

Abstract
A multiresistant clonal Escherichia coli O78:H10 strain qualifying molecularly as enteroaggregative Escherichia coli (EAEC) was recently shown to be the cause of a community-acquired outbreak of urinary tract infection (UTI) in greater Copenhagen, Denmark, in 1991. This marks the first time EAEC has been associated with an extraintestinal disease outbreak. Importantly, the outbreak isolates were recovered from the urine of patients with symptomatic UTI, strongly implying urovirulence. Here, we sought to determine the uropathogenic properties of the Copenhagen outbreak strain and whether these properties are conferred by the EAEC-specific virulence factors. We demonstrated that through expression of aggregative adherence fimbriae, the principal adhesins of EAEC, the outbreak strain exhibited pronouncedly increased adherence to human bladder epithelial cells compared to prototype uropathogenic strains. Moreover, the strain was able to produce distinct biofilms on abiotic surfaces, including urethral catheters. These findings suggest that EAEC-specific virulence factors increase uropathogenicity and may have played a significant role in the ability of the strain to cause a community-acquired outbreak of UTI. Thus, inclusion of EAEC-specific virulence factors is warranted in future detection and characterization of uropathogenic E. coli.
INTRODUCTION
Enteroaggregative Escherichia coli (EAEC) is best known for causing acute and persistent diarrheal illness in developing countries as well as in travelers and immunocompromised individuals (1). Moreover, several EAEC-associated outbreaks have been reported worldwide (2–4). Most notably, a Shiga toxin-producing EAEC strain caused a major food-borne outbreak in Germany in May-June 2011 infecting over 4,000 individuals and leading to 53 deaths (5), thus emphasizing the need for an increased understanding of EAEC pathogenesis and its epidemic potential.
Conventionally, EAEC and other diarrheagenic E. coli isolates are thought to be distinct from extraintestinal pathogenic E. coli (ExPEC) (6). However, recent studies have demonstrated the presence of EAEC-defining genes in collections of uropathogenic E. coli (UPEC) isolates (7–9). Most interesting is the recent discovery of a clonal EAEC strain responsible for a cluster of predominantly community-acquired urinary tract infections (UTI) in greater Copenhagen, Denmark, in 1991 (10, 11). During this outbreak, 19 isolates of a multiresistant E. coli O78:H10 strain were collected, 18 of which were urine culture isolates from patients with symptomatic UTI; the remaining fecal isolate was recovered from a patient with diarrhea (10).
The phylogenetic origin, clonal background, and virulence characteristics of these 19 isolates were recently characterized and compared to those of archived nonoutbreak O78:H10 strains. All of the outbreak isolates were found to (i) be multidrug resistant and (ii) represent a single clonal group within phylogenetic group A (sequence type 10), as well as (iii) fulfill molecular criteria for the EAEC pathotype (11). Notably, except aerobactin, none of the isolates harbored ExPEC-defining traits (P fimbriae, S/F1C fimbriae, Dr-binding adhesins, or group 2 capsule), thereby failing to fulfill the operational molecular criteria currently defining ExPEC (12). They did, however, contain ExPEC-associated virulence genes, including fyuA (yersiniabactin receptor) and traT (serum resistance associated), as well as the serine protease autrotransporters of Enterobacteriaceae (SPATEs) Pic and Sat, both of which are also associated with diarrheagenic E. coli such as EAEC (13).
EAEC-specific virulence factors present in all 19 Copenhagen outbreak isolates included aggregative adherence fimbria (AAF) variant I; AggR, a global regulator of EAEC virulence (14, 15); dispersin, required for proper dispersal of AAFs on the bacterial surface (16); and the Aat transporter system, which mediates dispersin secretion (17). AAFs play a central role in EAEC pathogenesis by facilitating adherence to human intestinal mucosa and formation of a thick biofilm within the mucus layer covering the epithelium, thus allowing the organism to persistently colonize the intestinal tract and cause infection (18). Notably, AAFs have also been shown to mediate adherence to human urethral mucosa in vitro (19), implying a role for these multifunctional adhesins in conferring uropathogenic fitness. Finally, AAF-mediated adherence also appears to trigger host inflammatory responses, including the release of proinflammatory cytokines and recruitment and infiltration of neutrophils (20–22).
As described by Olesen et al. (11), the facts that the Copenhagen outbreak isolates originated from patients with symptomatic UTI and that all of them possessed ExPEC-associated genes imply extraintestinal virulence properties. Moreover, unlike the commensal E. coli strain MG1655, a representative outbreak isolate (C555-91) was shown to be highly lethal in an established mouse model of subcutaneously induced sepsis (11, 23). Here, we sought to further characterize the extraintestinal potential of the outbreak strain, focusing on the role of the EAEC-specific virulence factors.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The 19 outbreak isolates used in this study were collected in 1991 during a predominantly community-acquired outbreak of a multiresistant clonal E. coli strain (O78:H10) in greater Copenhagen, Denmark, in 1991 (10). All other strains used are listed in Table 1.
Table 1.
Strains used in this study
| Strain | Description | Reference |
|---|---|---|
| C555-11 | Representative isolate from the 1991 Copenhagen UTI outbreak | 10 |
| C555-91 ΔaggDCBA | C555-91 in which a kanamycin cassette has been inserted into the aggDCBA gene cluster encoding AAF/I | This study |
| C555-91 ΔaggR | C555-91 in which a kanamycin cassette has been inserted into the aggR gene | This study |
| 042 | Prototype EAEC strain expressing AAF/II | 29 |
| JM221 | Prototype EAEC strain expressing AAF/I | 30 |
| CFT073 | Prototype UPEC strain | 28 |
| J96 | Prototype UPEC strain | 27 |
| MG1655 | Commensal E. coli K-12 strain | 24 |
| F-18 | Commensal E. coli strain | 26 |
| HB101 | Nonfimbriated E. coli K-12 strain | 25 |
Bacteria were routinely cultured at 37°C on Luria-Bertani (LB) agar and in LB broth. For cell adhesion experiments, EAEC overnight cultures grown in LB broth were subcultured into Dulbecco's modified Eagle medium (DMEM) with 0.45% glucose.
Construction of isogenic aggDCBA and aggR mutants.
