

Abstract
During vertebrate locomotion, spinal neurons act as oscillators when initiated by glutamate release from descending systems. Activation of NMDA receptors initiates Ca2+-mediated intrinsic membrane potential oscillations in central pattern generator (CPG) neurons. NMDA receptor-dependent intrinsic oscillations require Ca2+-dependent K+ (KCa2) channels for burst termination. However, the location of Ca2+ entry mediating KCa2 channel activation, and type of Ca2+ channel – which includes NMDA receptors and voltage-gated Ca2+ channels (VGCCs) – remains elusive. NMDA receptor-dependent Ca2+ entry necessitates presynaptic release of glutamate, implying a location at active synapses within dendrites, whereas VGCC-dependent Ca2+ entry is not similarly constrained. Where Ca2+ enters relative to KCa2 channels is crucial to information processing of synaptic inputs necessary to coordinate locomotion. We demonstrate that Ca2+ permeating NMDA receptors is the dominant source of Ca2+ during NMDA-dependent oscillations in lamprey spinal neurons. This Ca2+ entry is synaptically located, NMDA receptor-dependent, and sufficient to activate KCa2 channels at excitatory interneuron synapses onto other CPG neurons. Selective blockade of VGCCs reduces whole-cell Ca2+ entry but leaves membrane potential and Ca2+ oscillations unaffected. Furthermore, repetitive oscillations are prevented by fast, but not slow, Ca2+ chelation. Taken together, these results demonstrate that KCa2 channels are closely located to NMDA receptor-dependent Ca2+ entry. The close spatial relationship between NMDA receptors and KCa2 channels provides an intrinsic mechanism whereby synaptic excitation both excites and subsequently inhibits ventral horn neurons of the spinal motor system. This places the components necessary for oscillation generation, and hence locomotion, at glutamatergic synapses.
Introduction
Throughout the central nervous system oscillatory activity requires the precise temporal activation and inactivation of neurons. This may be achieved through intercellular inhibition, but also through mechanisms intrinsic to individual neurons. The intrinsic neuronal and synaptic properties that are utilized during oscillatory activity shape the behaviors neurons subserve at the network level. Rhythmic behaviors, such as locomotion, require the transformation of an excitatory command from brainstem neurons into an oscillatory output patterns of motoneuron bursting [1]. The precise rhythmic activity emerges by using both synaptic and intrinsic neuronal properties in concert: intrinsic coupling of excitatory and inhibitory conductances within neurons [2] and reciprocally inhibiting synaptic coupling between neurons [3]–[5].
The underlying properties that drive rhythmicity in neurons of the spinal CPG can directly inform how cellular information processing impacts systems level behaviors. The coupling of depolarization-induced Ca2+ entry to activation of an outward Ca2+-activated K+ channel (KCa2 [6] (formerly SK)) [7] contributes to bursting behavior of spinal motor neurons [8]–[11] and profoundly impacts dendritic integration [12], [13] and plasticity [14]–[16] by tempering excitability [7], [12], [17]. Behaviorally, KCa2 channels mediate the inactivation of depolarizing drive to an agonist muscle group. These channels regulate the frequency and stability of the neural rhythm of the CPG by causing a late after-hyperpolarization following action potentials. While this Ca2+-mediated regulation is common across vertebrates [2]–[5], [11], [18]–[23], the route of Ca2+ entry, its cellular location, and any synapse-specificity remains undetermined within the spinal motor system. Dendritic Ca2+-dependent K+ channels are activated by both VGCCs [24], [25] and NMDA receptors [17], [26]–[28] in multiple cell types and brain regions. In lamprey spinal neurons, the contributing routes of Ca2+ entry include VGCCs and NMDA receptors, but not release from internal stores [29]. VGCC-mediated Ca2+ entry has been shown to activate KCa2 currents involved in the action potential late after-hyperpolarization [30], while progressive Ca2+ entry through NMDA receptors may terminate motor burst plateau potentials [8] in non-spiking conditions [2] by activation of KCa2 channels [31].
Determining the route, location, synapse and corresponding proximity of Ca2+ entry to KCa2 channels leading to the repolarization of the oscillation is important for understanding how information processing occurs in single neurons [24], [32], particularly within the context of motor control. During NMDA-evoked fictive locomotion, intracellular Ca2+ oscillations in lamprey spinal neurons occur in phase with membrane potential oscillations and ventral root bursting [2]–[5], [11], [18]–[23], [33], [34]. These Ca2+ oscillations can occur subthreshold to action potential activation and are of greatest amplitude in distal dendrites [33] where NMDA receptors may show greater activation during locomotion [30], [35]. Furthermore, spiking in the same neurons, and presumably VGCC activation, selectively increases Ca2+ in the soma, but not in the dendrites [33]. Although KCa2 currents mediated by action potential-driven VGCC activation have profound effects on locomotion [8], [11], [20], [30], [36]–[38], they may be activated in a spatiotemporally different context that does not contribute to oscillations. Thus, we hypothesize that NMDA receptor dependent Ca2+ entry underlies repolarization during membrane potential oscillations.
NMDA receptor-dependent Ca2+ entry coupled to KCa2 activation [12], [17], [26] will provide a substantially different computational outcome than if activated by VGCCs [25] due to differing cellular function, distribution, response to neuromodulation [12], [39], current generation, and kinetics [40]. VGCCs are invariably activated in response to depolarization, and may be distributed in all regions of the neuronal membrane while varying in subtype [41]–[46] and function [13], [47], [48]. In contrast, NMDA receptors can initiate depolarization and are only physiologically active where synaptically driven by released glutamate. Consequently, NMDA receptors can restrict Ca2+ entry to the specific synapse or input dendrite, acting locally as coincidence detectors, coupled to presynaptic activity. Lastly, NMDA receptor-dependent synaptic plasticity allows for the modulation of dendritic signaling. We now demonstrate that the synaptic coupling of NMDA receptor-dependent Ca2+ entry to KCa2 activation underlies oscillatory properties of ventral horn neurons in the lamprey spinal cord responsible for locomotor pattern generation.
Methods
The Lamprey Preparation
Experiments were performed on isolated spinal cords of late stage larval (voltage clamp step experiments) and recently transformed lampreys (Petromyzon marinus). The animals were anesthetized with tricaine methanesulfonate (MS-222; 100 mg/l; Sigma, St. Louis, MO), decapitated, and dissected in cold saline solution of the following composition (in mM): 130 NaCl, 2.1 KCl, 2.6 CaCl2, 1.8 MgCl2 or 1.8 MgSO4, 4 glucose, 5 HEPES, adjusted to a pH of 7.60 with NaOH and to a final osmolarity of 270±5 mOsm. The spinal cord was isolated, removed from the protective meninx primitiva and placed in a cooled, small-volume chamber with a sylgard (Dow Corning, Midland, MI) floor that is inserted onto the stage of an upright microscope. At minimum, a 15-segment section of spinal cord was pinned to the sylgard. In experiments involving whole-cell patch recording, a 10–20 µm slice of tissue was removed from the surface of the spinal cord over the ventral horn using a vibratome tissue slicer. Patch pipettes were then readily introduced to the cut ventral surface [49].The recording chamber was continually superfused with cold (8–10°C), oxygenated solution. Solutions of pharmacological agents were bath-applied at a perfusion rate of ∼1 ml/min. TTX, AP5, NBQX, UCL 1684, BAPTA and apamin were purchased from Tocris (Bristol, UK), ω-conotoxin MVIIC was purchased from Alomone labs (Jerusalem, Israel), and all other chemicals were purchased from Sigma (St. Louis, MO). UCL1684 was dissolved in DMSO prior to a 1000-fold dilution (at minimum) in recording solution, and all other drugs were diluted in water to the appropriate stock concentration before achieving final concentration in the perfusion solution. For peptide toxins, bovine serum albumin was added to the solution to a final concentration of 0.1% w/v to prevent binding to the perfusion tubing and facilitate drug delivery to the tissue. During experiments involving NMDA receptor activation, glycine was added as co-agonist for the NMDA receptor to compensate for washout, while strychnine was used to prevent glycine receptor activation.
Electrophysiology
Ventral horn neurons (motoneurons or interneurons) were recorded in whole-cell voltage clamp configuration, blind to cell identity. The patch pipette solution contained (in mM): 102.5 cesium or potassium methane sulfonate, 1 NaCl, 1 MgCl2, 5 EGTA or 9.5 BAPTA, 5 HEPES, 0.3 GTP and 0.3 ATP, pH adjusted to 7.2 with CsOH (for voltage clamp experiments) or KOH (for oscillation and evoked EPSC experiments) to a final osmolarity of 250±2 mOsm. In voltage clamp experiments, 1 mM phosphocreatine (Sigma; St. Louis, MO) was added to the patch solution to prevent rundown of VGCCs. Cell types were identified by their location in the tissue and the electrode positioned on individual neurons under visual guidance. Pipettes had open-tip resistances of 5–10 MΩ. Series resistance compensation was performed for voltage step experiments and was monitored continuously by giving a 10 mV voltage step before each episode. Cells were discarded if the series resistance changed by more than 10%. Voltage steps were given at 5 s intervals to minimize current rundown. After whole-cell access was achieved, 10 min was allowed for diffusion of Ca2+ buffers from the pipette (unless stated otherwise) or replacement of Ca2+ with Ba2+ in the extracellular solution to allow currents to reach maximum amplitude before drug application. Due to the large size of ventral horn neurons in recently transformed lampreys, neurons from larval lampreys were used to minimize space clamp error generated from voltage steps. As an exception, transformer neurons were used for all experiments where membrane NMDA receptor-dependent membrane potential oscillations were induced in current clamp prior to switching to voltage clamp to perform step protocols. In adult transformer lampreys, paired recordings were made between presynaptic reticulospinal (RS) axons recorded with sharp microelectrodes and postsynaptic spinal neurons recorded under whole-cell voltage clamp in Mg2+-free ringer (to circumvent NMDA receptor block) while clamping between −70 mV and −60 mV. Sharp microelectrode were made conventionally with thin-walled glass with tip resistances of 20–50 MΩ. The pipette solution for recording and stimulating RS axons was 3 M KCl unless otherwise stated. Action potentials were evoked in the presynaptic axons at 15 s intervals to prevent postsynaptic current rundown. Drugs were only added after a stable baseline was achieved (10–15 min).
Extracellular Stimulation
Selective, extracellular excitatory interneuron (EIN) stimulation was achieved using custom made tungsten stimulating electrodes. Tungsten wire (A-M Systems, Inc., Sequim, WA) was electrochemically etched to make fine tips, inserted into a glass capillary tube and then secured in place by glue. The glass was then pulled using an electrode puller to expose the tips and sylgard was used for added insulation, only exposing the very tip of the wire. EPSCs were evoked using an isolated stimulator (A360; World Precision Instruments, Sarasota, FL) to deliver low intensity (5–10 µA), highly localized stimulation, 1–2 segments rostral and ipsilateral to the whole-cell voltage clamped cell. Strychnine was simultaneously applied to prevent glycinergic inhibition from coincident stimulation of inhibitory interneurons. Selective EIN excitation and reticulospinal axon avoidance was achieved through several factors: (1) reticulospinal axons have a very high membrane capacitance and require substantial focal current injection to induce an action potential. This means that the axons require more charge than the tungsten electrode delivers at low stimulation intensity, (2) extracellular stimulation is insufficient to cause spikes in reticulospinal axons recorded with a microelectrode, and (3) the majority of reticulospinal axons passing over cell somata are removed from the slice preparation, while remaining medial or lateral axons can be visually avoided. Stimuli were given at 15 s intervals to prevent postsynaptic current rundown and drugs were only added after a stable baseline was achieved (10–15 min).
