

Abstract
The endocannabinoid signaling system regulates diverse physiologic processes and has attracted considerable attention as a potential pharmaceutical target for treating diseases, such as pain, anxiety/depression, and metabolic disorders. The principal ligands of the endocannabinoid system are the lipid transmitters N-arachidonoylethanolamine (anandamide) and 2-arachidonoylglycerol (2-AG), which activate the two major cannabinoid receptors, CB1 and CB2. Anandamide and 2-AG signaling pathways in the nervous system are terminated by enzymatic hydrolysis mediated primarily by the serine hydrolases fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively. In this review, we will discuss the development of FAAH and MAGL inhibitors and their pharmacological application to investigate the function of anandamide and 2-AG signaling pathways in preclinical models of neurobehavioral processes, such as pain, anxiety, and addiction. We will place emphasis on how these studies are beginning to discern the different roles played by anandamide and 2-AG in the nervous system and the resulting implications for advancing endocannabinoid hydrolase inhibitors as next-generation therapeutics.
I. Introduction to the Endocannabinoid System
The endocannabinoid system is a neuromodulatory network and the target of the psychoactive component of marijuana, Δ9-tetrahydrocannabinol (THC) (Ahn et al., 2008). Preparations of Cannabis plants have been used for medicinal and recreational purposes for thousands of years and are known to affect appetite, pain, mood, motor control, memory, cognition, and perception. In the early 1990s, THC was found to activate two G-protein-coupled receptors that were subsequently named cannabinoid receptors CB1 (Matsuda et al., 1990) and CB2 (Munro et al., 1993). CB1 is the primary cannabinoid receptor in the central nervous system (CNS) and is widely distributed throughout the brain and at lower levels in peripheral tissues (Herkenham, 1995). Activation of CB1 accounts for most of the neurobehavioral effects of THC as CB1(−/−) mice exhibit none of the classic signs of cannabinoid intoxication in rodents—hypomotility, analgesia, hypothermia and catalepsy—following THC or synthetic cannabinoid administration (Ledent et al., 1999; Zimmer et al., 1999). CB2 is expressed primarily by immune cells, including microglia in the brain, and is thought to mediate THC’s immunosuppressive effects (Cabral et al., 2008), although evidence has emerged for a supporting role for CB2 in neurologic processes such as anxiety and addiction (Onaivi, 2006). The principal endogenous ligands of the cannabinoid receptors are the lipid transmitters N-arachidonoylethanolamine (anandamide) (Devane et al., 1992) and 2-arachidonoylglycerol (2-AG) (Mechoulam et al., 1995; Sugiura et al., 1995).
Endocannabinoid signaling regulates numerous aspects of mammalian neurophysiology, including pain perception, feeding, emotional state, learning and memory, and reward behaviors (Pacher et al., 2006; Ahn et al., 2008; Di Marzo, 2008). That the endocannabinoid system modulates such varied processes is perhaps to be expected when considering the widespread expression of CB1 in the brain (Herkenham et al., 1990; Tsou et al., 1998) and the diversity of effects elicited by THC and other exogenous cannabinoids.
A. Mechanism of Endocannabinoid Signaling
The mechanisms of endocannabinoid signaling in the nervous system differ considerably from those of the classic neurotransmission systems (e.g., cholinergic, aminoacidergic, and monoaminergic). In the classic model of neurotransmission, depolarization of the presynaptic neuron by an action potential results in the release of neurotransmitters, which then traverse the synaptic cleft to bind and activate their cognate receptors on the postsynaptic neuron (Siegel et al., 1999). In contrast, endocannabinoid signaling appears to occur via a retrograde mechanism (Fig. 1), where stimulation of the postsynaptic neuron triggers the biosynthesis of endocannabinoids, which are released and transported by poorly understood mechanisms to activate CB1 receptors expressed primarily on the presynaptic terminal (Alger and Kim, 2011). CB1 activation of Gi/o proteins initiates a signaling cascade that regulates calcium and potassium channels and ultimately suppresses further neurotransmitter release (Howlett, 2005). In this model, endocannabinoid signaling modulates transmission efficiency by facilitating communication from the postsynaptic to the presynaptic neuron. As CB1 activation acts to inhibit neurotransmission, the ultimate outcome of endocannabinoid signaling depends on the nature of the participating cells. If CB1 is activated on glutamatergic neurons, for instance, endocannabinoid signaling will be overall inhibitory, whereas if CB1 activation takes place on GABAergic neurons, the net result will be “disinhibitory” (or excitatory).
Fig. 1.

Schematic of retrograde endocannabinoid signaling in the nervous system. The endocannabinoid transmitters anandamide (AEA) and 2-AG are thought to be biosynthesized postsynaptically. Anandamide is produced from NAPE precursors, which are generated by a still uncharacterized CDTA enzyme. The release of anandamide from NAPEs is also an incompletely understood reaction pathway that likely involves one or more phospholipase A and/or D enzymes. 2-AG is synthesized from phosphatidylinositol (PI) lipid precursors by the sequential action of PLC and the DAGLα and DAGLβ enzymes. DAGLα is the major 2-AG biosynthetic enzyme in the brain. Following activity-dependent biosynthesis/mobilization, endocannabinoids traverse the synaptic cleft where they activate presynaptically localized CB1 receptors. CB1 signaling through Gi/o proteins eventually results in the inhibition of neurotransmitter release. Anandamide and 2-AG signaling is terminated by enzymatic hydrolysis, which, in the CNS, proceeds primarily through FAAH and MAGL.
B. Regulation of Endocannabinoid Signaling Tone
The distinct physical properties—specifically differences in aqueous solubility—of the endocannabinoids versus most other neurotransmitters influence their respective signaling mechanisms. Classic neurotransmitters are water-soluble metabolites that are packaged and stored in synaptic vesicles (Stephenson and Hawkins, 2001). Following release of vesicular contents into the extracellular space and postsynaptic receptor activation, neurotransmitter signaling is terminated by cellular reuptake and enzymatic degradation. Pharmacological inhibition of these processes can amplify signaling by extending neurotransmitter half-life in the synaptic cleft (Fon and Edwards, 2001). In fact, disruption of neurotransmitter clearance is a mechanism of action for both neuropharmaceuticals (e.g., selective serotonin reuptake inhibitors and monoamine oxidase inhibitors) and drugs of abuse (e.g., cocaine) (Brodal, 2004). Anandamide and 2-AG, in contrast, are lipid messengers, and their hydrophobicity would seem to preclude storage in synaptic vesicles. Instead, they are thought to be mobilized from membrane phospholipid precursors and/or storage sites in an activity-dependent manner, often referred to as “on demand” biogenesis (Min et al., 2010; Alger and Kim, 2011). After activating CB1 receptors on presynaptic membranes, anandamide and 2-AG are removed from the extracellular milieu and inactivated by rapid enzymatic hydrolysis. The mechanisms of endocannabinoid neuronal reuptake are not completely understood, but putative endocannabinoid transporters have been reported and chemical agents that modulate their function have been described (Di Marzo, 2008; Fu et al., 2012). Pharmacological inhibition of endocannabinoid degradative enzymes has been found to enhance endocannabinoid signaling in rodents and is considered a promising strategy for harnessing the therapeutic potential of the endocannabinoid system (Ahn et al., 2008; Fowler, 2008; Petrosino et al., 2009).
C. Endocannabinoid Ligand Diversification
For the major neurotransmission systems, receptor diversification allows the system to mediate diverse physiologic processes (Schofield et al., 1990). Endocannabinoid signaling in the nervous system, in contrast, proceeds in large part through a single receptor, CB1, and seems to gain functionality and flexibility through ligand diversity. Although the distinct signaling actions of anandamide and 2-AG in vivo are not well understood, they are recognized to differ in a few key aspects. Similar to THC, anandamide displays partial agonism toward CB1 in vitro, whereas 2-AG acts as a full agonist (Hillard, 2000). Bulk 2-AG levels in the brain are approximately three orders of magnitude higher than anandamide levels, although the relevance of this difference on their signaling actions is unclear, especially considering that their basal extracellular levels, as measured by in vivo microdialysis, are within 2- to 5-fold (Béquet et al., 2007; Caillé et al., 2007). The endocannabinoids also differ in their ability to impact synaptic plasticity in electrophysiological paradigms. 2-AG has been implicated as the mediator of the major forms of CB1-dependent synaptic plasticity, including depolarization-induced suppression of inhibition (DSI) and excitation (DSE), two models of retrograde neurotransmission (Kano et al., 2009). Inhibition of 2-AG degradation enhanced DSI and DSE in rodent slice cultures from multiple brain regions (Makara et al., 2005; Kano et al., 2009; Pan et al., 2009). Inversely, genetic ablation of 2-AG biosynthetic pathways virtually eliminated DSI and DSE (Gao et al., 2010; Tanimura et al., 2010). Anandamide has been found to regulate long-term depression in multiple brain regions by acting on postsynaptic transient receptor potential cation channel V1 (TRPV1) receptors (Chávez et al., 2010; Grueter et al., 2010; Puente et al., 2011) and presynaptic CB1 receptors (Grueter et al., 2010). Additionally, anandamide was shown to mediate homeostatic synaptic plasticity in hippocampal slice cultures and act as a tonic retrograde messenger at CB1 (Kim and Alger, 2010). Another distinguishing property of the endocannabinoids that is especially relevant for this review is their regulation by distinct sets of metabolic and catabolic enzymes in the nervous system. Not only does the independent regulation of anandamide and 2-AG have implications for their signaling actions (i.e., differential cellular and subcellular distribution of these enzymes may impart strict anatomic and temporal control over endocannabinoid activity), but it means that selective pharmacological or genetic manipulation of these enzymes can be used to dissect the functions of each endocannabinoid in vivo.
D. Endocannabinoid Biosynthesis
The mechanisms of anandamide biosynthesis in the nervous system are incompletely understood. It is generally accepted that anandamide is generated by calcium-dependent enzymatic transfer of arachidonic acid from the sn-1 position of membrane phospholipids to the primary amine of phosphatidylethanolamine (PE) to form N-arachidonoyl phosphatidylethanolamine (NArPE; Fig. 2A, step 1), followed by hydrolysis to give anandamide (Natarajan et al., 1983; Di Marzo et al., 1994; Cadas et al., 1996) (FIG. 2A, steps 2–8). However, the calcium-dependent transacylase enzyme (CDTA) that forms NArPE has not been molecularly identified, and the route by which NArPE is converted into anandamide remains incompletely understood. Multiple mechanisms and putative anandamide biosynthetic enzymes have been suggested, including 1) direct liberation of anandamide by NAPE-PLD, an N-acyl phosphatidylethanolamine (NAPE)-selective phospholipase D (PLD) enzyme (Fig. 2, step 2) (Okamoto et al., 2004); 2) sequential O-deacylation of NArPE by the lyso(NAPE)-lipase α-β hydrolase 4 (ABHD4; Fig. 2A, steps 3–4) (Simon and Cravatt, 2006) and cleavage of the phosphodiester bond by the glycerophosphodiesterase GDE1 (Fig. 2A, step 5) (Simon and Cravatt, 2008); 3) O-deacylation of NArPE by phospholipase A2 (Fig. 2A, step 3) and hydrolysis of the phosphodiester bond by a lyso-PLD enzyme (Fig. 2A, step 6) (Sun et al., 2004); and finally, 4) conversion of NArPE to phospho-anandamide by a phospholipase C (PLC)-like enzyme (Fig. 2A, step 7) followed by dephosphorylation by the tyrosine phosphatase PTPN22 (Liu et al., 2006) or the inositol 5′−phosphatase SHIP (Fig. 2A, step 8) (Liu et al., 2008). In brain metabolomes of mice lacking NAPE-PLD (Leung et al., 2006), GDE1 (Simon and Cravatt, 2010b), and PTPN22 (Liu et al., 2008); however, basal anandamide levels were unchanged, suggesting either that these enzymes do not control anandamide biogenesis in the CNS under normal conditions or that their constitutive disruption causes upregulation of alternate, compensatory pathways.
Fig. 2.
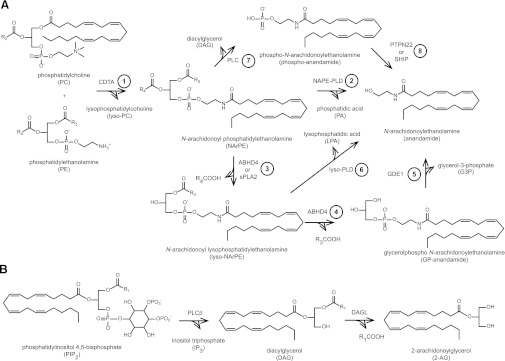
Endocannabinoid biosynthesis. (A) Anandamide biosynthesis begins with the formation of NArPE by the transfer of AA from phosphatidylcholine (PC) to the primary amine of PE by a molecularly uncharacterized CDTA enzyme (step 1). Multiple pathways have been postulated for the liberation of anandamide from NArPE (steps 2–8). (B) 2-AG is generated by hydrolysis of phosphatidylinositol 4,5-bisphosphate by PLCβ followed by cleavage of DAG by DAGLα and DAGLβ.
2-AG is synthesized from arachidonoyl-containing diacylglycerol (DAG) species by sn-1-specific diacylglycerol lipase-α and -β (DAGLα and DAGLβ) (Fig. 2B) (Bisogno et al., 2003). Characterization of DAGL(–/–) mice confirmed a primary role for DAGLα in 2-AG formation in the brain and DAGLβ in peripheral tissues such as the liver (Gao et al., 2010; Tanimura et al., 2010). DAG precursors are themselves synthesized from membrane phospholipids with most evidence suggesting that the major 2-AG biosynthetic pathway is hydrolysis of sn-2 arachidonoyl phosphatidylinositol 4,5-bisphosphate (PIP2) species by PLCβ (Fig. 2B) (Hashimotodani et al., 2005; Maejima et al., 2005).
E. Endocannabinoid Degradation
Despite considerable evidence that direct pharmacological activation of the endocannabinoid system by THC and other synthetic cannabinoids can elicit therapeutically beneficial effects on pain, sleep, appetite, and nausea (Russo et al., 2007; Pertwee, 2009; Rahn and Hohmann, 2009), the concomitant detrimental effects of CB1 agonists on cognition and motor control limit their broad use as pharmaceuticals. To minimize the problems associated with CB1 agonists, amplifying the actions of anandamide and 2-AG by inhibiting their enzymatic degradation has emerged as a potential strategy to exploit the endocannabinoid system for medicinal purposes. In the nervous system, anandamide and 2-AG are degraded primarily by the serine hydrolase enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively (Fig. 3). Pharmacological inhibition of FAAH and MAGL has been found to reduce pain, inflammation, anxiety, and depression in rodent models without the gross changes in motility and behavior observed with direct CB1 agonists (Ahn et al., 2008; Petrosino and Di Marzo, 2010). In addition to the potential translational implications of these findings, development of chemical agents to independently perturb FAAH or MAGL has allowed investigations of enhanced anandamide or 2-AG signaling, respectively. These studies have revealed functional differences between elevated levels of each endocannabinoid and have provided insights into the individual roles of anandamide and 2-AG in endocannabinoid-mediated physiology. In this review, we will focus on what the results of these studies suggest about the nature of anandamide versus 2-AG signaling and the potential of endocannabinoid hydrolase inhibitors as therapies for human disorders of the CNS.