The AAF/I gene cluster (aggDCBA) and the aggR gene in C555-91, a representative Copenhagen outbreak isolate, were deleted by allelic exchange with a kanamycin resistance-encoding cassette flanked by regions homologous to sequences on either side of the aggDCBA gene cluster and the aggR gene, respectively. All primers used are listed in Table 2. The aggDCBA-flanking cassette was generated by PCR amplification from a previously constructed JM221 ΔaggDCBA mutant strain using primers B-aggD-F and E-aggA-R (31). The aggR-flanking cassette was generated by amplification of the kanamycin resistance-encoding gene (kan) from pKD4 (32) using primers aggR-Kn1 and aggR-Kn2 containing 30-bp overhangs from the upstream and downstream regions of aggR, respectively. The purified PCR products were transformed into C555-91 harboring the thermosensitive plasmid pKOBEGApra encoding the λ Red recombinase (33). Both mutants were selected by growth on LB plates containing 50 μg/ml kanamycin at 37°C. Loss of the pKOBEGApra plasmid was verified by the inability of the mutants to grow on LB plates containing 30 μg/ml apramycin. Correct allelic exchange was verified by PCR analysis using combinations of primers inside the kan gene (K1 and K2) and primers B-aggD-F and E-aggA-R (for the aggDCBA mutant) or primers B-aggR-F and E-aggR-R (for the aggR mutant).
Table 2.
Primers used in this study
| Primer | Sequence (5′→3′) |
|---|---|
| B-aggD-F | TTTTTAGCGTTATATGATTTG |
| E-aggA-R | TATGATCCTTTCCGTTTTTATG |
| aggR-Kn1 | AAATATGATGTACTGGAAAATCCTATCATAGTGTAGGCTGGAGCTGCTTC |
| aggR-Kn2 | TACTATATACATCTAATTGTACAATCGATGATGGGAATTAGCCATGGTCC |
| K1 | CAGTCATAGCCGAATAGCCT |
| K2 | CGGTGCCCTGAATGAACTGC |
| B-aggR-F | TCAAGAATTGTTTTGGTGTTATGC |
| E-aggR-R | AAAACAAAACATCGAAAAAGAGA |
Biofilm formation in microtiter plates.
Overnight cultures of bacteria were diluted 1:50 in DMEM with 0.45% glucose and grown in 96-well microtiter plates (Nunc) for 24 h with mild shaking, washed to remove unbound bacteria, and then stained with 0.1% crystal violet (CV). Biofilm formation was quantified by dissolving the CV in 1 ml of 96% ethanol, and optical density at 595 nm (OD595) was measured.
Cell adhesion assays.
The human larynx cancer-derived epithelial cell line HEp-2 and the human bladder cancer-derived epithelial cell line 5637 were both maintained in RPMI 1640 medium (Invitrogen) containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. The human colon cancer-derived epithelial cell line T84 was maintained in DMEM–F-12 medium (Invitrogen) containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin.
For the HEp-2 adhesion assay, cells were grown to 50% confluence in Lab-Tek chamber slides (Nunc), washed in 0.9% NaCl, supplied with 1 ml DMEM with 0.45% glucose, 10% FBS, and 1% mannose, and infected with 10 μl of bacterial suspensions (1 × 108 bacteria) at 37°C for 3 h. Following washing, cells and adhering bacteria were fixed in 4% formalin in phosphate-buffered saline (PBS) for 10 min, washed, stained in 0.1% CV for 5 min, and washed again, and the slides were mounted using Vectashield solution (Vector).
For the T84 and 5637 adhesion assays, cells were grown to 90 to 100% confluence in 24-well plates (Nunc), washed, supplied with 1 ml fresh culture medium without antibiotics, and infected with 25 μl of bacterial suspensions (2 × 106 bacteria) at 37°C for 3 h. For quantification of the total number of bacteria, Triton X-100 (0.5% final concentration) was added to wells containing both cell-associated and nonadhering bacteria, followed by a 10-min lysis step. For quantification of cell-associated bacteria using other wells, nonadhering bacteria were removed by washing and the cell monolayers lysed as described above. Serial dilutions of lysed cells and bacteria were plated and colony counted the following day. The data shown are numbers of cell-associated bacteria relative to the total numbers of bacteria recovered.
Mouse model of ascending urinary tract infection.
Single- and coinfection studies using 6- to 8-week-old female OF-1 (Charles River Laboratories) mice were carried out as previously described (34, 35). Briefly, the mice were anesthetized and inoculated transurethrally with 50 μl of bacterial suspension containing approximately 5 × 108 CFU of wild-type and mutant strains, either separately or mixed 1:1, through a sterile catheter. The mice were sacrificed 3 days after inoculation. For the recovery of bacteria, bladders and kidneys were collected in 1 ml 0.9% NaCl and homogenized, and serial dilutions were plated on MacConkey medium. All animal experiments were conducted under the auspices of the Danish Animal Experiments Inspectorate, the Danish Ministry of Justice.
Biofilm on catheters under hydrodynamic conditions.
Biofilm formation under hydrodynamic conditions on size 14 sterile Foley all-silicone catheters (Bard) was carried out as previously described by Stahlhut et al. with some modifications (36–38). Briefly, catheters were cut sterilely to 4.5-cm-long pieces and connected to a Watson-Marlow 205S pump using silicone tubing. The system was sterilized with 0.5% sodium hypochlorite for 3 to 4 h and washed extensively with sterile water prior to connection of the sterile catheter pieces. After connection of the catheter, the system was filled with prewarmed (37°C) sterile artificial urine (39) and the flow was left on for 2 h to precondition the catheter surface. Overnight cultures of EAEC subcultured into DMEM with 0.45% glucose for 4 h and then diluted to an OD600 of 0.05 were then inoculated in each channel. The bacteria were allowed to become established in catheters for 1 h before the flow was turned on at a rate of 1 ml/h. Following incubation at 37°C for 48 h, the catheter pieces were disconnected from the system and connected to 1-ml syringes. Cells attached to the catheter surfaces were stained by aspiration with 0.1% CV through the catheters by the syringes and left for 20 min. The catheters were then washed three times in PBS to remove excess CV and subsequently dried. Finally, CV was dissolved in 1 ml of 96% ethanol and the OD595 was measured. A catheter piece treated the same way without any contact with bacterial cells served as a blank.
Catheterized bladder model.