Selective Retrograde Labeling of Motoneurons from the Muscle Wall
Recently transformed lampreys were anesthetized. The dextran-conjugated Ca2+-sensitive dye, 10,000 MW Oregon Green 488 BAPTA-1 dextran (Life Technologies (Invitrogen), Grand Island, NY), was injected into the body musculature (5 mM, 5 µl) to allow uptake into motoneuron axon terminals. The animal was then allowed to recover in a cooled aquarium, allowing the dye time to be retrogradely transported into somata and dendrites. After 48 h, the animal was decapitated under anesthesia, and the spinal cord adjacent to the injection site was removed. Imaging revealed Ca2+-sensitive dye labeling selectively in motoneurons of corresponding hemisegments.
Imaging
Recently transformed lampreys were used for all imaging experiments. Confocal imaging was performed using a modified Bio-Rad (Hercules, CA) MRC 600 confocal microscope. Two laser excitation wavelengths were used (488 nm argon ion and 568 nm krypton–argon) through an acousto-optic tunable filter-coupled fiber optic launch (Prairie Technologies, Madison, WI). Excitation was applied through a custom dichroic mirror with sharp excitation bands matching the two laser wavelengths (Omega Optical, Brattleboro, VT). Two detectors were placed after a second dichroic, with a transmission band from 500–560 nm and long-pass reflection from 580 nm. Emission filters were bandpass (500–560 nm) and long pass (above 580 nm). The photomultiplier outputs were amplified with low-noise current amplifiers (Stanford Research Systems, Sunnyvale, CA) and digitized to 12 bits with a National Instruments (Austin, TX) board and custom software written under Matlab (Mathworks; Natick, MA). The scan-head mirrors were driven though the MRC 600 scan-head amplifiers with the same custom software, available on our website (http://alford.bios.uic.edu/Research/software.html).
Simultaneous fluorescence imaging combined with electrophysiological recordings were performed with a CCD system (ORCA; Hamamatsu, Hamamatsu City, Japan) mounted onto a compound microscope (Olympus BX50WI; Olympus, Tokyo, Japan) equipped with a rapidly switchable Xenon source (Sutter Lambda DG4, Sutter Instrument Company, Novato, CA) and emission filter wheel (Sutter Lambda 10–2, Sutter Instrument Company, Novato, CA). A 490 nm or 560 nm bandpass excitation filter and 510 nm long pass or 645 nm bandpass emission filter was used for 488 nm Ca2+-sensitive dyes, OGB1 and Fluo-5F, or a 568 nm inert dye, respectively. All dyes were purchased from Life Technologies (Invitrogen). Acquisition was performed using µManager software [50], binning images at 1×1, 2×2, and 4×4 as needed.
Data Analysis
Electrophysiological analysis was performed offline using Axograph X software (Sydney, AU). Imaging data analysis was performed using ImageJ (NIH; Bethesda, MD). For analysis of Oregon Green 488 BAPTA-1 and Fluo-5F, fluorescence intensities after background subtraction were normalized to the baseline (prestimulus), giving a baseline value of ΔF/F = 0. Baseline was defined as the fluorescence at the location analyzed prior to electrical stimulation. For oscillation imaging experiments, this was defined as the fluorescence prior to NMDA application. For figures displaying the location of changes in fluorescence, the entire image sequence was divided by the average of the prestimulus images. This generated a relative fluorescence sequence, the peak of which was averaged and then contrast- and brightness-adjusted to display a black background. For all experiments, cells were discarded if bleaching or response amplitude rundown occurred for the entirety of the control condition. Data are given as means ± SEM and n = number of cells. Student’s paired, one-tailed t test was used to calculate the significance between control and drug conditions.
Ethics Statement
All experiments on animals were performed in accordance with Institutional and national guidelines as laid down by AALAC and under an approved University of Illinois at Chicago Animal Care Committee protocol issued to SA (Permit #: 12–023). All surgery was performed under MS-222 anesthesia, and all efforts were made to minimize suffering.
Results
BAPTA Disrupts NMDA-dependent TTX-resistant Membrane Potential Oscillations
NMDA-dependent, TTX-resistant oscillations are Ca2+-dependent [2], as Ca2+ is necessary for the repolarization of the cell via KCa2 channel activation [8]. However, the route of Ca2+ entry is unclear. We hypothesize that oscillations are driven through local, NMDA receptor-dependent synaptic activity where Ca2+ entry is coupled to KCa2 channels. To determine if the site of Ca2+ entry responsible for repolarization is closely apposed to KCa2 channels, we used whole-cell recordings to dialyze neurons with either BAPTA or EGTA, fast and slow Ca2+ buffers, respectively [51], [52]. After 30 min of diffusion following whole-cell access, NMDA (100 µM) and TTX (0.5 µM) were superfused over the spinal cord, depolarizing the cell, which eventually reached a stable plateau (∼ 2 min). Cells were monitored in the depolarized state for several minutes before injecting the minimal amount of negative current necessary to cause a fast hyperpolarization. Cells recorded with EGTA in the patch solution would then autonomously depolarize and subsequently exhibit repetitive membrane potential oscillations (Fig. 1Ai). In contrast, BAPTA (5 mM) within the patch pipette prevents membrane repolarization, abolishing oscillations – cells remain trapped at a depolarized membrane potential (∼−40 mV, Fig. 1Aii; n = 3). Thus, BAPTA out-competes endogenous buffers to bind entering Ca2+ before activating KCa2 channels to repolarize the membrane. In contrast, NMDA (50 µM) invariably induced repetitive oscillations with inclusion of EGTA in the pipette, indicating that EGTA does not bind Ca2+ before it can mediate KCa2 channel-dependent repolarization (Fig. 1Ai; 5 mM, n = 3). However, immediately after whole-cell access, EGTA (0.5–5 mM) diffusion progressively prolongs oscillations (>1 min), requiring increasing levels of current injection to repolarize (n = 8, Fig. 1B). This indicates that slow Ca2+ buffering reduces Ca2+ available for KCa2 channel activation, demonstrating that KCa2 channels further away from the site of Ca2+ entry can impact the termination of the oscillation. Despite this, the striking effect of BAPTA suggests that KCa2 channels are positioned close to the site of Ca2+ entry [51], [52] – BAPTA binds Ca2+ rapidly to block KCa2 channel activation whereas EGTA, which binds more slowly, does not. BAPTA’s fast buffering effect is mimicked by blockade of KCa2 channels with the potent inhibitor, UCL 1684 (100 nM). KCa2 channel blockade significantly and reversibly prolonged the oscillation duration (43±18% increase from control; P<0.05, n = 5) and in an additional 2 cells reversibly prevented repolarization (Fig. 1C), confirming that repolarization recorded under whole-cell recording conditions requires activation of KCa2 channels which has previously been shown with apamin in neurons recorded using sharp microelectrodes [8].
Figure 1. Impairment of NMDA-dependent TTX-resistant oscillations.

Following whole-cell access, cells were dialyzed with either EGTA (5 mM) or BAPTA (5 mM) before induction of membrane potential oscillations in NMDA (50 µM), TTX (0.5 µM), glycine (1 µM), and strychnine (5 µM). A A ventral horn neuron was dialyzed for 30 minutes prior to NMDA application (black bar). After depolarization, the cell remained at a plateau for several minutes before the minimum amount of negative current necessary for repolarization was injected (bottom trace) and then held constant thereafter. Repetitive oscillations were either induced (EGTA, Ai top trace) or prevented (BAPTA, Aii middle trace). B EGTA (5 mM) perfusion immediately following whole-cell access prolongs the oscillation duration, necessitating increasing negative current injection (bottom trace) for repolarization. C In a neuron recorded in current clamp with a patch pipette containing EGTA (5 mM), block of KCa2 channels with UCL 1684 (100 nM) prevented NMDA receptor-induced oscillations, which recovered after washout.
NMDA Modifies the Current-voltage Relationship of Ventral Horn Neurons
Whether the Ca2+ responsible for repolarization during oscillations enters the cytosol through NMDA receptors or through VGCCs [34] as the cell is depolarized may be determined by examining the voltage profile of its activation. We determined the membrane potential range over which oscillatory activity occurs and the voltage activation range of channels governing Ca2+ influx during oscillations within the same neurons. NMDA (100 µM) and TTX (1 µM) were bath-applied to induce oscillations in whole-cell current clamp (Fig. 2A), determining the peak and trough membrane potentials. Despite differences in oscillation frequency, duration, and trough potential, the peak value reached during the plateau showed little variation between cells (peak = −38.4±1.4 mV, n = 34), establishing the most depolarized limits of the oscillation membrane potential range.
Figure 2. NMDA receptor-induced current entry dominates within the oscillation range.
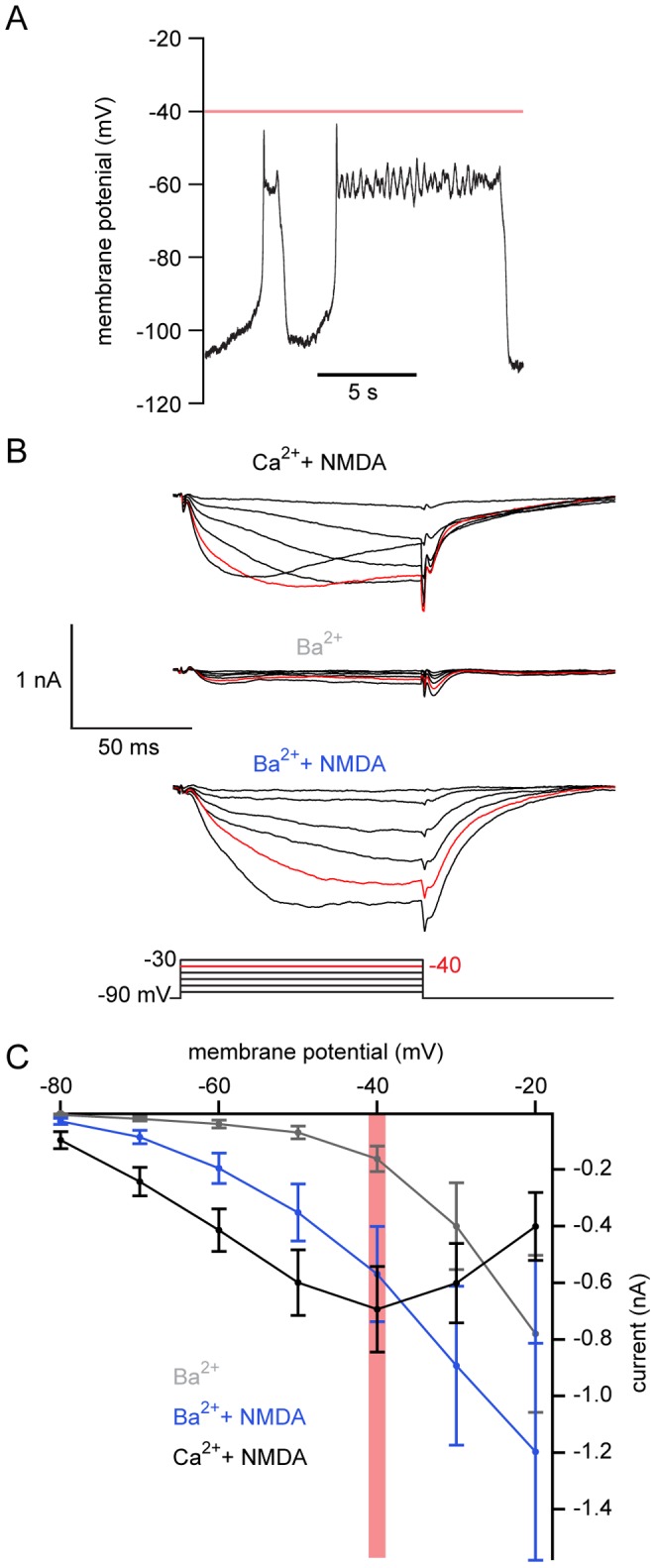
A. Oscillations were induced in whole-cell current clamp in a ventral horn neuron (100 µM NMDA, 1 µM TTX) from a recently transformed animal. The average plateau potential in this neuron was −60 mV. B The recording mode was switched to voltage clamp in the same cell as A. TEA (5 mM) and 4-AP (1 mM) were added to the solution. A step protocol (10 mV increments from a holding potential of −90 mV) shows the voltage-gated Ca2+ current in NMDA (black, 100 µM), following NMDA washout and exchange of Ca2+ (2.6 mM) for Ba2+ (grey, 2.6 mM). NMDA (100 µM) was then reapplied with Ba2+ present (blue). C Average I-V plot of voltage-gated currents for Ba2+ (grey), Ba2++NMDA (100 µM, blue) and Ca2++NMDA (100 µM, black). Red line and red traces denote −40 mV, the average peak of membrane potential oscillations. Error bars express ± SEM.
