

Abstract
β2 (CD18) integrins with α-chains CD11a, -b, -c, and -d are important adhesion molecules necessary for leukocyte migration and cellular interactions. CD18 deficiency leads to recurrent bacterial infections and poor wound healing due to reduced migration of leukocytes to inflammatory sites. CD8 T cells also upregulate CD11a, CD11b, and CD11c upon activation. However, the role these molecules play for CD8 T cells in vivo is not known. To determine the function of individual β2 integrins, we examined CD8 T cell responses to Listeria monocytogenes infection in CD11a-, CD11b-, and CD11c-deficient mice. The absence of CD11b or CD11c had no effect on the generation of antigen-specific CD8 T cells. In contrast, the magnitude of the primary CD8 T cell response in CD11a-deficient mice was significantly reduced. Moreover, the response in CD11a−/− mice exhibited reduced differentiation of short-lived effector cells (KLRG1hi CD127lo), although cytokine and granzyme B production levels were unaffected. Notably, CD11a deficiency resulted in greatly enhanced generation of CD62L+ central memory cells. Surprisingly, CD8 T cells lacking CD11a mounted a robust secondary response to infection. Taken together, these findings demonstrated that CD11a expression contributes to expansion and differentiation of primary CD8 T cells but may be dispensable for secondary responses to infection.
INTRODUCTION
Integrins are heterodimeric adhesion molecules comprised of α- and β-subunits that participate in immune cell interactions as well as in immune cell-extracellular matrix interactions. The β2 integrin (CD18) family consists of four members, based on α-chain pairings: CD11a (also called LFA-1 or αLβ2), CD11b (Mac-1 or αMβ2), CD11c (CR4 or αXβ2), and CD11d (αDβ2). Although numerous myeloid lineage cells, including dendritic cells (DC), neutrophils, and macrophages constitutively express CD11b and CD11c, lymphocytes do not, but all resting leukocytes express CD11a. Upon activation, CD8 T cells upregulate not only CD11a but also CD11b and CD11c (1, 2). Ligands for these molecules include members of the immunoglobulin superfamily (intercellular adhesion molecule 1 [ICAM-1], ICAM-2, and vascular cellular adhesion molecule 1) as well as fibrinogen and inactivated C3b (3). Patients with lymphocyte adhesion deficiency type I, who lack β2 integrins, suffer from recurrent infections and impaired wound healing (4–6). Similarly, CD18-deficient mice display increased rates of spontaneous infections and a reduced ability to induce graft-versus-host disease (7, 8). Nevertheless, the precise role of each individual β2 integrin in vivo in most cases is unclear.
Possible roles for CD11b and CD11c expressed by myeloid cells have been implicated in a number of infections, and their absence may lead to an increased microbial burden (9), which is attributed to their roles in adhesion-mediated phagocytosis and trafficking and clearance of the infectious agent by neutrophils and other innate immune cells (10–13). However, the importance of CD11b and CD11c expression levels in activated T cells during infection has not been explored in detail, and little is known about the roles of these molecules in antigen-specific T cell activation and function. A recent report showed that in the absence of CD11c, CD8 T cell responses to herpes simplex virus infection were enhanced, but whether this effect was a result of CD11c function on activated CD8 T cells or antigen-presenting cells (APC) is not known (14). Conversely, CD11b or CD11d deficiency, but not CD11c deficiency, resulted in a diminished T cell response to staphylococcus enterotoxin challenge, which was hypothesized to be due to defects in T cell development mediated by integrin-expressing non-T cells (15). In contrast to the relative paucity of literature regarding CD11b and CD11c function for T cells, the immunological relevance of CD11a expression by T cells has been characterized in some detail. Studies using planar bilayers have shown that the adhesive interaction of CD11a to ICAM-1 leads to stabilization of the immunological synapse (16). This adhesive property may also allow CD11a to perform costimulatory functions by augmenting T cell proliferation and cytokine production in vitro (17, 18). CD11a is also critical for lymphocyte entry into the lymph nodes (19). Interestingly, lymphocyte migration to the spleen in the absence of CD11a remains intact (20).
Establishing a role for CD11a in T cell priming in vivo has proven to be more complex. Prior studies illustrated an important role for CD11a in T cell activation in colitis, delayed-type hypersensitivity (DTH) responses, and tumor rejection (21, 22). In contrast, T cell priming in viral infections is apparently unaffected in the absence of CD11a (21). CD11a and CD11b are essential for protection against Streptococcus pneumoniae infection. Although the T cell response was not measured, early bacterial overgrowth suggested that the defects lie with the inability of innate immune cells to clear the infection (23). In the case of pulmonary Mycobacterium tuberculosis infection, CD11a deficiency also leads to loss of protection (24). T cell priming is delayed and fewer antigen-specific T cells are found in the lungs after infection, due to additional defects in T cell migration. Care must be taken when interpreting results related to situations where lymph node (LN) priming is required, since CD11a plays an important role in migration of naïve T cells to the LN (20, 25). Thus, the paucity of T cells in the CD11a−/− LN due to the migration defect could lead to ineffective T cell priming, rather than indicating a direct role for CD11a in T cell activation. In the case of Listeria monocytogenes infection, β2 integrin-deficient and CD11a−/− mice exhibit enhanced resistance to L. monocytogenes infection, potentially due to neutrophilia, particularly in the liver, as well as enhanced interleukin-12 (IL-12) and granulocyte colony-stimulating factor production (26–28). In fact, while neutrophils are important for L. monocytogenes clearance from the liver, they play much less of a role in splenic L. monocytogenes clearance, and inflammatory monocytes are essential for protection against L. monocytogenes infection (29–31). Thus far, the role of β2 integrins in the T cell response to L. monocytogenes infection has not been examined. Here, we assess T cell activation following L. monocytogenes infection of mice deficient for specific β2 integrins and demonstrate a T cell intrinsic requirement for CD11a in CD8 T cell activation and function independent of migration.
MATERIALS AND METHODS
Mice.
C57BL/6J, CD11b−/− (B6.129S4-Itgamtm1Myd/J) (32) and CD11a−/− (B6.129S7-Itgaltm1Bll/J) (15) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). CD11c−/− mice (15) and CD45.1+/CD45.2+ B6 mice were bred in our facility. All animal protocols were carried out in accordance with NIH guidelines and approved by the UCHC Animal Care Committee.
Infection.
Mice were infected intravenously (i.v.) with L. monocytogenes expressing ovalbumin (Ova) (33). A dose of 1 × 103 CFU or 5 × 104 CFU was used in primary infections, and a dose of 1 × 104 CFU was used for secondary infections. In some imaging experiments, 1 × 106 CFU of the attenuated ActA strain of L. monocytogenes Ova was used in order to generate sufficient endogenous Ova-specific CD8 T cells. Bacterial burdens, the bacterial titers within tissues, were determined by homogenizing the tissue samples in phosphate-buffered saline (PBS) containing 1% saponin, plating serial dilutions of the homogenate on brain heart infusion agar containing 5 μg/ml erythromycin, and incubating for 2 days at 37°C.
Flow cytometry.