Fig. 3.

Endocannabinoid hydrolysis. In the nervous system, anandamide and 2-AG are degraded primarily by FAAH and MAGL, respectively.
II. Identification and Molecular Characterization of Endocannabinoid Hydrolases in the Nervous System
A. Anandamide Hydrolysis by Fatty Acid Amide Hydrolase
When administered exogenously, anandamide elicits cannabimimetic “tetrad” effects in mice—antinociception, hypothermia, hypomotility, and catalepsy—but with a much shorter duration of action and less potency than THC (Smith et al., 1994). Rapid degradation of anandamide in vivo was considered a likely cause for this discrepancy and fueled the search for the catabolic enzyme(s) that regulate its bioavailability.
Before the discovery of anandamide as the first endocannabinoid messenger (Devane et al., 1992), an amidohydrolase activity was identified in rat liver membranes that hydrolyzed N-acyl ethanolamine (NAE) species containing unsaturated (C12–C18:0) and monounsaturated (C18:1) acyl chains into ethanolamine and the respective fatty acid (Schmid et al., 1985). Subsequently, anandamide hydrolase activity was found in cultured neuroblastoma and glioma cells (Deutsch and Chin, 1993) as well as rat (Desarnaud et al., 1995; Hillard et al., 1995) and porcine (Ueda et al., 1995) brain preparations. Efforts to determine the molecular identify of the anandamide hydrolase merged with those for a structurally related bioactive lipid, the sleep-inducing fatty acid amide oleamide (Cravatt et al., 1995), when the anandamide and oleamide hydrolysis activities of N18 neuroblastoma cells were found to be equivalent (Maurelli et al., 1995). Isolation, cloning, and expression of the rat brain oleamide hydrolase confirmed that anandamide was indeed an additional substrate of this enzyme, which was therefore named fatty acid amide hydrolase (FAAH) to convey its broad catalytic activity toward acyl amide species (Cravatt et al., 1996).
FAAH is an ∼60-kDa integral membrane protein that is highly expressed in the mammalian brain where it localizes to intracellular membranes of postsynaptic somata and dendrites (Gulyas et al., 2004). In many brain regions, including the neocortex, cerebellar cortex, and hippocampus, FAAH and CB1 exhibit complementary subcellular distributions with FAAH-expressing cell bodies or dendrites surrounded by CB1-expressing fibers (Egertová et al., 1998). FAAH is both a serine hydrolase, an enzyme class that utilizes a nucleophilic serine for catalysis, and a member of the amidase signature enzyme family. Recombinant expression and purification of full-length and transmembrane-truncated FAAH variants have enabled extensive biochemical characterization of its mechanism (McKinney and Cravatt, 2005). These studies revealed that unlike most serine hydrolases, which use a histidine residue as a catalytic base, FAAH enlists a lysine for this function, a distinction that enables FAAH to hydrolyze both amides and esters at equivalent rates (Patricelli and Cravatt, 1999). Structural determination of the transmembrane-truncated rat enzyme bound to the nonselective inhibitor methyl arachidonoylfluorophosphonate confirmed the presence of a serine-serine-lysine (Ser241-Ser217-Lys142) catalytic triad, typical for amidase signature enzymes and different from the serine-histidine-aspartatic acid motif common to most serine hydrolases (Bracey et al., 2002).
B. 2-AG Hydrolysis by Monoacylglycerol Lipase
After the discovery of 2-AG as a second endocannabinoid (Mechoulam et al., 1995; Sugiura et al., 1995), its inactivation in the nervous system was hypothesized to proceed through monoacylglycerol lipase (MAGL), a soluble serine hydrolase that peripherally associates with cell membranes (Dinh et al., 2002). MAGL was originally isolated and cloned from adipose tissue, where it was characterized as an enzyme responsible for the last step of triglyceride catabolism (Tornqvist and Belfrage, 1976; Karlsson et al., 1997). Multiple subsequent studies implicated MAGL as a key mediator of 2-AG degradation in the nervous system. Viral overexpression of MAGL in rat cortical neurons reduced the activity-dependent accumulation of 2-AG (Dinh et al., 2002). MAGL immunodepletion in soluble rat brain proteomes decreased 2-AG hydrolysis by 50% (Dinh et al., 2004). Functional profiling of serine hydrolases assigned 85% of the 2-AG hydrolase activity in mouse brain membranes to MAGL (Blankman et al., 2007). Additionally, first-generation MAGL inhibitors, although nonselective, were found to decrease brain 2-AG hydrolysis activity, increase brain 2-AG levels, and produce CB1-dependent antihyperalgesia in rats (Hohmann et al., 2005; Saario et al., 2005). Confirmation of MAGL as the primary brain 2-AG hydrolase was achieved by the generation of a selective and in vivo active MAGL inhibitor, JZL184 (Fig. 5B), which when administered to mice, reduced brain 2-AG hydrolase activity by ∼85%, dramatically elevated brain 2-AG levels, and elicited a select subset of cannabinoid behaviors (Long et al., 2009a). Genetic studies using MAGL(–/–) mice also support annotation of MAGL as the principal 2-AG hydrolase in the nervous system and several peripheral tissues (Chanda et al., 2010; Schlosburg et al., 2010; Taschler et al., 2011).
Fig. 5.

Structures of FAAH (A), MAGL (B), and dual FAAH/MAGL (C) inhibitors.
In the rat brain, MAGL expression is highest in the cerebellum, cortex, thalamus, and hippocampus (Dinh et al., 2002) where, like CB1, it is primarily localized to presynaptic axon terminals (Gulyas et al., 2004). MAGL contains the serine-histidine-aspartatic acid catalytic triad typical of most serine hydrolase enzymes (Karlsson et al., 1997). Murine MAGL is expressed as a single ∼33-kDa enzyme in most tissues except brain and testis, where multiple MAGL isoforms can be resolved by SDS-polyacrylamide gel electrophoresis analysis (Dinh et al., 2002; Long et al., 2009b). Whether these species represent alternate translation start sites, alternative RNA splicing, and/or post-translational modification of MAGL has yet to be fully elucidated. MAGL is highly selective for MAGs with negligible activity against diacylglycerols, triacylglycerols, phospholipids, or cholesterol esters (Tornqvist and Belfrage, 1976).
C. Additional Endocannabinoid Hydrolases
1. Fatty Acid Amide Hydrolase 2 and N-Acylethanolamine-Hydrolyzing Acid Amidase.
Although FAAH represents the primary anandamide hydrolase in the mammalian nervous system and many peripheral tissues, at least two other enzymes with this activity have been characterized: fatty acid amide hydrolase 2 (FAAH-2) and NAE-hydrolyzing acid amidase (NAAA). FAAH-2 is a ∼60-kDa amidase signature enzyme expressed in certain peripheral tissues in higher mammals, but not rodents (Wei et al., 2006). Unlike FAAH, FAAH-2 displays substrate preference for primary fatty acid amides, such as oleamide, over NAEs, such as anandamide. NAAA is an ∼40-kDa enzyme of the choloylglycine hydrolase family expressed by both rodents and humans in peripheral tissues and cell types, including macrophages, where it has been found to participate with FAAH in NEA degradation (Sun et al., 2005). Unlike FAAH, NAAA prefers N-palmitoylethanolamine to anandamide as a substrate (Tsuboi et al., 2005).
2. α/β-Hydrolases 6 and 12.
Studies with MAGL inhibitors confirmed the primacy of this enzyme in brain 2-AG degradation but found that a modest portion of 2-AG hydrolase activity persists in brain homogenates after MAGL inhibition (Hohmann et al., 2005; Saario et al., 2005; Long et al., 2009a). Additionally, the murine microglial BV2 cell line was shown to effectively hydrolyze 2-AG despite lacking MAGL expression (Muccioli et al., 2007).
By using a functional proteomics approach to survey all mouse brain 2-AG hydrolases, we discovered that the 2-AG hydrolysis activity of mouse brain membranes insensitive to MAGL inhibition (∼15% of total activity) was performed by the previously uncharacterized enzymes α/β-hydrolases 6 and 12 (ABHD6 and ABHD12) (Blankman et al., 2007). ABHD6 is an ∼30-kDa integral membrane serine hydrolase predicted to adopt an intracellular orientation (Blankman et al., 2007). Murine ABHD6 expression is abundant in brain and multiple peripheral tissues and cell types (Su et al., 2002; Bachovchin et al., 2010; Marrs et al., 2010). In the mouse brain, ABHD6 is highly expressed in cortical areas, where it preferentially localizes to postsynaptic dendrites that are often juxtaposed to presynaptic CB1 receptors (Marrs et al., 2010). ABHD12 is an ∼45-kDa membrane glycoprotein predicted to contain a single-pass transmembrane domain and face the extracellular/luminal cellular space (Blankman et al., 2007). High ABHD12 mRNA expression has been detected in mouse brain, bladder, prostate, white adipose, macrophages, and microglia (Su et al., 2002).
Despite their limited contribution to bulk 2-AG degradation, it is possible that the different subcellular or cellular localizations occupied by ABHD6 and ABHD12 compared with MAGL might allow each enzyme access to distinct pools of 2-AG in the nervous system. Indeed, ABHD6 has recently been shown to regulate 2-AG degradation and signaling in murine primary neurons and cortical slices (Marrs et al., 2010) and control 2-AG accumulation in the Neuro2A cell line, which lacks MAGL (Hsu et al., 2012). Additionally, ABHD6 inhibition in vivo produced CB1- and CB2-mediated anti-inflammatory and neuroprotective effects in a mouse model of traumatic brain injury (Tchantchou and Zhang, 2012). Human genetics has implicated ABHD12 as a critical regulator of neurologic function; loss-of-function mutations in ABHD12 cause the human neurodegenerative disease PHARC (polyneuropathy, hearing loss, ataxia, retinosis pigmentosa, and cataract) (Fiskerstrand et al., 2010), although whether ABHD12-disruption leads to neuronal death by dysregulation of 2-AG or an alternate mechanism remains to be determined.
III. Development of Endocannabinoid Hydrolase Inhibitors
Selective and in vivo active FAAH inhibitors have been available for nearly 10 years and have prompted numerous investigations into the metabolic and physiologic effects of augmented anandamide signaling. More recently, pharmacological agents were developed that potently and selectively block MAGL activity and elevate 2-AG levels in vivo, enabling, for the first time, direct comparison between the enhanced actions of each endocannabinoid. Multiple recent reviews have described the generation of endocannabinoid hydrolase inhibitors in detail (Minkkilä et al., 2010; Petrosino and Di Marzo, 2010; Otrubova et al., 2011; Feledziak et al., 2012). Here, we will focus our discussion on inhibitors that have been most widely used to probe endocannabinoid function in vivo.
The discovery and characterization of many of these inhibitors has benefited from the use of competitive activity-based protein profiling (ABPP), a chemoproteomic platform in which the enzymatic activity of mechanistically related enzymes is assessed in complex proteomes following treatment with small molecule inhibitors (Simon and Cravatt, 2010a) (Fig. 4). This strategy has proven especially conducive to endocannabinoid hydrolase inhibitor development, because nearly all of the enzymes involved in endocannabinoid degradation are serine hydrolases that can be readily assayed simultaneously using fluorophosphonate-containing reagents. Competitive ABPP allows the direct measurement of inhibitor potency and selectivity against all other serine hydrolases both in vitro and in vivo and has been used to characterize FAAH, MAGL, and dual FAAH/MAGL inhibitors.
Fig. 4.
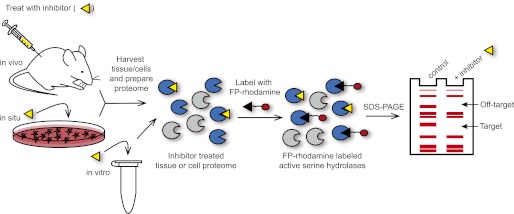
Competitive activity-based protein profiling (ABPP) serves as a chemoproteomic platform for assessing the potency and selectivity of endocannabinoid hydrolase inhibitors in vitro and in living systems. Animal models, cultured cells, or tissue/cell homogenates are treated with a small-molecule inhibitor (yellow triangle) for a specified time, and then proteomes are prepared and labeled with the activity-based probe fluorophosphonate (FP)-rhodamine, which reacts with active serine hydrolase enzymes. Serine hydrolases that are inactivated by the small-molecule inhibitor can be visualized by a loss of fluorescence signal following SDS-polyacrylamide gel electrophoresis analysis.
A. FAAH Inhibitors
Early FAAH inhibitors included the general serine protease inhibitor phenylmethylsulfonyl fluoride (PMSF) (Desarnaud et al., 1995) and fatty acid derivatives of sulfonylfluorides (Deutsch et al., 1997a), trifluoromethyl ketones (Koutek et al., 1994; Boger et al., 1999), and fluorophosphonates (Deutsch et al., 1997b). Although these first-generation FAAH inhibitors proved invaluable for biochemical and structural studies of FAAH in vitro (Bracey et al., 2002), they lacked the selectivity to be used to probe FAAH function in vivo. Several classes of FAAH inhibitors with improved selectivity and in vivo activity have since been reported and will be described briefly here.
1. α-Ketoheterocycles, e.g., OL-135.
One of the first classes of selective FAAH inhibitors to be explored were α-ketoheterocycle compounds that reversibly block FAAH activity (Otrubova and Boger, 2012). The archetype member of this compound class, OL-135 (Fig. 5A), has been shown to inhibit FAAH in vivo causing ∼3-fold increases in brain anandamide levels and concomitant CB1-dependent antinociceptive effects (Lichtman et al., 2004; Boger et al., 2005). Although selective for FAAH in the brain, OL-135 exhibited a fairly short duration of action (<4 hour) (Lichtman et al., 2004). Subsequent structure-activity relationship studies have extensively explored the potency, selectivity, and in vivo half-life of OL-135 derivatives (Otrubova et al., 2011), and recently oral administration of an α-keto-oxazole OL-135 derivative was found to significantly elevate brain anandamide levels (approximately 5-fold) and produce antinociceptive effects for over 9 hours (Ezzili et al., 2011).