To examine the relative contribution of Ca2+ current from VGCCs or NMDA receptors over the range of membrane potentials observed during oscillations, the same neurons were then switched to voltage clamp at a holding potential of −90 mV while 4-AP (1 mM) and TEA (5 mM) were applied to block voltage-gated K+ currents. Depolarizing steps were used to activate voltage-dependent currents in 10 mV increments. Despite blocking K+ channels, the true threshold for activation and magnitude of step-evoked inward currents was not measurable with Ca2+ in the Ringer’s solution and EGTA as a Ca2+ buffer in the recording pipette. A late, secondarily activated, outward current was recorded overlaying the inward current. We hypothesized that this is due to KCa2 channel activation [2], [8], [11], [38], [53]. These currents were prevented by replacing Ca2+ (2.6 mM) with Ba2+ (1 mM) in the superfusate (Fig. 2B). In Ba2+ (Fig. 2B–C; n = 23) the threshold for VGCC activation was between −40 and −30 mV, which is similar to experiments performed in dissociated lamprey spinal neurons [8], [33], [38], [53]–[55]. In 10 out of 15 cells where oscillations and step protocols were performed in the same cell, VGCC activation thresholds were more depolarized than the average oscillation plateau potential (i.e. −38.4±1.4 mV). The remaining 5 cells activated at −40 mV (n = 4) or at −50 mV (n = 1). This indicates that most cells oscillate in a voltage range subthreshold (Fig. 2A), or just approaching (Fig. 3A) the activation threshold of VGCCs.
Figure 3. BAPTA prevents an NMDA-induced outward current.
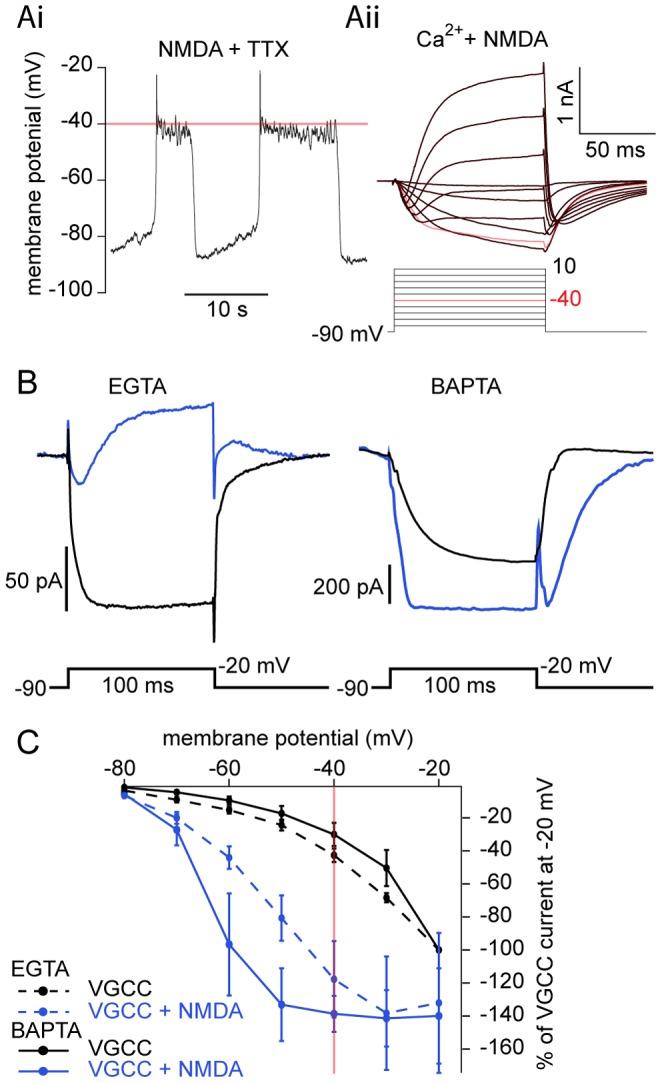
Comparison of step-evoked currents between two Ca2+ buffers. A NMDA (100 µM) and TTX (1 µM) induced oscillations in current clamp (Ai) from a recently transformed lamprey ventral horn neuron dialyzed with EGTA (5 mM). Step-evoked NMDA-receptor dependent current is shown in Aii in the same cell after switching to voltage clamp. K+ channels were blocked with bath-application of TEA (5 mM) and 4-AP (1 mM), with Cs+ in the patch pipette. With progressive depolarizing steps, NMDA-dependent inward current activation (Aii) becomes biphasic: initial inward currents lead to a subsequent late outward current during a single step. Red line and red traces denote −40 mV, the average peak of membrane potential oscillations. B Ventral horn neurons from larval lamprey were whole-cell patched and voltage clamped at −90 mV and then stepped to −20 mV in TTX (1 µM), TEA (5 mM), 4-AP (1 mM), glycine (1 µM), and strychnine (5 µM) before (black) and after the application of NMDA (50 µM, blue). Average current traces in EGTA (5 mM) demonstrate that NMDA leads to the activation of an inward Ca2+ current followed by an outward current compared to a purely inward VGCC current. In a different cell, dialysis with BAPTA (9.5 mM) abolishes outward current activation in NMDA, instead potentiating the inward current amplitude relative to VGCCs. C Average I-V relationship of cells from B, before (black) and after NMDA (50 µM, blue) application, normalized to peak VGCC current generated at the −20 mV step in EGTA (dotted line) or BAPTA (solid line). NMDA significantly potentiated voltage-dependent currents in BAPTA (−80 to −60 mV) and EGTA (−80 to −40 mV). Red line denotes −40 mV, the average peak of membrane potential oscillations. Error bars express ± SEM.
Addition of NMDA (50–100 µM) to the superfusate hyperpolarized the threshold for current activation to membrane potentials as negative as −80 mV in cells perfused in Ba2+ (Fig. 2 B–C; n = 23) or Ca2+ (Fig. 2 C; n = 14). Due to voltage-dependent unblocking of Mg2+ from the NMDA receptor [33], [34], [56], current magnitude increased as the cell was depolarized in NMDA, peaking at −20 mV in Ba2+ (Fig. 2C) compared to −40 mV in Ca2+ (Fig. 2C). NMDA significantly increased step-induced currents from −80 to −30 mV in Ba2+ (P<0.05 at all voltages, n = 23).
During NMDA-dependent TTX-resistant oscillations (Fig. 3Ai), NMDA receptor activation causes substantial voltage-dependent currents within the oscillation range of ventral horn neurons (Fig. 3Aii). Depolarization to positive potentials leads to progressive outward current activation (Fig. 3Aii). This outward current activation may be due to either Ca2+ entry via VGCCs or NMDA receptors. Thus, to further test the hypothesis that NMDA receptor-dependent current activates KCa2 channels within the oscillation range, a comparison was made between cells recorded with EGTA in the patch solution and those recorded with BAPTA. Cells were whole-cell voltage clamped with electrodes containing EGTA (5 mM) at −90 mV and depolarizing steps applied in TTX, 4AP and TEA as above. Again, addition of NMDA leads to a biphasic current in which the initial inward current is followed by activation of an outward current (Fig. 3Aii,B). Depolarizing steps within the NMDA-dependent oscillation range (<−40 mV) failed to activate an outward current without NMDA (i.e. solely VGCCs; Fig. 3B).
With no NMDA present, step-evoked VGCC currents only activated outward currents at very depolarized potentials (more depolarized than −20 mV). Addition of NMDA caused an outward current with steps to −20 mV (Fig. 3B). In contrast, this NMDA-dependent outward current activation was not present in cells dialyzed with BAPTA (9.5 mM; Fig. 3B, n = 4). NMDA receptor currents are significantly potentiated both in BAPTA (P<0.05; from −80 to −60 mV and −40 to −30 mV; n = 4) and EGTA (P<0.05, −80 to −40 mV; n = 6) relative to VGCC-generated current (Fig. 3C). Furthermore, NMDA receptor-dependent currents with BAPTA were more potentiated than with EGTA at more hyperpolarized test potentials (−70 mV to −50 mV). Thus, BAPTA prevents the activation of KCa2 channels in both NMDA-induced membrane potential oscillations in current clamp (Fig. 1Aii) and by step-evoked NMDA receptor activation in voltage clamp (n = 9; Fig. 3C). This demonstrates that NMDA receptor currents – but not VGCC-dependent currents – are sufficient to activate KCa2 channels in the membrane potential oscillation range.
NMDA Potentiates Step-evoked Ca2+ Signals
Whole-cell voltage clamp was combined with Ca2+ imaging to determine sites and magnitude of Ca2+ entry during voltage steps. Ventral horn neurons were whole-cell clamped with Ca2+-sensitive dye (either Oregon Green 488 BAPTA-1 (OGB1) 50 µM; or Fluo-5F, 100–200 µM combined with 50 µM of the inert dye, Alexa Fluor 568 hydrazide) included in the patch solution. Dye was allowed to diffuse into the cell for 30 minutes following whole-cell access and then imaged using a confocal microscope. From a holding potential of −90 mV in TTX (1 µM), 4-AP (1 mM) and TEA (5 mM), cells were stepped to membrane potentials between −70 mV and 0 mV to define the relationship between voltage-dependent currents and Ca2+ dye fluorescence (Fig. 4–5). In all dendrites imaged (n = 15 cells), voltage steps were sufficient to induce fluorescence changes. Thresholds for fluorescence responses evoked by voltage steps are indicated in Table 1. Thus, step-evoked Ca2+ signals are activated reliably between −40 and −30 mV via VGCCs, similar to step-evoked currents (Fig. 4A). The ability of OGB1 to detect fluorescence at more hyperpolarized step potentials (e.g., −50 mV, Fig. 4B–C) reflects its higher affinity for Ca2+ relative to Fluo 5F (Kd = 185 nM and 2.3 µM, respectively) and presumably intracellular Ca2+ (Ca2+ i) signals that are too small to be detected by voltage clamp current recording alone in these spatially complex neurons.
