

Abstract
Burkholderia pseudomallei is a tier 1 select agent, and the causative agent of melioidosis, a disease with effects ranging from chronic abscesses to fulminant pneumonia and septic shock, which can be rapidly fatal. Autotransporters (ATs) are outer membrane proteins belonging to the type V secretion system family, and many have been shown to play crucial roles in pathogenesis. The open reading frame Bp1026b_II1054 (bcaA) in B. pseudomallei strain 1026b is predicted to encode a classical autotransporter protein with an approximately 80-kDa passenger domain that contains a subtilisin-related domain. Immediately 3′ to bcaA is Bp11026_II1055 (bcaB), which encodes a putative prolyl 4-hydroxylase. To investigate the role of these genes in pathogenesis, large in-frame deletion mutations of bcaA and bcaB were constructed in strain Bp340, an efflux pump mutant derivative of the melioidosis clinical isolate 1026b. Comparison of Bp340ΔbcaA and Bp340ΔbcaB mutants to wild-type B. pseudomallei in vitro demonstrated similar levels of adherence to A549 lung epithelial cells, but the mutant strains were defective in their ability to invade these cells and to form plaques. In a BALB/c mouse model of intranasal infection, similar bacterial burdens were observed after 48 h in the lungs and liver of mice infected with Bp340ΔbcaA, Bp340ΔbcaB, and wild-type bacteria. However, significantly fewer bacteria were recovered from the spleen of Bp340ΔbcaA-infected mice, supporting the idea of a role for this AT in dissemination or in survival in the passage from the site of infection to the spleen.
INTRODUCTION
Burkholderia pseudomallei is a Gram-negative saprotroph that causes melioidosis, a disease that ranges from chronic abscesses to fulminant pneumonia and septic shock and which can be rapidly fatal. B. pseudomallei has an approximately 7.2-Mb genome divided into two chromosomes (1), affording this bacterium the metabolic repertoire necessary to adapt to and survive in a variety of different habitats, including soil, the rhizosphere of plants, and a wide variety of human and nonhuman mammals (2). Melioidosis, first reported by A. Whitmore, an army pathologist working in Burma, in 1912 (3), emerged as an infectious disease of serious public health concern in the latter part of the 20th century, manifesting as a rapidly progressing septicemia, with or without pneumonia; a localized soft tissue infection; or a subclinical infection with delayed evidence of clinical infection (4, 5). Southeast Asia and northern Australia are areas of B. pseudomallei endemicity; however, sporadic B. pseudomallei infections have been reported worldwide (6), including but not limited to the United States, Puerto Rico, El Salvador, and Brazil (4, 7–9). B. pseudomallei is classified as an NIH category B priority pathogen and select agent (10), and has recently been reclassified as a CDC tier 1 select agent due to its virulence in animals, low infectious dose, robust environment stability, and possible delay in diagnosis, since the United States is not an area of B. pseudomallei endemicity (http://www.selectagents.gov).
Autotransporters (ATs) are outer membrane proteins belonging to the type V secretion system (T5SS) family, the largest family of extracellular proteins in Gram-negative bacteria (11). These proteins have been shown to function as adhesins, degradative enzymes, and cytotoxins, as well as having roles in cell-to-cell spread and serum resistance (12). The AT family is divided into classical and trimeric proteins. The C-terminal 250 to 300 amino acids (aa) of classical ATs form a β-barrel that is inserted into the outer membrane, where it facilitates the translocation of the N-terminal passenger domain to the cell surface. Trimeric ATs require three proteins to form a functional unit, with the 67 to 76 C-terminal amino acids of each monomer contributing one-third of the barrel (13). The β-barrel domains of AT proteins are well conserved, while the passenger domains can differ substantially and have distinct functions (14).
To date, little is known about how B. pseudomallei causes disease (15). It is able to invade and replicate in phagocytic and nonphagocytic cells, and following type III secretion system (T3SS)-mediated endosomal escape, B. pseudomallei replicates in the cytoplasm of eukaryotic cells and spreads from cell to cell without leaving the cytoplasm (16, 17). One B. pseudomallei type VI secretion system (T6SS) has been shown to mediate multinucleated giant cell (MNGC) formation (18), allowing the bacteria to freely spread from cell to cell by actin-mediated motility, similar to Shigella flexneri and Listeria monocytogenes (19). B. pseudomallei actin-dependent motility requires the trimeric autotransporter BimA, which contains proline-rich motifs and WH2-like domains that are believed to be responsible for actin polymerization at the pole of the bacterial cell where BimA is localized (20).
The genome of B. pseudomallei strain 1026b contains genes predicted to encode nine trimeric ATs and two classical ATs. The purpose of this study was to determine the role of the predicted classical AT encoded by Bp1026b_II1054 in pathogenesis.
MATERIALS AND METHODS
Bacterial strains.
All manipulations of B. pseudomallei were conducted in a CDC/USDA-approved animal biosafety level 3 (ABSL3) facility at the University of North Carolina at Chapel Hill. The bacterial strains used in this study are listed in Table 1. B. pseudomallei strains were cultured in low-salt lysogeny broth (LSLB) or on low-salt lysogeny broth agar (LSLBA) (Sigma-Aldrich, St. Louis, MO) for 24 h at 37°C. Escherichia coli strains were cultured on LB or LB agar (LBA). When appropriate, culture media were supplemented with kanamycin (Km; 125 μg/ml for B. pseudomallei and 50 μg/ml for E. coli) or with Zeocin (Zeo; 100 μg/ml for B. pseudomallei and 35 μg/ml for E. coli). LB agar was supplemented with 400 μg/ml of diaminopimelic acid (DAP [ll-, dd-, and mesoisomers]; Sigma-Aldrich, St. Louis, MO) to support growth of RHO3 cells. Yeast extract-tryptone (YT) medium containing 10 g/liter of yeast extract (Difco, Detroit, MI) and 10 g/liter of tryptone (Fisher Scientific, Fairlawn, NJ) supplemented with 15% sucrose and X-Gluc (5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid; GoldBio, St. Louis, MO) was used for counterselection during the construction of B. pseudomallei deletion mutation strains.
