass coverslips were transfected with 2.5 μg of plasmid DNA expressing EGFP alone, EGFP-InlCwt, or EGFP-InlC.K173A. Transfections were performed using Lipofectamine 2000 as described previously (14). Approximately 24 h after addition of plasmid DNA, cells were stimulated with 25 ng/ml TNF-α for 30 min, followed by fixation and labeling with mouse anti-NF-κB, anti-mouse-Alexa Fluor 647, phalloidin-Alexa Fluor 555, and DAPI.
Labeled samples subjected to bacterial infection or transfection with EGFP-based plasmids were analyzed by confocal laser scanning microscopy in order to quantify effects on NF-κB nuclear localization. Images were acquired with a Zeiss LSM 710 confocal microscope equipped with diode (405-nm), multiline argon (458-, 488-, and 514-nm), helium-neon 1 (543-nm), and helium-neon 2 (633-nm) lasers. In the case of bacterial infection experiments (Fig. 1), only HeLa cells containing one or more intracellular bacteria decorated with F-actin were analyzed. In experiments involving EGFP-tagged protein expression (Fig. 2A), only cells positive for EGFP were evaluated. Analysis was performed by using ImageJ (version 1.43r) software to quantify integrated pixel densities corresponding to NF-κB in the nucleus and throughout the cell. Nuclear or cell borders were identified by DAPI or phalloidin labeling, respectively. The fraction of NF-κB in the nucleus was calculated as the amount (integrated pixel density) of NF-κB in the nucleus divided by the amount in the entire cell. Approximately 30 cells were used to quantify the fraction of nuclear NF-κB for each condition in each experiment. Data from three independent experiments were used to obtain the means ± standard errors of the means shown in Fig. 1B and 2Aii.
Fig 1.

Suppression of nuclear translocation of NF-κB by wild-type and inlC.K173A strains of L. monocytogenes. HeLa cells were either left uninfected or were infected for 5 h with the wild-type (wt), ΔinlC, or inlC.K173A bacterial strain. After infection, cells were treated or not for 30 min with 25 ng/ml TNF-α. After fixation and labeling, cells were analyzed by confocal laser scanning microscopy, as described in Materials and Methods. (A) Representative images of HeLa cells subjected to various infection conditions. Asterisks indicate cells containing intracellular L. monocytogenes, as indicated by decoration with F-actin. Note that TNF-α treatment caused an increase in the amount of nuclear NF-κB in uninfected cells and that this increase was inhibited by infection with the wild-type or inlC.K173A bacterial strain. (B) Quantification of the effect of bacterial infection on nuclear translocation of NF-κB. The fraction of NF-κB in the nucleus of uninfected or infected cells was quantified as described in Materials and Methods. Statistical analysis by ANOVA indicated that P was <0.0001. *, P < 0.05 relative to condition b (uninfected cells treated with TNF-α).
Fig 2.

Inhibition of NF-κB by ectopic expression of wild-type or K173A mutant InlC. HeLa cells were transfected with plasmids expressing EGFP alone or EGFP-tagged wild-type (wt) InlC or InlC.K173A. About 24 h posttransfection, cells were stimulated for 30 min with 25 ng/ml TNF-α. (A) Effect of wild-type or K173A mutant InlC on nuclear translocation of NF-κB. After fixation and labeling of samples, confocal microscopy was used to analyze nuclear distribution of NF-κB in EGFP-expressing cells. (i) Representative images of HeLa cells expressing the various EGFP-tagged proteins. Note that expression of EGFP-InlCwt or EGFP-InlC.K173A each inhibited nuclear accumulation of NF-κB compared to expression of EGFP alone. (ii) Quantification of the effect of ectopic InlC expression on NF-κB nuclear localization. The fraction of NF-κB in the nucleus of cells expressing the various EGFP-tagged proteins was quantified as described in Materials and Methods. (B) Effect of wild-type or K173A mutant InlC on activity of an NF-κB-dependent reporter gene. Statistical analysis by ANOVA indicated that P was <0.0001. *, P < 0.05 relative to EGFP-expressing cells treated with TNF-α.