The catheterized bladder model was performed as previously described by Stahlhut et al. with some modifications (38, 40). Briefly, two-compartment (inner and outer compartment) glass chambers were maintained at 37°C by circulating water through the outer compartment. A catheter was inserted into the inner compartment of the glass chambers through an outlet at the base, followed by inflation of the catheter retention balloon with 10 ml sterile water. The catheters were connected to standard drainage bags, and 15 ml of sterile artificial urine (39) was pumped into the inner chambers.
For inoculation, overnight bacterial cultures grown in LB were diluted 1:100 in DMEM with 0.45% glucose, incubated for 6 h, and then diluted to an OD600 of 0.2 in prewarmed (37°C) artificial urine. A 5-ml sample of bacterial suspension (approximately 1 × 108 CFU) was added to the artificial urine in the inner chambers. After 1 h, the artificial urine supply was resumed for 24 h at a constant rate of 30 ml/h. For quantification of biofilms on catheters, the artificial urine supply was stopped and a sample of the bladder (inner compartment) was collected. The inserted catheters were carefully removed and cut aseptically. The tip of the catheter (including the eyehole) was transferred to 1 ml 0.9% NaCl, and the retention balloon section was removed. The next 1-cm catheter section, serving as the catheter sample, was cut sterilely down the middle, transferred to 1 ml 0.9% NaCl, and scraped thoroughly to release catheter-associated biofilms. All samples were sonicated for 5 min (50% power) and vortexed for 2 min and then serially diluted, plated, and counted the next day.
Statistical analysis.
Student's t test was used for statistical evaluation, and P values of <0.05 were considered statistically significant.
RESULTS
Biofilm formation in microtiter wells by the Copenhagen outbreak isolates.
To assess the pathogenic potential of the 19 Copenhagen outbreak isolates, we initially screened all of them for their ability to form biofilms in microtiter plates using DMEM with 0.45% glucose as the medium (AggR-inducing conditions [41]). The majority of the 19 isolates were found to be modest to good biofilm producers (data not shown). Strain C555-91 was selected as a representative outbreak isolate for further examination in this study.
Biofilm formation in microtiter plates by C555-91 is AAF dependent.
We sought to determine the role of AAFs in the ability of the Copenhagen outbreak strain to form biofilms. The aggDCBA gene cluster (encoding AAF/I) in C555-91 was deleted by allelic replacement of the entire cluster with a kanamycin resistance-encoding cassette. The same technique was also used to generate a C555-91 isogenic AggR mutant strain. Confirming correct mutant constructions, we observed that, like other AAF-producing EAEC strains (42), wild-type C555-91 was capable of agglutinating human erythrocytes, whereas both the AAF and AggR mutants failed to do so (data not shown).
We next examined the ability of these strains to form biofilms in the microtiter plate assay in comparison with those of prototype EAEC and UPEC strains. Notably, wild-type C555-91 produced biofilms at levels comparable to those of EAEC prototype strains 042 and JM221 and at levels significantly higher than those of UPEC prototype strains CFT073 and J96 or commensal E. coli strains MG1655 and F-18 (Fig. 1). However, deletions of AAF or AggR strikingly attenuated the ability of C555-91 to form biofilms (P < 0.001), revealing that the ability of C555-91 to form a strong biofilm is mediated by AAF fimbriae. Moreover, the reduced biofilm-forming capacity of the AggR mutant strain is consistent with lack of AAF expression.
Fig 1.
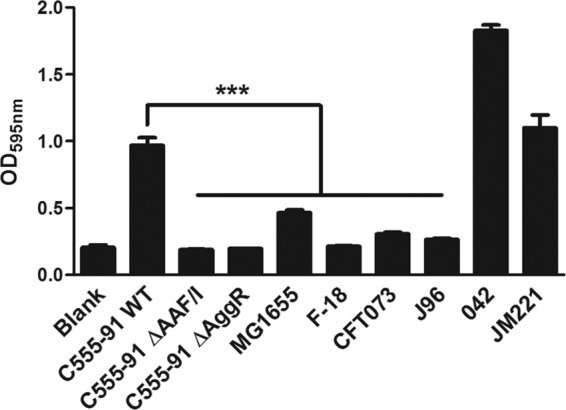
C555-91 forms AAF/I-dependent biofilms in microtiter plates. Wild-type (WT) C555-91, the AAF/I and AggR mutants, commensal E. coli strains MG1655 and F-18, UPEC prototype strains CFT073 and J96, and EAEC prototype strains 042 and JM221 were grown at 37°C in microtiter plates containing DMEM with 0.45% glucose for 24 h under shaking conditions, after which biofilm formation was quantified as described in Materials and Methods. The results are presented as the means ± standard errors of the means for eight replicates and represent one of three independent experiments performed with similar results. ***, P < 0.001.
AAF mediates aggregative adherence in C555-91.
To further characterize the C555-91 isolate, we assessed its ability to adhere to HEp-2 epithelial cells in an aggregative pattern, the gold standard for identifying EAEC (43). Similarly to prototype EAEC strain 042, C555-91 exhibited the characteristically stacked-brick aggregative pattern of adherence to the HEp-2 cells (Fig. 2). As expected, the aggregative phenotype was mediated by AAF/I fimbriae, as the AAF/I mutant was unable to adhere to the HEp-2 cells (Fig. 2).
Fig 2.

C555-91 exhibits AAF/I-dependent aggregative adherence to HEp-2 cells. Bacterial aggregative patterns of adherence (arrows) after 3 h of incubation of EAEC 042 or wild-type (WT) C555-91 with HEp-2 cells. The C555-91 AAF/I mutant strain failed to adhere to the HEp-2 cells or to the substratum.
AAF mediates adherence of C555-91 to human colonic and bladder epithelial cells.
We next sought to determine the ability of C555-91 to adhere to intestinal and bladder epithelial cells. The T84 human colonic epithelial cell line has previously been described as a model for EAEC adherence (44). We found that wild-type C555-91 adhered to T84 cells to nearly the same extent as EAEC strains 042 and JM221 (Fig. 3A). In contrast, deletions of AAF/I or AggR almost entirely abolished the cell-adhesive properties of C555-91 (P < 0.01) to levels comparable to HB101, a nonfimbriated E. coli strain incapable of adherence to epithelial cells.
Fig 3.