Antigen-specific CD8 T cells were identified by using an H-2Kb tetramer containing the Ova-derived peptide SIINFEKL, and listeriolysin O-specific CD4 T cells were identified with an LLO-I-Ab tetramer (34), generously provided by Marc Jenkins (University of Minnesota). Tetramer+ cells were characterized by using monoclonal antibodies reactive with the indicated antigens. All antibodies were purchased from BD Biosciences, Ebioscience, or Biolegend. For intracellular cytokine staining, splenocytes isolated from infected mice were cultured with or without 2 to 20 μg/ml SIINFEKL or LLO peptide for 5 h in the presence of GolgiPlug (BD Biosciences) and stained as directed (BD Biosciences). Viability was analyzed using LIVE/DEAD cell stain (Invitrogen). Samples were collected using an LSRII apparatus (BD Biosciences) and analyzed with FlowJo software (Tree Star, Ashland, OR).
Confocal microscopy.
Spleens were isolated from naïve or infected mice and prepared as previously described (35). Antibodies reactive to the indicated antigens were purchased from BD Biosciences, Invitrogen, or Ebioscience. Stained samples were mounted using Immumount, imaged using an LSM 780 microscope (Zeiss), and analyzed using Imaris Suite (Bitplane Inc.).
Lytic assay.
Spleens from L. monocytogenes ActA Ova-infected mice were harvested on day 7. Splenocytes were enriched for CD8 T cells by negative selection with a CD8 T cell isolation kit from Miltenyi Biotec. The CD8-enriched fraction was stained for Ova tetramer to determine the absolute number of effector T cells specific for SIINFEKL. EL4 cells were used as target cells and pulsed with 1 μg/ml SIINFEKL peptide for 45 min at 37°C. Pulsed or unpulsed EL4 cells were labeled with high (5 μM) or low (1 μM) concentrations of carboxyfluorescein succinimidyl ester (CFSE), respectively. Serial dilutions of effector cells were then incubated with 104 pulsed and 104 unpulsed EL4 cells for 4 H. Cells were stained with viability dye and examined by flow cytometry. After gating on live EL4 cells, specific lysis was calculated as follows: percent specific lysis = 100 − {100 × [(percent CFSEhigh infected mice)/(percent CFSElow infected mice)]/[(percent CFSEhigh target alone)/(percent CFSElow target alone)]}. Effector:target (E:T) ratios were calculated for each mouse based on the Ova tetramer number per well.
BrdU incorporation.
Mice were injected at days 5, 6, and 7 postinfection (p.i.) with 1 mg of bromodeoxyuridine (BrdU) intraperitoneally. Eight days postinfection, splenocytes were stained for surface markers as described above and for BrdU incorporation according to the manufacturer's protocol (BD Biosciences).
Tetramer decay assay.
T cell avidity was determined by tetramer decay analysis as described previously (36). Briefly, splenocytes of infected mice were stained using Ova-Kb tetramer, and subsequently Fab fragments of anti-H-2Kb monoclonal antibody (clone Y-3) were added to a final concentration of 5 μM. Stained aliquots of cells were removed at various time points and fixed immediately in 2% paraformaldehyde–PBS solution. Tetramer mean fluorescence intensity (MFI) was determined by flow cytometry, and the half-life was calculated by loss of tetramer reactivity over time.
Tetramer enrichment.
Splenocytes pooled from 3 infected mice were enriched for Ova-specific CD8 T cells as described previously (37). Briefly, splenocytes were first labeled with phycoerythrin (PE) and allophycocyanin-labeled Ova-Kb tetramers at room temperature for 1 h. Samples were then washed and incubated with anti-PE beads (Miltenyi Biotec) at 4°C for 30 min before positive selection using an autoMACS apparatus (Miltenyi Biotec). The cells were then stained with additional antibodies as described above and analyzed by flow cytometry.
Bone marrow chimeras.
Bone marrow was isolated from femurs and tibias of wild-type (WT; CD45.2+/CD45.1+) and CD11a−/− (CD45.2) mice. These cells were then mixed and injected i.v. into lethally irradiated (1,000 rads) CD45.1 hosts. After 6 to 8 weeks of reconstitution, chimeras were immunized as described above. Donor populations were separated based on CD45 expression.
Statistical analysis.
Statistical significance was determined by using Prism 5 software (GraphPad) and the indicated functions. Error bars indicate standard errors of the means.
RESULTS
Primary CD8 T cell responses to bacterial infection are CD11b and CD11c independent.
Previous studies have reported the induction of myeloid markers CD11b and CD11c on activated antigen-specific CD8 T cells during lymphocytic choriomeningitis virus (LCMV) infection (38) and the graft-versus-host disease response (2). We tested whether a similar phenomenon occurred in response to L. monocytogenes infection. CD8 T cell priming after intravenous L. monocytogenes infection occurs primarily in the spleen (39) and thus circumvents any potential effects of the role of β2 integrins in LN migration. At the peak of the primary CD8 T cell response, CD11a, CD11b, and CD11c were substantially upregulated by Ova-Kb-specific CD8 T cells (Fig. 1A). The selective upregulation of these markers on activated antigen-specific CD8 T cells suggested that engagement of these receptors may play an important role in the CD8 T cell responses to infection. To test this possibility, we examined Ova-Kb-specific CD8 T cell responses after L. monocytogenes infection in mice deficient for CD11b or CD11c. Neither CD11b−/− nor CD11c−/− mice exhibited any defects in generation of Ova-Kb-specific CD8 T cell responses in the spleen (Fig. 1B). In the lung, CD11b−/− mice generated a modest but statistically significant increase in the antigen-specific CD8 T cell response.
Fig 1.

Absence of CD11b or CD11c does not affect priming of CD8 T cells during Listeria infection. (A) Representative plots show gated CD8 T cells from spleens of WT C57BL/6 mice that were infected 8 days prior with L. monocytogenes Ova. Histograms show expression of CD11a, CD11b, and CD11c by tetramer+ CD8 T cells and endogenous naïve CD8 T cells (tetramer− CD44low). (B) Graphs show frequencies of Ova-Kb-specific CD8 T cells in WT versus CD11b−/− and CD11c−/− mice in spleen and lung at day 8 p.i. (C and D) Graphical representations of various effector subsets (C) and granzyme B production (D) of Ova-Kb-specific CD8 T cells. (E and F) Eight days p.i., splenocytes were restimulated in vitro with peptide to assess cytokine production from CD8 (E) and CD4 (F) T cells. These data are representative of two individual experiments with four to five mice per group. Statistical significance was determined using a one-way analysis of variance and Bonferroni post test. *, P < 0.05.
After infection, a heterogeneous pool of CD8 T cell effectors is formed that can be identified based on the expression of various cell surface markers, including KLRG1 and CD127 (IL7 receptor [IL-7R]) (40). The early effector CD8 T cells (EECs) that are KLRG1low CD127low are the first effector subset to emerge after L. monocytogenes infection (41). However, at the peak of the CD8 T cell response to L. monocytogenes infection, the terminally differentiated short-lived effector cells (SLEC) constitute the majority of the responding CD8 T cells and are identified by KLRG1hi and CD127low expression. The long-lived memory precursors (MPEC) are KLRG1low and CD127hi and constitute a smaller fraction of responding CD8 T cells, along with the double-positive effector cells (DPECs), that express both KLRG1 and CD127. An examination of effector cell heterogeneity in the absence of CD11b or CD11c further indicated that these integrins did not affect the CD8 T cell response to L. monocytogenes infection (Fig. 1C). Similarly, granzyme B and cytokine production levels among responding CD8 T cells in CD11b- or CD11c-deficient or wild-type mice were equivalent (Fig. 1D to F). Thus, despite their induction on responding CD8 T cells, CD11b and CD11c were not required for the generation of optimal primary antigen-specific CD8 T cell responses during L. monocytogenes infection.
