

Abstract
Subclones homozygous for JAK2V617F are more common and larger in patients with polycythemia vera compared to essential thrombocythemia, but their role in determining phenotype remains unclear. We genotyped 4564 erythroid colonies from 59 patients with polycythemia vera or essential thrombocythemia to investigate whether the proportion of JAK2V617F -homozygous precursors, compared to heterozygous precursors, is associated with clinical or demographic features. In polycythemia vera, a higher proportion of homozygous-mutant precursors was associated with more extreme blood counts at diagnosis, consistent with a causal role for homozygosity in polycythemia vera pathogenesis. Larger numbers of homozygous-mutant colonies were associated with older age, and with male gender in polycythemia vera but female gender in essential thrombocythemia. These results suggest that age promotes development or expansion of homozygous-mutant clones and that gender modulates the phenotypic consequences of JAK2V617F homozygosity, thus providing a potential explanation for the long-standing observations of a preponderance of men with polycythemia vera but of women with essential thrombocythemia.
Introduction
The JAK2V617F mutation is found in over 95% of patients with polycythemia vera (PV) and approximately 60% of those with essential thrombocythemia (ET).1 but the additional mechanisms which determine their distinct clinical phenotypes remain unclear. Circumstantial evidence suggests that JAK2V617F homozygosity may have a role in determining the PV phenotype: i) homozygous-mutant hematopoietic precursors are isolated from a higher proportion of patients and form larger clones in PV compared to ET;2–4 ii) mouse models have demonstrated PV-like, rather than ET-like, phenotypes associated with higher JAK2V617F gene dosage;5,6 iii) higher JAK2V617F allele burdens in granulocyte DNA have been associated with higher hemoglobin levels and lower platelet counts in PV.7,8
However, several lines of evidence indicate that the role of JAK2V617F homozygosity in PV pathogenesis is not simple. Clonal analyses indicate that, in fact, small homozygous-mutant clones arise frequently and recurrently in both PV and ET, suggesting that the mere presence of homozygosity is insufficient for a PV phenotype, and additional factors are required.4 Studies of some,9–11 but not other12 knock-in mouse models have reported that heterozygosity for JAK2V617F is associated with marked erythrocytosis, with no further increase in hemoglobin levels in homozygous mice.10 Associations of JAK2V617F allele burden with blood counts show inconsistencies between studies13 and have not determined the extent to which allele burdens reflect the relative proportions of heterozygous and homozygous-mutant cells. Moreover, reduced levels of STAT1 activation have been identified in heterozygous-mutant erythroblasts from patients with PV, compared to those with ET, and may contribute causally to the PV phenotype.14 It is, therefore, unclear whether JAK2V617F homozygosity makes a significant contribution to the pathogenesis of human PV.
Here we used a clonal approach to test the hypothesis that JAK2V617F homozygosity has a causal role in the PV phenotype, and investigated its associations with demographic features, by utilizing genotype data from 4564 erythroid colonies from 59 patients with PV or ET.
Design and Methods
Patients were recruited from Addenbrooke’s Hospital, Cambridge, UK. Demographic and clinical features, together with methods used for colony culture and genotyping, have been previously described.4 Patients met diagnostic criteria for JAK2V617F-positive PV or ET according to the British Committee for Standards in Haematology.15,16 The study was approved by the Cambridge and Eastern Region Ethics Committee, patients gave written informed consent, and research was carried out in accordance with the Declaration of Helsinki.
To test the associations of the relative proportions of JAK2V617F-homozygous and heterozygous colonies with hematologic or demographic features, Poisson’s regression analyses were performed for the count of JAK2V617F-homozygous colonies, with total JAK2V617F-mutant colonies as an offset. P<0.05 was considered significant.