2. Carbamates, e.g., URB597.
a. O-Aryl carbamates
Investigation into FAAH’s function in vivo was greatly advanced by the development of URB597 (Fig. 5A) (Kathuria et al., 2003), a carbamate compound that blocks FAAH activity through irreversible carbamylation of the enzyme’s catalytic serine nucleophile (Alexander and Cravatt, 2005). In rodents, URB597 reduced brain anandamide hydrolysis activity and elevated brain anandamide and other NAE levels without altering 2-AG levels (Kathuria et al., 2003). Numerous studies in rodents have since demonstrated that FAAH inhibition by URB597 represents a pharmacological model of enhanced anandamide signaling and have used URB597 to probe anandamide-regulated physiology in vivo. As will be described in more detail in the next section, URB597 administration has been shown to reduce pain, anxiety, depression, and nausea in rodents (Ahn et al., 2008). Importantly, these effects occurred without altering motility, body temperature, or appetite (Kathuria et al., 2003).
Although selective for FAAH in the brain, URB597 has been shown to inhibit multiple carboxylesterase (CES) enzymes in the liver (Lichtman et al., 2004; Zhang et al., 2007). Additionally, its relatively short half-life in vivo (Fegley et al., 2004; Lichtman et al., 2004) has limited its use in certain paradigms, including chronic dosing studies. Second-generation carbamate FAAH inhibitors have been reported, including URB694 (Fig. 5A), that display decreased activity toward CES enzymes and increased in vivo life span compared with URB597 (Clapper et al., 2009).
Bristol-Myers Squibb also reported a series of bisarylimidazole-containing carbamate inhibitors of FAAH including BMS-1 (Fig. 5A), which demonstrated antinociceptive effects in the formalin test and antiallodynic effects in the spinal nerve ligation model of neuropathic pain (Sit and Xie, 2002; Sit et al., 2007). ABPP analysis revealed that BMS-1 was relatively nonselective for FAAH, with multiple serine hydrolase off-targets in the brain and peripheral tissues (Lichtman et al., 2004; Zhang et al., 2007).
More recently, a peripherally restricted, p-hydroxyphenyl derivative of URB597 was developed. This compound, URB937 (Fig. 5A), although unable to access the CNS, retained CB1-dependent antinociceptive activity in mice in models of neuropathic, inflammatory, visceral, and arthritic pain, presumably by activating CB1 receptors on peripheral nociceptors (Clapper et al., 2010a; Moreno-Sanz et al., 2012; Sasso et al., 2012). In a model of arthritis induced by interplantar injection of complete Freund's adjuvant, URB937 administration reduced mechanical and thermal hyperalgesia to a greater extent than the systemically active FAAH inhibitors URB597 and PF-3845 (Sasso et al., 2012).
b. Oxime and enol carbamates.
Oxime carbamates have been reported as reversible FAAH inhibitors in the patent (Sit et al., 2003) and academic (Gattinoni et al., 2010b; Sit et al., 2010) literature. One member of this class, BMS-469908 (Fig. 5A), developed by Bristol-Myers Squibb, potentiated the antinociceptive effects of exogenously applied anandamide in mice, although this compound did not exhibit analgesic activity on its own (Sit et al., 2010)
A series of enol carbamates were also recently described as novel reversible FAAH inhibitors with high selectivity against other endocannabinoid targets (Gattinoni et al., 2010a). In vivo administration of one such compound, ST4070 (Fig. 5A), elicited anxiolytic effects in mice (Gattinoni et al., 2010a) and antiallodynic effects in several rodent models of neuropathic pain (Caprioli et al., 2012). FAAH activity in mouse brain tissue was reduced ∼65–75% by ST4070 treatment (10–100 mg/kg p.o.) and endogenous anandamide levels in mice were modestly (<2-fold) elevated by high doses of ST4070 (100 mg/kg p.o.) (Caprioli et al., 2012).
3. Alkylsufonylfluorides, e.g., AM3506.
Several analogs of PMSF with increased potency toward FAAH have been described, including the palmitoylsulfonyl fluoride AM374 (Fig. 5A) (Deutsch et al., 1997a) and the hydroxyl substituted phenylalkylsulfonylfluoride AM3506 (Fig. 5A) (Godlewski et al., 2010). AM3506 potently blocked FAAH in vivo and was found to display a unique anatomic activity profile; rodents treated with a single, systemic dose of AM3506 (1 mg/kg i.p. or p.o.) displayed a near complete loss of FAAH activity in the brain but retained wild-type FAAH activity in the liver due to rapid hepatic drug metabolism. This same dose of AM3506 partially inhibited the 2-AG hydrolases MAGL and ABHD6 in the mouse brain as determined by ABPP analysis, but selectively elevated brain anandamide levels ∼3-fold without affecting brain 2-AG in rats. AM3506 treatment (1 mg/kg i.p.) reduced blood pressure, heart rate, and cardiac contractibility in spontaneous hypertensive rats without inducing hypotension or bradycardia in control animals. The improvements in cardiovascular function elicited by AM3506 were similar to those observed following FAAH inhibition by URB597 (Batkai et al., 2004), but occurred without evidence of metabolic effects such as hyperglycemia or insulin resistance previously linked to reduced FAAH activity and increased hepatic anandamide (Osei-Hyiaman et al., 2005b, 2008; Tam et al., 2010; Touriño et al., 2010).
4. Aryl Ureas, e.g., PF-3845.
One of the earliest urea-based FAAH inhibitors, LY-2183240 (Fig. 5A), was originally described as an inhibitor of the putative anandamide transporter (Moore et al., 2005), but was later found to potently block FAAH and several other brain serine hydrolases by ABPP (Alexander and Cravatt, 2006) and substrate (Ortar et al., 2008) assays. Treatment of rats with LY-2183240 raised brain anandamide levels ∼5-fold and alleviated formalin-induced pain behaviors without affecting motor coordination (Moore et al., 2005).
In 2006, piperazinyl and piperidinyl urea FAAH inhibitors were disclosed by Takeda and Johnson & Johnson, including JNJ-1661010 (Fig. 5A) (Apodaca et al., 2006). One dose of JNJ-1661010 (20 mg/kg i.p.) significantly blunted FAAH activity in the rat brain for up to 24 hours and caused a modest ∼1.5-fold increase in brain anandamide levels (Keith et al., 2008; Karbarz et al., 2009). JNJ-1661010 administration elicited therapeutic analgesic effects in rat models of neuropathic, inflammatory, and acute thermal pain (Karbarz et al., 2009).
Pfizer has developed a series of covalent and irreversible urea-based FAAH inhibitors that display exceptional potency, selectivity, and duration of action in vivo (Ahn et al., 2007; Ahn et al., 2009; Meyers et al., 2011). Of these compounds, PF-3845 (Fig. 5A) has been most widely used in preclinical studies, and its remarkable selectivity for FAAH has been demonstrated not only by competitive ABPP but also by creating alkyne analogs, enabling direct detection of labeled proteins in vivo by coupling to azide-reporter tags (Speers et al., 2003; Wang et al., 2003; Speers and Cravatt, 2004) using copper-catalyzed azide-alkyne cycloaddition chemistry (Rostovtsev et al., 2002; Tornøe et al., 2002). A single, systemic dose of PF-3845 (10 mg/kg i.p.) has been shown selectively to block FAAH activity in the mouse brain for up to 24 hours and maximally elevate anandamide concentrations for 7–12 hours (Ahn et al., 2009). Medicinal chemistry efforts to improve the potency and pharmaceutical properties of PF-3845 for suitability in human clinical trials led to development of PF-04457845 (Fig. 5A), a FAAH inhibitor with excellent potency, selectivity, oral availability, and efficacy in a rat models of inflammatory and noninflammatory pain (Ahn et al., 2011; Johnson et al., 2011). In phase I trials, PF-04457845 nearly completely inhibited FAAH activity in blood leukocytes, elevated plasma anandamide and other NAE levels 3.5- to 10-fold, and was well tolerated (Li et al., 2012). Recently, however, PF-04457845 failed to show efficacy in Phase II trials in patients with osteoarthritic knee pain (Huggins et al., 2012).
Pfizer has also developed a series of benzothiophene piperazine/piperidine urea FAAH inhibitors including the substituted pyridyl PF-465 (Fig. 5A). ABPP analysis confirmed that PF-465 completely blocks FAAH activity in mouse and human proteomes with no off-target activity observed up to 100 μM (Johnson et al., 2009). PF-465 was active in vivo and effectively reduced allodynic responses in a rat model of inflammatory pain.
5. Comparison with FAAH(–/–) Mice.
Generation and characterization of mice bearing targeted disruption of the Faah gene [FAAH(–/–) mice] confirmed FAAH’s role as the principal anandamide hydrolase in vivo (Cravatt et al., 2001). FAAH(–/–) mice are viable, fertile, and largely indistinguishable from wild-type littermates. Brains from FAAH(–/–) mice have been found to lack anandamide hydrolytic activity and exhibit dramatically elevated (>10-fold) anandamide levels (Cravatt et al., 2001) but maintain wild-type 2-AG levels (Osei-Hyiaman et al., 2005a) and CB1 expression (Lichtman et al., 2002). FAAH(–/–) mice displayed robust CB1-dependent “tetrad” effects—analgesia, hypomotility, hypothermia, and catalepsy—in response to exogenously administered anandamide. Naively, FAAH KO mice have been shown to exhibit antinociceptive, anti-inflammatory, anxiolytic, and antidepressive phenotypes without evidence of motor or cognitive defects (Ahn et al., 2008).
B. MAGL Inhibitors
Selective pharmacological tools to disrupt the activity of MAGL in vivo have only become available within the last few years, but already they have been used to demonstrate the role of this enzyme in 2-AG signaling termination and the potential translational use of targeting MAGL in the treatment of nervous system disorders such as pain, anxiety, drug addiction, nausea, and neuroinflammation (Minkkilä et al., 2010; Petrosino and Di Marzo, 2010; Nomura et al., 2011b).
MAGL activity is sensitive to general serine hydrolase inhibitors such as methyl arachidonoylfluorophosphonate, PMSF, arachidonoyl trifluoromethylketone, and hexadecysulfonylfluoride (Dinh et al., 2002; Saario et al., 2004). These compounds also inhibit FAAH and are therefore not suitable to distinguish the function of these enzymes. Unlike most serine hydrolases, MAGL is also inhibited by general sulfhydryl-specific agents, including p-chloromercuribenzoic acid, mercury chloride, and N-ethlymaleimide, indicating the presence of a critical free cysteine residue (Tornqvist and Belfrage, 1976; Sakurada and Noma, 1981; Saario et al., 2005).
1. First-Generation MAGL Inhibitors: URB602, NAM, and OMDM169.
The carbamate compound URB602 (Fig. 5B) was reported as a selective MAGL inhibitor with relatively low potency (Hohmann et al., 2005). When microinjected into the rat periaqueductal gray matter, URB602 produced a modest (<2-fold) increase in the concentration of 2-AG, but not of anandamide, in this region and enhanced stress-induced analgesia (Hohmann et al., 2005). URB602 was also found to increase 2-AG content and prolong DSI, a form of CB1-mediated synaptic plasticity, in rat forebrain cultures (Makara et al., 2005). URB602’s relatively weak potency complicates its systemic administration in vivo and its selectivity is a matter of debate because it has been shown to be equally potent against FAAH in vitro (Muccioli et al., 2007; Vandevoorde et al., 2007). However, URB602 administration has been shown to attenuate nociception in rodent models of acute, inflammatory, and neuropathic pain (Comelli et al., 2007; Guindon et al., 2007, 2011; Desroches et al., 2008).
N-Arachidonoyl maleimide (NAM; Fig. 5B) is an irreversible MAGL inhibitor that was found to decrease 2-AG hydrolase activity of rat cerebellar membranes by ∼90% (Saario et al., 2005) and potentiate the effects of exogenously administered 2-AG (Burston et al., 2008). Although relatively selective for MAGL against other serine hydrolase enzymes, including FAAH (Blankman et al., 2007), NAM's maleimide group is a general thiol-reactive electrophile that will likely react with many cysteine-containing proteins in vivo, limiting its use in physiologic studies.
The tetrahydrolipostatin derivative OMDM169 (Fig. 5B) was shown to inhibit MAGL and pancreatic lipase with approximately 10-fold selectivity over FAAH and the 2-AG biosynthetic enzyme DAGLα in vitro (Bisogno et al., 2009). Systemic administration of OMDM169 (5 mg/kg i.p.) in mice reduced the expression of nociceptive behavior to the second phase of the formalin test and modestly elevated (<2-fold) 2-AG levels in the formalin-treated paw, but not in the brain.
2. O-Aryl Carbamates, e.g., JZL184.
In 2009, we reported a selective and in vivo-active MAGL inhibitor, JZL184 (Fig. 5B) (Long et al., 2009a), which has been used to demonstrate MAGL's role in 2-AG signaling termination and the potential translational use of targeting MAGL in the treatment of nervous system disorders such as pain, anxiety, drug addiction, and nausea (Minkkilä et al., 2010; Petrosino and Di Marzo, 2010). JZL184 is a piperidine carbamate that was shown to inhibit MAGL through irreversible carbamoylation of the enzyme's catalytic serine (Long et al., 2009b) and is another prime example of the carbamate reactive group as a privileged scaffold for serine hydrolase inhibitor design. As assessed by competitive ABPP, JZL184 was quite selective for MAGL against other mouse brain serine hydrolases and displayed >100-fold selectivity toward this enzyme over FAAH (Long et al., 2009a, 2010). Consistent with previous findings (Saario et al., 2005; Blankman et al., 2007), MAGL inhibition by JZL184 decreased 2-AG hydrolysis in mouse brain membranes by ∼85% (Long et al., 2009a). A single injection of JZL184 (16 mg/kg i.p., PEG vehicle) inhibited MAGL in the mouse brain for up to 24 hours and produced maximally elevated brain 2-AG levels (8-fold) for at least 8 hours without altering anandamide levels. In the initial report, JZL184-treated mice were found to display CB1-dependent analgesia, hypomotility, and hypothermia (Long et al., 2009a). However, core body temperature was not affected when JZL184 (40 mg/kg i.p.) was delivered in an optimized vehicle of an 18:1:1 solution of saline/emulphor/ethanol (Long et al., 2009b). As the PEG vehicle, but not the saline/emulphor/ethanol vehicle, caused a transient drop in core temperature, the discrepancy between JZL184’s hypothermic effects might suggest that MAGL inhibition disrupts temperature regulation following a hypothermic challenge (Long et al., 2009b). In vitro, JZL184 was ∼10-fold more potent toward murine or human MAGL than the rat ortholog (Long et al., 2009b).