Table 1.
Strains and plasmids used in this study
| B. pseudomallei strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| RHO3 | Kms; SM10(λpir) Δasd::FRT ΔaphA::FRT | 21 |
| Bp340 | 1026b with Δ(amrRAB-oprA) | 22 |
| Bp340ΔbcaA | Bp340 with ΔbcaA | This study |
| Bp340ΔbcaB | Bp340 with ΔbcaB | This study |
| Bp340ΔbcaA::attTn7bcaA | Bp340 with ΔbcaA::att Tn7 bcaA | This study |
| Bp340ΔbcaB::attTn7bcaB | Bp340 with ΔbcaB::att Tn7 bcaB | This study |
| Bp340::pCCS12HA1 | Bp340 with pCCS12HA1::bcaA | This study |
| Plasmids | ||
| pCCX1 | Kmr; pEXKm5 derivative | 21 |
| pCCX2 | Kmr; pEXKm5 derivative | 21 |
| pCCZ1 | Ampr, Kmr; pUC18T-mini-Tn7-Zeo derivative | 23 |
| pMBZ2 | Ampr, Kmr; pUC18T-mini-Tn7-Zeo derivative | 23 |
| pTNS2 | Ampr; plasmid expressing tnsABCD from Plac | 23 |
| pCCS12HA1 | Kmr; pCC derivative (pRE118 derivative), suicide plasmid for B. pseudomallei | This study |
Amp, ampicillin.
Construction of B. pseudomallei bcaA and bcaB mutant strains and plasmids.
Deletion of the bcaA and bcaB genes from B. pseudomallei strain Bp340 [a derivative of strain 1026b containing a Δ(amrRAB-oprA) mutation; the strain was shown by Herbert Schweizer to be as virulent as 1026b in the BALB/c acute model of infection] (22, 24) was carried out by allelic exchange using pEXKm5 (21) derivatives. The DNA fragments used to construct pCCX1 and pCCX2 (Table 1) were generated using a two-step, overlap PCR method and cloned into pEXKm5. DNA fragments contained approximately 500 bp 5′ to the gene, including the first three codons, and 500 bp 3′ to the gene, including the last three codons. pCCX1 and pCCX2 were transformed into E. coli RHO3 cells, and were delivered to Bp340 by conjugation. For Bp340ΔbcaA and Bp340ΔbcaB complementation strains, the gene and promoter region were cloned into pUC18T-mini-Tn7-Zeo and delivered as previously described (25). All plasmids were verified to be correct and to contain no unintended nucleotide changes by DNA sequence analysis.
Bacterial conjugations.
Matings between B. pseudomallei and E. coli strain RHO3 were performed by incubating Bp340 with RHO3 cells carrying the appropriate allelic exchange plasmid (Table 1) on LSLB-DAP agar plates overnight. Cointegrants were selected on LSLB-Km. PCR-confirmed cointegrants were grown overnight in LSLB without selection, allowing for a second recombination event and the loss of the allelic exchange plasmid. Colonies were selected on YT agar supplemented with 15% sucrose and X-Gluc (21), as previously described. Colonies were screened by PCR for the deletion mutation, and the strains were confirmed by DNA sequencing. To generate the complementation strains, RHO3 cells harboring pCCZ1 or pCCZ2 (Table 1) were mated with the Bp340ΔbcaA strain and the Bp340ΔbcaB strain, respectively, and cointegrants were selected on LSLB-Zeo (25). Integration at the correct location was confirmed by PCR. All DNA regions encompassing about 2 kb across the deletion junction were PCR amplified and verified to be correct and to contain no unintended nucleotide changes by DNA sequence analysis. DNA sequences inserted into the att Tn7 site in complementation strains were also PCR amplified and verified by DNA sequence analysis.
Total RNA isolation and cDNA synthesis.
Total RNA was isolated in TRIzol (Invitrogen, Grand Island, NY) from Bp340 grown overnight in LSLB at 37°C according to the manufacturer's protocol. For the reverse transcription (RT) step, 5 ng of total RNA was transcribed using SuperScript III reverse transcriptase (Invitrogen, Grand Island, NY) with oligo(dT) and random primers according to the manufacturer's instructions. Transcripts were determined by the use of PCR primers described in Table 2.
Table 2.
Primers used in this study
| Primer name | Primer sequence |
|---|---|
| CBcaART_F | TTC GACAGC TTC CAT CTC GGC |
| CBcaART_R | GTT CTT CAG ATG CAC ATA CGC GAC |
| CBcaBRT_F | TTT CGC AGA CGT ACT TGA CGC AGC |
| CBcaBRT_R | TTG AAC ATC AGC GTG ATC CGC ATC GTC |
| CBcaA/BRT_F | CTC GGC AAG AAC GGA TGG CTG |
| CBcaA/BRT_R | CAG AAA CCG GTG GAT CTG CGC |
Immunoblot analysis.