Analysis of NF-κB reporter activity.
A Cignal NF-κB luciferase reporter kit (SA Biosciences; catalog no. CCS-013L) was used to assess NF-κB transcriptional activity in HeLa cells expressing EGFP alone, EGFP-InlCwt, or EGFP-InlC.K173A. This kit permits the dual detection of an NF-κB-dependent firefly luciferase reporter and a constitutively expressed Renilla luciferase construct. A total of 4 × 104 HeLa cells were seeded in wells of 24-well plates and grown for approximately 24 h, followed by cotransfection with 250 ng of a mixture of the firefly (NF-κB-responsive) and Renilla (constitutive) luciferase constructs and also between 10 and 500 ng of plasmid DNA expressing EGFP alone, EGFP-InlCwt, or EGFP-InlC.K173A. Carrier plasmid DNA was included in order to maintain the same total amount of DNA for each transfection. Each transfection under each condition was performed in triplicate. In order to determine background firefly luciferase activity, cells were cotransfected with 250 ng of a mixture of a promoterless firefly luciferase construct and the constitutive Renilla luciferase plasmid. Transfection was performed using Lipofectamine 2000. Approximately 24 h after addition of plasmid DNA, cells were stimulated or not with 10 ng/ml TNF-α for 6 h. Cells were then washed in PBS and lysed. Samples were transferred to 96-well plates and processed using a dual-luciferase assay system kit (Promega; catalog no. E1910). A Polarstar Optima multimode microplate reader was used to measure chemiluminescence produced by the firefly and Renilla luciferase constructs. The data were expressed as the ratio of NF-κB-responsive firefly luciferase activity to constitutive Renilla luciferase activity, and background activity was subtracted. Relative expression of the NF-κB reporter presented was obtained by normalizing absolute luciferase activity values to that of EGFP-expressing cells treated with TNF-α. Data in Fig. 2B are means ± SEM of three independent experiments.
Animal infections.
Six-week-old male C57BL/6 mice purchased from Charles River were infected intravenously in tail veins with the wild-type L. monocytogenes strain EGD, the ΔinlC mutant, or the inlC.K173A mutant. The bacterial doses in LD50 studies were 1 × 104, 1 × 105, 1 × 106, and 1 × 107 CFU/mouse. Mice were assessed 14 days after inoculation. If animals exhibited the following symptoms, they were euthanized by cervical dislocation: lethargy, shaking, tachypnea, severe hunching, and ruffled fur. Pooled data from four separate experiments was used to determine LD50 values. In total, 10 to 17 mice were analyzed for each bacterial strain and dose. Fifty percent lethal doses (LD50s) were determined by probit analysis, using StatPlus for MacIntosh (version 2009). For studies measuring bacterial colonization of the liver, mice were infected with the EGD or inlC.K173A strain using a dose of 5 × 104 bacteria. At each of the 6-, 24-, 48-, or 96-h time points postinoculation, four mice infected with each L. monocytogenes strain were sacrificed. The livers of these mice were homogenized, and numbers of bacteria were determined through enumeration of CFU. Values were expressed as CFU per g of liver tissue. For histology studies, mice were infected for 24 h with the EGD, ΔinlC, or inlC.K173A strain, using a dose of 1 × 107 bacteria. Mice were then sacrificed, and liver samples were fixed in formalin. All procedures with mice were performed in accordance with the guidelines of the ethics committees at the University of Otago and the University of Toronto (U.S. Public Health Service animal welfare assurance no. A5608-01 and A5013-01, respectively).
Detection of Tuba and InlC expression.