3. O-Hexafluorisopropyl Carbamates, e.g., KML29.
Although JZL184 has proven to be a powerful tool to probe the function of MAGL in vivo, it displayed partial cross-reactivity with FAAH when used at high doses (Long et al., 2009a,b) and modestly elevated anandamide levels when administered chronically (Schlosburg et al., 2010). In peripheral tissues, JZL184, like many other carbamate agents, also blocked the activity of multiple CES enzymes (Long et al., 2009b). Very recently, next-generation MAGL inhibitors based on an O-hexafluoroisopropyl carbamate scaffold have been developed that possess superior selectivity toward MAGL versus other serine hydrolases in the brain and peripheral tissues (Chang et al., 2012). The O-hexafluoroisopropyl analog of JZL184, KML29 (Fig. 5B), maintained equivalent potency toward MAGL, but displayed greatly improved selectivity over FAAH and CES enzymes both in vitro and in vivo. In mice, KML29 dose dependently inhibited MAGL and dramatically elevated brain 2-AG levels without disrupting FAAH activity. KML29 exhibited complete selectivity against FAAH even when dosed chronically (6 days, 40 mg/kg p.o.). Additionally, KML29 retained much of its potency and selectivity for both rat and human MAGL orthologs. KML29 treatment (40 mg/kg i.p.) in rats nearly completely (>90%) inhibited MAGL and increased brain 2-AG levels ∼10-fold without affecting anandamide levels—KML29, therefore, represents a versatile inhibitor that can be used to investigate MAGL function in human cell and rodent models under both acute and chronic dosing regimens. Scientists at Sanofi-Aventis have independently reported a distinct set of O-hexafluoroisopropyl carbamates as MAGL inhibitors, although the selectivity and in vivo activity of these compounds were not disclosed (Bartsch et al. 2011).
4. Comparison with MAGL(–/–) Mice.
We and others have described mouse models bearing genetic disruption of the Mgll gene [MAGL(–/–) mice] (Chanda et al., 2010; Schlosburg et al., 2010; Taschler et al., 2011). Similar to JZL184-treated mice, MAGL(–/–) mice displayed dramatic reductions in 2-AG hydrolase activity and elevations in 2-AG and other MAGs in the brain and many peripheral tissues. Surprisingly however, MAGL(–/–) mice did not exhibit any cannabimimetic behaviors naively and displayed reduced sensitivity, or tolerance, to exogenous cannabinoids. This phenotype was replicated following chronic dosing of JZL184 (40 mg/kg i.p., 6 days) (Schlosburg et al., 2010) and could be attributed to CB1 downregulation in multiple brain areas (Chanda et al., 2010; Schlosburg et al., 2010). That chronic elevations in 2-AG led to functional antagonism of CB1 (Fig. 6) stands in stark contrast to the sustained agonism induced by long-term elevations in anandamide following chronic pharmacological or genetic FAAH disruption (Cravatt et al., 2001; Lichtman et al., 2002; Schlosburg et al., 2010).
Fig. 6.

Chronic MAGL disruption causes functional antagonism of the central endocannabinoid system. (A) In a wild-type brain, MAGL serves to limit the magnitude and duration of 2-AG signaling at CB1 by hydrolyzing this lipid to arachidonic acid. (B) Chronic pharmacological or genetic MAGL inactivation results in prolonged elevations in 2-AG that in turn cause the desensitization and downregulation of CB1 receptors. The net result of these adaptations is functional antagonism or reduced CB1 signaling.
C. Dual FAAH/MAGL Inhibitors
One curious observation following FAAH or MAGL inhibition in rodents is that neither completely reconstitutes the robust behavioral phenotypes of direct CB1 agonists like THC. This discrepancy begged the question of whether elevation of both endocannabinoids by simultaneous inhibition of FAAH and MAGL might evoke responses that more closely mirror those of exogenous cannabinoids. This hypothesis was tested by the development and characterization dual FAAH/MAGL inhibitors.
1. Organophosphates, e.g., Isopropyldodecylfluorophosphonate.
The organophosphorus nerve agent Isopropyldodecylfluorophosphonate (IDFP; Fig. 5C) was found to be a potent inhibitor of both FAAH and MAGL (IC50 values of 3 and 0.8 nM, respectively) as well as many additional serine hydrolases, including the alternate 2-AG hydrolase ABHD6, neuropathy target esterase, and the ether-lipid metabolic enzyme KIAA1363 (Nomura et al., 2008a). Treatment of mice with IDFP (10 mg/kg i.p.) caused dramatic elevations in brain endocannabinoid levels (∼10-fold) and CB1-mediated antinociception, hypomotility, hypothermia, and catalepsy. Mice treated acutely with IDFP died within 48 hours by a CB1-independent mechanism.
2. O-Aryl Carbamates, e.g., JZL195.
JZL195 (Fig. 5C) is a piperazine carbamate compound that was rationally designed to inhibit both MAGL and FAAH (Long et al., 2009c, 2010). JZL195 exhibits equipotency toward FAAH and MAGL (IC50 values of 2 and 4 nM, respectively) with substantial inhibitory activity toward the minor 2-AG hydrolase ABHD6. Mice treated with JZL195 (20 mg/kg i.p.) showed a near complete loss of brain anandamide and 2-AG hydrolysis activity, dramatic elevations in brain anandamide and 2-AG levels (∼10-fold) that persisted for at least 10 hours, and three out of four tetrad behaviors: antinociception, hypomotility, and catalepsy. Importantly, mice treated chronically with JZL195 for 6 days displayed no signs of toxicity.
3. O-Hydroxyacetamide Carbamates, e.g., SA-57.
O-Hydroxyacetamide carbamates disclosed in patents filed by Sanofi-Aventis (Abouabdellah et al., 2005) were profiled for their ability to inhibit endocannabinoid hydrolases and one such compound, SA-57 (Fig. 5C), was found to selectively block FAAH, MAGL, and ABHD6 activity in vitro and in vivo (Niphakis et al., 2012). Unlike JZL195, which displays similar potency toward FAAH, MAGL, and ABHD6, SA-57 is considerably more active toward FAAH. In mice, SA-57 selectively inhibited FAAH at low doses (0.05–0.25 mg/kg i.p.) and cross-reacted with MAGL and ABHD6 at higher doses (1.25–12.5 mg/kg i.p.). This selectivity profile allows SA-57 to act simultaneously as a complete FAAH inhibitor and partial MAGL/ABHD6 inhibitor in vivo. Considering that chronic, full MAGL inhibition induces CB1 desensitization/downregulation (Schlosburg et al., 2010) while chronic, partial MAGL inhibition has been found to maintain efficacy at CB1 in multiple preclinical models without causing receptor impairment (Schlosburg et al., 2010; Busquets-Garcia et al., 2011; Kinsey et al., 2011; Sciolino et al., 2011; Sticht et al., 2011), SA-57 should prove to be a useful tool to probe the effects of prolonged agonism of both endocannabinoids at CB1.
IV. Comparison of the Neurobehavioral Effects of FAAH and MAGL Inhibition
A. Tetrad Effects
The “tetrad test” for assessing cannabimimetic activity consists of assays for four of the most robust phenotypes displayed by THC-intoxicated rodents: antinociception, catalepsy, hypomotility, and hypothermia (Little et al., 1988). Rodents treated with exogenous CB1 agonists exhibit dramatically different behavior from vehicle-treated animals in each of these paradigms, while enhanced anandamide or 2-AG signaling caused by FAAH or MAGL inhibition, respectively, has been found to elicit more modest effects in a distinct subset of tests. FAAH inhibition by OL-135, URB597, or PF-3845 has been shown to induce antinociception, but not hypomotility, catalepsy, or hypothermia—effects phenocopied in FAAH(–/–) mice (Ahn et al., 2008). MAGL inhibition by JZL184 (40 mg/kg i.p., saline/emulphor/ethanol vehicle) elicited antinociception and hypomotility, but not hypothermia or catalepsy, although JZL184-treated mice did exhibit hyper-reflexia or “popcorning” behavior when presented with the catalepsy bar apparatus (Long et al., 2009c). Importantly, all of the tetrad behaviors induced by FAAH or MAGL inhibition were completely blocked by pretreatment with the CB1 antagonist rimonabant (Long et al., 2009c). These data indicate that although both endocannabinoids can facilitate CB1-mediated antinociception, CB1-dependent hypomotility and hyper-reflexia are driven primarily by the 2-AG/MAGL pathway.
Simultaneous blockade of FAAH and MAGL achieved by treatment of wild-type mice with the dual FAAH/MAGL inhibitor JZL195, co-treatment of wild-type mice with JZL184 and PF-3845, or treatment of FAAH(−/−) mice with JZL184, induced robust CB1-dependent antinociception, hypomotility, hyper-reflexia, and catalepsy (Long et al., 2009c). The effects of dual FAAH/MAGL inhibition on locomotor activity and hyper-reflexic behavior were equivalent to that observed following MAGL blockade, indicating that 2-AG is the major mediator of these behaviors. The analgesic effects of dual inhibition were synergistic and substantially higher than those elicited by single enzyme inhibition. Catalepsy was only observed following blockade of both enzymes. That dual FAAH/MAGL inhibition elicited a behavioral profile distinct from single enzyme inhibition and more akin to direct CB1 agonism suggests that anandamide and 2-AG signal through distinct modes that engage in significant cross-talk in the nervous system. Whether these interactions occur at shared CB1 receptors (i.e., intrasynaptically) or between distinct neuronal circuits (i.e., intersynaptically) (Fig. 7) remains to be determined.
Fig. 7.

Potential mechanisms for anandamide and 2-AG cross-talk in the nervous system. Whether anandamide and 2-AG produce differential effects through distinct modes of activation of shared CB1 receptors (“intrasynaptic,” A) or by activating separate sets of CB1 receptors in the brain (“intersynaptic,” B) is unknown. It is also possible that both intra- and intersynaptic mechanisms occur in vivo.
B. Pain
A considerable body of research has demonstrated that pharmacological blockade of FAAH reduces nociceptive and hyperalgesic behavior in a wide range of acute, inflammatory, and neuropathic pain models (Schlosburg et al., 2009b). Despite their relatively recent development, chemical tools that selectively inactivate MAGL in vivo have already demonstrated efficacy in multiple preclinical pain paradigms (Hohmann et al., 2005; Kinsey et al., 2009, 2010; Long et al., 2009a,c; Schlosburg et al., 2010; Busquets-Garcia et al., 2011; Guindon et al., 2011, 2013; Khasabova et al., 2011; Ghosh et al., 2012). Not only have these studies implicated MAGL as an additional therapeutic target for the treatment of pain, they have revealed mechanistic differences between anandamide and 2-AG-mediated antinociception.
Interestingly, complete FAAH (PF-3845, 10 mg/kg i.p.) or MAGL (JZL184, 40 mg/kg i.p.) inhibition, resulting in maximal endocannabinoid elevations (>10-fold), produced antinociceptive effects of similar magnitude in tests of acute thermal pain, visceral pain, neuropathic pain, and inflammatory pain, despite the fact that anandamide and 2-AG are present in the brain at dramatically different basal levels (Kinsey et al., 2009, 2010; Long et al., 2009c; Schlosburg et al., 2010; Ghosh et al., 2012).
Multiple lines of evidence suggest that despite bearing equivalent analgesic potential in certain paradigms, anandamide and 2-AG regulate pain processing through distinct signaling pathways. Multiple groups have reported that the analgesic effects of FAAH and MAGL inhibition proceed via different cannabinoid receptor mechanisms, and the responsible receptors appear to depend on the models and behavioral assays examined.
In a model of neuropathic pain in mice caused by chronic constriction injury of the sciatic nerve, acute, systemic FAAH (PF-3845, 10 mg/kg i.p.) and MAGL (JZL184, 40 mg/kg i.p.) blockade reduced mechanical and cold allodynia by similar magnitudes (Kinsey et al., 2009, 2010). However, the efficacy of FAAH inhibition was mediated by CB1 and CB2 receptors, whereas the antiallodynic effects of MAGL inhibition were solely dependent on CB1 activation. Cannabinoid receptor dependency was determined by sensitivity to both pharmacological (Kinsey et al., 2009) and genetic (Kinsey et al., 2010) disruption of CB1/2.
In a peripheral model of pain induced by local injection of capsaicin to the rat paw, the hypersensitivities observed were distinctly modulated by local inhibition of FAAH (URB597, 75 μg i.p.l.) or MAGL (JZL184, 100 μg i.p.l.) (Spradley et al., 2010). JZL184 attenuated the thermal hyperalgesia and nocifensive behavior caused by capsaicin injection, while URB597 reduced capsaicin-induced mechanical allodynia. URB597-mediated effects were blocked by CB1 antagonists, whereas JZL184-induced antinociception was prevented by CB1 and CB2 antagonists. CB1 and CB2 receptors also mediated the antinociceptive effects of local MAGL inhibition in the formalin test in rats (Guindon et al., 2011) and the anti-allodynic effects of systemic MAGL blockade in mice (Ghosh et al., 2012). Peripheral blockade of FAAH (URB597, 9 μg i.p.l.) (Khasabova et al., 2008) or MAGL (JZL184, 10 μg i.p.l.) (Khasabova et al., 2011) reduced mechanical hyperalgesia in a mouse model of bone cancer pain, and these antihyperalgesic phenotypes were found to be CB1 or CB2 dependent, respectively. Presumably the differences in cannabinoid receptor mechanisms reported in these studies stem from the pain paradigms examined, the route of drug administration, or even the species, but regardless, they all support the notion that enhanced anandamide and 2-AG act to alleviate pain via distinct signaling mechanisms.
Another difference between the analgesic effects of FAAH and MAGL inactivation is evident upon chronic drug treatment. We determined that the duration of FAAH or MAGL blockade affects the maintenance of analgesic behavior (Schlosburg et al., 2010). Whereas complete, acute disruption of FAAH or MAGL elicited a similar degree of antinociception in the tail immersion test of thermal pain or the chronic constriction injury model of neuropathic pain, chronic disruption of each enzyme produced opposing effects. After chronic drug treatment, mice became tolerant to the antinociceptive effects of JZL184 (40 mg/kg i.p., 6 days) but maintained sensitivity to the analgesic effects of PF-3845 (10 mg/kg i.p., 6 days). The lack of analgesic phenotype observed in chronic JZL184-treated mice was phenocopied in MAGL(–/–) mice, which displayed equivalent thermal pain sensitivity as their wild-type littermates (Chanda et al., 2010; Schlosburg et al., 2010). The tolerance observed in chronic MAGL-disrupted mice was accompanied by a heterogeneous reduction in CB1 expression and function throughout the brain, including the periaqueductal gray, a region of the brain involved in cannabinoid-mediated antinociception (Lichtman et al., 1996). The cellular and behavioral adaptations caused by chronic elevations in endogenous 2-AG are reminiscent of those observed following chronic THC administration in both rodents and humans (Lichtman and Martin, 2005). These results suggest that maximally elevated anandamide and 2-AG brain levels exert strikingly different effects on the integrity of the central endocannabinoid system, with chronic anandamide causing sustained agonism and chronic 2-AG inducing functional antagonism. Additionally, we found that chronic JZL184 treatment also induced cross-tolerance to the antiallodynic effects of acute PF-3845 treatment, indicating that the pain circuits regulated by 2-AG and anandamide interact at some level (Schlosburg et al., 2010). These results may have clinical implications for the use of FAAH or MAGL inhibitors in treating chronic pain. However, our study did not address the possibility that partial MAGL inhibition might retain analgesic efficacy without causing tolerance or CB1 disruption.