CD11a is crucial for the optimal expansion of antigen-specific CD8 and CD4 T cells.
Although CD11a plays a prominent role in lymphocyte migration to LN (42), the role of this molecule in CD8 T cell priming during infection in vivo is not well understood. Thus, we tested CD11a function in vivo by infecting CD11a-deficient mice with L. monocytogenes Ova and then evaluating antigen-specific CD8 T cell responses. At the peak of the CD8 T cell response, CD11a−/− mice displayed a significant defect in accumulation of splenic Ova-Kb-specific CD8 T cells (Fig. 2A), which was also evident in liver and lungs (data not shown). The reduced T cell response was not due to reduced antigen availability or altered bacterial load, since there were no significant differences in bacterial loads in the spleens of WT and CD11a−/− mice during the peak of L. monocytogenes replication at 3 days postinfection (see Fig. S1 in the supplemental material). Since previous reports had indicated that CD11a−/− mice are more resistant to L. monocytogenes infection (26, 27), we also challenged mice with a higher L. monocytogenes dose (5 × 104 CFU). In this case, bacterial burden in the spleen was decreased in CD11a−/− mice compared to control animals (see Fig. S1). At the higher dose, the CD8 T cell response in the WT animals was less than that observed when a low dose was given (see Fig. S2 in the supplemental material). Nevertheless, the CD8 T cell response was not reduced in the CD11a−/− mice, suggesting that increasing antigen and/or inflammation could overcome the defect. Interestingly, the CD4 T cell response was not reduced in CD11a−/− mice when a low L. monocytogenes dose was used, but it was decreased at the higher dose (see Fig. S2).
Fig 2.

CD11a drives optimal proliferation of responding CD8 T cells. (A) Dot plot and graph showing the magnitude of the Ova-Kb-specific CD8 T cell response in WT and CD11a−/− mice in the spleen at day 8 p.i. (B) Data plot and graph showing the magnitude of the Ova-Kb-specific response in tetramer-enriched splenocytes from at day 4 p.i. Each representative dot plot and data point on the graph is representative of 3 pooled mice from two individual experiments. (C) Histogram analysis and graphs of BrdU incorporation among tetramer+ CD8 T cells (open histogram) or naïve (CD44low) CD8 T cells (gray filled histograms) in individual spleens at day 8 p.i. Indicated numbers on dot plots and histograms signify the percentages of cells that were within each gated region of representative samples. Data in panels A and C are representative of two or more individual experiments, which included 4 to 5 mice per group. Student's t test was used to determine statistical significance. *, P < 0.05.
To examine the reason for the reduced magnitude of the CD8 T cell response, we utilized tetramer enrichment to track small populations of antigen-specific CD8 T cells early after infection (day 4). At this time, markedly reduced numbers of Ova-Kb-specific CD8 T cells were present in CD11a−/− mice (Fig. 2B). Next, we measured the proliferation of CD11a−/− CD8 T cells based on BrdU uptake on days 5 to 7 p.i. Fewer antigen-specific CD8 T cells in CD11a−/− mice incorporated BrdU (Fig. 2C), and the amount of BrDU incorporated into the CD11a−/− CD8 T cells was also significantly lower than that of control cells (Fig. 2C). Thus, optimal early expansion and continued proliferation both required CD11a.
Differential requirement for CD11a in cytokine production and the lytic activity of antigen-specific CD8 T cells.
Early data implied that CD11a plays a compulsory role in cytotoxic functions of CD8 T cells in vitro (43). We investigated whether the engagement of CD11a impacted the function of responding CD8 T cells after L. monocytogenes infection in vivo. Surprisingly, Ova-Kb-specific CD8 T cells in WT and CD11a−/− mice at day 8 postinfection exhibited similar levels of granzyme B ex vivo. Moreover, after antigen stimulation in vitro, CD11a−/− and CD8 T cells expressed similar levels of LAMP-1 (CD107a), a marker of degranulation (44) (Fig. 3A). Despite these results, the direct ex vivo lytic activity of CD11a−/− CD8 T cells was ∼10-fold less than that of WT CD8 T cells (Fig. 3B). We next determined whether in vivo-generated CD11a−/− Ova-Kb-specific CD8 T cells produce normal levels of gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) at day 8 p.i. Concomitant with the reduced frequency of tetramer+ cells in CD11a−/− mice (Fig. 2A), we observed a reduced frequency of IFN-γ-producing CD8 T cells (Fig. 3C, upper panel), although IFN-γ levels between WT and CD11a−/− cells were similar. In addition, the frequency of TNF-α producers among the IFN-γ-positive cells was similar in WT and CD11a−/− mice (Fig. 3C, lower panel). Thus, the absence of CD11a led to an overall reduction in the magnitude of the CD8 T cell response and reduced lytic activity but did not impact the cytokine-producing capacity of the responding CD8 T cells.
Fig 3.

Normal effector functions of responding CD8 T cells in CD11a-deficient mice. (A) Splenocytes from infected mice at day 8 were isolated and directly stained for granzyme B expression or restimulated in vitro for 5 h with SIINFEKL peptide before analysis for LAMP-1 (CD107a). Bar graphs show granzyme expression among Ova-Kb-specific CD8 T cells and LAMP expression among IFN-γ-producing CD8 T cells. (B) In vitro killing activity of CD11a−/− and WT CD8 T cells. Each line represents one mouse. (C) Representative dot plots of TNF-α producers (bottom panel and bar graph) gated on CD8 T cells producing IFN-γ (top panel) after in vitro restimulation with peptide. (D) Tetramer binding decay assay of Ova-Kb-specific CD8 T cells of day 8 p.i. splenocytes. Data are representative of combined results of two individual experiments with a total of 9 to 10 mice per group. The half-life of Ova-Kb tetramer binding (t½) was defined as the amount of time required to lose 50% of the maximal tetramer binding activity. The mean t½ is indicated for each group on the graph. Indicated numbers on the dot and zebra plots signify the percentages of cells that were within each gated region of representative samples.
It is possible that the absence of CD11a could alter the selection of CD8 T cells that contribute to the response whereby only those CD8 T cell clones that posses a higher avidity for major histocompatibility complex (MHC)-peptide complexes are equipped to aptly respond to infection. Although the fluorescence intensity of tetramer staining was similar between CD11a−/− and WT CD8 T cells (Fig. 2A), we wanted to directly test the avidity of WT and CD11a−/− CD8 T cells by using a tetramer decay assay that compared the dissociation kinetics of peptide-MHC and T cell receptor (TCR) interactions (36). In this assay, the half-life is defined by the time required to lose 50% of the maximal tetramer binding for each group. Combined results from two individual experiments indicated that the half-life of tetramer binding of WT and CD11a−/− T cells was similar; the WT averaged 9.04 min, while CD11a−/ T cells averaged 10.2 min (Fig. 3D). These data indicated that although the magnitude of the CD8 T cell response was blunted in the absence of CD11a, there was no evidence that the responding cells represented an atypical population made up of a subset of clones with distinct avidities.