Strain Bp340::pCCS12HA1 was constructed using pCCS12HA1, a suicide plasmid containing the constitutively active ribosomal S12 subunit promoter (PS12) up to and including the S12 ribosomal binding site fused to the first 329 codons of bcaA, starting at the ATG. The hemagglutinin (HA) epitope-encoding sequence was introduced between codons 58 and 59. The plasmid was verified by PCR and sequence analysis to have integrated into the Bp340 strain 3′ to the HA epitope-encoding sequence, yielding a chromosomal HA-tagged copy of bcaA driven by the S12 promoter. Whole-cell lysates were prepared from overnight cultures grown in LSLB-Km. SDS-PAGE was performed by the method of Laemmli (26) using denaturing 10% SDS polyacrylamide gels. Gels were transferred to a nitrocellulose membrane (Scheleicher and Schuell Bioscience, Dassel, Germany) and were probed with an anti-HA antibody (diluted 1:5,000) followed by an IR800-conjugated secondary antibody (Rockland, Gilbertsville, PA) (diluted 1:20,000). Antigen-antibody complexes were visualized using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE).
Plaque assay.
B. pseudomallei strains were grown overnight in LSLB at 37°C. Each well of a 6-well plate was seeded with A549 human lung cells such that confluent monolayers contained approximately 1 × 106 cells per well. Cells were incubated in F12K (Cellgro, Circle Westwood, MA) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY) at 37°C with 5% CO2. Bacterial strains were diluted to an optical density at 600 nm (OD600) of 0.1 in fresh tissue culture medium and further diluted 1:10, and 25 μl of the diluted culture was added to each well (multiplicity of infection [MOI] of 0.1). Plates were incubated for 2 h, and each well was washed thoroughly with fresh culture medium and overlaid with a mixture containing 1.2% low-melting agarose (Fisher Scientific, Fairlawn, NJ), F12K with 10% FBS, gentamicin (90 μg/ml), and 0.01% Neutral red (Fisher Scientific, Waltham, MA). Plates were incubated for 24 h at 37°C with 5% CO2, and plaques were enumerated in each well. Experiments were performed three times in duplicate, and the results were combined. The combined results were analyzed using a one-way analysis of variance (ANOVA) with Tukey's posttest at a 95% confidence interval.
Adherence and invasion assay.
Bacterial strains and A549 cells were grown as described above. Bacteria were diluted to an OD600 of 0.1 in fresh tissue culture medium, and 250 μl of the diluted cultures was added to each well (MOI of 100). Plates were incubated for 2 h, and each well was washed thoroughly with fresh culture medium. For the adherence assay, cells were immediately lysed using 1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) and were diluted and plated to determine the total CFU in each well. For the invasion assay, cells were incubated an additional 1.5 h with gentamicin (90 μg/ml), washed with fresh culture medium, and lysed using 1% Triton X-100. Lysates were diluted and plated to determine the total CFU in each well. To calculate the percentage of adherent or invading bacteria, the number of adherent or invading bacteria was divided by the total number of bacteria in the inoculum and multiplied by 100. Experiments were performed three times in duplicate, and the results were combined. The combined results were analyzed using a one-way ANOVA with Tukey's posttest at a 95% confidence interval.
Animal experiments.
All animal experiments were approved by the Animal Studies Committee of the University of North Carolina at Chapel Hill (protocol 10-165). Six- to 8-week-old female BALB/c mice (The Jackson Laboratory, Bar Harbor, ME) were allowed free access to sterilized food and water. Animals were anesthetized prior to infection with Avertin (140 mg/kg of body weight) by intraperitoneal injection. For all infections, the desired inoculum of B. pseudomallei was suspended in sterile phosphate-buffered saline (PBS). Mice were inoculated intranasally (i.n.) with 500 CFU (the 50% lethal dose [LD50] for strain 1026b has been determined to be around 900 CFU [24]), and at the indicated time points were euthanized by CO2 overdose. Organs were aseptically harvested and homogenized, and the bacterial burden of each organ was determined by plating serial dilutions of the homogenates. Animal experiments were performed twice, with three to four animals per strain per time point, and the results were combined. Animal experiments were terminated at 48 h, at which time all animals had become moribund.
Pathology.
Lungs, livers, and spleens of 6- to 8-week-old female BALB/c mice intranasally infected with 500 CFU of Bp340 or the Bp340ΔbcaA mutant were harvested and fixed in 10% neutral buffered formalin for 24 h. Samples were stored in 70% ethanol until processed into paraffin using routine methods. Paraffin-embedded tissues were divided into sections 3 to 5 μm thick, captured onto glass slides, and stained with hematoxylin and eosin (HE). Sections of liver, spleen, and lung were analyzed using light microscopy by a single pathologist (L. Borst) who was blinded with respect to the identity of the group. Microscopic lesions consisting primarily of necrosis and acute inflammation (neutrophils, macrophages, apoptotic bodies, and fibrin) were quantified as follows. In the lung, the affected bronchi per 20 bronchiolar profiles were counted starting with an affected bronchiole. In the liver and spleen, inflammatory foci were enumerated in 10 consecutive fields at 20× magnification, starting with an area of inflammation identified at low magnification.
RESULTS
Bioinformatic analysis of the bcaA and bcaB genes of B. pseudomallei strain 1026b.
Bp1026b_II1054 is a 3,393-bp gene, predicted to encode a classical AT, that we named bcaA for Burkholderia classical autotransporter A. Forty base pairs 3′ to bcaA is Bp11026_II1055 (which we named bcaB), a 687-bp gene predicted to encode a hypothetical protein. There is a 480-bp gene predicted to encode a hypothetical protein 1,522 bp 5′ to bcaA, and 198 bp 3′ to bcaB is a 1,593-bp gene oriented in the opposite direction, predicted to encode a periplasmic solute-binding protein (Fig. 1A).