C57BL/6 mice were either left uninfected or were infected intravenously with 1 × 107 EGD, ΔinlC, or inlC.K173A bacteria for 24 h. The livers of mice were homogenized and solubilized in lysis buffer containing 50 mM Tris (pH 8), 1% Triton X-100, 5 mM EDTA, and 150 mM NaCl with protease inhibitors. The numbers of viable bacteria were determined by enumeration of CFU. In order to detect expression of Tuba, 15 μg of liver lysate from an uninfected mouse was migrated on 10% SDS-polyacrylamide gels, transferred to polyvinylidene difluoride (PVDF) membranes, and probed with affinity-purified antibodies directed against the SH36 domain of Tuba (6). In order to detect InlC expression, volumes of liver lysates containing equal CFU (∼2 × 105) of each of the three bacterial strains were loaded on 12% SDS-PAGE gels. As a control, 15 μg of a lysate from an uninfected mouse was loaded. After migration, proteins were transferred to PVDF membranes and probed with rabbit polyclonal antibodies against InlC (6).
Histological analysis.
Fixed liver samples were stained with anti-L. monocytogenes antibodies and secondary antibodies coupled to peroxidase, followed by counterstaining with hematoxylin. Samples were inspected using an inverted bright-field microscope, and digital images of foci of infection (regions of heavily infected tissue) were acquired. ImageJ software (version 1.43r) was used to quantify surface areas of foci. For each bacterial strain, about 100 foci were analyzed in each of three independent experiments. Absolute focus sizes were converted to relative sizes by normalization to absolute values for the wild-type strain.
Statistical analysis.
Statistical analysis was performed using Prism (version 5.0a; GraphPad software). For comparison of data with three or more conditions, analysis of variance (ANOVA) was performed. The Tukey-Kramer test was used as a posttest. For comparison of two conditions, a Student t test was used. A P value of ≤0.05 was considered significant.
RESULTS AND DISCUSSION
The inlC.K173A mutation does not affect the NF-κB pathway.
In addition to interacting with Tuba, InlC is known to inhibit activation of the host transcription factor NF-κB (6, 10). Since we were interested in using the InlC.K173A mutation to specifically investigate the role of InlC-Tuba interaction in virulence, we performed experiments to determine if this mutation affects NF-κB.
NF-κB activity was assessed by measuring nuclear translocation of the transcription factor (Fig. 1 and 2A) and also by monitoring expression of an NF-κB-dependent reporter gene (Fig. 2B). It was previously reported that infection of HeLa cells with the wild-type L. monocytogenes strain EGD inhibits the nuclear translocation of NF-κB that normally occurs in response to TNF-α (10). Infection with a bacterial mutant deleted for inlC (ΔinlC) does not affect NF-κB nuclear localization, indicating an important role for InlC in this event (10). We reproduced these findings with EGD and the ΔinlC mutant by measuring the fraction of total cellular NF-κB that is present in the nucleus of HeLa cells infected with either of these two bacterial strains for 5 h followed by TNF-α stimulation (Fig. 1). Furthermore, we found that the inlC.K173A mutant was indistinguishable from the isogenic wild-type strain EGD in terms of its ability to suppress nuclear translocation of NF-κB (Fig. 1). To complement these experiments involving bacteria, we compared the ability of ectopically expressed wild-type InlC or InlC.K173A to inhibit nuclear translocation of NF-κB or expression of an NF-κB-dependent reporter gene in uninfected HeLa cells (Fig. 1B and 2). The results indicated that EGFP-tagged wild-type InlC and InlC.K173A caused similar impairments in NF-κB translocation or NF-κB-mediated gene expression. Taken together, the findings in Fig. 1 and 2 indicate that the InlC.K173A protein is not compromised in its ability to suppress NF-κB activity. These results suggest that the inlC.K173A mutant bacterial strain is an appropriate tool to specifically investigate the function of InlC-Tuba interaction in infection.
The inlC.K173A mutation reduces virulence in mice.