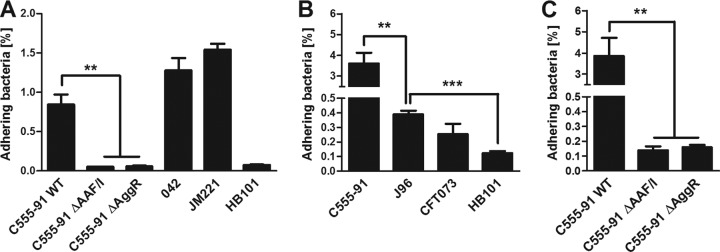
AAF/I facilitates adherence of C555-91 to colonic and bladder epithelial cells. (A) T84 colonic epithelial cell monolayers were infected with wild-type (WT) C555-91, the C555-91 AAF/I or AggR mutant, EAEC prototype strain 042 or JM221, or E. coli strain HB101 (negative control). (B and C) 5637 bladder epithelial cell monolayers were infected with wild-type C555-91, the C555-91 AAF/I or AggR mutant, UPEC prototype strain CFT073 or J96, or HB101 (negative control). The number of cell-adhering bacteria was quantified 3 h later as described in Materials and Methods. The results are presented as the means ± standard errors of the means for at least triplicate samples and represent one of three independent experiments performed with similar results. ***, P < 0.001; **, P < 0.01.
We next examined the ability of C555-91 to adhere to cell line 5637 human bladder epithelial cells. We found that wild-type C555-91 exhibited a remarkable high adherence capacity to the bladder cell line. Thus, the percentages of adhering C555-91 cells were 10- to 20-fold higher than those of the UPEC prototype strains J96 and CFT073 (P < 0.01) (Fig. 3B). In line with the observations from the T84 cell assays, the C555-91 AAF/I and AggR mutants both failed to adhere to the 5637 monolayers almost entirely (P < 0.01) (Fig. 3C). Thus, adherence of C555-91 to both intestinal and bladder epithelial cells requires AAF expression, and AggR is required for AAF expression.
C555-91 is virulent in a UTI mouse model.
To investigate the ability of C555-91 to cause UTI and assess the influence of EAEC virulence factors, a previously described mouse model of ascending UTI was applied (34). Groups of 7 or 8 mice were inoculated transurethrally with C555-91, C555-91 ΔAAF/I, or C555-91 ΔAggR, either separately or mixed 1:1 for competition experiments (wild-type versus either one of the two mutant strains). Indeed, C555-91 was able to cause UTI in the mouse model, as all bladder samples were found to be infected, with a median bacterial count of approximately 1 × 106 CFU per bladder (Fig. 4). However, the bacterial counts of the AAF/I and the AggR mutant were similar to that of the wild-type strain (Fig. 4). Also, from the kidney and urine samples similar levels of wild-type and both mutant strains were recovered (Fig. 4). In an attempt to detect more subtle differences in virulence, competition experiments were applied; however, even when in direct competition with the wild type, the AAF/I and AggR mutants were as virulent as the wild-type strain (data not shown). Thus, these findings show that C555-91 is capable of establishing a UTI but that AAF and AggR do not appear to influence the urovirulence in mice.
Fig 4.
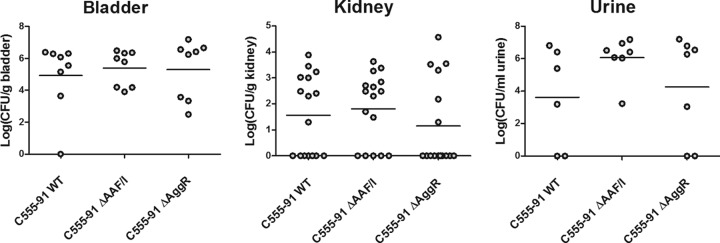
C555-91 causes urovirulence in a UTI mouse model without the requirement for AAF/I or AggR expression. Groups of eight mice were inoculated with wild-type (WT) C555-91, the AAF/I mutant, or the AggR mutant. Three days following inoculation, the mice were sacrificed and the bladders (n = 8) and kidneys (n = 16) collected. Urine samples (n = 6 or 7) were collected where possible. Bacteria present in the samples were enumerated as described in Materials and Methods. Values of 0 represent CFU counts below the lower detection limit of 50 CFU/g sample or CFU/ml sample.
AAF promotes biofilm formation on urethral catheters.
We next refocused our attention on the biofilm-forming properties of the Copenhagen outbreak strain. In particular, we sought to address its ability to form biofilms on urinary catheters, a common trait of uropathogens such as Klebsiella pneumoniae and UPEC strains, which frequently cause biofilm-related catheter-associated UTIs (CAUTIs) (45, 46).
We first investigated the ability of C555-91 to form biofilms on urethral catheters under hydrodynamic conditions using a previously described in vitro model system (38). Wild-type C555-91 and its isogenic AAF/I mutant were inoculated in catheters at 37°C under continuous-flow conditions employing artificial urine as the growth medium. After 48 h, the catheters were emptied and the catheter-associated bacteria were quantified by CV staining. Wild-type C555-91 produced extensive amounts of catheter-associated biofilm in this model system (Fig. 5). In contrast, the AAF/I mutant strain showed a 4-fold attenuation in biofilm formation compared to the wild type (P < 0.001).
Fig 5.
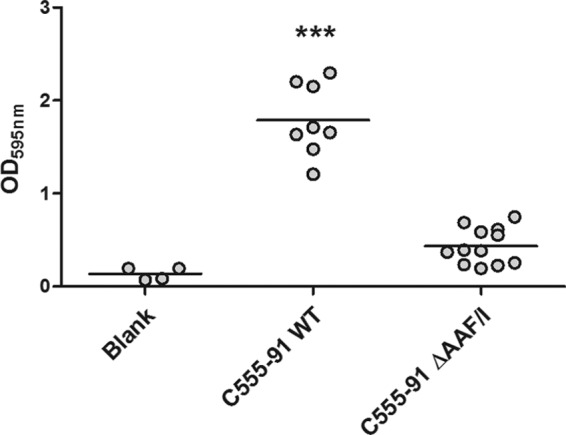
AAF/I plays a key role in promoting biofilm formation by C555-91 on urethral catheters under hydrodynamic conditions. Wild-type (WT) C555-91 and the C555-91 AAF/I mutant were inoculated in catheters under hydrodynamic conditions for 48 h, after which catheter-associated biofilm formation was quantified by CV staining (see Materials and Methods). All treatments were repeated at least 4 times. ***, P < 0.001.