Results and Discussion
In order to assess the importance of JAK2V617F homozygosity in hematologic phenotype, we first investigated the relationship between relative proportions of JAK2V617F-homozygous and heterozygous hematopoietic precursors and hematologic features in PV and ET. We used BFU-E colonies because overactive erythropoiesis is a feature of both PV and JAK2V617F-positive ET1 and is important in distinguishing between the two. Moreover, the proportions of colony genotypes are similar between BFU-E and CFU-G in PV patients.3 Colony genotypes are summarized in Online Supplementary Table S1. Homozygous-mutant erythroid precursors were identified in 24 of 30 patients with PV and 15 of 29 patients with ET. The ratios of homozygous-mutant to heterozygous colonies were generally higher in PV than ET, but varied significantly within each subgroup, and were reproducible for individual patients.4
A causal relationship between JAK2V617F homozygosity and PV would predict an association between a higher proportion of JAK2V617F-homozygous colonies and more extreme PV-like clinical features, even within a cohort of PV patients. We used Poisson’s regression analyses to ask whether the relative proportion of homozygous-mutant precursors, as a proportion of all mutant precursors, was associated with hematologic features at diagnosis. This was a retrospective analysis since blood counts at diagnosis were being compared with the proportions of homozygous- and heterozygous-mutant precursors at the time of colony assays. In univariable analyses of the combined PV and ET cohort, a larger proportion of homozygous precursors were associated with higher hemoglobin, higher white cell count and lower platelet count at diagnosis (all P<0.0001). In a multivariable analysis, controling for diagnosis, age, gender, disease duration, presence of cytoreductive therapy and erythropoietin concentration in colony assays, an increased proportion of homozygous colonies remained significantly associated with higher hemoglobin, higher white cell count and lower platelet count at diagnosis in the whole patient group and the PV subgroup, but not in the ET subgroup (Table 1). A larger proportion of homozygous colonies were, therefore, independently associated with more extreme PV-like features at diagnosis in the whole JAK2V617F-positive cohort and in the PV subgroup.
Table 1.
Associations between the number of homozygous-mutant colonies (as a proportion of all mutant colonies) and full blood count parameters at diagnosis, in JAK2V617F-positive patients.
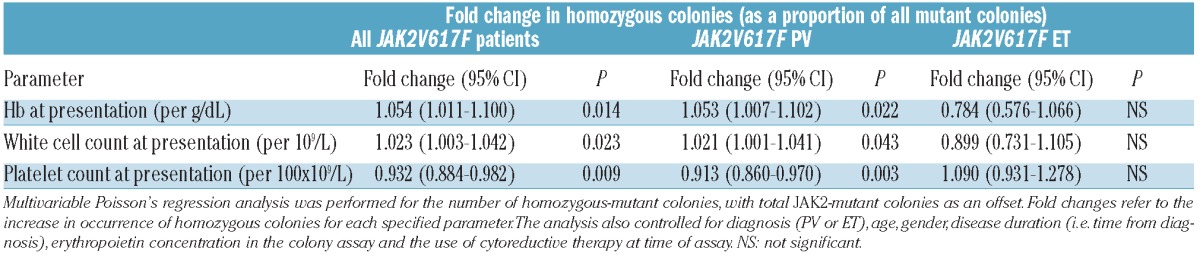
We considered the possibility that expansion of homozygous clones is secondary to the abnormal blood counts in PV, for example, through erythrocytosis suppressing erythropoietin levels and selecting for JAK2V617F-homozygous progenitors. However, multivariable analyses showed that a higher proportion of homozygous colonies was not associated with higher hemoglobin, higher white cell count or lower platelet count at the time of colony assays (data not shown). A higher proportion of homozygous-mutant colonies were, therefore, associated with more extreme PV-like blood counts at diagnosis but not at the time of colony assay, consistent with the concept that JAK2V617F homozygosity contributes causally to a PV phenotype.
In order to assess whether other clinical features predispose to or interact with JAK2V617F-homozygous clones, we next used Poisson’s regression analysis to test whether the number of homozygous-mutant precursors, as a proportion of all mutant precursors, was associated with demographic features or disease duration. In univariable analyses of the whole cohort, a larger proportion of homozygous colonies were associated with a diagnosis of PV (rather than ET), older age at assay, male sex and shorter time from diagnosis to assay (all P<0.0001). In a multivariable analysis, a larger proportion of homozygous colonies were associated with a diagnosis of PV and older age at time of assay (both P<0.0001, Table 2). A surprising interaction was identified between gender and diagnosis (P=0.003): an increased proportion of homozygous colonies were associated with male gender in PV (P=0.0006) but with female gender in ET (P=0.033). A significant interaction was also found between disease duration and diagnosis (P<0.0001), with an increased proportion of homozygous colonies associated with shorter disease duration in PV but with longer disease duration in ET (both P<0.0001). These associations were confirmed to be significant in multivariable analyses within the PV and ET subgroups (Table 2).