Subsequent to our study, chronic low doses of JZL184 (8 mg/kg i.p. 6 days) and URB597 (1 mg/kg i.p., 6 days) were found to retain efficacy in the acetic acid stretching model of visceral pain (Busquets-Garcia et al., 2011). Brain CB1 expression in these mice was unaltered, whereas mice treated with THC (10 mg/kg i.p.) under the same dosing schedule had significantly reduced hippocampal CB1 expression. In this study, brain endocannabinoid levels were differentially increased—anandamide was elevated <2-fold following URB597 treatment, whereas 2-AG was elevated ∼5-fold by JZL184—and MAGL inhibition was found to be more efficacious than FAAH inhibition. Tolerance to the anti-inflammatory effects of high (16 or 40 mg/kg i.p.) but not low (4 mg/kg i.p.) doses of JZL184 was also observed following chronic dosing in the carrageenan model (Ghosh et al., 2012).
Recently, peripheral FAAH blockade by the CNS-impermeant inhibitor URB937 (Clapper et al., 2010b) was found to elicit CB1-mediated antinociceptive effects in mouse models of neuropathic, inflammatory, and arthritic pain (Clapper et al., 2010b; Moreno-Sanz et al., 2012; Sasso et al., 2012). In a model of arthritis induced by interplantar injection of complete Freund’s adjuvant, URB937 reduced mechanical and thermal hyperalgesia to a greater extent than the systemically active FAAH inhibitors URB597 and PF-3845 (Sasso et al., 2012).
C. Anxiety and Depression
Anandamide was named after the Sanskrit word for “bliss” (Devane et al., 1992) in reference to the euphoric high elicited by Cannabis derivatives in humans. Direct CB1 agonism produces diverse psychotropic effects that can also include feelings of anxiety, panic, and paranoia. These biphasic effects on mood and emotionality have also been observed in rodent models where exogenous cannabinoids can elicit anxiolytic-like or anxiogenic-like responses depending on dose and context (Moreira and Lutz, 2008). To selectively harness the anxiolytic potential of the endocannabinoid system, enhancing endocannabinoid levels has emerged as a promising strategy. FAAH inhibitors, for instance, have demonstrated CB1-dependent anxiolytic effects in a number of rodent models of anxiety and depression without the negative side effects associated with direct CB1 agonists and are under consideration for the treatment of anxiety-related disorders (Gaetani et al., 2009). Less is known about the consequences of heightened 2-AG signaling on emotional status, but recent studies with JZL184 suggest that MAGL could represent a target for next-generation anxiolytic drugs.
Mice treated acutely with low doses of URB597 (1 mg/kg i.p.) or JZL184 (8 mg/kg i.p.) displayed similar anxiolytic-like responses in the elevated zero maze and the elevated plus maze, and these effects persisted following chronic drug treatment (6 days) (Busquets-Garcia et al., 2011). As previously demonstrated (Moreira et al., 2008), the anxiolytic-like effects of URB597 were mediated by CB1 and prevented by genetic or pharmacological CB1 disruption. Rather surprisingly, however, the anxiolytic-like effects of JZL184 were mediated via CB2, because JZL184 lacked efficacy in CB2(–/–) mice and mice pretreated with the CB2 antagonists SR144528 and JWH-133. JZL184 (8 mg/kg i.p.) also produced anxiolytic-like effects in rats in the elevated plus maze under high levels of environmental aversiveness, effects that were reversed by the CB1 antagonist rimonabant (Accomplia) and maintained after chronic administration (8 mg/kg day for 6 days) (Sciolino et al., 2011).
In the marble burying assay, a model of obsessive-compulsive behaviors, FAAH inhibition by URB597 (0.1–1 mg/kg i.p.) or PF-3845 (10 mg/kg i.p.) and MAGL inhibition by JZL184 (16 and 40 mg/kg i.p.) decreased marble burying behavior in mice through a CB1-dependent mechanism (Gomes et al., 2011; Kinsey et al., 2011). Importantly, the anxiolytic-like effects of JZL184 could be decoupled from its locomotor effects, because low doses of JZL184 (16 mg/kg i.p.) reduced marble burying without causing hypomotility (Kinsey et al., 2011).
D. Nausea and Emesis
Purified THC (dronabinol, Marinol [Abbott Laboratories, Abbott Park, IL]) and the synthetic cannabinoid nabilone (Cesemet; Meda Pharmaceuticals, Somerset, NJ) are licensed medicines approved for treating nausea and vomiting caused by chemotherapy and loss of appetite and weight loss in AIDS patients (Pertwee, 2009). Although effective as antiemetic agents, these direct CB1 agonists can also produce undesirable side effects, including dizziness, dysphoria, paranoia, and hallucinations, which limit their widespread use (Tramèr et al., 2001). FAAH and MAGL inhibitors exhibit antinausea/antiemetic effects in preclinical models, and enhancement of endocannabinoid signaling may also prove to be an effective method for treating nausea and vomiting in humans (Parker et al., 2011).
Both FAAH and MAGL inhibition suppress vomiting in house musk shrews (Suncus murinus) to various emetic stimuli. URB597 (0.9 mg/kg) treatment reduced cisplatin and nicotine-induced emetic episodes, and the effects on nicotine vomiting were CB1-dependent and reversed by rimonabant (Parker et al., 2009). JZL184 (16 and 40 mg/kg i.p.) treatment suppressed lithium chloride-induced vomiting, which was reversed by the CB1 antagonist AM251 (Sticht et al., 2011).
In rats, which do not have an emetic reflex, URB597 (0.3 mg/kg i.p.) treatment suppressed conditioned gaping, a model of nausea, in response to odors (Rock et al., 2008) or tastes (Cross-Mellor et al., 2007) that had been previously paired with the emetic stimulant lithium chloride (LiCl) by CB1-dependent mechanisms. In contrast, JZL184 treatment (40 mg/kg i.p.) did not affect LiCl-induced conditional gaping on its own, but did potentiate the antinausea-like effects of exogenously administered 2-AG partially through CB1 activation (Sticht et al., 2011).
E. Addiction and Withdrawal
Activation of the endocannabinoid system has been implicated in many aspects of addiction. Marijuana possesses mild addictive qualities, and its chronic use can lead to physical dependence (Haney et al., 1999; Budney et al., 2003; Maldonado et al., 2011). CB1 activation can also influence the addictive potential of other drugs of abuse, including opioids, cocaine, alcohol, and nicotine (Kogan and Mechoulam, 2007). Additionally, CB1 agonists can alleviate the severe physical symptoms induced by withdrawal from opioid drugs in preclinical models (Frederickson et al., 1976). For therapeutic purposes, modulation of the endocannabinoid system to harness the palliative effects of cannabinoids without concomitant abuse potential would be ideal.
1. Opioid Withdrawal.
THC is known to reduce the severity of opioid withdrawal in humans and rodents, and recently endocannabinoid hydrolase inhibitors were also found to alleviate symptoms of precipitated and spontaneous withdrawal in opioid-dependent mice (Ramesh et al., 2011). Treatment of morphine-dependent mice with the mu opioid receptor antagonist nalaxone induces a profound withdrawal phenotype consisting of jumping, paw tremors, diarrhea, and weight loss. Acute MAGL inhibition (JZL184, 4–40 mg/kg i.p.) dose-dependently reduced all of these effects through a CB1-dependent mechanism and, at the highest dose, nearly completely blocked all nalaxone-induced behaviors. Although FAAH(–/–) mice displayed partial protection from all four withdrawal behaviors, acute FAAH inhibition (PF-3845, 10 mg/kg i.p.) attenuated nalaxone-induced jumps and paw flutters, without effecting diarrhea or weight loss, effects also mediated by CB1 activation. JZL184 (40 mg/kg i.p.) also reduced opioid withdrawal signs in a model of spontaneous withdrawal where the morphine pellet was removed from morphine-dependent mice.
2. Precipitated THC Withdrawal.
Compared with other drugs of abuse, marijuana is generally considered to possess a reduced risk of dependency in part because cessation of chronic marijuana use in humans is not accompanied by a dramatic physical withdrawal syndrome like that of highly addictive drugs such as opioids, cocaine, and methamphetamine. Nevertheless, heavy cannabis use has been show to provoke a distinct abstinence syndrome consisting of disruptions in mood, appetite, sleep, and physical comfort (Haney et al., 1999; Budney et al., 2003). Mice treated chronically with THC also exhibit physical withdrawal symptoms, most commonly paw tremors and head twitches, when withdrawal is precipitated by treatment with a CB1 antagonist such as rimonabant (Lichtman and Martin, 2002). The severity of rimonabant-induced withdrawal symptoms in THC-dependent mice was attenuated by acute inhibition of FAAH (URB597, 10 mg/kg i.p.) or MAGL (JZL184, 16 mg/kg i.p.), suggesting that endocannabinoid hydrolases might represent a novel therapeutic strategy to treat cannabis dependence in humans (Schlosburg et al., 2009a).
3. Abuse Potential of FAAH and MAGL Inhibitors.
Although the findings presented in this review suggest that modulating FAAH and MAGL activity might have therapeutic potential, there is concern that FAAH, MAGL, or dual FAAH/MAGL inhibitors might exhibit abuse potential. FAAH inhibition did not produce rewarding effects in rats as assessed by conditioned place preference (URB597, 0.03-0.3 mg/kg i.p.) or THC-like psychotropic effects as determined by the drug-discrimination test (URB597, 0.1–3 mg/kg i.p.) (Gobbi et al., 2005). Similarly, URB597 (0.3 mg/kg i.v.) did not produce reinforcing effects in monkeys (Justinova et al., 2008). A high dose of JZL184 (40 mg/kg i.p.) produced partial generalization in the drug discrimination paradigm in mice, where it substituted for THC ∼50% of the time (Long et al., 2009c). In contrast, the dual FAAH/MAGL inhibitor JZL195 fully substituted for THC, producing >80% THC-appropriate responses. As the drug discrimination test serves as a model of THC intoxication in the rodent (Balster and Prescott, 1992), these results suggest that dual MAGL/FAAH blockers and perhaps high doses of MAGL inhibitors might produce THC-like psychotropic effects and abuse potential in humans.
Rimonabant-precipitated withdrawal has been used to assess whether FAAH or MAGL inhibition produces physical dependence. Mice treated chronically with URB597 (2 doses of 10 mg/kg for 5.5 days) (Schlosburg et al., 2009a) or PF-3845 (10 mg/kg i.p., 6 days) (Schlosburg et al., 2010) did not display any withdrawal-like symptoms when challenged with rimonabant. However, rimonabant did precipitate paw flutters in mice treated chronically with a high dose of JZL184 (40 mg/kg i.p., 6 days) (Schlosburg et al., 2010).
Although FAAH inhibitors have been found to lack abuse potential in preclinical models, a missense polymorphism in human FAAH, P129T, has been linked to problem drug use (Sipe et al., 2002) and obesity (Sipe et al., 2005). FAAH expression and activity are reduced in T-lymphocytes derived from patients homozygous for the P129T mutation versus control lymphocytes (Chiang et al., 2004). Although the factors that influence vulnerability to drug addiction and dependence are complex, these findings suggest that lifelong alterations in FAAH in humans may enhance the addictive properties of drugs or food in certain contexts.
V. Non-CB-Mediated Effects
Our discussion of pharmacological modulation of the anandamide/FAAH and 2-AG/MAGL pathways has focused on effects on endocannabinoid signaling. Importantly, however, anandamide and 2-AG interact with targets outside of the cannabinoid system, and both FAAH and MAGL regulate nonendocannabinoid metabolites in vivo. Chemical inhibition of FAAH and MAGL can therefore exert CB1/2-independent effects, which can be identified by their insensitivity to CB1/2 antagonists.
A. Additional FAAH-Regulated Metabolites
FAAH exhibits a broad substrate selectivity in vitro and regulates multiple classes of bioactive fatty acid amide species in vivo, including anandamide and its NAE congeners and N-acyl taurines (Saghatelian et al., 2004; Long et al., 2011). N-Acyl taurines are TRPV1 agonists in vitro (Saghatelian et al., 2006) and the NAEs palmitoylethanolamide and oleoylethanolamide have been found to display, among other effects, anti-inflammatory and anorexic properties, respectively (Hansen, 2010). Although many of the therapeutic effects of FAAH blockade are reversed by CB1/2 antagonists and can therefore be attributed to enhanced endocannabinoid signaling, noncannabinoid effects observed in FAAH-disrupted mice [e.g., anti-inflammatory phenotypes (Cravatt et al., 2004; Sagar et al., 2008)] might be due to elevations in additional FAAH substrates or to anandamide activity at non-CB1/2 targets, as discussed below.
B. Additional MAGL-Regulated Metabolites
In addition to elevating brain 2-AG, MAGL inhibition also stoichiometrically decreased brain arachidonic acid (AA) (Nomura et al., 2008a,b; Long et al., 2009a). This result indicated that 2-AG is a major source for AA in the brain and implicated MAGL as a potential regulator of AA derivatives such as prostaglandins. Subsequent studies confirmed that MAGL controls basal and neuroinflammatory prostaglandin levels in the brain and demonstrated that MAGL inactivation is neuroprotective in mouse models of Parkinson’s (Nomura et al., 2011b) and Alzheimer’s (Chen et al., 2012; Piro et al., 2012) diseases.
MAGL also influences energy metabolism by catalyzing the final step of triglyceride metabolism. Pharmacological (Long et al., 2009a,b) or genetic (Chanda et al., 2010; Schlosburg et al., 2010; Taschler et al., 2011) MAGL disruption elevated MAG levels in multiple peripheral tissues including adipose and liver. MAGL(–/–) mice displayed partial protection from high-fat diet-induced insulin resistance, although whether these effects were due to impaired lipolysis or CB1 downregulation is unclear (Taschler et al., 2011).
In aggressive ovarian, breast, and melanoma cancer cells, increased MAGL expression promoted pathogenesis by cleaving MAGs and liberating a free fatty acid pool that was converted by other enzymes into oncogenic lipid signals (Nomura et al., 2010). In these cancer cell types, pharmacological or RNA-interference disruption of MAGL impaired pathogenicity independent of CB1/2 activation; however, in prostate cancer cells, MAGL blockade impaired aggressiveness through both endocannabinoid and fatty acid-dependent mechanisms (Nomura et al., 2011a).