To evaluate the potential of C555-91 to cause CAUTIs under conditions mimicking the conditions in the patients, an in vitro model of a catheterized bladder was applied. This model mirrors all the physicochemical properties of a CAUTI patient except for the presence of a bladder epithelium (38, 40). Using artificial urine as the growth medium, wild-type C555-91 and the AAF/I mutant were inoculated in the artificial bladder lumen at an initial concentration of ∼2 × 107 CFU/ml, mimicking an established CAUTI. After 24 h of artificial urine supply, bacterial counts were monitored in the bladder lumen, on the catheter tip (located in the bladder), and on the lower part of the catheter (below the retention balloon of the catheter). The two strains grew to the same extent in the bladder (Fig. 6), suggesting that deletion of AAF did not alter the growth rate in the bladder and thus did not affect the number of bacteria transferred from the bladder into the catheter. Extensive biofilm formation was observed on the catheter tips located in bladders infected with wild-type C555-91, whereas the AAF mutant showed an 8-fold attenuation in biofilm formation on the catheter tips (P < 0.001). On the catheter itself, the biofilm-promoting effect of AAF was even more striking, as the AAF mutant showed more than a 100-fold attenuation in biofilm formation on the catheter compared to the wild-type strain (P < 0.001) (Fig. 6).
Fig 6.
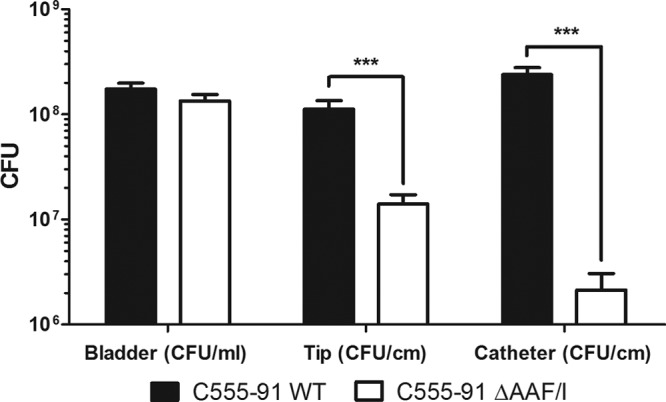
AAF/I-enhanced biofilm formation by C555-91 on urethral catheters in a catheterized bladder model. Biofilm formation by wild-type (WT) C555-91 (black bars) and the C555-91 AAF/I mutant (white bars) was quantified from the bladder, the catheter tip (located in the bladder), and the catheter as described in Materials and Methods. Both treatments were repeated 9 times. Means ± standard errors of the means are shown. ***, P < 0.001.
In conclusion, these results clearly demonstrate that C555-91 is capable of extensive biofilm formation on urethral catheters and that AAF expression plays a key role in facilitating this event.
DISCUSSION
The 1991 community-acquired UTI outbreak caused by a multiresistant clonal E. coli O78:H10 strain is quite unique since, unlike previously reported multiresistant clonal E. coli groups associated with UTI outbreaks (47–49), the Copenhagen outbreak strain did not fulfill the molecular criteria for ExPEC (11). Instead, it qualified molecularly as EAEC, thus for the first time associating a clonal EAEC strain with an extraintestinal disease outbreak. Importantly, 18 of the 19 collected outbreak isolates were from the urine of patients with symptomatic UTI, strongly implying an extraintestinal virulence potential (11).
The objective of this study was to characterize the uropathogenic properties of the Copenhagen outbreak strain, focusing on the role of the EAEC virulence factors. Notably, all 19 outbreak isolates harbored AAF variant I whereas none of the ExPEC-defining adherence factors P fimbriae, S/F1C fimbriae, or Dr-binding adhesin were found in any of the isolates (11). As adherence to epithelial surfaces is expected to be a critical first step in the pathogenesis of most diarrheagenic and uropathogenic E. coli strains (6), we therefore focused our initial attention on the role of the AAF adhesin.
C555-91 was chosen as a representative Copenhagen outbreak isolate. Of particular interest, this isolate originated from a 4-month-old boy with recurrent UTIs as well as diarrhea, suggesting that the Copenhagen outbreak strain exhibits both diarrheagenic and uropathogenic properties. In support of the diarrheagenic properties, the outbreak strain was also isolated from an AIDS patient with diarrhea (10).
As expected, the EAEC-characteristic pattern of adherence of C555-91 to HEp-2 cells was shown to require AAF expression. Consistent with EAEC prototype strain findings (44), C555-91 furthermore exhibited AAF-dependent adherence to human T84 colonic epithelial cells. More intriguingly, the AAF adhesin was shown also to mediate extensive adherence of C555-91 to human bladder epithelial cells, notably to a much greater extent than prototype UPEC strains, thus illustrating the potential of the Copenhagen outbreak strain to both colonize the intestinal tract and infect the bladder.
Olesen et al. evaluated the performance of C555-91 in a subcutaneous mouse sepsis model and found it to be highly lethal compared to the commensal MG1655 strain (11). While these in vivo observations point to extraintestinal pathogenic capabilities of the Copenhagen outbreak strain, this model system bypasses key early steps in uropathogenesis, including the requirement for adherence to urinary epithelial cells as well as entry and persistence within the urinary tract (23).
To address the urovirulence of the outbreak strain in vivo, we tested C555-91 in a mouse model of ascending UTI. The strain was indeed found to be virulent in the mouse UTI model, as all bladder samples were found to be infected. As C555-91 exhibited a remarkable AAF-mediated ability to adhere to human epithelial bladder cells, it was surprising that similar levels of bacteria were detected in the bladders of mice infected with the C555-91 wild-type or AAF or AggR mutant strain. Thus, in the mouse model, the ability of C555-91 to infect the bladder does not appear to correlate with AAF expression. Notably, in addition to regulating the machinery required for AAF expression (i.e., the AAF genes themselves, dispersin, and the dispersin transporter system), AggR has recently been shown to regulate a large number of additional EAEC genes (50). Thus, while further studies are needed to clarify the role of these potential virulence genes, our findings here suggest that none of them impact the uropathogenic properties of EAEC.