Table 2.
Associations between the number of homozygous-mutant colonies (as a proportion of all mutant colonies) and demographic features.

The association between an increased proportion of homozygous progenitors and increasing age, identified in both PV and ET, was also found in a cohort of 18 patients with JAK2 exon 12-mutated PV4 (P=0.024 in multivariable analysis) and was independent of disease duration. These data raise the possibility that age-dependent changes in DNA damage17 and/or DNA repair mechanisms18,19 contribute to an increased rate of mitotic recombination, and thus acquisition of JAK2 mutation homozygosity, in older patients. Alternatively, expansion of JAK2V617F-homozygous subclones may require additional mutations4 which accumulate with age.
The associations of an increased proportion of homozygous-mutant colonies with shorter disease duration in PV, but with increasing disease duration in ET, are likely to reflect distinct mechanisms in the two diseases. In ET, homozygous-mutant clones are present in approximately 50% of patients and are small.4 Our results indicate that, over time, mitotic recombination is more likely to occur and/or that an increasing proportion of ET patients develop clones above the detection threshold. By contrast, most PV patients have large homozygous-mutant clones.4 The association of an increased proportion of homozygous-mutant colonies with shorter time from diagnosis suggests that the size of homozygous-mutant clones, relative to heterozygous clones, decreases with time. This observation does not merely reflect suppressed erythropoietin levels and selection for homozygous-mutant precursors soon after diagnosis, since the association remained significant (P<0.0001) when we removed 3 patients with hemoglobin levels or hematocrit above the normal range at the time of colony assay, or when hemoglobin at time of assay was incorporated as a variable. Instead, we favor the possibility that hydroxycarbamide suppresses JAK2V617F-homozygous progenitors more than their heterozygous counterparts. This effect would be more evident in PV given the large number of homozygous-mutant colonies in most PV patients, and is consistent with evidence that hydroxycarbamide reduces granulocyte JAK2V617F allele burden especially in patients with PV and higher initial allele burdens.20,21
An association between male gender and increasing granulocyte JAK2V617F allele burden has been reported in PV, and was suggested to reflect gender-related differences in the frequency of mitotic recombination.22 Our data, however, indicate that the proportion of homozygous-mutant colonies was higher in males with PV than in females with ET, and do not favor this interpretation (Figure 1A). An alternative model is that JAK2V617F homozygosity occurs in both sexes, but that the phenotypic consequences of homozygosity are modulated by gender, so that the occurrence of a dominant JAK2V617F-homozygous clone is more likely to lead to development of PV in a male than in a female (Figure 1B). This would predict an excess of males with homozygosity in PV but an excess of females with homozygosity in ET, consistent with our data. The effect of JAK2V617F homozygosity on erythropoiesis could be modulated by gender-specific factors including a permissive effect of androgens in men and a constraining effect of iron depletion in pre-menopausal women. Importantly, this model provides an explanation for the long-standing observations that PV is more common in men23 whereas ET is more common in women.24
Figure 1.

Possible models to explain the associations of JAK2V617F homozygosity with gender in PV and ET. Each symbol represents one patient. Starting from a pool of individuals with subclinical JAK2V617F heterozygous mutations (pink symbols), there are three possible outcomes: acquisition of a significant JAK2V617F-homozygous clone (red symbols); acquisition of other mechanisms that can drive PV (green symbols); or maintenance of JAK2V617F heterozygosity alone. (A) JAK2V617F homozygosity occurs at a higher rate in males compared to females, but the phenotypic consequences of homozygosity are equivalent between males and females: most individuals develop PV and a minority develop ET. Of those individuals who remain JAK2V617F-heterozygous, some develop PV as a consequence of mechanisms other than JAK2V617F homozygosity, and others develop ET. (B) JAK2V617F homozygosity occurs at an equivalent rate in males and females but the phenotypic consequences are different: homozygosity in males is more likely to lead to PV than homozygosity in females, because of other factors that facilitate erythropoiesis in males and/or constrain erythropoiesis in females. The relative numbers of individuals with each genotype are approximations for illustrative purposes, and are not derived directly from the colony analysis.