We examined the ability of the inlC.K173A mutant L. monocytogenes strain to cause lethal disease in mice. C57BL/6 mice were inoculated intravenously with increasing doses of either the wild-type L. monocytogenes strain EGD, the isogenic inlC.K173A mutant strain, or a mutant strain deleted for the inlC gene (ΔinlC). LD50s were determined by probit analysis (Table 1). The LD50 values for the ΔinlC or inlC.K173A strains were approximately 4- or 6-fold higher than that of the wild-type strain, respectively. A graph of the relationship between the doses of the three bacterial strains and percentages of mortality in mice is shown in Fig. 3. The increased LD50s of the two inlC mutant strains relative to the wild-type strain indicate a role for InlC in pathogenesis. The effect of the inlC.K173A mutation on LD50 supports the notion that interaction of InlC with host Tuba enhances virulence.
Table 1.
Median lethal doses of wild-type and inlC mutant strains of L. monocytogenes
| Bacterial strain | LD50 (CFU)a | LD50 (CFU) 95% confidence limit |
|
|---|---|---|---|
| Lower | Upper | ||
| Wild type | 2.44 × 105 | 7.50 × 104 | 5.12 × 105 |
| ΔinlC mutant | 9.05 × 105 | 4.55 × 105 | 1.76 × 106 |
| inlC.K173A mutant | 1.53 × 106 | 8.30 × 105 | 2.90 × 106 |
LD50 values were determined from data involving 15 to 17 mice for each bacterial strain and dose (CFU).
Fig 3.
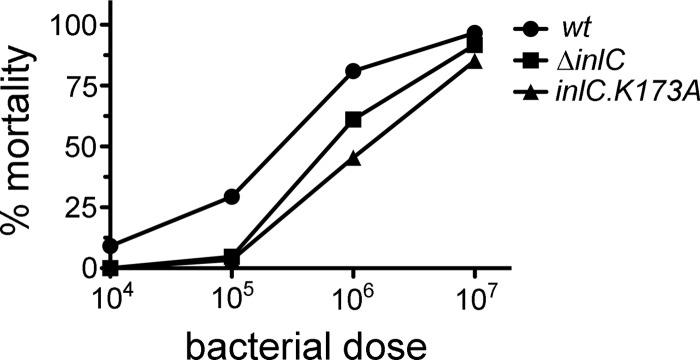
Effect of inlC mutations on mortality in mice. C57BL/6 mice were infected intravenously with the indicated doses of the wild-type (wt), ΔinlC, or inlC.K173A strain of L. monocytogenes. Viability of mice was assessed 14 days after inoculation. The graph depicts the percentages of mice surviving infection at various bacterial doses.
It is noteworthy that the 4- or 6-fold increase in LD50 due to the ΔinlC or inlC.K173A mutation is much smaller than the ∼1,000-fold increase in LD50 caused by a null mutation in actA (4). We conclude that InlC enhances virulence, rather than being essential for virulence like ActA. The relatively small effect of the two inlC mutations on LD50 is not surprising, given that the ΔinlC and inlC.K173A mutants are only partly defective in cell-to-cell spread in cultured enterocytes (6). In addition, ActA is likely required for spreading in all cell types, whereas the role of InlC might be restricted to a subset of cell types encountered during infection (e.g., polarized cells) (6). Finally, in addition to its role in cell-to-cell spread, ActA mediates escape from autophagy (16). Autophagic avoidance could potentially contribute to the critical role for ActA in virulence.
It is also pertinent to note that two previous studies reported larger effects of inlC-null mutations on virulence than those observed in our work (10, 12). In these studies, mutants deleted for inlC had LD50 values that were 40- to 50-fold higher than those of the isogenic wild-type strain. The previous studies and our experiments involved intravenous inoculation of the same mouse background. One of the earlier studies used wild-type and ΔinlC strains identical to those in our work (12). The reason for the quantitatively different virulence defects observed for inlC mutants in our experiments and previous work is unclear.
The inlC.K173A mutation affects bacterial infection of the liver.