Figure 4. NMDA potentiates step-activated Ca2+ currents.

Current and fluorescence measurements made from ventral horn neurons filled with Ca2+-sensitive dyes administered via the patch pipette. A–B In whole-cell voltage clamp, a cell was filled with 200 µM Fluo 5F and 50 µM Alexa Fluor 568 hydrazide. From a holding potential of −90 mV, the cell was depolarized using varying step pulse amplitudes while simultaneously imaging a selected dendritic region using a confocal microscope. VGCC currents (Ai bottom trace, enlarged in Aii) were isolated using TTX (1 µM), TEA (5 mM), and 4-AP (1 mM) before application of NMDA (Bi bottom trace, enlarged in Bii; 20 µM). The corresponding depolarization-induced increase in Ca2+ dye fluorescence is shown (top traces, Ai–Bi) on the same time scale as bottom traces (grey box, Ai-Bi). The corresponding changes in fluorescence (displayed as peak/prestimulus fluorescence) at each step are shown below (Aiii, Biii). C Pooled step-evoked fluorescence (positive y-axis, expressed as ΔF/F+1) and current (negative y-axis, expressed as a ratio relative to the peak VGCC response at −30 mV) are compared against each voltage step (x-axis) for VGCCs before (black) and after addition of NMDA (blue, 10–100 µM) using either Fluo 5F (top, 100–200 µM) or OGB1 (bottom, 50 µM) as a Ca2+-sensitive dye. Error bars express ± SEM. D Average z-stack of the cell from A–B filled with Fluo 5F (200 µM) imaged after NMDA application. The inset (blue box) designates the region of analysis; scale bars = 10 µm.
Figure 5. Discrete regions of increased Ca2+ fluorescence emerge in NMDA.

A A cell was whole-cell voltage clamped and dialyzed with Fluo 5F (200 µM) and Alexa Fluor 568 hydrazide (50 µM) and imaged using an epifluorescence CCD system. The cell was then stepped in TTX (1 µM), TEA (5 mM), and 4-AP (1 mM) from −90 mV to −20 mV (Aii, black) to activate VGCC-dependent fluorescence (Ai, black and grey) and current (Aiii, black). After NMDA (50 µM) application, step-evoked (Aii, blue) NMDA receptor-dependent Ca2+ fluorescence (Ai, dark and light blue) and current (Aiii, blue) is present at hyperpolarized potentials (Aii, −30 mV, blue). Fluorescent measurements were taken from both left (L; Ai, B, C) and right (R; Ai, B,C) dendritic branches shown in B before (black, grey) and after (blue, light blue) NMDA application. Images showing the location of fluorescent increases taken from the prestimulus (Aiv, 1), peak (Aiv, 2), and decay (Aiv, 3) of the Ca2+ signal for VGCCs (Aiv, top row, black) and after NMDA application (Aiv, bottom row, blue) from the same cell expressed as the peak/prestimulus. B Raw, unaltered image of the cell in A showing 2 prominent dendritic branches. Scale bar = 10 µm. C Profile of Ca2+ signal at response peak along left (L, grey) and right (R, black) dendritic branches (B) from VGCC step to −20 mV (Aii). After NMDA application and step to −30 mV (Aii, blue), left (light blue) and right (dark blue) show both a greater overall fluorescent signal and discrete regions of increased fluorescence along the length of dendrite. 0 µm indicates top of image for both left and right branches.
Table 1. Percent of Cells Showing VGCC-Evoked Ca2+ Dye Fluorescence.
| Response threshold | −60 mV | −50 mV | −40 mV | −30 mV |
| Fluo-5F | 0% (0/7) | 25% (2/8) | 63% (7/10) | 100% (5/5) |
| OGB1 | 0% (0/4) | 63% (5/8) | 100% (4/4) | 100% (3/3) |
Similarly to when currents were recorded alone, NMDA (10–100 µM) lowered the threshold for step-evoked Ca2+ fluorescence and current. Fluorescent Ca2+ signals (Fig. 4B) were evoked at steps to −70 mV with both dyes, subthreshold to VGCC activation (Fig. 4C) in all neurons examined (n = 15). NMDA also substantially and significantly enhanced step-evoked fluorescence transients previously observed at membrane potentials of −50 mV or more depolarized (Fig. 4C; P<0.05, n = 8). Using an epifluorescence CCD system, Ca2+ signals were also examined in many dendritic regions simultaneously (Fig. 5). Again, NMDA led to a profound increase in both step-evoked fluorescence (Fig. 5Ai) and current magnitude (Fig. 5Aiii). Additionally, NMDA led to discrete regions of enhanced fluorescence along the length of dendrites (Fig. 5B-C). Thus, Ca2+-sensitive dyes demonstrate that NMDA receptor activation leads to greater increases in Ca2+ entry compared to VGCCs over membrane potential ranges observed during TTX-resistant membrane potential oscillations.
Effect of Cd2+ on Step-evoked VGCC and NMDA Receptor Currents
To investigate whether VGCC-dependent Ca2+ entry is necessary for KCa2-dependent repolarization during membrane potential oscillations recorded in NMDA and TTX, we pharmacologically isolated NMDA receptor-mediated currents. Cd2+ was applied as a non-specific high voltage-activated VGCC antagonist during the step protocol. Cd2+ completely blocks VGCCs at concentrations of 50–200 µM [35], [57]. However, Cd2+ (>50 µM) reduces NMDA receptor currents in cultured rat hippocampal neurons [58], [59]. Thus, to confirm that Cd2+ blocks VGCCs at 25 µM in situ, neurons were whole-cell voltage clamped (in 1 µM TTX, 1 mM 4-AP, 5 mM TEA) with BAPTA in the patch pipette (9.5 mM) to isolate VGCC currents evoked by stepping from a holding potential of −90 mV as before. Cd2+ (25 µM) significantly inhibited VGCC current (Fig. 6Ai–ii,B; inhibited to 25±8% of peak conductance of VGCC at 0 mV, P<0.05, n = 5). NMDA was then applied to the preparation following Cd2+-mediated block. This caused a significant, voltage-dependent potentiation of step-evoked currents (Fig. 6 Aiii,B; −80 to −50 mV, P<0.05, −40 to −30 mV, P<0.01; n = 5), increasing current magnitude as the cell was depolarized. The resultant current has a similar voltage activation profile to purely NMDA receptor-mediated current. Thus, Cd2+ can be used to isolate NMDA receptor-mediated currents with substantial block of VGCCs.
Figure 6. Effect of Cd2+ on step-evoked VGCC and NMDA receptor currents.

Step protocols performed in cells dialyzed with BAPTA (9.5 mM) demonstrating the effect of Cd2+ application to VGCC and NMDA receptor-induced currents. A A larval lamprey ventral horn neuron was whole-cell voltage clamped and stepped in TTX (1 µM), TEA (5 mM), and 4-AP (1 mM) from −90 mV to profile VGCC currents (displayed in 20 mV step increments). VGCC currents (Ai, black) are nearly abolished in Cd2+ (Aii, teal, 25 µM), but potentiated within the oscillation range (<−40 mV, red line) after NMDA (Aiii, grey, 50 µM) application. Red traces denote −40 mV, the average peak of membrane potential oscillations. B I–V plot of cell from A showing voltage-gated Ca2+ currents (Bi, black) in the presence of Cd2+ (Bi, teal) and NMDA+Cd2+ (Bi, grey). An I-V plot of cells pooled from A showing significant potentiation of evoked current in NMDA (Bii, grey) relative to Cd2+-depressed currents within the oscillation range, expressed relative to the peak VGCC current at 0 mV. Red line denotes −40 mV, the average peak of membrane potential oscillations. Error bars express ± SEM. C A neuron was whole-cell voltage clamped at −70 mV. Ringer’s solution containing NMDA (500 µM) was pressure-ejected (black bar) from a glass pipette (∼5 MΩ) in the approximate vicinity of the dendritic arbor. NMDA receptor EPSCs were recorded before (black), during (teal), and after (grey) the application of Cd2+ (25 µM).
To confirm that this concentration of Cd2+ did not cause an inhibition of NMDA receptor- mediated currents [59] in lamprey ventral horn neurons, NMDA (500 µM) was pressure-ejected from a pipette onto neurons recorded in whole-cell voltage clamp at −70 mV to evoke an NMDA receptor-dependent EPSC (Fig. 6C). Bath-applied Cd2+ (25 µM) had no effect on either the peak current or area under the curve (P>0.05, n = 6). At this reduced concentration, Cd2+ is thus selective for VGCCs, having little effect on NMDA receptors.
NMDA-dependent TTX-resistant Oscillations Persist after VGCC Blockade
Cd2+ was then applied to neurons in which TTX-resistant NMDA receptor-activated membrane potential oscillations were recorded [2] to determine whether VGCCs contribute to the oscillation. Ventral horn neurons were whole-cell current clamped during NMDA (50–100 µM) and TTX (1 µM) application. After dialysis with low concentrations of buffer (25 µM OGB1) and achieving stable induction of membrane potential oscillations (∼5–10 min; Fig. 7A), Cd2+ (25 µM) was applied to the superfusate. Cd2+ caused a significant and reversible reduction in the duration of the depolarized phase (duration reduced to 62±6.4% of control, P<0.001, n = 12; Fig. 7A,B). In contrast, neither the duration of the hyperpolarized trough period (P>0.05, n = 8) nor the oscillation frequency showed significant changes (P>0.05, n = 10) – both measures were variable. Thus, 25 µM Cd2+ nearly abolishes step-induced VGCC activation and reduces the duration of depolarization during NMDA receptor-activated membrane potential oscillations. VGCCs may contribute to the depolarizing phase of the oscillation, but do not activate KCa2 channels to influence repolarization.
Figure 7. Membrane potential oscillations persist after VGCC blockade.
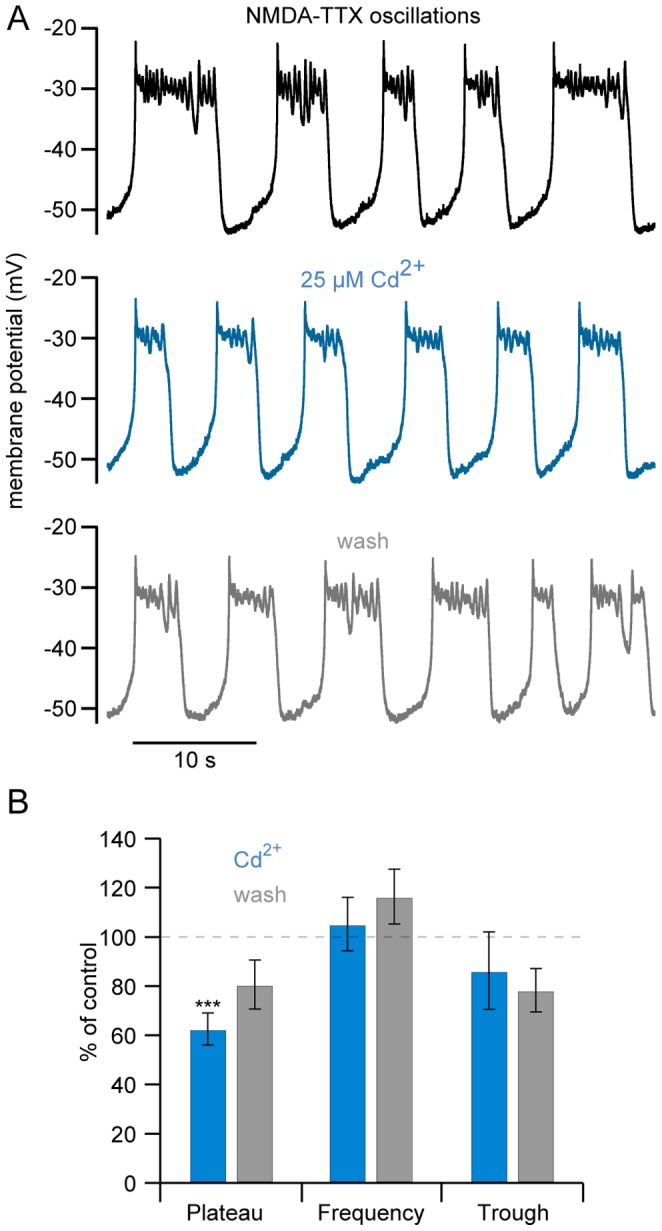
A Membrane potential oscillations were induced in a ventral horn neuron in whole-cell current clamp by application of NMDA (50 µM) and TTX (1 µM), buffered with OGB1 (25 µM). Repetitive oscillations (control, black) persisted in the presence of Cd2+ (teal; 25 µM) with a small reduction in plateau duration, partially recovering upon washout (grey). B Summary of Cd2+ effects on oscillation plateau duration, frequency, and trough period duration.