Fig 1.

bcaA and bcaB appear to form an operon. (A) Genetic organization of the bcaA and bcaB locus and description of the strains used in this study. A visual representation of the primer pairs used for RT-PCR is shown. (B) The putative domains of BcaA and BcaB proteins drawn to scale. (C) RT-PCR analysis of the operon structure of bcaA and bcaB. gDNA, genomic DNA. Primer pairs 1 and 2, 3 and 4, and 5 and 6 were used as indicated in panel A.
SignalP identified a signal peptide and a cleavage site on the predicted 1,133-aa BcaA protein between aa 40 and 41 (27). The Simple Modular Architecture Research Tool (SMART) predicts BcaA to have a serine protease domain, from aa 64 to 380, belonging to the peptidase S8 or subtilase family, and a classical AT β-domain from aa 857 to 1121. BcaB is a 255-aa protein predicted to contain a prolyl 4-hydroxylase domain from aa 41 to 225 (Fig. 1B) (28).
BLAST indicated that homologues of bcaA are present in B. pseudomallei, B. mallei, B. thailandensis, and B. gladioli strains but not in Ralstonia or Cupriavidus species, close relatives of the Burkholderia genus. All B. pseudomallei and B. mallei strains for which genome sequences are available are predicted to encode proteins with 99% identity with BcaA, and 89% identity is found in B. thailandensis and 82% identity in B. oklahomensis. Predicted homologues are not found in bacteria outside the Burkholderia genus, suggesting that this gene is unique to Burkholderia.
To investigate the role of bcaA and bcaB in the pathogenesis of B. pseudomallei, we constructed strains of Bp340 containing in-frame deletion mutations in each gene (Fig. 1A) by allelic exchange (21). Complementation of these deletion mutations was accomplished by delivering a full-length copy of the gene, along with its presumed promoter region (the approximately 1-kb region immediately 5′ to the bcaA translation start site), to Tn7 att sites in the respective deletion strains using pUC18T-mini-Tn7-Zeo (25). The same presumed promoter region, approximately 1 kb preceding the translation start site of bcaA, was fused to the full-length copy of bcaB for the construction of the complementation of the Bp340ΔbcaB strain.
bcaA and bcaB appear to form an operon.
RT-PCR was used to determine if bcaA and bcaB are cotranscribed. cDNA was prepared from strain Bp340 grown overnight at 37°C in LSLB, and used as the template for PCR using the primer pairs (Table 2) shown in Fig. 1A. Products of expected sizes were obtained for each primer pair (Fig. 1C), including the pair spanning the intergenic region between bcaA and bcaB, suggesting that these genes are cotranscribed. No product was observed in the mock RT samples, demonstrating the lack of DNA contamination in the isolated RNA samples and, consequently, the cDNA samples.
BcaA appears to be proteolyzed to smaller polypeptides.
To visualize the production of BcaA protein, we constructed Bp340 derivatives in which nucleotides encoding HA epitopes were inserted either immediately following the protein's predicted signal sequence cleavage site or between codons 58 and 59. Both strains were grown overnight in LSLB at 37°C, but no polypeptide was recognized by Western blotting using an anti-HA antibody. After several unsuccessful attempts to visualize the BcaA protein when the gene was expressed from its native promoter, we constructed a strain in which bcaA was expressed from a strong, constitutively active promoter (the promoter for the rpsL gene from Burkholderia pseudomallei 1026b). First, we inserted the HA epitope-encoding codons immediately following the predicted signal sequence cleavage site, but again were unsuccessful at visualizing BcaA. Finally, we inserted the HA epitope-encoding codons between codons 58 and 59 (Bp340::pCCS12HA1) and the protein was then successfully visualized by Western blot analysis. Polypeptides approximately 21 kDa and 28 kDa in size as well as a large smear between 250 and 75 kDa were observed in whole-cell lysates of the Bp340::pCC12HA1 strain, but not in whole-cell lysates of the wild-type strain lacking the HA epitope tag (Fig. 2). These data suggest that BcaA is proteolyzed to smaller polypeptides. The smaller polypeptides were not detected in Western blots of concentrated supernatant fractions (data not shown), suggesting that they remain associated with the bacterial cell.
Fig 2.
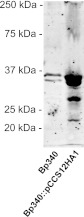
Immunoblot of whole-cell lysates of strain Bp340 with no tag and the Bp340::pCCS12HA1 strain. Strains were separated by SDS-PAGE and stained with anti-HA antibody.
bcaA and bcaB are required for efficient plaque formation.
B. pseudomallei can spread from cell to cell without exiting the cytoplasm and can form plaques in a cell monolayer (29). To determine if bcaA or bcaB contributes to plaque formation, A549 respiratory epithelial cell monolayers were infected at an MOI of 0.1 and plaques were counted after 24 h. The Bp340 strain formed an average of 100 plaques/well, while the Bp340ΔbcaA strain (Fig. 3A) and the Bp340ΔbcaB strain (Fig. 3B) each formed only about 50 to 60 plaques/well. Complementation of both mutations restored plaque-forming ability to wild-type levels, indicating that both bcaA and bcaB are required for efficient plaque formation.
Fig 3.

Average numbers of plaques formed in an A549 monolayer by Bp340, the Bp340ΔbcaA strain (A), the Bp340ΔbcaB strain (B), and the respective complementation strains. Assays were performed three times in duplicate, and the results were combined. Data are means ± standard errors of the means (SEM). **, P < 0.0080; ***, P < 0.0001 (by one-way ANOVA with Tukey's posttest at a 95% confidence interval).
bcaA and bcaB are required for efficient invasion of A549 cells.