C. Additional Endocannabinoid Targets
Anandamide and 2-AG interact with protein targets outside of the endocannabinoid system. These targets include receptors such as the TRPV1, the G protein-coupled receptor 55, and peroxisome proliferator-activated receptors (De Petrocellis and Di Marzo, 2010; Pertwee et al., 2010), as well as oxidative cyclooxygenase and lipoxygenase enzymes (Kozak and Marnett, 2002; Woodward et al., 2008). FAAH and MAGL inhibitors may elicit CB1/2-independent effects by increasing the flux of endocannabinoids through these alternative pathways, which, depending on the specific targets involved, could potentiate or dampen CB receptor-mediated effects.
VI. Conclusions, Clinical Implications, and Future Directions
The generation of chemical probes that selectively disrupt the endocannabinoid hydrolases FAAH and MAGL in vivo is a vital first step to dissect the different functional roles of anandamide and 2-AG signaling and assess the potential therapeutic value of endocannabinoid hydrolase inhibitors in human pathologies. Mounting evidence supports the notion that 2-AG and anandamide have distinct functions in vivo and point to ligand diversification as a major mechanism by which the endocannabinoid system regulates physiology and behavior.
Pharmacological inhibition of FAAH and MAGL has demonstrated that heightened anandamide and 2-AG produce discrete, but overlapping, subsets of cannabinoid-mediated behaviors. For instance, although acute elevation of brain 2-AG or anandamide produces analgesic, anxiolytic, and antiemetic effects, only increases in 2-AG elicit motility defects and hyper-reflexia. Additionally, FAAH inhibitors have been repeatedly shown to lack abuse potential or physical dependence in preclinical models, whereas high doses of the MAGL inhibitor JZL184 partially substitute for THC and elicit a mild precipitated withdrawal phenotype in mice.
The mechanisms by which FAAH and/or MAGL disruption induce distinct subsets of CB1-mediated phenotypes remain unknown. One hypothesis is that each endocannabinoid interacts with physically distinct receptor pools in separate cells and circuits—in this model, the unique distribution of anandamide and 2-AG would be controlled by differential expression of endocannabinoid metabolic and catabolic enzymes. Another theory is that anandamide and 2-AG differentially activate shared receptor pools, a concept known as “biased agonism.” Regardless of their respective signaling mechanisms, the fact that simultaneous elevation of anandamide and 2-AG by dual FAAH/MAGL inhibition produces novel and synergistic CB1-mediated effects compared with elevation of either endocannabinoid alone strongly suggests that these two pathways cross-talk in the nervous system to regulate mammalian behavior.
Comparing the consequences of chronic FAAH or MAGL inhibition revealed that sustained maximal elevations in anandamide or 2-AG result in agonism or functional antagonism of CB1, respectively. Again, mechanistic explanations for why chronic elevations in 2-AG, but not anandamide, disrupt CB1 expression and function are lacking. One hypothesis is that anandamide and 2-AG may possess intrinsic differences in their ability to promote CB1 desensitization and/or downregulation, which has been previously demonstrated in Xenopus laevis oocytes, where anandamide was found to desensitize CB1 more slowly than 2-AG (Luk et al., 2004). In FAAH(–/–) mice, chronic exogenous anandamide administration, despite inducing full cannabimimetic behaviors, was found to cause less behavioral tolerance and cellular adaptations than chronic THC in these animals (Falenski et al., 2010). Minimal propensity to desensitize CB1 may be critical to the function of anandamide under physiologic conditions. Anandamide was recently proposed to act in the hippocampus as a tonic messenger at CB1 as a mechanism to maintain its responsiveness to activity-dependent 2-AG flux (Kim and Alger, 2010).
Future investigation into the effects of chronic, dual inhibition of MAGL and FAAH could potentially be informative here. Sustained elevations in both endocannabinoids might induce equivalent or greater CB1 downregulation than 2-AG alone, suggesting that endogenous 2-AG can uniquely disrupt CB1 function or that CB1 desensitization/downregulation is another example of a process regulated by endocannabinoid cross-talk, respectively. Alternatively, chronic increases in both endocannabinoids might induce less CB1 downregulation than elevations of 2-AG alone, which would suggest that anandamide can act to “protect” CB1 from 2-AG-induced downregulation.
That complete, systemic MAGL inhibition has been shown to induce hypomotility and hyper-reflexia, carry some abuse liability, and eventually disrupt the integrity of the central endocannabinoid system could suggest that FAAH inhibitors, which lack these negative properties, may be more suitable as human therapeutics. However, several studies have reported that lower doses of JZL184 that partially inhibit brain MAGL and cause submaximal elevations in brain 2-AG (Long et al., 2009b) retain efficacy in rodent models of pain (Busquets-Garcia et al., 2011; Ghosh et al., 2012), anxiety (Busquets-Garcia et al., 2011; Kinsey et al., 2011; Sciolino et al., 2011), emesis (Sticht et al., 2011), and opioid withdrawal (Ramesh et al., 2011). Importantly, the antinociceptive and anxiolytic-like effects elicited by a low dose of JZL184 (8 mg/kg i.p.) that elevated brain 2-AG levels ∼5-fold were maintained following chronic administration (8 mg/kg per day for 6 days) without evidence of CB1 impairment (Busquets-Garcia et al., 2011). Similarly, repeated administration of a low dose of JZL184 (4 mg/kg per day for 6 days i.p.) maintained efficacy in the carrageenan model of inflammatory pain (Ghosh et al., 2012). Anxiolytic-like effects were also preserved in rats treated chronically with a low dose of JZL184 (8 mg/kg per day for 6 days i.p.) (Sciolino et al., 2011). Significantly, multiple groups have reported doses of JZL184 that reduced anxiety-like behavior in rodents without affecting locomotor activity (Kinsey et al., 2011; Sciolino et al., 2011), demonstrating that the anxiolytic and motility effects of MAGL blockade can be pharmacologically separated. These findings indicate that partial MAGL inhibition may serve as a general strategy to dissociate the therapeutic and undesirable effects of enhanced 2-AG signaling.
The generation of selective chemical probes for the endocannabinoid hydrolases FAAH and MAGL has facilitated investigation into the function of endocannabinoid signaling in mammalian biology and dissection of the unique activities regulated by anandamide versus 2-AG in the nervous system. From a translational perspective, these same tools have demonstrated the potential therapeutic value of FAAH and MAGL inhibitors for a wide host of human pathologies.
Acknowledgments
The authors thank the Cravatt laboratory for helpful discussions.
Abbreviations
- AA
arachidonic acid
- 2-AG
2-arachidonoylglycerol
- ABHD
α-β hydrolase
- ABPP
activity-based protein profiling
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- CDTA
calcium-dependent transacylase
- CES
carboxylesterase
- CNS
central nervous system
- DAG
diacylglycerol
- DAGLα and DAGLβ
sn-1-specific diacylglycerol lipase-α and -β
- DSE
depolarization-induced suppression of excitation
- DSI
depolarization-induced suppression of inhibition
- FAAH
fatty acid amide hydrolase
- FAAH-2
fatty acid amide hydrolase 2
- IDFP
isopropyldodecylfluorophosphonate
- i.p.l.
intraplantar
- MAGL
monoacylglycerol lipase
- NAAA
N-acyl ethanolamine-hydrolyzing acid amidase
- NAE
N-acyl ethanolamine
- NAM
N-arachidonoyl maleimide
- NAPE
N-acyl phosphatidylethanolamine
- NArPE
N-arachidonoyl phosphatidylethanolamine
- PE
phosphatidylethanolamine
- PLC
phospholipase C
- PLD
phospholipase D
- PMSF
phenylmethylsulfonyl fluoride
- ΤΗC
Δ9-tetrahydrocannabinol
- TRPV1
transient receptor potential cation channel V1
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Blankman, Cravatt.
Footnotes
This work was supported by grants from the National Institutes of Health [Grants DA017259, DA033760, DA032933, DA009789]. J.L.B. was also supported by a Ruth L. Kirschstein National Institutes of Health Predoctoral Fellowship [Grant DA026261].
References
- Abouabdellah A, Almario GA, Hoornaert C, Lardenois P, Marguet F. (2005) inventors; Sanofi-Aventis, assignee. Aryl- and heteroaryl-piperidinecarboxylate derivatives, their preparation and use as fatty acid amido hydrolase (FAAH) inhibitors for treating FAAH-related pathologies. World Patent WO 2005/090347. 2005 Sep 29. [Google Scholar]
- Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, et al. (2007) Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry 46:13019–13030 [DOI] [PubMed] [Google Scholar]
- Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, et al. (2009) Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol 16:411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, McKinney MK, Cravatt BF. (2008) Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev 108:1687–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, Young T, Wren P, Zhang Y, Swaney S, et al. (2011) Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther 338:114–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JP, Cravatt BF. (2005) Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol 12:1179–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JP, Cravatt BF. (2006) The putative endocannabinoid transport blocker LY2183240 is a potent inhibitor of FAAH and several other brain serine hydrolases. J Am Chem Soc 128:9699–9704 [DOI] [PubMed] [Google Scholar]
- Alger BE, Kim J. (2011) Supply and demand for endocannabinoids. Trends Neurosci 34:304–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apodaca R, Breitenbucher JG, Pattabiraman K, Seierstad M, Xiao W. (2006), inventors; Janssen Pharmaceutica, assignee. Piperazinyl and piperidinyl ureas as modulators of fatty acid amide hydrolase. World Patent WO 2006/074025. 2006 Jul 13.
- Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. (2010) Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci USA 107:20941–20946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balster RL, Prescott WR. (1992) Δ 9-tetrahydrocannabinol discrimination in rats as a model for cannabis intoxication. Neurosci Biobehav Rev 16:55–62 [DOI] [PubMed] [Google Scholar]
- Bartsch R, Cheuret D, Even L, Hoornaert C, Jeunesse J, Marguet F. (2011), inventors; Sanofi-Aventis, assignee. Hexafluoroisopropyl carbamate derivatives, their preparation and their therapeutic application. World Patent WO 2011/151808. 2011 Dec 8. [Google Scholar]
- Bátkai S, Pacher P, Osei-Hyiaman D, Radaeva S, Liu J, Harvey-White J, Offertáler L, Mackie K, Rudd MA, Bukoski RD, et al. (2004) Endocannabinoids acting at cannabinoid-1 receptors regulate cardiovascular function in hypertension. Circulation 110:1996–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béquet F, Uzabiaga F, Desbazeille M, Ludwiczak P, Maftouh M, Picard C, Scatton B, Le Fur G. (2007) CB1 receptor-mediated control of the release of endocannabinoids (as assessed by microdialysis coupled with LC/MS) in the rat hypothalamus. Eur J Neurosci 26:3458–3464 [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, et al. (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163:463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Ortar G, Petrosino S, Morera E, Palazzo E, Nalli M, Maione S, Di Marzo V, Endocannabinoid Research Group (2009) Development of a potent inhibitor of 2-arachidonoylglycerol hydrolysis with antinociceptive activity in vivo. Biochim Biophys Acta 1791:53–60 [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14:1347–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, et al. (2005) Discovery of a potent, selective, and efficacious class of reversible α-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J Med Chem 48:1849–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, Cravatt BF. (1999) Trifluoromethyl ketone inhibitors of fatty acid amide hydrolase: a probe of structural and conformational features contributing to inhibition. Bioorg Med Chem Lett 9:265–270 [DOI] [PubMed] [Google Scholar]
- Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. (2002) Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 298:1793–1796 [DOI] [PubMed] [Google Scholar]
- Brodal P, ed (2004) The central nervous system: Structure and function, Oxford University Press, Oxford [Google Scholar]
- Budney AJ, Moore BA, Vandrey RG, Hughes JR. (2003) The time course and significance of cannabis withdrawal. J Abnorm Psychol 112:393–402 [DOI] [PubMed] [Google Scholar]
- Burston JJ, Sim-Selley LJ, Harloe JP, Mahadevan A, Razdan RK, Selley DE, Wiley JL. (2008) N-arachidonyl maleimide potentiates the pharmacological and biochemical effects of the endocannabinoid 2-arachidonylglycerol through inhibition of monoacylglycerol lipase. J Pharmacol Exp Ther 327:546–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busquets-Garcia A, Puighermanal E, Pastor A, de la Torre R, Maldonado R, Ozaita A. (2011) Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol Psychiatry 70:479–486 [DOI] [PubMed] [Google Scholar]
- Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F. (2008) CB2 receptors in the brain: role in central immune function. Br J Pharmacol 153:240–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. (1996) Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci 16:3934–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillé S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH. (2007) Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci 27:3695–3702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli A, Coccurello R, Rapino C, Di Serio S, Di Tommaso M, Vertechy M, Vacca V, Battista N, Pavone F, Maccarrone M, et al. (2012) The novel reversible fatty acid amide hydrolase inhibitor ST4070 increases endocannabinoid brain levels and counteracts neuropathic pain in different animal models. J Pharmacol Exp Ther 342:188–195 [DOI] [PubMed] [Google Scholar]
- Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu P, Piesla MJ, Zhang M-Y, Bingham B, Uveges A, et al. (2010) Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol Pharmacol 78:996–1003 [DOI] [PubMed] [Google Scholar]
- Chang JW, Niphakis MJ, Lum KM, Cognetta AB, 3rd, Wang C, Matthews ML, Niessen S, Buczynski MW, Parsons LH, Cravatt BF. (2012) Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem Biol 19:579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Chiu CQ, Castillo PE. (2010) TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci 13:1511–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang Y-P, Teng Z, Chen C. (2012) Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep 2:1329–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang KP, Gerber AL, Sipe JC, Cravatt BF. (2004) Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: evidence for a link between defects in the endocannabinoid system and problem drug use. Hum Mol Genet 13:2113–2119 [DOI] [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, et al. (2010a) Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci 13:1265–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, et al. (2010b) Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci 13:1265–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, Sanchini S, Tarzia G, Mor M, Piomelli D. (2009) A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem 4:1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comelli F, Giagnoni G, Bettoni I, Colleoni M, Costa B. (2007) The inhibition of monoacylglycerol lipase by URB602 showed an anti-inflammatory and anti-nociceptive effect in a murine model of acute inflammation. Br J Pharmacol 152:787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. (2001) Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA 98:9371–9376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87 [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Prospero-Garcia O, Siuzdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. (1995) Chemical characterization of a family of brain lipids that induce sleep. Science 268:1506–1509 [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. (2004) Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc Natl Acad Sci USA 101:10821–10826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Mellor SK, Ossenkopp K-P, Piomelli D, Parker LA. (2007) Effects of the FAAH inhibitor, URB597, and anandamide on lithium-induced taste reactivity responses: a measure of nausea in the rat. Psychopharmacology (Berl) 190:135–143 [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Di Marzo V. (2010) Non-CB1, non-CB2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: Focus on G-protein-coupled receptors and transient receptor potential channels. J Neuroimmune Pharmacol 5:103–121 [DOI] [PubMed] [Google Scholar]