During systemic infection of mice with L. monocytogenes, the liver serves as a major site of bacterial replication (17–20). Western blot analysis indicated that the mouse liver expresses Tuba (Fig. 4A). In addition, InlC was expressed by the wild-type or inlC.K173A strain in the liver of infected mice (Fig. 4A). We therefore examined if mutations in inlC affect bacterial infection of the liver.
Fig 4.
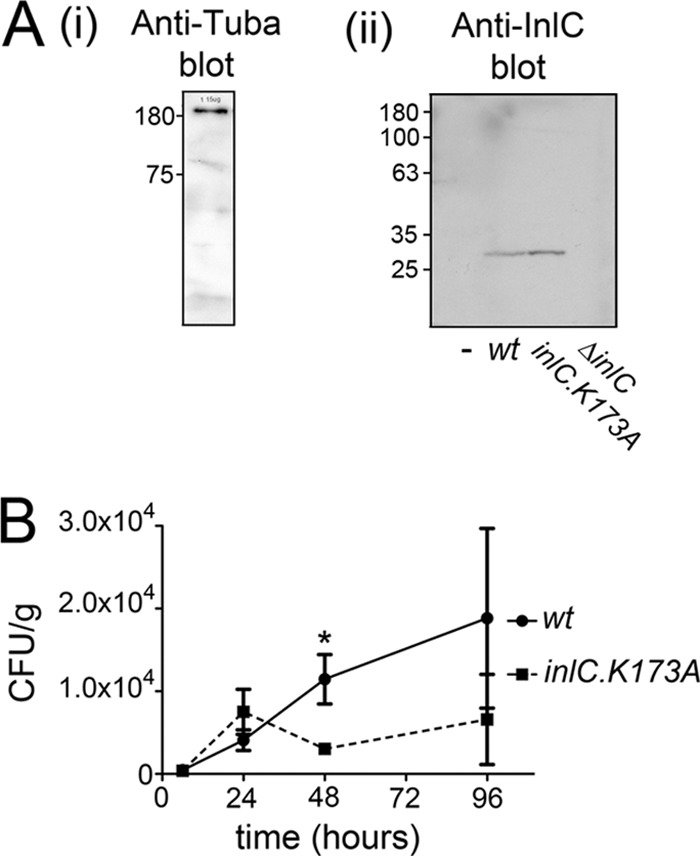
Ability of the wild-type (wt) or inlC.K173A L. monocytogenes strain to replicate in the liver of infected mice. (A, i) Expression of Tuba in the livers of C57BL/6 mice was confirmed by Western blotting of liver tissue lysates. (ii) Expression of InlC in the livers of infected mice was detected by Western blotting of solubilized liver tissue obtained from mice inoculated intravenously with the wild-type (wt), ΔinlC, or inlC.K173A strain for 24 h. As a control, a liver sample from uninfected (−) mice was included. (B) Growth of the wild-type or inlC.K173A bacterial strain in the livers of mice. C57BL/6 mice were inoculated intravenously with 5 × 104 wild-type or inlC.K174 mutant bacteria. At the indicated time points, 4 mice infected with each Listeria strain were sacrificed. Livers were homogenized, and the numbers of bacteria in these organs were determined through enumeration of CFU. Means ± SEM are presented. Statistical analysis was performed with Student's posttest. The difference in CFU between the wild-type and inlC.K173A mutant strain at 48 h (*) is statistically significant (P = 0.0337).
We determined the numbers of viable bacteria in the livers of mice infected with a sublethal dose (5 × 104) of the wild-type L. monocytogenes strain EGD or of the inlC.K173A mutant strain (Fig. 4B). Forty-eight hours after intravenous inoculation, bacterial counts in the livers of mice infected with the inlC.K173A mutant were approximately 25% of those in mice infected with wild-type L. monocytogenes. These results indicate that the inlC mutant is partly defective in infection of the liver. Interestingly, no differences in numbers of the wild-type and inlC.K173A mutant strains were observed before 48 h postinoculation. The reduction in numbers of the inlC.K173A mutant observed at 48 h, but not at earlier time points, is consistent with the possibility that this mutant may be defective in cell-to-cell spread in the liver.