As C555-91 and its AAF mutant express type 1 fimbriae, as shown by the ability to agglutinate yeast cells in a mannose-sensitive manner, we speculate that in the mouse bladder adherence is primarily mediated by type 1 fimbriae. Notably, using the same model, we have previously demonstrated that K. pneumoniae adheres to the mouse bladder by virtue of type 1 fimbriae (35). The fact that AAF did not influence infection of the mouse bladder is consistent with a previous study demonstrating that AAF expression has no effect on the ability of EAEC to colonize the mouse intestinal tract (51). These observations likely reflect the general assumption that EAEC pathogenesis is highly adapted to human tissue and may result from the inability of AAF adhesins to bind to different receptors present on the mucosal surfaces of animal species compared with that in humans. Thus, it is likely that mice do not express AAF-specific receptors. In contrast, clear signs of disease and host inflammatory responses were observed when EAEC pathogenesis was examined in a model using human intestinal xenografts in immunodeficient mice in which the bacteria interact with epithelium of human origin (21).
In addition to our studies related to epithelial adherence, we also assessed the potential biofilm formation properties of the Copenhagen outbreak strain. Formation of thick layers of biofilms covering the intestinal mucosa is a defining feature of EAEC pathogenesis and likely contributes profoundly to the ability of the pathogen to persistently colonize the intestinal tract (18). Biofilm formation is also a common characteristic trait of uropathogens such as K. pneumoniae and UPEC strains and is believed to play a significant role in uropathogenicity. Biofilm formation is of particular concern for catheterized patients since indwelling urinary catheters provide an inert surface on which uropathogens can attach and establish CAUTIs. Moreover, the frequent recurrence of CAUTIs shortly after antibiotic treatment is believed to be caused by recolonization of organisms having survived in the biofilms (45, 46).
In line with AAF-producing EAEC prototype strains (41, 42), we found that the majority of the Copenhagen outbreak isolates were excellent producers of biofilms in the microtiter plate assay. Moreover, using the C555-91 wild-type and mutant strains we demonstrated that this event is AAF dependent. More intriguingly, using a catheterized bladder model, we found that the C555-91 wild-type strain was capable of producing extensive biofilms on catheters. Again, the remarkable ability of the outbreak strain to form these biofilms was mediated by AAF, as the C555-91 AAF/I mutant showed more than a 100-fold attenuation in biofilm formation on catheters compared to the wild type.
Whereas our study reveals that EAEC-specific virulence factors may promote the urovirulence of the outbreak strain, the fact that no previous reports of EAEC-associated UTI outbreaks have been reported raises an important question: are the uropathogenic properties of the Copenhagen outbreak strain attributable to a unique combination of ExPEC-associated and EAEC-specific virulence factors present in this particular strain? Or are future UTI-associated outbreaks caused by other strains with the EAEC-specific virulence background likely to occur? Our findings in this study show that EAEC virulence factors, in particular the AAF adhesins, have the capacity to confer uropathogenic virulence, e.g., uroepithelial adhesion and biofilm formation. Thus, EAEC strains appear capable of spreading from the human distal colon, their natural reservoir, to the urinary tract, where AAF may promote increased adhesion to and colonization of the bladder epithelium. In addition, the fact that the AAF organelle mediates extensive catheter-associated biofilm formation indicates a strong potential for urinary EAEC strains to cause CAUTIs. Collectively, we suggest that the Copenhagen outbreak strain was spread via a fecal-oral route and that the UTIs were a secondary consequence in some infected individuals.
In addition to AggR and AAF/I, other EAEC-associated virulence factors may also contribute to the urovirulence of the outbreak strain. Notably, the strain also possessed the SPATEs Sat and Pic, both of which are commonly found in EAEC but also in ExPEC strains (13, 52, 53). Sat functions as a vacuolating cytotoxin (54), whereas Pic exhibits mucinolytic properties causing modulation of host immune responses (55). Both Sat and Pic are also present in UPEC strain CFT073, but neither of the two SPATEs appears to effect colonization of this strain in the mouse model of ascending UTI (53, 54). However, Sat did cause histopathological changes to the mouse kidneys (54), whereas Pic appeared to decrease infiltration of neutrophils into the bladder lumen (53). The role of these SPATEs in uropathogenesis in humans warrants further investigation.
The current lack of global routine surveillance systems for detecting EAEC, likely rendering EAEC underreported, makes it difficult to address the extent to which this pathogen is associated with extraintestinal disease (56). Focus on EAEC in the context of diarrheal disease has increased since the 2011 major German outbreak (5), but only a few studies have investigated the prevalence of EAEC virulence factors in strains causing extraintestinal infections. However, the fact that EAEC strains were found with relatively high prevalence in collections of UTI isolates (7–9, 11) supports that EAEC-related virulence factors may likely play a role in uropathogenicity and illustrates the need for including these genes in future detection and characterization of uropathogenic E. coli. Moreover, given our findings that EAEC-specific fimbriae promote extensive biofilm formation on catheters, it will be highly relevant to screen CAUTI isolates for prevalence of EAEC-specific genes. Analysis of E. coli isolates from UTI patients will contribute to clarifying the potential role of EAEC in these important and very common infections.
ACKNOWLEDGMENTS
We thank Jakob Frimodt-Møller for verifying PCR results.
This work was supported by Danish Council for Strategic Research grant 2101-07-0023 to Karen A. Krogfelt.