The plaques formed by the Bp340ΔbcaA and Bp340ΔbcaB strains appeared to be of sizes similar to those formed by wild-type bacteria, suggesting that bcaA and bcaB are involved in the first steps of plaque formation and that they are therefore required for either adhesion or invasion. To determine if bcaA and bcaB are required for efficient adhesion or invasion, A549 cell monolayers were infected at an MOI of 100, and following a 2-hour incubation, cells were either washed, lysed, serially diluted, and plated to determine percent adhesion, or were washed, incubated with 90 μg/ml of gentamicin for an additional 1.5 h, and then lysed, serially diluted, and plated to determine percent invasion. No difference in adhesion between the Bp340 and Bp340ΔbcaA strains or between the Bp340 and Bp340ΔbcaB strains was observed (Fig. 4A). However, the Bp340ΔbcaA and Bp340ΔbcaB strains demonstrated significantly decreased invasion compared to Bp340 (Fig. 4B), suggesting that bcaA and bcaB are required for efficient invasion of A549 cells. Complementation of ΔbcaA and ΔbcaB at the Tn7 att site restored invasion to wild-type levels. C57BL/6 bone marrow-derived macrophages were also used in similar experiments, but no difference was observed between the Bp340, Bp340ΔbcaA, and Bp340ΔbcaB strains (data not shown). bcaA and bcaB, therefore, are required for invasion of nonphagocytic cells, but do not appear to affect uptake by phagocytic cells.
Fig 4.

CFU recovered after 2-h adherence and invasion assays with Bp340, the Bp340ΔbcaA strain (A), the Bp340ΔbcaB strain (B), and the respective complementation strains. Assays were performed three times in duplicate, and the results were combined. Data are means ± SEM. ***, P < 0.0001 (by one-way ANOVA with Tukey's posttest at a 95% confidence interval).
bcaA is required for efficient dissemination to or survival in the spleen.
To determine the contribution of bcaA and bcaB to B. pseudomallei pathogenesis, we used an acute intranasal (i.n.) mouse model of infection. Bp340 has been shown to have the same LD50 as 1026b (24) when delivered by the i.n. route, so 6- to 8-week-old female BALB/c mice were inoculated with 500 CFU of B. pseudomallei delivered in a 25-μl volume to the nose. All animals showed signs of respiratory distress by 48 h postinoculation, becoming moribund and marking the endpoint of our experiments. The numbers of CFU in the lungs, liver, and spleen were determined at 48 h postinoculation. The numbers of CFU recovered from the lungs and livers of animals inoculated with Bp340 and the Bp340ΔbcaA mutant were not significantly different. The number of CFU recovered from the spleen of strain Bp340ΔbcaA-infected mice, however, was significantly lower than the number recovered from mice infected with Bp340 (Fig. 5A). Complementation of ΔbcaA at the Tn7 att site restored bacterial numbers in the spleen to wild-type levels, suggesting that bcaA is required for efficient dissemination to or survival of B. pseudomallei infection in the spleen in this model. The numbers of bacteria recovered from the lungs, liver, and spleen of animals inoculated with the Bp340ΔbcaB mutant were not significantly different from those of animals infected with Bp340 (Fig. 5B).
Fig 5.

Six- to 8-week-old female BALB/c mice were inoculated with 500 CFU of B. pseudomallei Bp340 (white circles), the Bp340ΔbcaA strain (A), the Bp340ΔbcaB strain (B) (light gray circles), and the respective complementation strains (dark gray circles). Each circle represents the number of CFU recovered from a mouse. The horizontal lines represent the average numbers of CFU. The dashed line represents the lower limit of detection. ***, P < 0.0001. Animal experiments were performed twice for each strain, and the results were combined.
bcaA does not appear to contribute to B. pseudomallei-induced organ pathology.
To determine if bcaA contributes to tissue pathology, lungs, livers, and spleens were harvested at 48 h post-intranasal inoculation, and hematoxylin and eosin (H&E)-stained sections were prepared, coded for blind scoring, and examined for histopathological changes. Inflammation was observed in the lungs, livers, and spleens of all mice infected with the Bp340 or Bp340ΔbcaA strain, differing only in the degree of severity. Approximately the same number of affected bronchi per bronchiolar profile was found in the mice infected with both strains, and the numbers of inflammatory foci found in the livers and spleens were also similar.
Lungs showed suppurative bronchointerstitial pneumonia, with small airways partially to completely filled with mostly degenerative neutrophils. Adjacent and randomly scattered alveolar septa were moderately expanded by degenerative neutrophils and fibrin (microthrombi). Livers showed multifocoal necrosuppurative hepatitis with random hepatocellular necrosis and inflammatory infiltrate (neutrophils), as well as fibrin hemorrhage. Spleens showed multifocal necrosuppurative splenitis with scattered (red pulp) areas of necrosis and inflammatory infiltrate (neutrophilic predominant, rare macrophages, apoptotic bodies), as well as mild fibrin hemorrhage.
The overall histological picture was of bronchopneumonia followed by acute sepsis as evidenced by interstitial pulmonary involvement and multifocal distribution of inflammation and necrosis in the spleens and livers (Fig. 6); however, there was no significant difference in histological scoring between organs harvested from animals inoculated with strain Bp340 and those from animals inoculated with strain Bp340ΔbcaA.
Fig 6.