- Desarnaud F, Cadas H, Piomelli D. (1995) Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem 270:6030–6035 [DOI] [PubMed] [Google Scholar]
- Desroches J, Guindon J, Lambert C, Beaulieu P. (2008) Modulation of the anti-nociceptive effects of 2-arachidonoyl glycerol by peripherally administered FAAH and MGL inhibitors in a neuropathic pain model. Br J Pharmacol 155:913–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch DG, Chin SA. (1993) Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 46:791–796 [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Lin S, Hill WA, Morse KL, Salehani D, Arreaza G, Omeir RL, Makriyannis A. (1997a) Fatty acid sulfonyl fluorides inhibit anandamide metabolism and bind to the cannabinoid receptor. Biochem Biophys Res Commun 231:217–221 [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A. (1997b) Methyl arachidonyl fluorophosphonate: a potent irreversible inhibitor of anandamide amidase. Biochem Pharmacol 53:255–260 [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949 [DOI] [PubMed] [Google Scholar]
- Di Marzo V. (2008) Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov 7:438–455 [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J-C, Piomelli D. (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372:686–691 [DOI] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA 99:10819–10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Kathuria S, Piomelli D. (2004) RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol 66:1260–1264 [DOI] [PubMed] [Google Scholar]
- Egertová M, Giang DK, Cravatt BF, Elphick MR. (1998) A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci 265:2081–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzili C, Mileni M, McGlinchey N, Long JZ, Kinsey SG, Hochstatter DG, Stevens RC, Lichtman AH, Cravatt BF, Bilsky EJ, et al. (2011) Reversible competitive α-ketoheterocycle inhibitors of fatty acid amide hydrolase containing additional conformational constraints in the acyl side chain: orally active, long-acting analgesics. J Med Chem 54:2805–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falenski KW, Thorpe AJ, Schlosburg JE, Cravatt BF, Abdullah RA, Smith TH, Selley DE, Lichtman AH, Sim-Selley LJ. (2010) FAAH-/- mice display differential tolerance, dependence, and cannabinoid receptor adaptation after delta 9-tetrahydrocannabinol and anandamide administration. Neuropsychopharmacology 35:1775–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, Piomelli D. (2004) Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci USA 101:8756–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feledziak M, Lambert DM, Marchand-Brynaert J, Muccioli GG. (2012) Inhibitors of the endocannabinoid-degrading enzymes, or how to increase endocannabinoid’s activity by preventing their hydrolysis. Recent Patents CNS Drug Discov 7:49–70 [DOI] [PubMed] [Google Scholar]
- Fiskerstrand T, H’mida-Ben Brahim D, Johansson S, M’zahem A, Haukanes BI, Drouot N, Zimmermann J, Cole AJ, Vedeler C, Bredrup C, et al. (2010) Mutations in ABHD12 cause the neurodegenerative disease PHARC: An inborn error of endocannabinoid metabolism. Am J Hum Genet 87:410–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fon EA, Edwards RH. (2001) Molecular mechanisms of neurotransmitter release. Muscle Nerve 24:581–601 [DOI] [PubMed] [Google Scholar]
- Fowler CJ. (2008) “The tools of the trade”—an overview of the pharmacology of the endocannabinoid system. Curr Pharm Des 14:2254–2265 [DOI] [PubMed] [Google Scholar]
- Frederickson RC, Hewes CR, Aiken JW. (1976) Correlation between the in vivo and an in vitro expression of opiate withdrawal precipitated by naloxone: their antagonism by l-(-)-delta9-tetrahydrocannabinol. J Pharmacol Exp Ther 199:375–384 [PubMed] [Google Scholar]
- Fu J, Bottegoni G, Sasso O, Bertorelli R, Rocchia W, Masetti M, Guijarro A, Lodola A, Armirotti A, Garau G, et al. (2012) A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat Neurosci 15:64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetani S, Dipasquale P, Romano A, Righetti L, Cassano T, Piomelli D, Cuomo V. (2009) The endocannabinoid system as a target for novel anxiolytic and antidepressant drugs. Int Rev Neurobiol 85:57–72 [DOI] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang M-Y, Strassle BW, Lu P, et al. (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30:2017–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni S, De Simone C, Dallavalle S, Fezza F, Nannei R, Amadio D, Minetti P, Quattrociocchi G, Caprioli A, Borsini F, et al. (2010a) Enol carbamates as inhibitors of fatty acid amide hydrolase (FAAH) endowed with high selectivity for FAAH over the other targets of the endocannabinoid system. ChemMedChem 5:357–360 [DOI] [PubMed] [Google Scholar]
- Gattinoni S, Simone CD, Dallavalle S, Fezza F, Nannei R, Battista N, Minetti P, Quattrociocchi G, Caprioli A, Borsini F, et al. (2010b) A new group of oxime carbamates as reversible inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett 20:4406–4411 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Wise LE, Chen Y, Gujjar R, Mahadevan A, Cravatt BF, Lichtman AH. (2012) The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, et al. (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA 102:18620–18625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski G, Alapafuja SO, Bátkai S, Nikas SP, Cinar R, Offertáler L, Osei-Hyiaman D, Liu J, Mukhopadhyay B, Harvey-White J, et al. (2010) Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem Biol 17:1256–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes FV, Casarotto PC, Resstel LBM, Guimarães FS. (2011) Facilitation of CB1 receptor-mediated neurotransmission decreases marble burying behavior in mice. Prog Neuropsychopharmacol Biol Psychiatry 35:434–438 [DOI] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. (2010) Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci 13:1519–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Desroches J, Beaulieu P. (2007) The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br J Pharmacol 150:693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Guijarro A, Piomelli D, Hohmann AG. (2011) Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol 163:1464–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Lai Y, Takacs SM, Bradshaw HB, Hohmann AG. (2013) Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: Effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacol Res 67:94–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. (2004) Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci 20:441–458 [DOI] [PubMed] [Google Scholar]
- Haney M, Ward AS, Comer SD, Foltin RW, Fischman MW. (1999) Abstinence symptoms following smoked marijuana in humans. Psychopharmacology (Berl) 141:395–404 [DOI] [PubMed] [Google Scholar]
- Hansen HS. (2010) Palmitoylethanolamide and other anandamide congeners. Proposed role in the diseased brain. Exp Neurol 224:48–55 [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin H-S, Kano M. (2005) Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron 45:257–268 [DOI] [PubMed] [Google Scholar]
- Herkenham M. (1995) Localization of cannabinoid receptors in brain and periphery, in Cannabinoid Receptors (Pertwee RG, ed) pp 145–166, Academic Press, London [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. (1990) Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA 87:1932–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ. (2000) Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat 61:3–18 [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. (1995) Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta 1257:249–256 [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, et al. (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435:1108–1112 [DOI] [PubMed] [Google Scholar]
- Howlett AC. (2005) Cannabinoid receptor signaling. Handb Exp Pharmacol 168:53–79 [DOI] [PubMed] [Google Scholar]
- Hsu K-L, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF. (2012) DAGLβ inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol 8:999–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins JP, Smart TS, Langman S, Taylor L, Young T. (2012) An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153:1837–1846 [DOI] [PubMed] [Google Scholar]
- Johnson DS, Ahn K, Kesten S, Lazerwith SE, Song Y, Morris M, Fay L, Gregory T, Stiff C, Dunbar JB, Jr, et al. (2009) Benzothiophene piperazine and piperidine urea inhibitors of fatty acid amide hydrolase (FAAH). Bioorg Med Chem Lett 19:2865–2869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta MB, Smith SE, Dudley DT, et al. (2011) Discovery of PF-04457845: A highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med Chem Lett 2:91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, King AR, Redhi GH, Yasar S, Piomelli D, et al. (2008) Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiatry 64:930–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89:309–380 [DOI] [PubMed] [Google Scholar]
- Karbarz MJ, Luo L, Chang L, Tham C-S, Palmer JA, Wilson SJ, Wennerholm ML, Brown SM, Scott BP, Apodaca RL, et al. (2009) Biochemical and biological properties of 4-(3-phenyl-[1,2,4] thiadiazol-5-yl)-piperazine-1-carboxylic acid phenylamide, a mechanism-based inhibitor of fatty acid amide hydrolase. Anesth Analg 108:316–329 [DOI] [PubMed] [Google Scholar]
- Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. (1997) cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem 272:27218–27223 [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, et al. (2003) Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9:76–81 [DOI] [PubMed] [Google Scholar]
- Keith JM, Apodaca R, Xiao W, Seierstad M, Pattabiraman K, Wu J, Webb M, Karbarz MJ, Brown S, Wilson S, et al. (2008) Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett 18:4838–4843 [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Chandiramani A, Harding-Rose C, Simone DA, Seybold VS. (2011) Increasing 2-arachidonoyl glycerol signaling in the periphery attenuates mechanical hyperalgesia in a model of bone cancer pain. Pharmacol Res 64:60–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Khasabov SG, Harding-Rose C, Coicou LG, Seybold BA, Lindberg AE, Steevens CD, Simone DA, Seybold VS. (2008) A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. J Neurosci 28:11141–11152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. (2010) Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci 13:592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, Cravatt BF, Lichtman AH. (2010) Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. J Pain 11:1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. (2009) Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther 330:902–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, O’Neal ST, Long JZ, Cravatt BF, Lichtman AH. (2011) Inhibition of endocannabinoid catabolic enzymes elicits anxiolytic-like effects in the marble burying assay. Pharmacol Biochem Behav 98:21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan NM, Mechoulam R. (2007) Cannabinoids in health and disease. Dialogues Clin Neurosci 9:413–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutek B, Prestwich GD, Howlett AC, Chin SA, Salehani D, Akhavan N, Deutsch DG. (1994) Inhibitors of arachidonoyl ethanolamide hydrolysis. J Biol Chem 269:22937–22940 [PubMed] [Google Scholar]
- Kozak KR, Marnett LJ. (2002) Oxidative metabolism of endocannabinoids. Prostaglandins Leukot Essent Fatty Acids 66:211–220 [DOI] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Böhme GA, Imperato A, Pedrazzini T, Roques BP, et al. (1999) Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283:401–404 [DOI] [PubMed] [Google Scholar]
- Leung DD, Saghatelian AA, Simon GMGM, Cravatt BFBF. (2006) Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry 45:4720–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GL, Winter H, Arends R, Jay GW, Le V, Young T, Huggins JP. (2012) Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br J Clin Pharmacol 73:706–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Cook SA, Martin BR. (1996) Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J Pharmacol Exp Ther 276:585–593 [PubMed] [Google Scholar]
- Lichtman AH, Hawkins EG, Griffin G, Cravatt BF. (2002) Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J Pharmacol Exp Ther 302:73–79 [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. (2004) Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther 311:441–448 [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Martin BR. (2002) Marijuana withdrawal syndrome in the animal model. J Clin Pharmacol 42(11, Suppl)20S–27S [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Martin BR. (2005) Cannabinoid tolerance and dependence. Handb Exp Pharmacol 168:691–717 [DOI] [PubMed] [Google Scholar]
- Little PJ, Compton DR, Johnson MR, Melvin LS, Martin BR. (1988) Pharmacology and stereoselectivity of structurally novel cannabinoids in mice. J Pharmacol Exp Ther 247:1046–1051 [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Huang BX, Kim H-Y, Luquet S, Palmiter RD, Krystal G, Rai R, Mahadevan A, et al. (2008) Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 54:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim H-Y, et al. (2006) A biosynthetic pathway for anandamide. Proc Natl Acad Sci USA 103:13345–13350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Jin X, Adibekian A, Li W, Cravatt BF. (2010) Characterization of tunable piperidine and piperazine carbamates as inhibitors of endocannabinoid hydrolases. J Med Chem 53:1830–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, et al. (2009a) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5:37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, LaCava M, Jin X, Cravatt BF. (2011) An anatomical and temporal portrait of physiological substrates for fatty acid amide hydrolase. J Lipid Res 52:337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Cravatt BF. (2009b) Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol 16:744–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, et al. (2009c) Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci USA 106:20270–20275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk T, Jin W, Zvonok A, Lu D, Lin X-Z, Chavkin C, Makriyannis A, Mackie K. (2004) Identification of a potent and highly efficacious, yet slowly desensitizing CB1 cannabinoid receptor agonist. Br J Pharmacol 142:495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M. (2005) Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cbeta4 signaling cascade in the cerebellum. J Neurosci 25:6826–6835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makara JK, Mor M, Fegley D, Szabó SI, Kathuria S, Astarita G, Duranti A, Tontini A, Tarzia G, Rivara S, et al. (2005) Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci 8:1139–1141 [DOI] [PubMed] [Google Scholar]
- Maldonado R, Berrendero F, Ozaita A, Robledo P. (2011) Neurochemical basis of cannabis addiction. Neuroscience 181:1–17 [DOI] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS-J, Woodruff G, et al. (2010) The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci 13:951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564 [DOI] [PubMed] [Google Scholar]
- Maurelli S, Bisogno T, De Petrocellis L, Di Luccia A, Marino G, Di Marzo V. (1995) Two novel classes of neuroactive fatty acid amides are substrates for mouse neuroblastoma ‘anandamide amidohydrolase’. FEBS Lett 377:82–86 [DOI] [PubMed] [Google Scholar]
- McKinney MK, Cravatt BF. (2005) Structure and function of fatty acid amide hydrolase. Annu Rev Biochem 74:411–432 [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90 [DOI] [PubMed] [Google Scholar]
- Meyers MJ, Long SA, Pelc MJ, Wang JL, Bowen SJ, Schweitzer BA, Wilcox MV, McDonald J, Smith SE, Foltin S, et al. (2011) Discovery of novel spirocyclic inhibitors of fatty acid amide hydrolase (FAAH). Part 2. Discovery of 7-azaspiro[3.5]nonane urea PF-04862853, an orally efficacious inhibitor of fatty acid amide hydrolase (FAAH) for pain. Bioorg Med Chem Lett 21:6545–6553 [DOI] [PubMed] [Google Scholar]
- Min R, Di Marzo V, Mansvelder HD. (2010) DAG lipase involvement in depolarization-induced suppression of inhibition: does endocannabinoid biosynthesis always meet the demand? Neuroscientist 16:608–613 [DOI] [PubMed] [Google Scholar]