Footnotes
Published ahead of print 28 January 2013
REFERENCES
- 1. Harrington SM, Dudley EG, Nataro JP. 2006. Pathogenesis of enteroaggregative Escherichia coli infection. FEMS Microbiol. Lett. 254:12–18 [DOI] [PubMed] [Google Scholar]
- 2. Cobeljić M, Miljković-Selimović B, Paunović-Todosijević D, Velicković Z, Lepsanović Z, Zec N, Savić D, Ilić R, Konstantinović S, Jovanović B, Kostić V. 1996. Enteroaggregative Escherichia coli associated with an outbreak of diarrhoea in a neonatal nursery ward. Epidemiol. Infect. 117:11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Itoh Y, Nagano I, Kunishima M, Ezaki T. 1997. Laboratory investigation of enteroaggregative Escherichia coli O untypeable:H10 associated with a massive outbreak of gastrointestinal illness. J. Clin. Microbiol. 35:2546–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith HR, Cheasty T, Rowe B. 1997. Enteroaggregative Escherichia coll and outbreaks of gastroenteritis in UK. Lancet 350:814–815 [DOI] [PubMed] [Google Scholar]
- 5. Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Møller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. 2011. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365:709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140 [DOI] [PubMed] [Google Scholar]
- 7. Abe CM, Salvador FA, Falsetti IN, Vieira MA, Blanco J, Blanco JE, Blanco M, Machado AM, Elias WP, Hernandes RT, Gomes TA. 2008. Uropathogenic Escherichia coli (UPEC) strains may carry virulence properties of diarrhoeagenic E. coli. FEMS Immunol. Med. Microbiol. 52:397–406 [DOI] [PubMed] [Google Scholar]
- 8. Nazemi A, Mirinargasi M, Merikhi N, Sharifi SH. 2011. Distribution of pathogenic genes aatA, aap, aggR, among uropathogenic Escherichia coli (UPEC) and their linkage with StbA gene. Indian J. Microbiol. 51:355–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park HK, Jung YJ, Chae HC, Shin YJ, Woo SY, Park HS, Lee SJ. 2009. Comparison of Escherichia coli uropathogenic genes (kps, usp and ireA) and enteroaggregative genes (aggR and aap) via multiplex polymerase chain reaction from suprapubic urine specimens of young children with fever. Scand. J. Urol. Nephrol. 43:51–57 [DOI] [PubMed] [Google Scholar]
- 10. Olesen B, Kolmos HJ, Orskov F, Orskov I. 1994. Cluster of multiresistant Escherichia coli O78:H10 in Greater Copenhagen. Scand. J. Infect. Dis. 26:406–410 [DOI] [PubMed] [Google Scholar]
- 11. Olesen B, Scheutz F, Andersen RL, Menard M, Boisen N, Johnston B, Hansen DS, Krogfelt KA, Nataro JP, Johnson JR. 2012. Enteroaggregative Escherichia coli O78:H10, the cause of an outbreak of urinary tract infection. J. Clin. Microbiol. 50:3703–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson JR, Murray AC, Gajewski A, Sullivan M, Snippes P, Kuskowski MA, Smith KE. 2003. Isolation and molecular characterization of nalidixic acid-resistant extraintestinal pathogenic Escherichia coli from retail chicken products. Antimicrob. Agents Chemother. 47:2161–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boisen N, Ruiz-Perez F, Scheutz F, Krogfelt KA, Nataro JP. 2009. Short report: high prevalence of serine protease autotransporter cytotoxins among strains of enteroaggregative Escherichia coli. Am. J. Trop. Med. Hyg. 80:294–301 [PMC free article] [PubMed] [Google Scholar]
- 14. Dudley EG, Thomson NR, Parkhill J, Morin NP, Nataro JP. 2006. Proteomic and microarray characterization of the AggR regulon identifies a pheU pathogenicity island in enteroaggregative Escherichia coli. Mol. Microbiol. 61:1267–1282 [DOI] [PubMed] [Google Scholar]
- 15. Nataro JP, Yikang D, Yingkang D, Walker K. 1994. AggR, a transcriptional activator of aggregative adherence fimbria I expression in enteroaggregative Escherichia coli. J. Bacteriol. 176:4691–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheikh J, Czeczulin JR, Harrington S, Hicks S, Henderson IR, Le Bouguénec C, Gounon P, Phillips A, Nataro JP. 2002. A novel dispersin protein in enteroaggregative Escherichia coli. J. Clin. Invest. 110:1329–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nishi J, Sheikh J, Mizuguchi K, Luisi B, Burland V, Boutin A, Rose DJ, Blattner FR, Nataro JP. 2003. The export of coat protein from enteroaggregative Escherichia coli by a specific ATP-binding cassette transporter system. J. Biol. Chem. 278:45680–45689 [DOI] [PubMed] [Google Scholar]
- 18. Nataro JP. 2005. Enteroaggregative Escherichia coli pathogenesis. Curr. Opin. Gastroenterol. 21:4–8 [PubMed] [Google Scholar]
- 19. Yamamoto T, Endo S, Yokota T, Echeverria P. 1991. Characteristics of adherence of enteroaggregative Escherichia coli to human and animal mucosa. Infect. Immun. 59:3722–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boll EJ, Struve C, Sander A, Demma Z, Krogfelt KA, McCormick BA. 2012. Enteroaggregative Escherichia coli promotes transepithelial migration of neutrophils through a conserved 12-lipoxygenase pathway. Cell. Microbiol. 14:120–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boll EJ, Struve C, Sander A, Demma Z, Nataro JP, McCormick BA, Krogfelt KA. 2012. The fimbriae of enteroaggregative Escherichia coli induce epithelial inflammation in vitro and in a human intestinal xenograft model. J. Infect. Dis. 206:714–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harrington SM, Strauman MC, Abe CM, Nataro JP. 2005. Aggregative adherence fimbriae contribute to the inflammatory response of epithelial cells infected with enteroaggregative Escherichia coli. Cell. Microbiol. 7:1565–1578 [DOI] [PubMed] [Google Scholar]
- 23. Johnson JR, Porter SB, Zhanel G, Kuskowski MA, Denamur E. 2012. Virulence of Escherichia coli clinical isolates in a murine sepsis model in relation to sequence type ST131 status, fluoroquinolone resistance, and virulence genotype. Infect. Immun. 80:1554–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462 [DOI] [PubMed] [Google Scholar]
- 25. Boyer HW, Roulland-Dussoix D. 1969. A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41:459–472 [DOI] [PubMed] [Google Scholar]
- 26. Cohen PS, Rossoll R, Cabelli VJ, Yang SL, Laux DC. 1983. Relationship between the mouse colonizing ability of a human fecal Escherichia coli strain and its ability to bind a specific mouse colonic mucous gel protein. Infect. Immun. 40:62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson JR, Brown JJ. 1996. A novel multiply primed polymerase chain reaction assay for identification of variant papG genes encoding the Gal(alpha 1–4)Gal-binding PapG adhesins of Escherichia coli. J. Infect. Dis. 173:920–926 [DOI] [PubMed] [Google Scholar]