In order to more directly test the hypothesis that the inlC.K173A mutant is compromised for spreading in the liver, we performed histological analysis. L. monocytogenes is known to produce foci in the liver that result from infection of hepatocytes (17, 18, 20). The size of these foci may reflect the efficiency of bacterial spreading. Liver samples from mice infected with wild-type, inlC.K173A, or ΔinlC bacterial strain were fixed and stained with anti-L. monocytogenes antibodies. Foci of infection were visible as regions in liver tissue that reacted strongly with the antibodies, indicating extensive infection (Fig. 5A). The surface areas of these regions were measured using ImageJ (version 1.43r) software (Fig. 5B), as described in Materials and Methods. Results from three independent experiments indicate that the inlC.K173A or ΔinlC mutants produced infection foci whose mean sizes were 40 or 50% smaller than those made by wild-type L. monocytogenes (Fig. 5C). The findings in Fig. 5 are consistent with a role for InlC-Tuba interaction in spreading in the liver.
Fig 5.

InlC is needed for efficient spreading in the liver. C57BL/6 mice were infected intravenously for 24 h with 5 × 105 wild-type (wt), ΔinlC, or inlC.K173A bacteria. Liver samples were stained with anti-Listeria antibodies and secondary antibodies coupled to peroxidase. Bacteria appear brown. Samples were counterstained with hematoxylin, giving the nuclei a blue color. Scale bars indicate 100 μm. (A) Typical images of liver samples infected with wt, ΔinlC, or inlC.K173A bacteria. Foci with bacteria appear brown. (B, i) An example of how quantification of foci size is performed. ImageJ software was used to determine the surface areas of foci. The resulting areas are presented below the images. For example, focus 1 in the “wt” image has a surface area of 9,511 μm2. (C, ii) Mean size ± SEM from three experiments of foci made by the wt, ΔinlC, and inlC.K173A strains. About 100 foci were measured for each strain in each of the three experiments. Absolute focus sizes were converted to relative sizes by normalization to absolute values for the wild-type strain. Statistical analysis by ANOVA indicated that P was <0.0001.*, P < 0.05 relative to the wild-type control (P = <0.05).
Conclusions.
The data in this work indicate a role for InlC in virulence during invasive infection of mice and support the idea that at least one of the ways that InlC impacts disease is by controlling spreading in the liver. Furthermore, results with the inlC.K173A bacterial mutant provide evidence that full virulence of L. monocytogenes requires the ability of InlC to interact with its host cytoplasmic receptor Tuba. The K173A mutation in InlC causes a partial defect in binding of InlC to an SH3 domain in Tuba (6), but importantly, this mutation did not affect the ability of InlC to impair nuclear translocation or activation of NF-κB. Therefore, the inlC.K173A mutant appears to be an appropriate tool to probe the function of host Tuba in L. monocytogenes infection. In the future, it would be interesting to extend these studies by using mice genetically inactivated in Tuba. Such mutant mice are not currently available.
ACKNOWLEDGMENTS
We thank Tina Rajabian for constructing the EGFP-InlC plasmid, and Balramakrishna Gavicherla for performing site-directed mutagenesis.
This work was supported by grants from the National Institutes of Health (R01AI085072) and the Marsden Fund of the Royal Society of New Zealand (UOO1003) awarded to K.I.