- Minkkilä A, Saario SM, Nevalainen T. (2010) Discovery and development of endocannabinoid-hydrolyzing enzyme inhibitors. Curr Top Med Chem 10:828–858 [DOI] [PubMed] [Google Scholar]
- Moore SA, Nomikos GG, Dickason-Chesterfield AK, Schober DA, Schaus JM, Ying BP, Xu YC, Phebus L, Simmons RM, Li D, et al. (2005) Identification of a high-affinity binding site involved in the transport of endocannabinoids. Proc Natl Acad Sci USA 102:17852–17857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira FA, Kaiser N, Monory K, Lutz B. (2008) Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology 54:141–150 [DOI] [PubMed] [Google Scholar]
- Moreira FA, Lutz B. (2008) The endocannabinoid system: emotion, learning and addiction. Addict Biol 13:196–212 [DOI] [PubMed] [Google Scholar]
- Moreno-Sanz G, Sasso O, Guijarro A, Oluyemi O, Bertorelli R, Reggiani A, Piomelli D. (2012) Pharmacological characterization of the peripheral FAAH inhibitor URB937 in female rodents: interaction with the Abcg2 transporter in the blood-placenta barrier. Br J Pharmacol 167:1620–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli GG, Xu C, Odah E, Cudaback E, Cisneros JA, Lambert DM, López Rodríguez ML, Bajjalieh S, Stella N. (2007) Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J Neurosci 27:2883–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65 [DOI] [PubMed] [Google Scholar]
- Natarajan V, Schmid PC, Reddy PV, Zuzarte-Augustin ML, Schmid HHO. (1983) Biosynthesis of N-acylethanolamine phospholipids by dog brain preparations. J Neurochem 41:1303–1312 [DOI] [PubMed] [Google Scholar]
- Niphakis MJ, Johnson DS, Ballard TE, Stiff C, Cravatt BF. (2012) O-hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem Neurosci 3:418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Blankman JL, Simon GM, Fujioka K, Issa RS, Ward AM, Cravatt BF, Casida JE. (2008a) Activation of the endocannabinoid system by organophosphorus nerve agents. Nat Chem Biol 4:373–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Hudak CSS, Ward AM, Burston JJ, Issa RS, Fisher KJ, Abood ME, Wiley JL, Lichtman AH, Casida JE. (2008b) Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorg Med Chem Lett 18:5875–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, Hoover HH, Cravatt BF. (2011a) Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem Biol 18:846–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng S-W, Cravatt BF. (2010) Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MCG, Ward AM, Hahn YK, Lichtman AH, Conti B, et al. (2011b) Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334:809–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279:5298–5305 [DOI] [PubMed] [Google Scholar]
- Onaivi ES. (2006) Neuropsychobiological evidence for the functional presence and expression of cannabinoid CB2 receptors in the brain. Neuropsychobiology 54:231–246 [DOI] [PubMed] [Google Scholar]
- Ortar G, Cascio MG, Moriello AS, Camalli M, Morera E, Nalli M, Di Marzo V. (2008) Carbamoyl tetrazoles as inhibitors of endocannabinoid inactivation: a critical revisitation. Eur J Med Chem 43:62–72 [DOI] [PubMed] [Google Scholar]
- Osei-Hyiaman D, Depetrillo M, Harvey-White J, Bannon AW, Cravatt BF, Kuhar MJ, Mackie K, Palkovits M, Kunos G. (2005a) Cocaine- and amphetamine-related transcript is involved in the orexigenic effect of endogenous anandamide. Neuroendocrinology 81:273–282 [DOI] [PubMed] [Google Scholar]
- Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Bátkai S, Harvey-White J, Mackie K, Offertáler L, Wang L, et al. (2005b) Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest 115:1298–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong WI, Bátkai S, Marsicano G, Lutz B, Buettner C, et al. (2008) Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest 118:3160–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otrubova K, Boger DL. (2012) α-Ketoheterocycle-based inhibitors of fatty acid amide hydrolase (FAAH). ACS Chem Neurosci 3:340–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otrubova K, Ezzili C, Boger DL. (2011) The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH). Bioorg Med Chem Lett 21:4674–4685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, Kunos G. (2006) The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 58:389–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. (2009) Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) Enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther 331:591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker LA, Limebeer CL, Rock EM, Litt DL, Kwiatkowska M, Piomelli D. (2009) The FAAH inhibitor URB-597 interferes with cisplatin- and nicotine-induced vomiting in the Suncus murinus (house musk shrew). Physiol Behav 97:121–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker LA, Rock EM, Limebeer CL. (2011) Regulation of nausea and vomiting by cannabinoids. Br J Pharmacol 163:1411–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Cravatt BF. (1999) Fatty acid amide hydrolase competitively degrades bioactive amides and esters through a nonconventional catalytic mechanism. Biochemistry 38:14125–14130 [DOI] [PubMed] [Google Scholar]
- Pertwee RG. (2009) Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol 156:397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SPH, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, et al. (2010) International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB₁ and CB₂. Pharmacol Rev 62:588–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Di Marzo V. (2010) FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Curr Opin Investig Drugs 11:51–62 [PubMed] [Google Scholar]
- Petrosino S, Ligresti A, Di Marzo V. (2009) Endocannabinoid chemical biology: a tool for the development of novel therapies. Curr Opin Chem Biol 13:309–320 [DOI] [PubMed] [Google Scholar]
- Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM, Schwartz JW, Nomura DK, Samad TA. (2012) A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep 1:617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ. (2011) Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci 14:1542–1547 [DOI] [PubMed] [Google Scholar]
- Rahn EJ, Hohmann AG. (2009) Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics 6:713–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh D, Ross GR, Schlosburg JE, Owens RA, Abdullah RA, Kinsey SG, Long JZ, Nomura DK, Sim-Selley LJ, Cravatt BF, et al. (2011) Blockade of endocannabinoid hydrolytic enzymes attenuates precipitated opioid withdrawal symptoms in mice. J Pharmacol Exp Ther 339:173–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Mechoulam R, Piomelli D, Parker LA. (2008) The effect of cannabidiol and URB597 on conditioned gaping (a model of nausea) elicited by a lithium-paired context in the rat. Psychopharmacology (Berl) 196:389–395 [DOI] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. (2002) A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl 41:2596–2599 [DOI] [PubMed] [Google Scholar]
- Russo EB, Guy GW, Robson PJ. (2007) Cannabis, pain, and sleep: lessons from therapeutic clinical trials of Sativex, a cannabis-based medicine. Chem Biodivers 4:1729–1743 [DOI] [PubMed] [Google Scholar]
- Saario SM, Salo OMH, Nevalainen T, Poso A, Laitinen JT, Järvinen T, Niemi R. (2005) Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem Biol 12:649–656 [DOI] [PubMed] [Google Scholar]
- Saario SM, Savinainen JR, Laitinen JT, Järvinen T, Niemi R. (2004) Monoglyceride lipase-like enzymatic activity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem Pharmacol 67:1381–1387 [DOI] [PubMed] [Google Scholar]
- Sagar DR, Kendall DA, Chapman V. (2008) Inhibition of fatty acid amide hydrolase produces PPAR-α-mediated analgesia in a rat model of inflammatory pain. Br J Pharmacol 155:1297–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghatelian A, McKinney MK, Bandell M, Patapoutian A, Cravatt BF. (2006) A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry 45:9007–9015 [DOI] [PubMed] [Google Scholar]
- Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. (2004) Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry 43:14332–14339 [DOI] [PubMed] [Google Scholar]
- Sakurada T, Noma A. (1981) Subcellular localization and some properties of monoacylglycerol lipase in rat adipocytes. J Biochem 90:1413–1419 [DOI] [PubMed] [Google Scholar]
- Sasso O, Bertorelli R, Bandiera T, Scarpelli R, Colombano G, Armirotti A, Moreno-Sanz G, Reggiani A, Piomelli D. (2012) Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res 65:553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, et al. (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13:1113–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Carlson BL, Ramesh D, Abdullah RA, Long JZ, Cravatt BF, Lichtman AH. (2009a) Inhibitors of endocannabinoid-metabolizing enzymes reduce precipitated withdrawal responses in THC-dependent mice. AAPS J 11:342–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Kinsey SG, Lichtman AH. (2009b) Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J 11:39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid PC, Zuzarte-Augustin ML, Schmid HH. (1985) Properties of rat liver N-acylethanolamine amidohydrolase. J Biol Chem 260:14145–14149 [PubMed] [Google Scholar]
- Schofield PR, Shivers BD, Seeburg PH. (1990) The role of receptor subtype diversity in the CNS. Trends Neurosci 13:8–11 [DOI] [PubMed] [Google Scholar]
- Sciolino NR, Zhou W, Hohmann AG. (2011) Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol Res 64:226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD, eds, ed (1999) Basic neurochemistry, Lippincott-Raven, Philadelphia [Google Scholar]
- Simon GM, Cravatt BF. (2006) Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J Biol Chem 281:26465–26472 [DOI] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. (2008) Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J Biol Chem 283:9341–9349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. (2010a) Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J Biol Chem 285:11051–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. (2010b) Characterization of mice lacking candidate N-acyl ethanolamine biosynthetic enzymes provides evidence for multiple pathways that contribute to endocannabinoid production in vivo. Mol Biosyst 6:1411–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe JC, Chiang K, Gerber AL, Beutler E, Cravatt BF. (2002) A missense mutation in human fatty acid amide hydrolase associated with problem drug use. Proc Natl Acad Sci USA 99:8394–8399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe JC, Waalen J, Gerber A, Beutler E. (2005) Overweight and obesity associated with a missense polymorphism in fatty acid amide hydrolase (FAAH). Int J Obes (Lond) 29:755–759 [DOI] [PubMed] [Google Scholar]
- Sit SY, Conway C, Bertekap R, Xie K, Bourin C, Burris K, Deng H. (2007) Novel inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett 17:3287–3291 [DOI] [PubMed] [Google Scholar]
- Sit SY, Conway CM, Xie K, Bertekap R, Bourin C, Burris KD. (2010) Oxime carbamate—discovery of a series of novel FAAH inhibitors. Bioorg Med Chem Lett 20:1272–1277 [DOI] [PubMed] [Google Scholar]
- Sit S-Y, and Xie K (2002), inventors; Bristol-Myers Squibb, assignee. Bisarylimidazolyl fatty acid amide hydrolase inhibitors. World Patent WO 2002/087569. 2002 Nov 7.
- Sit S-Y, Xie K, and Deng H (2003), inventors; Bristol-Myers Squibb, assignee. (Oxime)carbamoyl fatty acid amide hydrolase inhibitors. World Patent WO 2003/065989. 2003 Aug 14.
- Smith PB, Compton DR, Welch SP, Razdan RK, Mechoulam R, Martin BR. (1994) The pharmacological activity of anandamide, a putative endogenous cannabinoid, in mice. J Pharmacol Exp Ther 270:219–227 [PubMed] [Google Scholar]
- Speers AE, Adam GC, Cravatt BF. (2003) Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc 125:4686–4687 [DOI] [PubMed] [Google Scholar]
- Speers AE, Cravatt BF. (2004) Profiling enzyme activities in vivo using click chemistry methods. Chem Biol 11:535–546 [DOI] [PubMed] [Google Scholar]
- Spradley JM, Guindon J, Hohmann AG. (2010) Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacol Res 62:249–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson FA, Hawkins LM. (2001) Neurotransmitter receptors in the postsynaptic neuron. eLS, John Wiley & Sons, Ltd., Hoboken, NJ. [Google Scholar]
- Sticht MA, Long JZ, Rock EM, Limebeer CL, Mechoulam R, Cravatt BF, Parker LA. (2011) The MAGL inhibitor, JZL184, attenuates LiCl-induced vomiting in the suncus murinus and 2AG attenuates LiCl-induced nausea-like behavior in rats. Br J Pharmacol 165:2425–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, et al. (2002) Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA 99:4465–4470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. (1995) 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 215:89–97 [DOI] [PubMed] [Google Scholar]
- Sun YX, Tsuboi K, Okamoto Y, Tonai T, Murakami M, Kudo I, Ueda N. (2004) Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase D. Biochem J 380:749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y-X, Tsuboi K, Zhao L-Y, Okamoto Y, Lambert DM, Ueda N. (2005) Involvement of N-acylethanolamine-hydrolyzing acid amidase in the degradation of anandamide and other N-acylethanolamines in macrophages. Biochim Biophys Acta 1736:211–220 [DOI] [PubMed] [Google Scholar]
- Tam J, Vemuri VK, Liu J, Bátkai S, Mukhopadhyay B, Godlewski G, Osei-Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, et al. (2010) Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest 120:2953–2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, et al. (2010) The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase α mediates retrograde suppression of synaptic transmission. Neuron 65:320–327 [DOI] [PubMed] [Google Scholar]
- Taschler U, Radner FPW, Heier C, Schreiber R, Schweiger M, Schoiswohl G, Preiss-Landl K, Jaeger D, Reiter B, Koefeler HC, et al. (2011) Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J Biol Chem 286:17467–17477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchantchou F, Zhang Y. (2012) Selective inhibition of α/β-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown and improves functional recovery in a mouse model of traumatic brain injury. J Neurotrauma, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornøe CW, Christensen C, Meldal M. (2002) Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 67:3057–3064 [DOI] [PubMed] [Google Scholar]
- Tornqvist H, Belfrage P. (1976) Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. J Biol Chem 251:813–819 [PubMed] [Google Scholar]
- Touriño C, Oveisi F, Lockney J, Piomelli D, Maldonado R. (2010) FAAH deficiency promotes energy storage and enhances the motivation for food. Int J Obes (Lond) 34:557–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramèr MR, Carroll D, Campbell FA, Reynolds DJM, Moore RA, McQuay HJ. (2001) Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ 323:16–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM. (1998) Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393–411 [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Sun Y-X, Okamoto Y, Araki N, Tonai T, Ueda N. (2005) Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem 280:11082–11092 [DOI] [PubMed] [Google Scholar]
- Ueda N, Kurahashi Y, Yamamoto S, Tokunaga T. (1995) Partial purification and characterization of the porcine brain enzyme hydrolyzing and synthesizing anandamide. J Biol Chem 270:23823–23827 [DOI] [PubMed] [Google Scholar]
- Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ. (2007) Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br J Pharmacol 150:186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. (2003) Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc 125:3192–3193 [DOI] [PubMed] [Google Scholar]
- Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. (2006) A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem 281:36569–36578 [DOI] [PubMed] [Google Scholar]
- Woodward DF, Carling RWC, Cornell CL, Fliri HG, Martos JL, Pettit SN, Liang Y, Wang JW. (2008) The pharmacology and therapeutic relevance of endocannabinoid derived cyclo-oxygenase (COX)-2 products. Pharmacol Ther 120:71–80 [DOI] [PubMed] [Google Scholar]
- Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, Lannoye GS, Richardson P, Stewart A, Rogers JC, et al. (2007) Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology 52:1095–1105 [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. (1999) Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci USA 96:5780–5785 [DOI] [PMC free article] [PubMed] [Google Scholar]