- 28. Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. 1990. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect. Immun. 58:1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nataro JP, Baldini MM, Kaper JB, Black RE, Bravo N, Levine MM. 1985. Detection of an adherence factor of enteropathogenic Escherichia coli with a DNA probe. J. Infect. Dis. 152:560–565 [DOI] [PubMed] [Google Scholar]
- 30. Nataro JP, Deng Y, Cookson S, Cravioto A, Savarino SJ, Guers LD, Levine MM, Tacket CO. 1995. Heterogeneity of enteroaggregative Escherichia coli virulence demonstrated in volunteers. J. Infect. Dis. 171:465–468 [DOI] [PubMed] [Google Scholar]
- 31. Strauman MC, Harper JM, Harrington SM, Boll EJ, Nataro JP. 2010. Enteroaggregative Escherichia coli disrupts epithelial cell tight junctions. Infect. Immun. 78:4958–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chaveroche MK, Ghigo JM, d'Enfert C. 2000. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 28:E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hvidberg H, Struve C, Krogfelt KA, Christensen N, Rasmussen SN, Frimodt-Møller N. 2000. Development of a long-term ascending urinary tract infection mouse model for antibiotic treatment studies. Antimicrob. Agents Chemother. 44:156–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Struve C, Bojer M, Krogfelt KA. 2009. Identification of a conserved chromosomal region encoding Klebsiella pneumoniae type 1 and type 3 fimbriae and assessment of the role of fimbriae in pathogenicity. Infect. Immun. 77:5016–5024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Christensen BB, Sternberg C, Andersen JB, Palmer RJ, Nielsen AT, Givskov M, Molin S. 1999. Molecular tools for study of biofilm physiology. Methods Enzymol. 310:20–42 [DOI] [PubMed] [Google Scholar]
- 37. Ferrières L, Hancock V, Klemm P. 2007. Specific selection for virulent urinary tract infectious Escherichia coli strains during catheter-associated biofilm formation. FEMS Immunol. Med. Microbiol. 51:212–219 [DOI] [PubMed] [Google Scholar]
- 38. Stahlhut SG, Struve C, Krogfelt KA, Reisner A. 2012. Biofilm formation of Klebsiella pneumoniae on urethral catheters requires either type 1 or type 3 fimbriae. FEMS Immunol. Med. Microbiol. 65:350–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martino PD, Fursy R, Bret L, Sundararaju B, Phillips RS. 2003. Indole can act as an extracellular signal to regulate biofilm formation of Escherichia coli and other indole-producing bacteria. Can. J. Microbiol. 49:443–449 [DOI] [PubMed] [Google Scholar]
- 40. Stickler DJ, Morris NS, Winters C. 1999. Simple physical model to study formation and physiology of biofilms on urethral catheters. Methods Enzymol. 310:494–501 [DOI] [PubMed] [Google Scholar]
- 41. Sheikh J, Hicks S, Dall'Agnol M, Phillips AD, Nataro JP. 2001. Roles for Fis and YafK in biofilm formation by enteroaggregative Escherichia coli. Mol. Microbiol. 41:983–997 [DOI] [PubMed] [Google Scholar]
- 42. Boisen N, Struve C, Scheutz F, Krogfelt KA, Nataro JP. 2008. New adhesin of enteroaggregative Escherichia coli related to the Afa/Dr/AAF family. Infect. Immun. 76:3281–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nataro JP, Kaper JB. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11:142–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nataro JP, Hicks S, Phillips AD, Vial PA, Sears CL. 1996. T84 cells in culture as a model for enteroaggregative Escherichia coli pathogenesis. Infect. Immun. 64:4761–4768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Macleod SM, Stickler DJ. 2007. Species interactions in mixed-community crystalline biofilms on urinary catheters. J. Med. Microbiol. 56:1549–1557 [DOI] [PubMed] [Google Scholar]
- 46. Wang X, Lünsdorf H, Ehrén I, Brauner A, Römling U. 2010. Characteristics of biofilms from urinary tract catheters and presence of biofilm-related components in Escherichia coli. Curr. Microbiol. 60:446–453 [DOI] [PubMed] [Google Scholar]
- 47. Johnson JR, Stell AL, O'Bryan TT, Kuskowski M, Nowicki B, Johnson C, Maslow JN, Kaul A, Kavle J, Prats G. 2002. Global molecular epidemiology of the O15:K52:H1 extraintestinal pathogenic Escherichia coli clonal group: evidence of distribution beyond Europe. J. Clin. Microbiol. 40:1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lau SH, Kaufmann ME, Livermore DM, Woodford N, Willshaw GA, Cheasty T, Stamper K, Reddy S, Cheesbrough J, Bolton FJ, Fox AJ, Upton M. 2008. UK epidemic Escherichia coli strains A-E, with CTX-M-15 beta-lactamase, all belong to the international O25:H4-ST131 clone. J. Antimicrob. Chemother. 62:1241–1244 [DOI] [PubMed] [Google Scholar]
- 49. Manges AR, Tabor H, Tellis P, Vincent C, Tellier PP. 2008. Endemic and epidemic lineages of Escherichia coli that cause urinary tract infections. Emerg. Infect. Dis. 14:1575–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morin N, Santiago AE, Ernst RK, Guillot SJ, Nataro JP. 2013. Characterization of the AggR regulon in enteroaggregative Escherichia coli. Infect. Immun. 81:122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harrington SM, Sheikh J, Henderson IR, Ruiz-Perez F, Cohen PS, Nataro JP. 2009. The Pic protease of enteroaggregative Escherichia coli promotes intestinal colonization and growth in the presence of mucin. Infect. Immun. 77:2465–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guyer DM, Henderson IR, Nataro JP, Mobley HL. 2000. Identification of sat, an autotransporter toxin produced by uropathogenic Escherichia coli. Mol. Microbiol. 38:53–66 [DOI] [PubMed] [Google Scholar]
- 53. Heimer SR, Rasko DA, Lockatell CV, Johnson DE, Mobley HL. 2004. Autotransporter genes pic and tsh are associated with Escherichia coli strains that cause acute pyelonephritis and are expressed during urinary tract infection. Infect. Immun. 72:593–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guyer DM, Radulovic S, Jones FE, Mobley HL. 2002. Sat, the secreted autotransporter toxin of uropathogenic Escherichia coli, is a vacuolating cytotoxin for bladder and kidney epithelial cells. Infect. Immun. 70:4539–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ruiz-Perez F, Wahid R, Faherty CS, Kolappaswamy K, Rodriguez L, Santiago A, Murphy E, Cross A, Sztein MB, Nataro JP. 2011. Serine protease autotransporters from Shigella flexneri and pathogenic Escherichia coli target a broad range of leukocyte glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 108:12881–12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chattaway MA, Dallman T, Okeke IN, Wain J. 2011. Enteroaggregative E. coli O104 from an outbreak of HUS in Germany 2011, could it happen again? J. Infect. Dev. Ctries. 5:425–436 [DOI] [PubMed] [Google Scholar]