Footnotes
Published ahead of print 12 February 2013
REFERENCES
- 1. Posfay-Barbe KM, Wald ER. 2009. Listeriosis. Semin. Fetal Neonatal. Med. 14:228–233 [DOI] [PubMed] [Google Scholar]
- 2. Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gouin E, Welch WD, Cossart P. 2005. Actin-based motility of intracellular pathogens. Curr. Opin. Microbiol. 8:35–45 [DOI] [PubMed] [Google Scholar]
- 4. Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. 1993. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 90:11890–11894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Monack DM, Theriot JA. 2001. Actin-based motility is sufficient for bacterial membrane protrusion formation and host cell uptake. Cell. Microbiol. 3:633–647 [DOI] [PubMed] [Google Scholar]
- 6. Rajabian T, Gavicherla B, Heisig M, Muller-Altrock S, Goebel W, Gray-Owen SD, Ireton K. 2009. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat. Cell Biol. 11:1212–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salazar MA, Kwiatkowski AV, Pellegrini L, Cestra G, Butler MH, Rossman KL, Serna DM, Sondek J, Gertler FB, De Camilli P. 2003. Tuba, a novel protein containing Bin/Amphiphysin/Rvs and Dbl homology domains, links dynamin to regulation of the actin cytoskeleton. J. Biol. Chem. 278:49031–49043 [DOI] [PubMed] [Google Scholar]
- 8. Alto NM, Weflen AW, Rardin MJ, Yarar D, Lazar C, Tonikian R, Koller A, Taylor SS, Boone C, Sidhu SS, Schmidt AL, Hecht FA, Dixon JE. 2007. The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. J. Cell Biol. 178:1265–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li SSC. 2005. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. J. Biochem. 390:641–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gouin E, Adib-Conque M, Balestrino D, Nahori MA, Villiers V, Colland F, Dramsi S, Dusserget O, Cossart P. 2010. The Listeria monocytogenes InlC protein interferes with innate immune responses by targeting the IκB kinase subunit IKKα. Proc. Natl. Acad. Sci. U. S. A. 107:17333–17338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vallabhapurapu S, Karin M. 2009. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27:693–733 [DOI] [PubMed] [Google Scholar]
- 12. Engelbrecht F, Chun S-K, Ochs C, Hess J, Lottspeich F, Goebel W, Sokolovic Z. 1996. A new PrfA-regulated gene of Listeria monocytogenes encoding a small, secreted protein which belongs to the family of internalins. Mol. Microbiol. 21:823–837 [DOI] [PubMed] [Google Scholar]
- 13. Ireton K, Payrastre B, Cossart P. 1999. The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositude-3-kinase. J. Biol. Chem. 274:17025–17032 [DOI] [PubMed] [Google Scholar]
- 14. Dokainish H, Gavicherla B, Shen Y, Ireton K. 2007. The carboxyl-terminal SH3 domain of the mammalian adaptor CrkII promotes internalization of Listeria monocytogenes through activation of host phosphoinositide 3-kinase. Cell. Microbiol. 10:2497–2516 [DOI] [PubMed] [Google Scholar]
- 15. Sun H, Shen Y, Dokainish H, Holgado-Madruga M, Wong A, Ireton K. 2005. Host adaptor proteins Gab1 and CrkII promote InlB-dependent entry of Listeria monocytogenes. Cell. Microbiol. 7:443–457 [DOI] [PubMed] [Google Scholar]
- 16. Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa Y, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C. 2009. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11:1233–1240 [DOI] [PubMed] [Google Scholar]
- 17. Conlan JW, North RJ. 1991. Neutrophil-mediated dissolution of infected host cells as a defense strategy against a facultative intracellular bacterium. J. Exp. Med. 174:741–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaillard JL, Jaubert F, Berche P. 1996. The inlAB locus mediates the entry of Listeria monocytogenes into hepatocytes in vivo. J. Exp. Med. 183:359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gregory SH, Barczynski LK, Wing EJ. 1992. Effector function of hepatocytes and Kupffer cells in the resolution of systemic bacterial infections. J. Leukoc. Biol. 51:421–424 [DOI] [PubMed] [Google Scholar]
- 20. Rosen H, Gordon S, North RJ. 1989. Exacerbation of murine listeriosis by a monoclonal antibody specific for the type 3 complement receptor of myelomonocytic cells. J. Exp. Med. 170:27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]