CD11a regulates effector subset development of CD8 T cells.
Several studies have shown that numerous cell extrinsic factors, such as antigen availability, precursor frequency, and the inflammatory milieu, are integrated to subsequently determine the differentiation pattern of effector and memory CD8 T cells after infection (37, 45–47). We hypothesized that the absence of CD11a may lead to reduced interaction of APCs and CD8 T cells and that this could result in a reduced “strength” of signal downstream of TCR engagement. To determine if CD11a deficiency affected differentiation of effector CD8 T cells, we examined the SLEC-MPEC differentiation profile of Ova-Kb-specific CD8 T cells. Interestingly, CD11a−/− mice had a reduced frequency of SLEC that was concomitant with a significant increase in the frequency of CD127 and KLRG1 DPEC compared to WT mice. We also observed a trend toward increased accumulation of MPEC, although this difference failed to reach statistical significance (Fig. 4A).
Fig 4.

CD11a regulates effector CD8 T cell differentiation. (A) KLRG1 and CD127 expression levels were used to subdivide Ova-Kb-specific CD8 T cells into EEC (KLRG1low CD127low), MPEC (KLRG1low CD127hi), DPEC (KLRG1hi CD127hi), and SLEC (KLRG1hi CD127low). (B and C) Representative zebra plots and bar graphs show CD62L versus PD-1 expression either among total Ova-Kb-specific CD8 T cells (B) or within specific effector subsets (C) in splenocytes of D8 infected mice. (D) Histogram displaying CD25 expression of antigen-specific CD8 T cells in tetramer-enriched splenocytes at day 4 from infected mice. Splenocytes from 3 mice were pooled per sample with a total of 12 mice per group. Bar graphs show frequenies and MFI values for CD25 expression in total tetramer-enriched CD8 T cells. The numbers on the zebra plots and histograms signify the percentages of cells that were within each gated region of representative samples. Data are representative of two or more experiments. Student's t test was used for statistical analysis. *, P < 0.05.
We previously demonstrated that programmed death 1 (PD-1) expression inversely correlates with CD62L expression on antigen-specific CD8 T cells after infection (47). Moreover, limiting antigen availability during infection resulted in increased expression of CD62L with a decrease in PD-1 expression. CD11a−/− antigen-specific CD8 T cells displayed reduced expression of PD-1, with a concurrent major increase in expression of CD62L (Fig. 4B). Our previous results showed that CD62L expression is restricted to the MPEC subset (48), leading to development of central memory CD8 T cells (TCM) (Fig. 4C, WT). TCM express the homing molecules CD62L and CCR7, while effector memory cells (TEM) lack expression of these molecules. However, although CD62L expression was greatly increased in the CD11a−/− MPEC, CD62L was also expressed by significant populations of CD11a−/− EEC and SLEC, which we have never observed in wild-type mice, even when TCR triggering is limited (48, 49). Because EEC are formed early after initial CD8 T cell activation, our findings suggested that CD11a may be important for the initial downregulation of CD62L.
Our earlier studies demonstrated that CD25 (IL-2R) expression by antigen-specific CD8 T cells correlates with antigen availability and is maximal at 4 days post-L. monocytogenes infection (47, 49). The frequency of CD25+ cells as well as the level of CD25 expression were significantly lower in CD11a−/− Ova-Kb-specific CD8 T cells (Fig. 4D). The reduced CD25 expression is likely a result of reduced TCR triggering, which we have linked to enhanced CD62L expression and increased MPEC generation (47, 49).
Effects of CD11a on splenic anatomy and CD8 T cell localization.
Recent data showed that the absence of chemokine receptors alters localization of CD8 T cells to inflammatory sites, resulting in changes in effector subset differentiation and memory generation (50). By using confocal microscopy, we tested whether CD11a was involved in the anatomical organization of the spleen in normal and infected mice. A comparison of spleen sections from WT and CD11a−/− mice did not reveal any gross differences in the localization of T, B, CD11c+, CD11b+, or Moma+ marginal zone macrophages (Fig. 5 and data not shown). After infection, we examined the localization of antigen-specific CD8 T cells by using in situ MHC class I tetramer staining and confocal microscopy (35) (Fig. 5). In order to enhance detection of Ova-Kb-specific CD8 T cells at early time points, we infected mice with a higher dose of an attenuated ActA-deficient strain of L. monocytogenes Ova (35) that generates a larger response. Both the CD4 and CD8 T cell responses were significantly reduced after ActA L. monocytogenes infection (see Fig. S3 in the supplemental material). At day 5 p.i., in both the WT and CD11a−/− spleens, a readily detectable population of tetramer+ CD8 T cells were located in T cell zones (PALS) as well as in the red pulp (RP) (Fig. 5). We also examined the spleens at 8 days after infection with WT L. monocytogenes Ova. Consistent with the flow cytometry data, the CD11a−/− spleens contained fewer Ova-specific CD8 T cells than did the WT spleens (Fig. 5). Moreover, in WT and CD11a−/− spleens, the majority of antigen-specific CD8 T cells were located in the RP, with a smaller percentage present in the B and T cell zones. Overall, these data indicated that CD11a expression was not required for localization or movement of CD8 T cells during priming in the spleen.
Fig 5.

Normal localization of Ova-Kb-specific CD8 T cells in CD11a−/− mice after L. monocytogenes infection. Thick sections of spleens were stained with Ova-Kb tetramer and antibodies to other surface markers to indicate B (B220) and T cell (CD8) zones and CD31 to identify blood vessels. (A) Images of uninfected spleens from WT mice were acquired with a 20× 0.75 numerical aperture (NA) objective. (B and C) Infection with 1 × 106 CFU ActA−/− L. monocytogenes Ova was used to assess the early T cell response (day 5 p.i.) in WT (B) and CD11a−/− (C) mice. (D and E) Sections from spleens of WT (D) and CD11a knockout (E) mice that had been infected with 1 × 103 CFU L. monocytogenes Ova 8 days prior. B, B cell zone; PALS, peri-arteriolar lymphoid sheath (T cell zone); RP, red pulp; CA, central arteriole.
CD11a requirement for CD8 T cell priming is cell intrinsic.
In CD11a−/− mice, all cells lack CD11a expression. In order to test whether T cells intrinsically require CD11a to mount a normal response, we generated mixed chimeras by reconstituting lethally irradiated WT mice with equal numbers of bone marrow cells from congenically distinct WT and CD11a−/− mice. This system also alleviates any concerns of potential differences in bacterial burdens or antigen loads, since WT and CD11a−/− cells are responding in the same host. Interestingly, 8 weeks after reconstitution, we observed an incomplete reconstitution of the T cell compartment from CD11a−/− donor cells that appeared to be due to a partial block in T cell development (T. O. Bose et al., unpublished data). To compensate for differences in reconstitution, we generated chimeras using a 1:3 ratio (WT:CD11a−/−) of bone marrow cells. Although reconstitution remained unequal (∼3:1, WT:CD11a−/−), it was nevertheless possible to accurately assess the CD8 T cell response after infection. The defect in CD8 T cell priming observed in CD11a−/− mice was recapitulated in CD11a−/− CD8 T cells in chimeras (Fig. 6A). Moreover, CD11a−/− CD8 T cells in chimeras also displayed altered effector subset development, characterized by fewer SLEC and an increase in EEC and MPEC compared to their WT counterparts (Fig. 6B). Cytokine production was also unaffected in the chimeras (Fig. 6C). Thus, CD11a expression by CD8 T cells was required for optimal CD8 T cell expansion and normal effector subset development in response to L. monocytogenes infection.