Ca2+ oscillations Persist after Selective VGCC Block
Results obtained using Cd2+ indicate that VGCCs do not contribute to repolarization during membrane potential oscillations. However, while Cd2+ blocks all VGCC subtypes, it lacks complete selectivity and interferes with Ca2+ imaging by binding Ca2+-sensitive dyes. Thus, to assess if there is also a reduction in Ca2+ entering through VGCCs during membrane potential oscillations, we examined the effect of the specific N- and P/Q-subtype VGCC blocker, ω-conotoxin MVIIC (ω-CgTxMVIIC), on NMDA-dependent TTX-resistant membrane potential and Ca2+ oscillations recorded with fluorescent dyes. ω-CgTxMVIIC (5 and 2 µM) inhibits both VGCCs in lamprey cultured ventral horn neurons [60] and the slow afterhyperpolarization in lamprey reticulospinal axons [61]. To confirm that ω-CgTxMVIIC (5 µM) blocks VGCC currents in our in vitro spinal cord preparation, we performed membrane potential step protocols as before (in 9.5 mM BAPTA, 1 µM TTX, 1 mM 4-AP, 5 mM TEA). VGCC currents were substantially inhibited to 28±13% of control (Fig. 8A; P<0.05, n = 3).
Figure 8. Ca2+ oscillations persist after selective VGCC blockade.

A A ventral horn neuron was whole-cell voltage clamped at −90 mV and stepped (10 mV increments) in TTX (1 µM), TEA (5 mM), and 4-AP (1 mM). Step-induced VGCC current (Ai, top) is blocked by ω-CgTxMVIIC (Ai, bottom; 5 µM). A pooled I-V plot shows the significant reduction of whole-cell VGCC current (Aii, solid line) after application of ω-CgTxMVIIC (Aii, dashed line, 5 µM). Red line and traces denote −40 mV, the average peak of membrane potential oscillations. Error bars express ± SEM. B A ventral horn neuron was whole-cell voltage clamped and filled with OGB1 (Biii, 25 µM) at −70 mV. In current clamp mode, application of NMDA (50 µM) and TTX (1 µM) induced simultaneous, repetitive oscillations in membrane potential (Bi) and Ca2+ dye fluorescence (Bii, black), which persisted in ω-CgTxMVIIC (Bi-ii; 5 µM). White box denotes region of interest from which measurements in Bii were taken. Red line denotes −40 mV, the average peak of membrane potential oscillations. Scale bar = 50 µm.
To determine whether ω-CgTxMVIIC affects membrane potential and Ca2+ oscillations, ventral horn neurons were recorded in current clamp during whole-cell recording with OGB1 (25 µM) in the patch solution. After dye diffusion (∼30 min after whole-cell access), oscillations were induced by bath application of NMDA (50–100 µM) and TTX (1 µM). Image sequences of Ca2+ fluorescence in dendrites were acquired while making simultaneous electrophysiological recordings before and during application of ω-CgTxMVIIC (5 µM, Fig. 8Bi). Individual dendritic branches were measured as a single region of interest, comparing mean fluorescence values between each peak and immediately preceding trough. ω-CgTxMVIIC had no effect on the amplitude of fluorescence oscillations (Fig. 8Bii; P>0.05, n = 5). Despite a change in the membrane potential oscillation waveform in Cd2+, there was no significant reduction in the duration due to CgTxMVIIC (P>0.05, n = 5). Thus, despite a profound block of VGCC current, ω-CgTxMVIIC does not impair membrane potential or Ca2+ oscillations.
Evoked NMDA Receptor EPSCs Lead to Localized Increases in Dendritic Ca2+
Intrinsic rhythmicity of individual cells requires KCa2 channel activation. Ca2+ entry through either NMDA receptors or VGCCs [33], [34] might activate KCa2 channels. During locomotion, NMDA receptors may be physiologically activated by glutamate release from either descending reticulospinal (RS) neurons [49], [62], or through oscillatory drive provided by local EINs [3], [63].
Ventral horn neurons were patched as before with OGB1 (50 µM) included in the patch solution. After dye diffusion, EIN-mediated synaptic responses were selectively recorded by placing a 2–5 µm stimulating electrode, over the ventro-lateral spinal cord (1–2 segments rostral to the patched cell) and using very low intensity stimulation (5–10 µA), which is insufficient to evoke RS axon stimulation (data not shown, see methods). In the presence of strychnine (5 µM), presumed EINs were repetitively stimulated (20–40 Hz; 5–10 stimuli, respectively) to evoke EPSCs recorded somatically in the patched cell while its dendrites were simultaneously imaged. Evoked monosynaptic EPSCs were recorded with an invariant delay from stimulation artifact to peak (Fig. 9A; 10.8±1.6 ms between cells, n = 6). Stimulation was performed while holding the cell between −70 mV and −35 mV, spanning the region of negative slope conductance of the NMDA receptor-mediated response (Fig. 9B). EPSCs were coincident with an increase in dye fluorescence that far outlasted the EPSC duration (Fig. 9Ci; n = 8). Low intensity stimulations reliably evoked fluorescent signals that were only found in dendrites, often in discrete locations within the arbor, and with considerable distance from the soma (>100 µm). In cells where the holding potential was varied, a voltage-dependent increase in evoked Ca2+ signals was found as the holding potential was increased from −80 mV to −30 mV (Fig. 9Ci). Evoked fluorescent changes at −35 mV were significantly greater than those at −80 mV (13.0±4.5% increase, P<0.05, n = 6). This confirms that EINs evoke localized Ca2+ signals in dendrites within values of membrane potential recorded during NMDA receptor-mediated oscillations.
Figure 9. Synaptically evoked Ca2+ signals mimic the voltage-dependency of the NMDA receptor current.

A Recording schematic: STIM = stimulating electrode, EIN = black, Ca2+ dye = green. B A cell was whole-cell voltage clamped in NBQX (5 µM), glycine (1 µM), strychnine (5 µM) to isolate evoked, NMDA receptor EPSCs, while the holding potential was varied between −80 mV and −40 mV. C A ventral horn neuron was whole-cell voltage clamped at −70 mV and filled with OGB1 (50 µM). An extracellular electrode was used to repetitively stimulate (25 Hz for 10 stimuli) excitatory interneurons (EINs) to evoke glutamatergic EPSCs at increasing holding potentials (denoted on right of trace). A transient increase in fluorescence (top) in the dendrite is coincident with the EPSC (bottom). Cii shows an enlargement and EPSC overlay of box in Ci. D Image of the ventral horn neuron in C (Di) demonstrating the localization (Dii; white) of evoked fluorescence at −35 mV. Evoked fluorescence changes relative to resting prestimulus fluorescence plotted against dendritic distance at each holding potential (Diii). Scale bar = 10 µm.
We confirmed that EIN-evoked, localized Ca2+ signals are NMDA receptor-dependent. Motoneurons were retrogradely labeled with the Ca2+-sensitive dye, OGB1 dextran applied to ventral roots (see methods). NMDA receptor-mediated responses were pharmacologically isolated with the AMPA receptor antagonist, NBQX (5 µM), and strychnine (5 µM). Single shocks were applied with tungsten microelectrodes (as before), while the extensively labeled dendritic tree of a single motoneuron was simultaneously imaged using a CCD camera. Highly localized, NMDA receptor-dependent Ca2+ transients were recorded. Application of the NMDA receptor antagonist, DL-AP5 (100 µM), reversibly abolished these evoked increases in Ca2+ fluorescence in all cells imaged (Fig. 10; P<0.05, n = 5). No reduction in Ca2+ transient amplitudes was recorded following NBQX application. Thus, EIN-evoked, localized Ca2+ signals are NMDA receptor-dependent.
Figure 10. EIN-evoked Ca2+ signals are NMDA receptor-dependent.

A EIN stimulation evoked fluorescent Ca2+ signals in a motor neuron dendrite retrogradely filled with the Ca2+-sensitive dye, OGB1-dextran (5 mM), in the presence of NBQX (5 µM), glycine (1 µM), and strychnine (5 µM) in Mg2+-free ringer. Ca2+ signals were recorded before (black), during (dark grey) and after (light grey) the application of D/L-AP5 (100 µM). Pooled responses (Aii) demonstrate that D/L-AP5 significantly and reversibly abolishes NMDA receptor-induced changes in Ca2+ fluorescence. Error bars express ± SEM. B Image of the dendrite recorded in Ai. Localized EIN-evoked fluorescent change is shown in Bii (arrow, white). Scale bar = 20 µm. C Recording schematic: STIM = stimulating electrode, EIN = black, Ca2+-dye filled motor neuron = green.
To determine whether RS axon-mediated excitation also leads to Ca2+ entry in ventral horn neuron dendrites, we retrogradely labeled motoneurons with OGB1 dextran to completely fill dendritic arbors (Fig. 11C). Mg2+ was removed from the ringer to facilitate NMDA receptor activation and potentiate Ca2+ signals for easier detection. A sharp microelectrode filled with the inert dye, Alexa Fluor 568 hydrazide, was used to penetrate, label (Fig. 11C), repetitively stimulate and record from RS axons (Fig. 11Aii) passing through the visualized dendritic arbors (Fig. 11Ci) imaged simultaneously with a CCD camera. Indeed, repetitive RS axon action potential firing is coincident with highly localized rises in dendritic fluorescence found at the axonal interface (Fig 11Ai, Cii-iii). However, to record a signal above background noise it was necessary to apply a more extended stimulus (30–45 Hz for 10–32 stimuli; Fig. 11Aii, n = 5).
Figure 11. RS axons evoke Ca2+ transients in motoneuron dendrites.