Histological analysis of B. pseudomallei-infected tissues. Representative tissue samples from BALB/c mice infected i.n. with Bp340 or the Bp340ΔbcaA and Bp340ΔbcaA::attTn7bcaA strains (∼500 CFU) or from mock (PBS)-infected control animals are shown.
DISCUSSION
The sequenced genome of B. pseudomallei contains 11 genes predicted to encode AT proteins. Only the trimeric AT BimA (20) has been well characterized, and it has been shown to play a role in pathogenesis. This work focused on the characterization of bcaA, a gene predicted to encode a classical AT. Our data indicate that bcaA and bcaB contribute to nonphagocytic cell invasion and bcaA to dissemination to or survival of B. pseudomallei in the spleen in a BALB/c intranasal model of infection.
The bcaA and bcaB genes appear to form an operon. RT-PCR supports this hypothesis by showing that bcaA and bcaB are cotranscribed from a common promoter upstream of bcaA. Our data do not rule out the presence of an additional bcaB promoter located within the bcaA gene. However, the DNA fragment used to complement the ΔbcaB strain contained approximately 1 kb preceding the translation start site of bcaA fused directly to the full-length copy of bcaB and this DNA fragment restored invasion of the ΔbcaB strain to wild-type levels, further suggesting that bcaA and bcaB are transcribed from a promoter 5′ to bcaA.
BcaA is predicted to be a 113-kDa classical AT protein with an approximately 80-kDa passenger domain containing a serine protease domain belonging to the peptidase S8 or subtilase family and a 30-kDa β-domain. Subtilisins are the second largest family of serine proteases, characterized by a conserved catalytic triad that functions in a charge relay system much like trypsin family proteases (30). Subtilisins use well-conserved Asp, Ser, and His catalytic residues for their protease activity (31), and their functions include contributing to cellular nutrition, mediating host cell invasion, and the maturation of other polypeptides (32). A few AT subtilases have been described, including Ssp from Serratia marcescens, a 112-kDa protein with an approximately 80-kDa passenger domain that is autoproteolyzed into a 41-kDa polypeptide that is released into the medium (33), and AasP from Actinobacillus pleuropneumoniae, a 104-kDa protein that cleaves another outer membrane protein (OmlA) into smaller polypeptides of about 30 kDa that are released into the medium (34). BcaA contains the same well-conserved catalytic triad (Asp 90, His 124, and Ser 329) found in other subtilisins. Western blot characterization studies using a HA-tagged BcaA detected 21-kDa and 28-kDa polypeptides that were present in whole-cell lysates but not in supernatants. Much like Ssp from S. marcescens, the BcaA 80-kDa passenger domain appears to be proteolyzed into smaller polypeptides; however, these small polypeptides do not appear to be released into the extracellular environment in the case of BcaA. Further studies will include characterization of these polypeptides and determining if these polypeptides are generated by autoproteolysis or if another protein is involved in processing.
We showed that bcaA and bcaB are required for invasion and plaque formation in epithelial cells but did not affect uptake in phagocytic cells. B. pseudomallei has been shown to invade and replicate in both phagocytic and nonphagocytic cells and to spread from cell to cell without leaving the cytoplasm (17). Although a substantial amount has been learned about the intracellular life cycle of B. pseudomallei, no proteins responsible for the initial invasion step have been identified previously. BcaA, therefore, represents the first identified invasin in B. pseudomallei.
In vivo, bcaA is required for efficient dissemination to or survival of B. pseudomallei in the spleen in a BALB/c intranasal model of infection. Although histology showed no difference in the amounts of inflammatory foci found in the spleens of animals infected with the Bp340ΔbcaA strain and in those of animals infected with the wild-type strain, significantly fewer bacteria were recovered from the spleens of animals infected with the Bp340ΔbcaA strain. How the invasion phenotype in vitro relates to the spleen dissemination or survival defect in vivo is unknown. Yersinia enterocolitica has been shown to have both invasin-dependent and invasin-independent routes of spleen colonization from the intestine of C57BL/6 mice; Handley et al. showed that a Y. enterocolitica invasin mutant was attenuated in its ability to disseminate from the intestine to the spleen (35). The same phenotype has also been described for Y. pseudotuberculosis after oral inoculation (36). It had been proposed that enteropathogenic Yersinia bacteria colonize the Payer's patch and then drain into the mesenteric lymph nodes and, in turn, enter tissues such as the spleen. The studies by Marra et al. and Handley et al., however, showed that there are several routes of spread, and how initial colonization of one site leads to the colonization of another appears to be more complicated than initially appreciated. To date, little is known about how B. pseudomallei disseminates from the initial site of infection (37). Inhalation, ingestion, and soft tissue abrasions or lacerations are routes of infection (6, 38); however, the steps the bacteria follow after the inoculation are still unknown. It is possible that bcaA functions like the Y. enterocolitica invasin, specifically aiding dissemination to the spleen. Further studies will include determining the route of spleen colonization, as well as determining if bcaA and bcaB affect survival once the bacteria have reached the spleen.
bcaB is predicted to encode a hypothetical protein with a prolyl 4-hydroxylase domain. Bacterial prolyl 4-hydroxylases are believed to hydroxylate peptidyl prolines, and although some such hydroxylases have been identified in bacteria, their substrates and therefore their function remain unknown (39). Prolyl 4-hydroxylase, which catalyzes the most prevalent posttranslational modification in humans, has as the substrate collagen, functioning by stabilizing the collagen triple-helix structure (40), elastin, where it affects the formation of elastin fibrils (41), and even prion proteins, although the consequences of the presence of the prion proteins are unknown (39). In bacteria, a Bacillus anthracis enzyme designated anthrax-P4H has been shown to hydroxylate peptidyl prolines; however, although it binds to collagen-like peptides in vitro, its physiological substrate and role remain unknown (42). Further studies should include determining if bcaB plays a role in hydroxylating any of the 52 prolines found in BcaA, since it is possible that BcaA is the physiological substrate for BcaB, which would be consistent with their operon structures.