Fig 6.
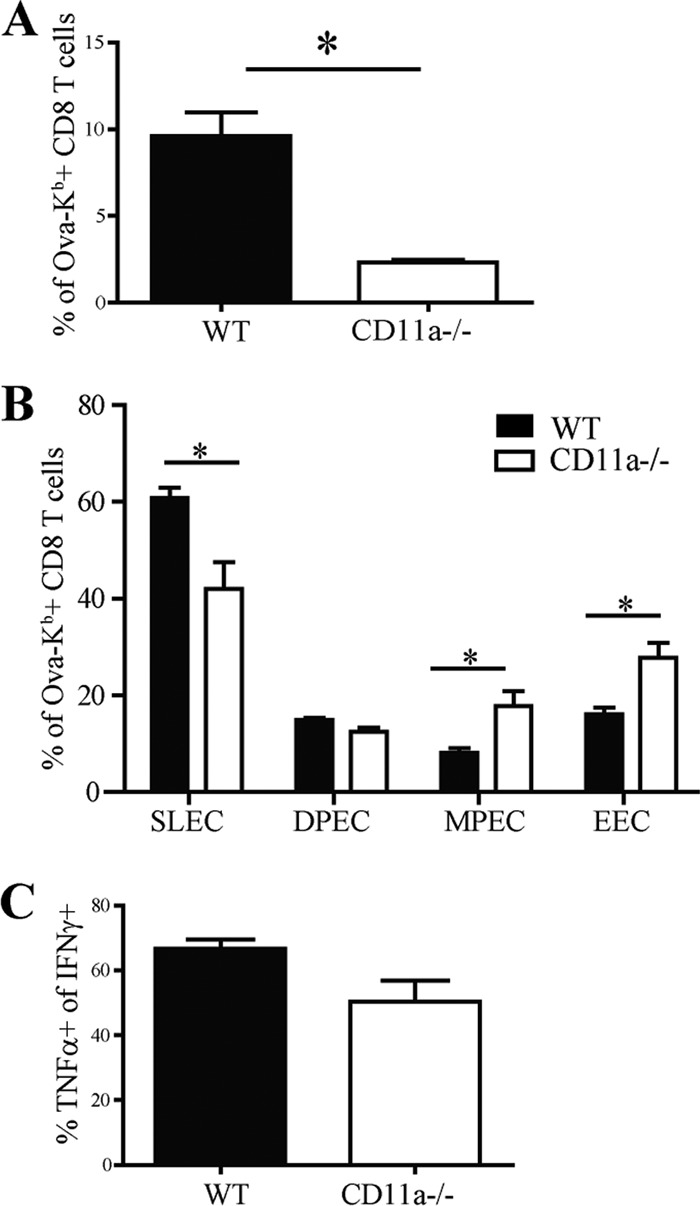
The CD11a requirement for CD8 T cell activation is cell intrinsic. Chimeric mice reconstituted with a mixture of bone marrow from WT and CD11a−/− mice were infected with 1 × 103 CFU L. monocytogenes Ova and assessed for T cell responses in the spleen 8 days postinfection. Bar graphs display overall frequencies of antigen-specific T cell responses (A) and differentiation among responding cells (B). (C) Splenocytes were used for in vitro peptide restimulation to determine the frequency of cells producing TNF-α among IFN-γ+ CD8 T cells. Data are representative of two individual experiments with 4 chimeric mice per experiment.
Generation and reactivation of memory CD8 T cells is CD11a independent.
A previous report demonstrated that in the absence of ICAM-1, the presumed ligand for CD11a, memory CD8 T cells did not develop, due to a drastic contraction of effector OT-I T cells (51). We asked whether a similar phenomenon occurs in the absence of CD11a, and we examined the kinetics of the antigen-specific CD8 T cell response to infection in the peripheral blood of WT and CD11a−/− mice. Although CD11a−/− mice had lower frequencies of Ova-Kb-specific CD8 T cells at all time points tested, the rate of contraction did not appear to be greater in CD11a−/− versus WT mice (Fig. 7A). Additionally, even at day 70 p.i., memory cells were present in the blood of CD11a−/− mice, albeit at a reduced frequency, concomitant with the reduced magnitude of the primary response. Moreover, memory CD8 T cells remained phenotypically distinct between CD11a−/− and WT mice with regard to KLRG1, CD127, and CD62L expression, which was again reflective of the primary response (Fig. 7C and D). Similar results were obtained in the spleen (data not shown). Thus, CD11a is not required for memory development per se. To determine whether CD11a plays a role in memory CD8 T cell reactivation, WT and CD11a−/− mice were first infected with 1 × 103 CFU of L. monocytogenes Ova and 70 days later were rechallenged with 1 × 104 CFU of L. monocytogenes Ova. Surprisingly, Ova-Kb-specific CD8 T cells in the spleens of both groups of mice underwent a massive expansion in the blood (Fig. 7E) and spleen (Fig. 7F) by day 6 after recall. CD8 T cells in WT mice underwent an ∼102-fold increase, while CD11a−/− CD8 T cells increased ∼184-fold in comparison to prechallenge memory cell levels (Fig. 7E). Effector subset composition in WT and CD11a−/− mice after recall was essentially identical (Fig. 7G). However, an increased proportion of CD11a−/− recalled CD8 T cells expressed CD62L, which may have been reflective of the heightened levels of CD62L expression prior to challenge (Fig. 7H). Analysis of cytokine production indicated similar frequencies of TNF-α+ CD8 T cells in WT and CD11a−/− mice (Fig. 7I). Overall, these findings indicated a more stringent CD11a requirement for primary versus memory CD8 T cell activation.
Fig 7.

Memory CD8 T cell reactivation is CD11a independent. (A) Graph showing the kinetics of Ova-Kb-specific CD8 T cells in the peripheral blood at various time points after infection with 1 × 103 CFU of L. monocytogenes Ova. (B and C) Representative dot plots show staining of Ova-Kb-specific CD8 T cells (B) and KLRG1/CD127 expression (C) among Ova-Kb-specific CD8 T cells in peripheral blood 70 days postinfection. (D) CD62L expression among Ova-Kb-specific MPEC (KLRG1low CD127hi) at day 70 in the peripheral blood. (E) Memory mice were challenged with 1 × 104 CFU of L. monocytogenes Ova, and recall responses were assessed in the blood before and after recall. (F to I) Splenocytes were harvested 6 days postrecall to determine the frequency of Ova-specific CD8 T cells (F) or SLEC/MPEC differentiation of antigen-specific CD8 T cells (G). (H) Bar graphs represent CD62L expression in total Ova-Kb-specific CD8 T cells or among the MPEC population. (I) In vitro restimulation of recalled splenocytes was used to assess the frequency of TNF-α producers (gated first on CD8 T cells producing IFN-γ). Numbers on dot plots and histograms signify percentages of cells that were within each gated region of representative samples. These data are representative of two individual experiments with a total of 5 mice per group.