A–C Motoneurons were retrogradely filled via ventral roots with OGB1 dextran (B, green; 5 mM) applied to the trunk musculature. An RS axon passing through the dendritic field was filled with Alexa Fluor 568 hydrazide (B, Ci; red) via pressure ejection and repetitively stimulated (Aii, 32 Hz for 60 s) while simultaneously imaging the postsynaptic dendritic arbor of the filled motor neuron (Ci, green) with a CCD camera in Mg2+-free ringer. RS axon action potential firing (Aii) is coincident with dendritic Ca2+ transients (Ai). Each color represents the average response from different loci (Cii). A merged image of the dendritic field (Ci; green) is shown with the stimulated axon in addition to another filled axon below (Ci; red). The dendritic region where the axon makes en passant synaptic connections is outlined in blue (Cii). The corresponding blue region from Cii shows the location and relative signal intensities measured (Ciii; white), displayed as the peak/prestimulus fluorescence. * Denotes signal truncation for display purposes. Scale bar = 20 µm.
Synaptically Localized Activation of KCa2 Channels
Both EIN and RS axons evoke Ca2+ transients in CPG neurons. Thus, we compared input from RS neurons to responses from EINs to determine whether these evoked Ca2+ signals lead to KCa2 channel activation. If Ca2+ entry via NMDA receptors at glutamatergic synapses is sufficient to activate KCa2 channels, then EPSCs will be augmented by apamin by blocking an evoked outward current caused by synaptically evoked Ca2+ entry. Paired cell recordings were made between a presynaptic RS axon using a sharp microelectrode and a postsynaptic neuron recorded in whole-cell voltage clamp. Postsynaptic neurons were held at −60 mV, Mg2+ was removed from the superfusate and NBQX (5 µM), and strychnine (5 µM) added to isolate NMDA receptor EPSCs and glycine (1 µM) to ensure NMDA receptor activation. Superfusion of apamin (5 µM) over the preparation had no significant effect on the amplitude of single, monosynaptic RS-evoked NMDA receptor EPSCs (Fig. 12B; P>0.05, n = 10).
Figure 12. Synapse-dependent KCa2 channel activation.
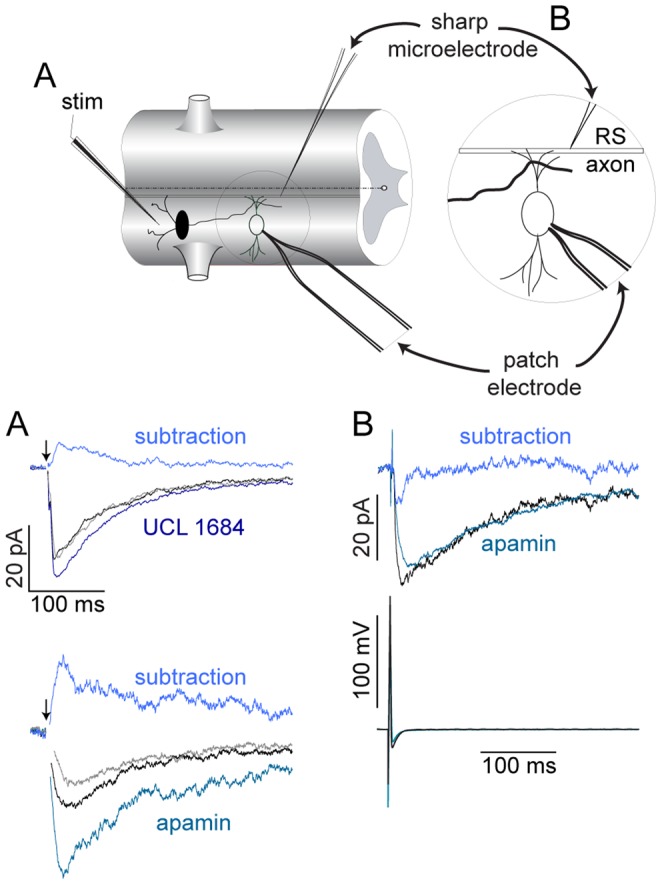
A–B Recording schematic depicting how responses were presynaptically evoked and recorded postsynaptically in ventral horn neurons in whole-cell patch clamp configuration using either extracellular EIN (A) or paired intracellular RS axon (B) stimulation. Average NMDA receptor EPSCs were recorded in voltage clamp at −60 mV (A, EIN) and −70 mV (B, RS) after removal of extracellular Mg2+ in control (black, 5 µM NBQX, 1 µM glycine and 5 µM strychnine), drug (100 nM UCL 1684, dark blue; 5 µM apamin, teal), and washout (grey) conditions. Corresponding subtractions for EIN-evoked responses reveal the underlying KCa2 current (A, top traces; light blue for UCL and apamin). Paired NMDA receptor EPSCs (B, top trace) from single RS axon action potentials (B, bottom trace) before (black) and after apamin (teal; 5 µM). Subtraction reveals no underlying KCa2 current (top trace, blue). Arrows denote time of single EIN stimulation. Stim = stimulating electrode.
To determine if EIN-evoked NMDA receptor EPSCs activate KCa2 channels. Evoked NMDA receptor EPSCs were achieved as before with an extracellular stimulating electrode in the presence of NBQX (5 µM), glycine (1 µM) and strychnine (5 µM) at a holding potential between −60 and −50 mV. Both apamin and the potent KCa2 channel inhibitor, UCL 1684, led to a significant and reversible increase of EPSC amplitude (Fig. 12A; 52±15%, P<0.01, n = 8 in 5 µM apamin; 9.1±4.3% from control, P<0.05, n = 7 in 100 nM UCL 1684). This demonstrates that EIN-evoked NMDA receptor EPSCs leads to Ca2+ entry sufficient to activate KCa2 channels necessary for the repolarization phase of the oscillation. Indeed, the components necessary for membrane potential oscillations are synaptically located.
Discussion
Nonlinear dynamics, such as oscillations, are found in all regions of the nervous system and underlie activities from encephalographic recordings of coordinated brainwave behavior [64], to bursting activity thought to underlie cognition [19]. Within neural networks, Ca2+ plays a vital role in the coordination of nonlinear membrane properties generating oscillatory behavior. In spinal premotor and motoneurons, Ca2+ entry through the plasma membrane contributes to both depolarization and indirectly to repolarization of neurons through the activation of Ca2+-dependent K+ currents (i.e. KCa2), tempering excitability and synaptic plasticity [14].
Nonlinear membrane properties in response to NMDA receptor activation and Ca2+ entry are central to the generation of membrane potential oscillations in lamprey ventral horn neurons which contribute to the precise rhythmic output of the spinal CPG [63]. Pattern generation requires both intrinsic neuron oscillatory properties [2], and precise synaptic connections for segmental [63], [65]–[67] and intersegmental [68] coordination. If intrinsic oscillatory properties activated by NMDA receptors contribute to locomotion, then synaptically released glutamate is required during rhythmic behavior. Both descending RS command neurons [69]–[71] and local EINs [3] lead to synaptic activation of NMDA receptors. By virtue of Mg2+ block [56], NMDA receptors also contribute a voltage-dependent depolarization in addition to Ca2+ entry as may VGCCs. However, depending on the location of KCa2 channels with respect to the NMDA receptor [2], VGCC [57] or internal store [72], [73], and the degree of depolarization, Ca2+ entry to the cytosol may subsequently activate an outward current. The relationship between this Ca2+ entry and secondary effectors will influence oscillatory properties extending from the cell to network level. Thus, it is important to determine the route of Ca2+ entry that drives K+ current needed to terminate bursting activity [8]. While Ca2+ release from internal stores has little or no effect on NMDA-induced fictive locomotion [29], both NMDA receptors and VGCCs are clearly necessary. We have focused on these latter routes of Ca2+ entry to determine their contribution to nonlinear properties of neurons during oscillatory activity.
Oscillations evoked in NMDA and TTX demonstrate a remarkably stereotypical value of peak depolarization very close to −40 mV. This is recorded regardless of the oscillation frequency, which is slower than locomotion. However, this slow oscillation frequency is reflective of NMDA-induced fictive swimming which is slower than either brainstem-evoked [1] or AMPA-induced [74] fictive swimming in isolated spinal cords. Additionally, in cells dialyzed with high concentrations of EGTA (5 mM; Fig. 1Ai, B, C), the oscillation is prolonged. When fast Ca2+ buffering occurs with BAPTA dialysis (Fig. 1Aii), or when KCa2 channels are blocked directly with UCL 1684 (Fig. 1C), subsequent repolarization may be mediated by voltage-dependent K+ conductances [75]. Thus, the variability of the oscillation duration depends on a complex cellular mechanism, which leads it to differ substantially from in vivo oscillations in which excitatory and inhibitory synaptic coupling are important.
In non-spiking conditions (i.e. in TTX or subthreshold oscillations during fictive swimming), NMDA receptors and VGCCs may be activated during the depolarization phase of ventral horn neuron oscillations, yet the relative thresholds for VGCC and NMDA receptor activation have not been examined in neurons within the spinal cord. Direct comparisons between the VGCC activation threshold (made by measuring Ba2+ currents) and the peak plateau membrane potential reached within the same cell during NMDA-evoked TTX-resistant oscillations revealed a sharp contrast (Fig. 2). Thresholds of voltage-activated Ba2+ currents were often more depolarized than the plateau peak (Fig. 2A), whereas NMDA application led to substantial current at less depolarized voltage steps within the membrane potential oscillation range (Fig. 2B, 3B). This demonstrates that VGCCs either remain largely inactive, or just begin to activate as the oscillation reaches its peak. While it is conceivable that even minimal activation of VGCCs would cause KCa2 channel activation due to either close coupling or the low threshold (EC50 ∼0.3 µM) for Ca2+-dependent KCa2 activation [76], our data suggest that NMDA receptor current is the dominant source of Ca2+ whereas VGCCs contribute very little Ca2+ within the oscillation range. Ventral horn neurons undergo phase-locked oscillations in Ca2+ i with the largest amplitudes in distal dendrites [33]. Thus, intracellular Ca2+ signals parallel NMDA-induced changes in membrane potential [77], localized to regions of the cell innervated by glutamatergic synapses [35]. Additionally, oscillations occur subthreshold to spiking, while action potentials in the same cells only increased somatic Ca2+, leaving dendritic Ca2+ oscillation amplitudes unaffected. For these reasons, we hypothesized that NMDA receptors provide the main source of Ca2+ underlying the repolarization phase of the oscillation via KCa2 channel activation, localized to dendritic synapses.
During behavior, NMDA receptors are necessarily synaptically activated and require depolarization to allow Ca2+ entry, whereas VGCCs can be activated independently of synaptic drive. NMDA receptors and VGCCs will differentially impact the dynamic range of intrinsic membrane properties [12]. Differences in location, voltage-sensitivity, activity-dependency, and kinetic profiles will correspondingly impact dendritic integration and computation [13], [47], [78], [79]. Furthermore, a number of other neuromodulatory systems [54] have been shown to modify these currents. The effectiveness of such metaplastic modulators will depend on their appropriate targeting in spinal neurons. In vertebrates [37] and specifically lamprey ventral horn neurons, VGCC activation following an action potential activates KCa2 channels to cause a late after-hyperpolarization [8], [30], [38], [80]. Yet, the route of Ca2+ entry that activates KCa2 currents at voltages subthreshold to action potentials during both locomotion and NMDA-dependent TTX-resistant oscillations has remained elusive. We now show that NMDA causes a profound hyperpolarized shift in the VGCC I-V relationship in ventral horn neurons, leading to activation at lower voltages observed during NMDA-dependent membrane potential oscillations (Figs. 2–5), indicating that primarily NMDA receptor-based Ca2+ entry occurs within the membrane potential oscillation range.