Both bcaA and bcaB are required for invasion and plaque formation, consistent with the idea that they function together. However, only bcaA showed a phenotype in vivo. It is possible that this simply reflects the limitations of the tools we used. Although the BALB/c model has been broadly used as an acute model for B. pseudomallei infection (43), it may not be sensitive enough to reveal phenotypes for all factors that contribute to disease, depending on the step and stage of disease in which they function. Our future experiments will include the development of additional animal models that will expand the repertoire of B. pseudomallei disease processes that we are able to study in the laboratory.
ACKNOWLEDGMENTS
We thank Herbert Schweizer for providing us with B. pseudomallei strains and genetic tools. We also thank Michael Henderson and Sharon Taft-Benz for their technical assistance in the ABSL3 facility.
This work was supported by a PSWRCE grant (U54 AI065359).
Footnotes
Published ahead of print 22 January 2013
REFERENCES
- 1. Holden MT, Titball RW, Peacock SJ, Cerdeno-Tarraga AM, Atkins T, Crossman LC, Pitt T, Churcher C, Mungall K, Bentley SD, Sebaihia M, Thomson NR, Bason N, Beacham IR, Brooks K, Brown KA, Brown NF, Challis GL, Cherevach I, Chillingworth T, Cronin A, Crossett B, Davis P, DeShazer D, Feltwell T, Fraser A, Hance Z, Hauser H, Holroyd S, Jagels K, Keith KE, Maddison M, Moule S, Price C, Quail MA, Rabbinowitsch E, Rutherford K, Sanders M, Simmonds M, Songsivilai S, Stevens K, Tumapa S, Vesaratchavest M, Whitehead S, Yeats C, Barrell BG, Oyston PC, Parkhill J. 2004. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc. Natl. Acad. Sci. U. S. A. 101:14240–14245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sprague LD, Neubauer H. 2004. Melioidosis in animals: a review on epizootiology, diagnosis and clinical presentation. J. Vet. Med. B Infect. Dis. Vet. Public Health 51:305–320 [DOI] [PubMed] [Google Scholar]
- 3. Whitmore A, Krisnaswami CS. 1912. An account of the discovery of a hitherto undescribed infective disease occurring among the population of Ragoon. Ind. Med. Gaz. 262-267 [PMC free article] [PubMed] [Google Scholar]
- 4. Inglis TJ, Sagripanti JL. 2006. Environmental factors that affect the survival and persistence of Burkholderia pseudomallei. Appl. Environ. Microbiol. 72:6865–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wiersinga WJ, Currie BJ, Peacock SJ. 2012. Melioidosis. N. Engl. J. Med. 367:1035–1044 [DOI] [PubMed] [Google Scholar]
- 6. Cheng AC, Currie BJ. 2005. Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18:383–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dorman SE, Gill VJ, Gallin JI, Holland SM. 1998. Burkholderia pseudomallei infection in a Puerto Rican patient with chronic granulomatous disease: case report and review of occurrences in the Americas. Clin. Infect. Dis. 26:889–894 [DOI] [PubMed] [Google Scholar]
- 8. Rolim DB, Vilar DC, Sousa AQ, Miralles IS, de Oliveira DC, Harnett G, O'Reilly L, Howard K, Sampson I, Inglis TJ. 2005. Melioidosis, northeastern Brazil. Emerg. Infect. Dis. 11:1458–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yabuuchi E, Kosako Y, Arakawa M, Hotta H, Yano I. 1992. Identification of Oklahoma isolate as a strain of Pseudomonas pseudomallei. Microbiol. Immunol. 36:1239–1249 [DOI] [PubMed] [Google Scholar]
- 10. Valvano MA, Keith KE, Cardona ST. 2005. Survival and persistence of opportunistic Burkholderia species in host cells. Curr. Opin. Microbiol. 8:99–105 [DOI] [PubMed] [Google Scholar]
- 11. Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala'Aldeen D. 2004. Type V protein secretion pathway: the autotransporter story. Microbiol. Mol. Biol. Rev. 68:692–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Henderson IR, Nataro JP. 2001. Virulence functions of autotransporter proteins. Infect. Immun. 69:1231–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cotter SE, Surana NK, St Geme JW., III 2005. Trimeric autotransporters: a distinct subfamily of autotransporter proteins. Trends Microbiol. 13:199–205 [DOI] [PubMed] [Google Scholar]
- 14. Nishimura K, Tajima N, Yoon YH, Park SY, Tame JR. 2010. Autotransporter passenger proteins: virulence factors with common structural themes. J. Mol. Med. (Berl) 88:451–458 [DOI] [PubMed] [Google Scholar]
- 15. Lazar Adler NR, Govan B, Cullinane M, Harper M, Adler B, Boyce JD. 2009. The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease? FEMS Microbiol. Rev. 33:1079–1099 [DOI] [PubMed] [Google Scholar]
- 16. French CT, Toesca IJ, Wu TH, Teslaa T, Beaty SM, Wong W, Liu M, Schroder I, Chiou PY, Teitell MA, Miller JF. 2011. Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc. Natl. Acad. Sci. U. S. A. 108:12095–12100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stevens MP, Haque A, Atkins T, Hill J, Wood MW, Easton A, Nelson M, Underwood-Fowler C, Titball RW, Bancroft GJ, Galyov EE. 2004. Attenuated virulence and protective efficacy of a Burkholderia pseudomallei bsa type III secretion mutant in murine models of melioidosis. Microbiology 150:2669–2676 [DOI] [PubMed] [Google Scholar]