DISCUSSION
Precursor frequency, inflammatory milieu, affinity, and duration of T cell-APC interactions are important determinants of T cell response kinetics and function (45, 52–54). Recently, two-photon microscopy revealed that interaction of DC with naïve T cells is complex. Initial transient DC-T cell interactions give way to more sustained synapse formation that can be hours long (55). Perturbation of DC-T cell interactions can lead to altered T cell responses (51). Thus, regulation of these interactions is crucial for the formation of efficient immune responses. In the current study, we highlighted the important role of CD11a in CD8 T cell priming and function.
CD11a reorganization is fundamental to the “bull's-eye”-like architecture of the immunological synapse (56). Several in vitro studies have provided evidence that the interaction of CD11a on T cells with ICAM-1 on APC facilitates stabilization of the immune synapse that promotes costimulatory function, leading to increased T cell proliferation and cytotoxicity (18, 57). Confirming a similar role for CD11a in vivo has proven to be more complicated, although studies have illustrated the importance of CD11a during induction of colitis, DTH responses, and tumor rejection (21, 22). Based on in vitro killing assays, the CD8 T cell response to vesicular stomatitis virus or LCMV infection in CD11a−/− mice is equivalent to that of WT mice (21), but quantitation of antigen-specific CD8 T cell numbers was not performed. In our studies, in which we took into account the magnitude of the CD8 T cell response, defective lytic activity but normal cytokine production by CD11a−/− CD8 T cells were observed. The role of CD11a in migration should also be taken into account in studies that use CD11a−/− or β2 integrin-deficient mice. Not only are β2 integrins involved in migration of T cells to LN, but also are involved in establishing the normal complement of immune components in certain tissues, such as the intestine (58). To circumvent LN involvement, we took advantage of the i.v. L. monocytogenes infection system, in which T cell priming occurs primarily in the spleen (39).
Our results indicated that CD11a deficiency significantly impacted the differentiation of the responding CD8 T cells. The development of effector subsets was skewed away from SLEC generation and favored EEC and MPEC formation. This result mirrored our previous findings, where similar patterns of effector subsets developed in the absence of CD4 T cell help or CD25 signaling (49). CD4 T cells play an important role in the CD25 upregulation by CD8 T cells that bestows IL-2 responsiveness, an important component of the help provided by CD4 T cells during L. monocytogenes infection (49). Moreover, sustained IL-2 signaling drives SLEC differentiation after infection (49). Yet, in the case of CD11a deficiency, the CD4 T cell response to L. monocytogenes infection was normal (data not shown). In addition, the mixed chimera studies showed that intrinsic CD11a expression by CD8 T cells was needed for an optimal response. Thus, CD11a appears to play a direct role in CD25 upregulation and effector CD8 T cell subset development, presumably through augmentation of T cell-APC interactions. The precise signaling mechanism by which CD11a regulates CD8 T cell responses remains unclear. β2 integrins lack immunoreceptor tyrosine-based activation motifs (ITAM), but activation of protein tyrosine kinases via CD11a may initiate outside-in signaling, which could then control activation and programming of CD8 T cells (6, 59). In addition, ligand-independent signaling via CD11a coupled to TCR triggering may also play a role in T cell activation (60). Thus, the absence of CD11a could lead to altered downstream signaling events that yield the observed results. In support of this possibility, a recent study showed that the proline-rich tyrosine kinase 2 (PYK2), which is important in CD11a-mediated adhesion and costimulation, regulates SLEC differentiation during LCMV infection (61). These findings along with ours point to CD11a as an important determinant of effector CD8 T cell differentiation.
The ligands for CD11a in vivo that drive CD8 T cell responses have not been well defined. ICAM-1 is thought to be a major counterreceptor for CD11a (17). The role of ICAM-1 in CD8 T cell priming after immunization with DC-targeted antigen and CD40 costimulation (51) was examined by intravital microscopy. ICAM-1 expression by DC enhanced long-lived stable interactions with responding OT-I TCR transgenic CD8 T cells. However, immunization of ICAM-1−/− hosts resulted in a numerically and functionally normal primary response of adoptively transferred OT-I cells, suggesting that, at least in this immunization scheme, ICAM-1 is not a major contributor to initial CD8 T cell activation in vivo. Our results indicated that CD11a was essential for optimal CD8 T cell expansion in response to L. monocytogenes infection, implying that ICAM-1 may not always play a dominant role in CD11a-mediated T cell activation or that initiation/amplification of different immune responses may involve distinct integrin-ligand combinations.
Our data also demonstrated an important function for CD11a in regulating the TCM/TEM ratio through control of CD62L expression. In the absence of CD11a, the number of CD62L+ MPEC was greatly increased, and unusual expression of CD62L by EEC and SLEC was noted. This correlated with decreased PD-1 expression, which was similar to our previous results where specifically limiting antigen availability resulted in a decrease in CD25 levels and an increase in CD62L by MPEC (47, 48). Coupled with these results, our findings with CD11a-deficient CD8 T cells suggest a differential regulation of effector subset development and the TCM/TEM ratio. Thus, CD62L expression is more sensitive to reductions in signal strength, while altering MPEC and SLEC development occurs only when signal transduction is more severely limited. Furthermore, the induction of aberrant CD62L expression by CD11a-deficient EEC and SLEC indicated an additional level of CD62L regulation related to signal strength, perhaps at an early activation stage when CD62L is enzymatically cleaved (62, 63).
Of particular note was the role of CD11a in CD8 memory T cell responses. Although the primary response of CD11a−/− CD8 T cells was diminished, development of memory was not overtly affected. Indeed, based on the maximum peak of each response, CD11a−/− mice tended to generate a larger proportion of memory cells than WT mice, perhaps as a result of enhanced TCM generation, since these cells undergo greater proliferation than do TEM (64, 65). In contrast, the aforementioned ICAM-1 study showed that memory is not generated in the absence of ICAM-1, despite a normal primary response (51). This is difficult to reconcile with our findings and other available literature and suggests that ICAM-1 is possibly involved in survival of cells during or after the contraction phase. Another possibility is that the adoptively transferred OT-I CD8 T cells upregulate ICAM-1 after activation and were rejected by the ICAM-1−/− host. Further studies will be needed to resolve these issues. Our results also showed that reactivation of memory CD8 T cells was largely CD11a independent. One interpretation of these findings is that long-lived stable interactions are not compulsory for memory CD8 T cell activation. This is in agreement with our previous findings that memory CD8 T cell reactivation requires a significantly shorter window of antigen availability than do naïve T cells (54).
Collectively, our results delineated several key features of CD11a-mediated costimulation that regulates the CD8 T cell response to infection. First, optimal initial priming for proliferation and induction of lytic activity, but not effector cytokine production, required CD11a. Second, effector subset ratios were controlled by CD11a. Third, CD11a played a major role in controlling CD62L expression and, therefore, the TCM/TEM ratio. Finally, memory CD8 T cell reactivation was CD11a independent. These data provide a comprehensive view of the important role of CD11a in the CD8 T cell response to infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sara Colpitts for critically reading the manuscript.