We demonstrate that Ca2+ entry is located physically close to the KCa2 channels that it activates. Step-evoked NMDA receptor-mediated currents activated a delayed outward current when neurons were dialyzed with EGTA (Fig. 3). Outward currents were prevented in Ba2+ (Fig. 1) or BAPTA (Fig. 3), which binds entering Ca2+ before binding endogenous buffers [52]. Similarly, BAPTA, but not EGTA, abolishes the NMDA-induced membrane potential repolarization (Fig. 1). These results indicate a close association between KCa2 channels and NMDA receptors and demonstrate the importance of proximity for achieving KCa2 channel activation.
Furthermore, Ca2+ imaging experiments corroborates the localized nature of synaptic activation and subsequent Ca2+ entry following stimulation of either local EINs (Fig. 10), or RS descending command neurons (Fig. 11). We demonstrate that Ca2+ increases are isolated to within short distances (∼10s of microns) around points of entry. However, such synaptic Ca2+ entry is capable of activating a substantial apamin- and UCL 1684-sensitive outward current following a single stimulus of presumed EINs (Fig. 12A), but not by RS axon activation (Fig. 12B). Repetitive and single RS axon stimulation during paired cell recording failed to evoke apamin-sensitive EPSPs [53] or currents (Fig. 12B), respectively. Ideally, paired recordings between EINs and ventral horn neurons would clarify whether expressly monosynaptic NMDA receptor EPSCs can activate KCa2 currents, but due to their small size, EINs are difficult to identify and individually stimulate [65].
EINs [3] and RS axons [69]–[71] are both sources of glutamatergic input onto ventral horn neuron dendrites, but serve different functions. RS axons provide descending excitation to spinal CPGs along the length of the spinal cord and thus cannot be phase-locked to multiple segments simultaneously. EINs, however, make local connections within a few consecutive CPG segments and phase-lock to corresponding ventral root discharges [65]. This lack of phase-locked excitation during RS neuron activation perhaps provides one explanation for why RS synaptic activation of Ca2+ entry does not directly couple to KCa2 current activation. Such a direct coupling will generate phase-inappropriate hyperpolarization, which would disrupt locomotion. Thus, the close spatial coupling of excitation-evoked Ca2+ entry and subsequent outward current activation will correspondingly impact is physiologically important for information processing and coordination of locomotor circuits.
The efficacy of BAPTA at blocking TTX-resistant oscillations and step-evoked late outward currents indicates a close spatial relationship between Ca2+ entry and KCa2 channel activation. We also show that EGTA is capable of interfering slightly with repolarization by prolonging the plateau duration during NMDA-dependent TTX-resistant oscillations (Fig. 1B). The latter finding implies that Ca2+ diffusing to KCa2 channels beyond the synaptic nanodomain may contribute to oscillatory properties of neurons. Nevertheless, the complete disruption of oscillations in BAPTA (Fig. 1A) demonstrates that the intimate spatial coupling of Ca2+ entry with KCa2 activation is necessary for the repetitive oscillations. Thus, oscillations may be achieved through a postsynaptic complex formed by NMDA receptors and KCa2 channels in ventral horn neuron dendrites to drive the cellular oscillatory rhythm. Similar structural arrangements have been reported elsewhere in the CNS [17] and may play an influential role in synaptic modification [14] underlying the short- and long-term modulation of the lamprey CPG.
Our imaging data also demonstrates that the majority of Ca2+ entering neurons during NMDA-induced oscillations requires NMDA receptor activation, but not VGCCs. Both Na+ and Ca2+ permeate the NMDA receptor to cause depolarization. Thus, step-evoked NMDA receptor-mediated currents may not directly reflect an increase in Ca2+ entry within the oscillation range. We used Ca2+-sensitive dyes, Fluo-5F and OGB1, as reporters of both increases in [Ca2+]i and location of Ca2+ entry. This allowed a direct comparison of Ca2+ entry between VGCCs and NMDA receptors. Over the membrane potential oscillation range (steps to −40mV), very little VGCC-dependent current is recorded and, correspondingly, very little Ca2+ dye fluorescence is detected at any somatic or dendritic location (Fig. 4A,C). However, NMDA caused both substantial voltage-dependent current and Ca2+ entry over the same voltage range (Fig. 4B–C). This demonstrates that NMDA-induced current correlates well to Ca2+ entry within the oscillation range. Inevitable voltage breakthrough resulting from uncorrectable space clamp errors may contribute to this enhancement in NMDA-evoked currents. However, block of VGCCs with ω-CgTxMVIIC, which selectively inhibits N- and P/Q-subtype VGCCs [60] accounting for over 70% of the total VGCC current [57], had little effect on the either membrane potential or Ca2+ oscillations (Fig. 8B). Similarly, Cd2+ abolishes VGCC current but did not prolong membrane potential oscillations. Thus, because oscillations persist and do not increase in duration, and Ca2+-sensitive dye fluorescence does not reduce after pharmacological block of VGCCs, it is unlikely that VGCCs contribute substantially to the repolarization. This suggests that the NMDA receptor, which is maximally activated by the peak of the oscillation, is the primary route of Ca2+ entry that subsequently activates KCa2 channels to drive the repolarizing phase of the oscillation.
Conclusion
Previously, VGCCs have been shown to activate KCa2 in lamprey spinal neurons [8], [30], [38], [80]. Our data demonstrates that there are most likely two sub-populations of apamin-sensitive KCa2 channels [81] which differ both spatially and functionally. One group is activated by ω-CgTxMVIIC-sensitive high voltage activated VGCCs following action potentials, and cause the action potential slow afterhyperpolarization [30], [53] that mediates spike frequency adaptation. The other group contains KCa2 channels located in dendrites, closely apposed to NMDA receptors activated by glutamate release from EINs. These KCa2 channels participate in the repolarization of membrane potential oscillations. The location and route of Ca2+ entry is central to the corresponding intrinsic membrane property and subsequent output.
It has proven difficult to sort the physiological roles of various Ca2+ ionophores whose activation is voltage-dependent in excitatory, synaptically driven neural behaviors. In the case of spinal motor oscillators, their function requires the activation of NMDA receptors, but Ca2+-activated outward currents might arise from a number of distinct systems. We demonstrate that the dominant Ca2+ signal for KCa2 channel activation responsible for nonlinear oscillations underlying rhythmogenesis in lamprey ventral horn neurons is entry through the NMDA receptor itself, particularly those receptors synaptically activated by local spinal EINs.
Funding Statement
National Institutes of Health Grant RO1 MH 084874; http://www.nimh.nih.gov/index.shtml. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sirota MG, Di Prisco GV, Dubuc R (2000) Stimulation of the mesencephalic locomotor region elicits controlled swimming in semi-intact lampreys. Eur J Neurosci 12: 4081–4092. [DOI] [PubMed] [Google Scholar]
- 2. Wallén P, Grillner S (1987) N-methyl-D-aspartate receptor-induced, inherent oscillatory activity in neurons active during fictive locomotion in the lamprey. J Neurosci 7: 2745–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buchanan JT, Grillner S (1987) Newly identified “glutamate interneurons” and their role in locomotion in the lamprey spinal cord. Science 236: 312–314. [DOI] [PubMed] [Google Scholar]
- 4. Alford S, Williams TL (1989) Endogenous activation of glycine and NMDA receptors in lamprey spinal cord during fictive locomotion. J Neurosci 9: 2792–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buchanan JT (2001) Contributions of identifiable neurons and neuron classes to lamprey vertebrate neurobiology. Prog Neurobiol 63: 441–466. [DOI] [PubMed] [Google Scholar]
- 6. Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, et al. (2005) International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacological Reviews 57: 463–472 doi:10.1124/pr.57.4.9. [DOI] [PubMed] [Google Scholar]
- 7. Faber ESL, Sah P (2003) Calcium-activated potassium channels: multiple contributions to neuronal function. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 9: 181–194. [DOI] [PubMed] [Google Scholar]
- 8. Manira El A, Tegnér J, Grillner S (1994) Calcium-dependent potassium channels play a critical role for burst termination in the locomotor network in lamprey. Journal of Neurophysiology 72: 1852–1861. [DOI] [PubMed] [Google Scholar]
- 9. Han P, Nakanishi ST, Tran MA, Whelan PJ (2007) Dopaminergic modulation of spinal neuronal excitability. Journal of Neuroscience 27: 13192–13204 doi:10.1523/JNEUROSCI.1279-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li X, Bennett DJ (2007) Apamin-sensitive calcium-activated potassium currents (SK) are activated by persistent calcium currents in rat motoneurons. Journal of Neurophysiology 97: 3314–3330 doi:10.1152/jn.01068.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wall MJ, Dale N (1995) A slowly activating Ca(2+)-dependent K+ current that plays a role in termination of swimming in Xenopus embryos. The Journal of Physiology 487 (Pt 3): 557–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Faber ESL (2010) Functional interplay between NMDA receptors, SK channels and voltage-gated Ca2+ channels regulates synaptic excitability in the medial prefrontal cortex. The Journal of Physiology 588: 1281–1292 doi:10.1113/jphysiol.2009.185645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bloodgood BL, Sabatini BL (2007) Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53: 249–260 doi:10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 14. Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, et al. (2002) Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. Journal of Neuroscience 22: 10163–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, et al. (2006) Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci 26: 1844–1853 doi:10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin MT, Luján R, Watanabe M, Adelman JP, Maylie J (2008) SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat Neurosci 11: 170–177 doi:10.1038/nn2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, et al. (2005) SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci 8: 642–649 doi:10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- 18. Grillner S (2006) Biological pattern generation: the cellular and computational logic of networks in motion. Neuron 52: 751–766 doi:10.1016/j.neuron.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 19. Wang X-J (2010) Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev 90: 1195–1268 doi:10.1152/physrev.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hill R, Matsushima T, Schotland J, Grillner S (1992) Apamin blocks the slow AHP in lamprey and delays termination of locomotor bursts. Neuroreport 3: 943–945. [DOI] [PubMed] [Google Scholar]
- 21. Harris-Warrick RM (2002) Voltage-sensitive ion channels in rhythmic motor systems. Curr Opin Neurobiol 12: 646–651. [DOI] [PubMed] [Google Scholar]
- 22. Wallén P, Buchanan JT, Grillner S, Hill RH, Christenson J, et al. (1989) Effects of 5-hydroxytryptamine on the afterhyperpolarization, spike frequency regulation, and oscillatory membrane properties in lamprey spinal cord neurons. Journal of Neurophysiology 61: 759–768. [DOI] [PubMed] [Google Scholar]
- 23. Hochman S, Jordan LM, Schmidt BJ (1994) TTX-resistant NMDA receptor-mediated voltage oscillations in mammalian lumbar motoneurons. Journal of Neurophysiology 72: 2559–2562. [DOI] [PubMed] [Google Scholar]