- 18. Burtnick MN, Brett PJ, Harding SV, Ngugi SA, Ribot WJ, Chantratita N, Scorpio A, Milne TS, Dean RE, Fritz DL, Peacock SJ, Prior JL, Atkins TP, Deshazer D. 2011. The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei. Infect. Immun. 79:1512–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pantaloni D, Le Clainche C, Carlier MF. 2001. Mechanism of actin-based motility. Science 292:1502–1506 [DOI] [PubMed] [Google Scholar]
- 20. Stevens MP, Stevens JM, Jeng RL, Taylor LA, Wood MW, Hawes P, Monaghan P, Welch MD, Galyov EE. 2005. Identification of a bacterial factor required for actin-based motility of Burkholderia pseudomallei. Mol. Microbiol. 56:40–53 [DOI] [PubMed] [Google Scholar]
- 21. López CM, Rholl DA, Trunck LA, Schweizer HP. 2009. Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl. Environ. Microbiol. 75:6496–6503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mima T, Schweizer HP. 2010. The BpeAB-OprB efflux pump of Burkholderia pseudomallei 1026b does not play a role in quorum sensing, virulence factor production, or extrusion of aminoglycosides but is a broad-spectrum drug efflux system. Antimicrob. Agents Chemother. 54:3113–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choi KH, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, Schweizer HP. 2008. Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Appl. Environ. Microbiol. 74:1064–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goodyear A, Kellihan L, Bielefeldt-Ohmann H, Troyer R, Propst K, Dow S. 2009. Protection from pneumonic infection with burkholderia species by inhalational immunotherapy. Infect. Immun. 77:1579–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi KH, DeShazer D, Schweizer HP. 2006. mini-Tn7 insertion in bacteria with multiple glmS-linked attTn7 sites: example Burkholderia mallei ATCC 23344. Nat. Protoc. 1:162–169 [DOI] [PubMed] [Google Scholar]
- 26. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 27. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783–795 [DOI] [PubMed] [Google Scholar]
- 28. Letunic I, Doerks T, Bork P. 2012. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40(Database issue):D302–D305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kespichayawattana W, Intachote P, Utaisincharoen P, Sirisinha S. 2004. Virulent Burkholderia pseudomallei is more efficient than avirulent Burkholderia thailandensis in invasion of and adherence to cultured human epithelial cells. Microb. Pathog. 36:287–292 [DOI] [PubMed] [Google Scholar]
- 30. Krem MM, Di Cera E. 2001. Molecular markers of serine protease evolution. EMBO J. 20:3036–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siezen RJ, Leunissen JA. 1997. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 6:501–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tripathi LP, Sowdhamini R. 2008. Genome-wide survey of prokaryotic serine proteases: analysis of distribution and domain architectures of five serine protease families in prokaryotes. BMC Genomics 9:549 doi:10.1186/1471-2164-9-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohnishi Y, Horinouchi S. 1996. Extracellular production of a Serratia marcescens serine protease in Escherichia coli. Biosci. Biotechnol. Biochem. 60:1551–1558 [DOI] [PubMed] [Google Scholar]
- 34. Ali T, Oldfield NJ, Wooldridge KG, Turner DP, Ala'Aldeen DA. 2008. Functional characterization of AasP, a maturation protease autotransporter protein of Actinobacillus pleuropneumoniae. Infect. Immun. 76:5608–5614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Handley SA, Newberry RD, Miller VL. 2005. Yersinia enterocolitica invasin-dependent and invasin-independent mechanisms of systemic dissemination. Infect. Immun. 73:8453–8455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marra A, Isberg RR. 1997. Invasin-dependent and invasin-independent pathways for translocation of Yersinia pseudotuberculosis across the Peyer's patch intestinal epithelium. Infect. Immun. 65:3412–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goodyear A, Bielefeldt-Ohmann H, Schweizer H, Dow S. 2012. Persistent gastric colonization with Burkholderia pseudomallei and dissemination from the gastrointestinal tract following mucosal inoculation of mice. PLoS One 7:e37324 doi:10.1371/journal.pone.0037324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dance DA. 2000. Ecology of Burkholderia pseudomallei and the interactions between environmental Burkholderia spp. and human-animal hosts. Acta Trop. 74:159–168 [DOI] [PubMed] [Google Scholar]
- 39. Gorres KL, Raines RT. 2010. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 45:106–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holmgren SK, Bretscher LE, Taylor KM, Raines RT. 1999. A hyperstable collagen mimic. Chem. Biol. 6:63–70 [DOI] [PubMed] [Google Scholar]
- 41. Dunn DM, Franzblau C. 1982. Effects of ascorbate on insoluble elastin accumulation and cross-link formation in rabbit pulmonary artery smooth muscle cultures. Biochemistry 21:4195–4202 [DOI] [PubMed] [Google Scholar]
- 42. Culpepper MA, Scott EE, Limburg J. 2010. Crystal structure of prolyl 4-hydroxylase from Bacillus anthracis. Biochemistry 49:124–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leakey AK, Ulett GC, Hirst RG. 1998. BALB/c and C57Bl/6 mice infected with virulent Burkholderia pseudomallei provide contrasting animal models for the acute and chronic forms of human melioidosis. Microb. Pathog. 24:269–275 [DOI] [PubMed] [Google Scholar]