This work was supported in part by NIH grants AI41576, AI78289, and AI76457 (to L.L.), T32 AI07080 (to T.O.B.), and F32 AI84328 (to E.R.J.).
Footnotes
Published ahead of print 28 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00749-12.
REFERENCES
- 1. McFarland HI, Nahill SR, Maciaszek JW, Welsh RM. 1992. CD11b(Mac-1): a marker for CD8+ cytotoxic T cell activation and memory in virus infection. J. Immunol. 149:1326–1333 [PubMed] [Google Scholar]
- 2. Huleatt JW, Lefrançois L. 1995. Antigen-driven induction of CD11c on intestinal intraepithelial lymphocytes and CD8(+) T cells in vivo. J. Immunol. 154:5684–5693 [PubMed] [Google Scholar]
- 3. Hynes RO. 1992. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69:11–25 [DOI] [PubMed] [Google Scholar]
- 4. Anderson DC, Springer TA. 1987. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu. Rev. Med. 38:175–194 [DOI] [PubMed] [Google Scholar]
- 5. Hogg N, Bates PA. 2000. Genetic analysis of integrin function in man: LAD-1 and other syndromes. Matrix Biol. 19:211–222 [DOI] [PubMed] [Google Scholar]
- 6. Evans R, Patzak I, Svensson L, De Filippo K, Jones K, McDowall A, Hogg N. 2009. Integrins in immunity. J. Cell Sci. 122:215–225 [DOI] [PubMed] [Google Scholar]
- 7. Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, Smith CW, Montgomery CA, Rich S, Beaudet AL. 1998. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 188:119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liang Y, Liu C, Djeu JY, Zhong B, Peters T, Scharffetter-Kochanek K, Anasetti C, Yu XZ. 2008. β2 integrins separate graft-versus-host disease and graft-versus-leukemia effects. Blood 111:954–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ren B, McCrory MA, Pass C, Bullard DC, Ballantyne CM, Xu Y, Briles DE, Szalai AJ. 2004. The virulence function of Streptococcus pneumoniae surface protein A involves inhibition of complement activation and impairment of complement receptor-mediated protection. J. Immunol. 173:7506–7512 [DOI] [PubMed] [Google Scholar]
- 10. Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, Smith CW, Ballantyne CM. 1999. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 163:5029–5038 [PubMed] [Google Scholar]
- 11. Borjesson DL, Simon SI, Hodzic E, DeCock HE, Ballantyne CM, Barthold SW. 2003. Roles of neutrophil beta 2 integrins in kinetics of bacteremia, extravasation, and tick acquisition of Anaplasma phagocytophila in mice. Blood 101:3257–3264 [DOI] [PubMed] [Google Scholar]
- 12. Borjesson DL, Simon SI, Hodzic E, Ballantyne CM, Barthold SW. 2002. Kinetics of CD11b/CD18 up-regulation during infection with the agent of human granulocytic ehrlichiosis in mice. Lab. Invest. 82:303–311 [DOI] [PubMed] [Google Scholar]
- 13. Kadioglu A, De FK, Bangert M, Fernandes VE, Richards L, Jones K, Andrew PW, Hogg N. 2011. The integrins Mac-1 and α4β1 perform crucial roles in neutrophil and T cell recruitment to lungs during Streptococcus pneumoniae infection. J. Immunol. 186:5907–5915 [DOI] [PubMed] [Google Scholar]
- 14. Allen SJ, Mott KR, Chentoufi AA, BenMohamed L, Wechsler SL, Ballantyne CM, Ghiasi H. 2011. CD11c controls herpes simplex virus 1 responses to limit virus replication during primary infection. J. Virol. 85:9945–9955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu H, Rodgers JR, Perrard XY, Perrard JL, Prince JE, Abe Y, Davis BK, Dietsch G, Smith CW, Ballantyne CM. 2004. Deficiency of CD11b or CD11d results in reduced staphylococcal enterotoxin-induced T cell response and T cell phenotypic changes. J. Immunol. 173:297–306 [DOI] [PubMed] [Google Scholar]
- 16. Dustin ML, Springer TA. 1989. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature 341:619–624 [DOI] [PubMed] [Google Scholar]
- 17. van Seventer GA, Shimizu Y, Horgan KJ, Shaw S. 1990. The LFA-1 ligand ICAM-1 provides an important costimulatory signal for T cell receptor-mediated activation of resting T cells. J. Immunol. 144:4579–4586 [PubMed] [Google Scholar]
- 18. Bachmann MF, Kall-Faienza K, Schmits R, Bouchard D, Beach J, Speiser DE, Mak TW, Ohashi PS. 1997. Distinct roles for LFA-1 and CD28 during activation of naive T cells: adhesion versus costimulation. Immunity 7:549–557 [DOI] [PubMed] [Google Scholar]
- 19. Park EJ, Peixoto A, Imai Y, Goodarzi A, Cheng G, Carman CV, von Andrian UH, Shimaoka M. 2010. Distinct roles for LFA-1 affinity regulation during T-cell adhesion, diapedesis, and interstitial migration in lymph nodes. Blood 115:1572–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berlin-Rufenach C, Otto F, Mathies M, Westermann J, Owen MJ, Hamann A, Hogg N. 1999. Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1-deficient mice. J. Exp. Med. 189:1467–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmits R, Kundig TM, Baker DM, Shumaker G, Simard JJL, Duncan G, Wakeman A, Shahinian A, van der Heiden A, Bachmann MF, Ohashi PS, Mak TW, Hickstein DD. 1996. LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J. Exp. Med. 183:1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marski M, Ye AL, Abraham C. 2007. CD18 is required for intestinal T cell responses at multiple immune checkpoints. J. Immunol. 178:2104–2112 [DOI] [PubMed] [Google Scholar]
- 23. Prince JE, Brayton CF, Fossett MC, Durand JA, Kaplan SL, Smith CW, Ballantyne CM. 2001. The differential roles of LFA-1 and Mac-1 in host defense against systemic infection with Streptococcus pneumoniae. J. Immunol. 166:7362–7369 [DOI] [PubMed] [Google Scholar]
- 24. Ghosh S, Chackerian AA, Parker CM, Ballantyne CM, Behar SM. 2006. The LFA-1 adhesion molecule is required for protective immunity during pulmonary Mycobacterium tuberculosis infection. J. Immunol. 176:4914–4922 [DOI] [PubMed] [Google Scholar]
- 25. Hamann A, Jablonski-Westrich D, Duijvestijn A, Butcher EC, Baisch H, Harder R, Thiele HG. 1988. Evidence for an accessory role of LFA-1 in lymphocyte-high endothelium interaction during homing. J. Immunol. 140:693–699 [PubMed] [Google Scholar]
- 26. Miyamoto M, Emoto M, Emoto Y, Brinkmann V, Yoshizawa I, Seiler P, Aichele P, Kita E, Kaufmann SH. 2003. Neutrophilia in LFA-1-deficient mice confers resistance to listeriosis: possible contribution of granulocyte-colony-stimulating factor and IL-17. J. Immunol. 170:5228–5234 [DOI] [PubMed] [Google Scholar]
- 27. Emoto M, Miyamoto M, Emoto Y, Yoshizawa I, Brinkmann V, van Rooijen N, Kaufmann SH. 2003. Highly biased type 1 immune responses in mice deficient in LFA-1 in Listeria monocytogenes infection are caused by elevated IL-12 production by granulocytes. J. Immunol. 171:3970–3976 [DOI] [PubMed] [Google Scholar]