- 24. Cai X, Liang CW, Muralidharan S, Muralidharan S, Kao JPY, et al. (2004) Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 44: 351–364 doi:10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 25. Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, et al. (1992) Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron 9: 1163–1173. [DOI] [PubMed] [Google Scholar]
- 26. Faber ESL, Delaney AJ, Sah P (2005) SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci 8: 635–641 doi:10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- 27. Isaacson JS, Murphy GJ (2001) Glutamate-mediated extrasynaptic inhibition: direct coupling of NMDA receptors to Ca(2+)-activated K+ channels. Neuron 31: 1027–1034. [DOI] [PubMed] [Google Scholar]
- 28. Shah MM, Haylett DG (2002) K+ currents generated by NMDA receptor activation in rat hippocampal pyramidal neurons. Journal of Neurophysiology 87: 2983–2989. [DOI] [PubMed] [Google Scholar]
- 29. Krieger P, Hellgren-Kotaleski J, Kettunen P, Manira El AJ (2000) Interaction between metabotropic and ionotropic glutamate receptors regulates neuronal network activity. 20: 5382–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wikström MA, Manira El A (1998) Calcium influx through N- and P/Q-type channels activate apamin-sensitive calcium-dependent potassium channels generating the late afterhyperpolarization in lamprey spinal neurons. Eur J Neurosci 10: 1528–1532. [DOI] [PubMed] [Google Scholar]
- 31.Nanou E, Alpert MH, Alford S, Manira El A (2013) Differential regulation of synaptic transmission by pre- and postsynaptic KCa channels in the spinal locomotor network. Journal of Neurophysiology : in press. [DOI] [PMC free article] [PubMed]
- 32. Spruston N, Schiller Y, Stuart G, Sakmann B (1995) Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 268: 297–300. [DOI] [PubMed] [Google Scholar]
- 33. Viana Di Prisco G, Alford S (2004) Quantitative investigation of calcium signals for locomotor pattern generation in the lamprey spinal cord. Journal of Neurophysiology 92: 1796–1806 doi:10.1152/jn.00138.2004. [DOI] [PubMed] [Google Scholar]
- 34. Bacskai BJ, Wallén P, Lev-Ram V, Grillner S, Tsien RY (1995) Activity-related calcium dynamics in lamprey motoneurons as revealed by video-rate confocal microscopy. Neuron 14: 19–28. [DOI] [PubMed] [Google Scholar]
- 35. Moore LE, Buchanan JT, Murphey CR (1995) Localization and interaction of N-methyl-D-aspartate and non-N-methyl-D-aspartate receptors of lamprey spinal neurons. Biophys J 68: 96–103 doi:10.1016/S0006–3495(95)80163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huss M, Lansner A, Wallen P, Manira El A, Grillner S, et al. (2007) Roles of Ionic Currents in Lamprey CPG Neurons: A Modeling Study. Journal of Neurophysiology 97: 2696–2711 doi:10.1152/jn.00528.2006. [DOI] [PubMed] [Google Scholar]
- 37. Guertin PA, Hounsgaard J (1998) NMDA-Induced intrinsic voltage oscillations depend on L-type calcium channels in spinal motoneurons of adult turtles. Journal of Neurophysiology 80: 3380–3382. [DOI] [PubMed] [Google Scholar]
- 38. Meer DP, Buchanan JT (1992) Apamin reduces the late afterhyperpolarization of lamprey spinal neurons, with little effect on fictive swimming. Neurosci Lett 143: 1–4. [DOI] [PubMed] [Google Scholar]
- 39. Sah P, Bekkers JM (1996) Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci 16: 4537–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Regehr WG, Tank DW (1992) Calcium concentration dynamics produced by synaptic activation of CA1 hippocampal pyramidal cells. J Neurosci 12: 4202–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Llinás R (1988) The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science 242: 1654–1664 doi:10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- 42. Isope P, Hildebrand ME, Snutch TP (2012) Contributions of T-type voltage-gated calcium channels to postsynaptic calcium signaling within Purkinje neurons. Cerebellum 11: 651–665 doi:10.1007/s12311–010–0195–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mills LR, Niesen CE, So AP, Carlen PL, Spigelman I, et al. (1994) N-type Ca2+ channels are located on somata, dendrites, and a subpopulation of dendritic spines on live hippocampal pyramidal neurons. J Neurosci 14: 6815–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, et al. (1992) Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron 9: 1099–1115. [DOI] [PubMed] [Google Scholar]
- 45. Westenbroek RE, Ahlijanian MK, Catterall WA (1990) Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature 347: 281–284 doi:10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 46. Llinás R, Yarom Y (1981) Properties and distribution of ionic conductances generating electroresponsiveness of mammalian inferior olivary neurones in vitro. The Journal of Physiology 315: 569–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Herlitze S, Xie M, Han J, Hümmer A, Melnik-Martinez KV, et al. (2003) Targeting mechanisms of high voltage-activated Ca2+ channels. J Bioenerg Biomembr 35: 621–637. [DOI] [PubMed] [Google Scholar]
- 48. Evans RM, Zamponi GW (2006) Presynaptic Ca2+ channels–integration centers for neuronal signaling pathways. Trends Neurosci 29: 617–624 doi:10.1016/j.tins.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 49. Takahashi M, Freed R, Blackmer T, Alford S (2001) Calcium influx-independent depression of transmitter release by 5-HT at lamprey spinal cord synapses. The Journal of Physiology 532: 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N (2010) Computer Control of Microscopes Using µManager. Current Protocols in Molecular Biology 92: 14.20.1–14.20.17 doi:10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Augustine GJ, Adler EM, Charlton MP (1991) The calcium signal for transmitter secretion from presynaptic nerve terminals. Annals of the New York Academy of Sciences 635: 365–381. [DOI] [PubMed] [Google Scholar]
- 52. Adler EM, Augustine GJ, Duffy SN, Charlton MP (1991) Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci 11: 1496–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cangiano L, Wallén P, Grillner S (2002) Role of apamin-sensitive k(ca) channels for reticulospinal synaptic transmission to motoneuron and for the afterhyperpolarization. Journal of Neurophysiology 88: 289–299. [DOI] [PubMed] [Google Scholar]
- 54. Alford S, Schwartz E, Viana Di Prisco G (2003) The pharmacology of vertebrate spinal central pattern generators. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 9: 217–228. [DOI] [PubMed] [Google Scholar]
- 55. Manira El A, Bussières N (1997) Calcium channel subtypes in lamprey sensory and motor neurons. Journal of Neurophysiology 78: 1334–1340. [DOI] [PubMed] [Google Scholar]
- 56. Mayer ML, Westbrook GL, Guthrie PB (1984) Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309: 261–263. [DOI] [PubMed] [Google Scholar]
- 57. Manira El A, Bussières N (1997) Calcium channel subtypes in lamprey sensory and motor neurons. Journal of Neurophysiology 78: 1334–1340. [DOI] [PubMed] [Google Scholar]
- 58. Legendre P, Westbrook GL (1990) The inhibition of single N-methyl-D-aspartate-activated channels by zinc ions on cultured rat neurones. The Journal of Physiology 429: 429–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mayer ML, Vyklicky L, Westbrook GL (1989) Modulation of excitatory amino acid receptors by group IIB metal cations in cultured mouse hippocampal neurones. The Journal of Physiology 415: 329–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bussières N, Manira El A (1999) GABA(B) receptor activation inhibits N- and P/Q-type calcium channels in cultured lamprey sensory neurons. Brain Res 847: 175–185. [DOI] [PubMed] [Google Scholar]
- 61. McClellan AD, Kovalenko MO, Benes JA, Schulz DJ (2008) Spinal cord injury induces changes in electrophysiological properties and ion channel expression of reticulospinal neurons in larval lamprey. J Neurosci 28: 650–659 doi:10.1523/JNEUROSCI.3840-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McClellan AD (1988) Brainstem command systems for locomotion in the lamprey: localization of descending pathways in the spinal cord. Brain Res 457: 338–349. [DOI] [PubMed] [Google Scholar]
- 63. Buchanan JT, Cohen AH (1982) Activities of identified interneurons, motoneurons, and muscle fibers during fictive swimming in the lamprey and effects of reticulospinal and dorsal cell stimulation. Journal of Neurophysiology 47: 948–960. [DOI] [PubMed] [Google Scholar]
- 64. Tognoli E, Kelso JAS (2009) Brain coordination dynamics: True and false faces of phase synchrony and metastability. Prog Neurobiol 87: 31–40 doi:10.1016/j.pneurobio.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Buchanan JT, Grillner S, Cullheim S, Risling M (1989) Identification of excitatory interneurons contributing to generation of locomotion in lamprey: structure, pharmacology, and function. Journal of Neurophysiology 62: 59–69. [DOI] [PubMed] [Google Scholar]
- 66. Buchanan JT (1982) Identification of interneurons with contralateral, caudal axons in the lamprey spinal cord: synaptic interactions and morphology. Journal of Neurophysiology 47: 961–975. [DOI] [PubMed] [Google Scholar]
- 67. Buchanan JT (1993) Electrophysiological properties of identified classes of lamprey spinal neurons. Journal of Neurophysiology 70: 2313–2325. [DOI] [PubMed] [Google Scholar]
- 68. Williams TL, Sigvardt KA, Kopell N, Ermentrout GB, Remler MP (1990) Forcing of coupled nonlinear oscillators: studies of intersegmental coordination in the lamprey locomotor central pattern generator. Journal of Neurophysiology 64: 862–871. [DOI] [PubMed] [Google Scholar]
- 69. Buchanan JT, Brodin L, Dale N, Grillner S (1987) Reticulospinal neurones activate excitatory amino acid receptors. Brain Res 408: 321–325. [DOI] [PubMed] [Google Scholar]
- 70. Brodin L, Grillner S, Dubuc R, Ohta Y, Kasicki S, et al. (1988) Reticulospinal neurons in lamprey: transmitters, synaptic interactions and their role during locomotion. Arch Ital Biol 126: 317–345. [PubMed] [Google Scholar]
- 71. Kasicki S, Grillner S, Ohta Y, Dubuc R, Brodin L (1989) Phasic modulation of reticulospinal neurones during fictive locomotion and other types of spinal motor activity in lamprey. Brain Res 484: 203–216. [DOI] [PubMed] [Google Scholar]
- 72. Kettunen P, Krieger P, Hess D, Manira El A (2002) Signaling mechanisms of metabotropic glutamate receptor 5 subtype and its endogenous role in a locomotor network. J Neurosci 22: 1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fiorillo CD, Williams JT (1998) Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Curr Opin Neurobiol 394: 78–82 doi:10.1038/27919. [DOI] [PubMed] [Google Scholar]
- 74. Alford S, Grillner S (1990) CNQX and DNQX block non-NMDA synaptic transmission but not NMDA-evoked locomotion in lamprey spinal cord. Brain Res 506: 297–302. [DOI] [PubMed] [Google Scholar]
- 75. Hess D, Manira El A (2001) Characterization of a high-voltage-activated IA current with a role in spike timing and locomotor pattern generation. Proc Natl Acad Sci USA 98: 5276–5281 doi:10.1073/pnas.091096198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, et al. (1998) Mechanism of calcium gating in small-conductance calcium-activated potassium channels : Article : Nature. Nature 395: 503–507 doi:10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 77. Moore LE, Chub N, Tabak J, O'Donovan M (1999) NMDA-induced dendritic oscillations during a soma voltage clamp of chick spinal neurons. Journal of Neuroscience 19: 8271–8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Branco T, Häusser M (2011) Synaptic integration gradients in single cortical pyramidal cell dendrites. Neuron 69: 885–892 doi:10.1016/j.neuron.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Llinás RR (1988) The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science 242: 1654–1664. [DOI] [PubMed] [Google Scholar]
- 80. Hounsgaard J, Kiehn O, Mintz I (1988) Response properties of motoneurones in a slice preparation of the turtle spinal cord. The Journal of Physiology 398: 575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hill RH, Brodin L, Grillner S (1989) Activation of N-methyl-D-aspartate (NMDA) receptors augments repolarizing responses in lamprey spinal neurons. Brain Res 499: 388–392. [DOI] [PubMed] [Google Scholar]