- 28. Wu H, Prince JE, Brayton CF, Shah C, Zeve D, Gregory SH, Smith CW, Ballantyne CM. 2003. Host resistance of CD18 knockout mice against systemic infection with Listeria monocytogenes. Infect. Immun. 71:5986–5993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi C, Hohl TM, Leiner I, Equinda MJ, Fan X, Pamer EG. 2011. Ly6G+ neutrophils are dispensable for defense against systemic Listeria monocytogenes infection. J. Immunol. 187:5293–5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carr KD, Sieve AN, Indramohan M, Break TJ, Lee S, Berg RE. 2011. Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection. Eur. J. Immunol. 41:2666–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Serbina NV, Shi C, Pamer EG. 2012. Monocyte-mediated immune defense against murine Listeria monocytogenes infection. Adv. Immunol. 113:119–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. 1996. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5:653–666 [DOI] [PubMed] [Google Scholar]
- 33. Pope C, Kim Marzo S-KA, Masopust D, Williams K, Jiang J, Shen H, Lefrançois L. 2001. Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J. Immunol. 166:3402–3409 [DOI] [PubMed] [Google Scholar]
- 34. Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, Cleary PP, Jenkins MK. 2010. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat. Immunol. 11:83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khanna KM, McNamara JT, Lefrancois L. 2007. In situ imaging of the endogenous CD8 T cell response to infection. Science 318:116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Turner MJ, Jellison ER, Lingenheld EG, Puddington L, Lefrancois L. 2008. Avidity maturation of memory CD8 T cells is limited by self-antigen expression. J. Exp. Med. 205:1859–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Obar JJ, Khanna KM, Lefrancois L. 2008. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity 28:859–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin Y, Roberts TJ, Sriram V, Cho S, Brutkiewicz RR. 2003. Myeloid marker expression on antiviral CD8+ T cells following an acute virus infection. Eur. J. Immunol. 33:2736–2743 [DOI] [PubMed] [Google Scholar]
- 39. Klonowski KD, Marzo AL, Williams KJ, Lee SJ, Pham QM, Lefrancois L. 2006. CD8 T cell recall responses are regulated by the tissue tropism of the memory cell and pathogen. J. Immunol. 177:6738–6746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198 [DOI] [PubMed] [Google Scholar]
- 41. Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham QM, Zickovich JM, Lefrancois L. 2011. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J. Immunol. 187:4967–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Varga G, Nippe N, Balkow S, Peters T, Wild MK, Seeliger S, Beissert S, Krummen M, Roth J, Sunderkotter C, Grabbe S. 2010. LFA-1 contributes to signal I of T-cell activation and to the production of T(h)1 cytokines. J. Invest. Dermatol. 130:1005–1012 [DOI] [PubMed] [Google Scholar]
- 43. Davignon D, Martz E, Reynolds T, Kurzinger K, Springer TA. 1981. Lymphocyte function-associated antigen 1 (LFA-1): a surface antigen distinct from Lyt-2,3 that participates in T lymphocyte-mediated killing. Proc. Natl. Acad. Sci. U. S. A. 78:4535–4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. 2003. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 281:65–78 [DOI] [PubMed] [Google Scholar]
- 45. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. 2007. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27:281–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cui W, Joshi NS, Jiang A, Kaech SM. 2009. Effects of Signal 3 during CD8 T cell priming: bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine 27:2177–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Obar JJ, Lefrancois L. 2010. Early signals during CD8 T cell priming regulate the generation of central memory cells. J. Immunol. 185:263–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Obar JJ, Lefrancois L. 2010. Early events governing memory CD8+ T-cell differentiation. Int. Immunol. 22:619–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, Lefrancois L. 2010. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc. Natl. Acad. Sci. U. S. A. 107:193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kohlmeier JE, Reiley WW, Perona-Wright G, Freeman ML, Yager EJ, Connor LM, Brincks EL, Cookenham T, Roberts AD, Burkum CE, Sell S, Winslow GM, Blackman MA, Mohrs M, Woodland DL. 2011. Inflammatory chemokine receptors regulate CD8+ T cell contraction and memory generation following infection. J. Exp. Med. 208:1621–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scholer A, Hugues S, Boissonnas A, Fetler L, Amigorena S. 2008. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity 28:258–270 [DOI] [PubMed] [Google Scholar]
- 52. Badovinac VP, Porter BB, Harty JT. 2004. CD8+ T cell contraction is controlled by early inflammation. Nat. Immunol. 5:809–817 [DOI] [PubMed] [Google Scholar]
- 53. Sarkar S, Teichgraber V, Kalia V, Polley A, Masopust D, Harrington LE, Ahmed R, Wherry EJ. 2007. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J. Immunol. 179:6704–6714 [DOI] [PubMed] [Google Scholar]
- 54. Blair DA, Turner DL, Bose TO, Pham QM, Bouchard KR, Williams KJ, McAleer JP, Cauley LS, Vella AT, Lefrancois L. 2011. Duration of antigen availability influences the expansion and memory differentiation of T cells. J. Immunol. 187:2310–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mempel TR, Henrickson SE, von Andrian UH. 2004. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427:154–159 [DOI] [PubMed] [Google Scholar]
- 56. Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. 1998. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395:82–86 [DOI] [PubMed] [Google Scholar]
- 57. Parameswaran N, Suresh R, Bal V, Rath S, George A. 2005. Lack of ICAM-1 on APCs during T cell priming leads to poor generation of central memory cells. J. Immunol. 175:2201–2211 [DOI] [PubMed] [Google Scholar]
- 58. Huleatt JW, Lefrançois L. 1996. β2-integrins and ICAM-1 are involved in establishment of the mucosal T cell compartment. Immunity 5:263–273 [DOI] [PubMed] [Google Scholar]
- 59. Sims TN, Dustin ML. 2002. The immunological synapse: integrins take the stage. Immunol. Rev. 186:100–117 [DOI] [PubMed] [Google Scholar]
- 60. Li D, Molldrem JJ, Ma Q. 2009. LFA-1 regulates CD8+ T cell activation via T cell receptor-mediated and LFA-1-mediated Erk1/2 signal pathways. J. Biol. Chem. 284:21001–21010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Beinke S, Phee H, Clingan JM, Schlessinger J, Matloubian M, Weiss A. 2010. Proline-rich tyrosine kinase-2 is critical for CD8 T-cell short-lived effector fate. Proc. Natl. Acad. Sci. U. S. A. 107:16234–16239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chao CC, Jensen R, Dailey MO. 1997. Mechanisms of L-selectin regulation by activated T cells. J. Immunol. 159:1686–1694 [PubMed] [Google Scholar]
- 63. Smalley DM, Ley K. 2005. L-selectin: mechanisms and physiological significance of ectodomain cleavage. J. Cell. Mol. Med. 9:255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4:225–234 [DOI] [PubMed] [Google Scholar]
- 65. Marzo AL, Klonowski KD, Le Bon A, Borrow P, Tough DF, Lefrancois L. 2005. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat. Immunol. 6:793–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.