Acknowledgments
The authors would like to thank the Cambridge Blood and Stem Cell Bank for sample collection.
Footnotes
The Online Version of this article has a Supplementary Appendix.
Funding
Work in ARG’s laboratory is supported by Leukemia and Lymphoma Research, the Wellcome Trust, the Medical Research Council, the Kay Kendall Leukaemia Fund, the Cambridge NIHR Biomedical Research Center, the Cambridge Experimental Cancer Medicine Centre and Leukemia and Lymphoma Society of America. ALG is a Kay Kendall junior clinical fellow. PJC is a Wellcome Trust senior clinical fellow.
Authorship and Disclosures
Information on authorship, contributions, and financial and other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Campbell PJ, Green AR. The myeloproliferative disorders. N Engl J Med. 2006;355(23):2452–66 [DOI] [PubMed] [Google Scholar]
- 2.Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006;108(7):2435–7 [DOI] [PubMed] [Google Scholar]
- 3.Dupont S, Masse A, James C, Teyssandier I, Lecluse Y, Larbret F, et al. The JAK2 V617F mutation triggers erythropoietin hypersensitivity and terminal erythroid amplification in primary cells from patients with polycythemia vera. Blood. 2007; 110(3):1013–21 [DOI] [PubMed] [Google Scholar]
- 4.Godfrey AL, Chen E, Pagano F, Ortmann CA, Silber Y, Bellosillo B, et al. JAK2V617F homozygosity arises commonly and recurrently in PV and ET, but PV is characterized by expansion of a dominant homozygous subclone. Blood. 2012;120(13):2704–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111(8):3931–40 [DOI] [PubMed] [Google Scholar]
- 6.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108 (5):1652–60 [DOI] [PubMed] [Google Scholar]
- 7.Vannucchi AM, Antonioli E, Guglielmelli P, Rambaldi A, Barosi G, Marchioli R, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110(3):840–6 [DOI] [PubMed] [Google Scholar]
- 8.Passamonti F, Rumi E, Pietra D, Elena C, Boveri E, Arcaini L, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010;24(9):1574–9 [DOI] [PubMed] [Google Scholar]
- 9.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010; 17(6):584–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010;115(17):3589–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marty C, Lacout C, Martin A, Hasan S, Jacquot S, Birling MC, et al. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood. 2010;116(5):783–7 [DOI] [PubMed] [Google Scholar]
- 12.Li J, Spensberger D, Ahn JS, Anand S, Beer PA, Ghevaert C, et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood. 2010;116(9):1528–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vannucchi AM, Antonioli E, Guglielmelli P, Pardanani A, Tefferi A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia. 2008;22(7):1299–307 [DOI] [PubMed] [Google Scholar]
- 14.Chen E, Beer PA, Godfrey AL, Ortmann CA, Li J, Costa-Pereira AP, et al. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell. 2010;18(5):524–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McMullin MF, Reilly JT, Campbell P, Bareford D, Green AR, Harrison CN, et al. Amendment to the guideline for diagnosis and investigation of polycythaemia/erythrocytosis. Br J Haematol. 2007; 138(6):821–2 [DOI] [PubMed] [Google Scholar]
- 16.Harrison CN, Bareford D, Butt N, Campbell P, Conneally E, Drummond M, et al. Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br J Haematol. 2010;149 (3):352–75 [DOI] [PubMed] [Google Scholar]
- 17.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–9 [DOI] [PubMed] [Google Scholar]
- 18.Preston CR, Flores C, Engels WR. Age-dependent usage of double-strand-break repair pathways. Curr Biol. 2006;16(20): 2009–15 [DOI] [PubMed] [Google Scholar]
- 19.McMurray MA, Gottschling DE. An age-induced switch to a hyper-recombinational state. Science. 2003;301(5641):1908–11 [DOI] [PubMed] [Google Scholar]
- 20.Ricksten A, Palmqvist L, Johansson P, Andreasson B. Rapid decline of JAK2V617F levels during hydroxyurea treatment in patients with polycythemia vera and essential thrombocythemia. Haematologica. 2008;93(8):1260–1 [DOI] [PubMed] [Google Scholar]
- 21.Besses C, Alvarez-Larran A, Martinez-Aviles L, Mojal S, Longaron R, Salar A, et al. Modulation of JAK2 V617F allele burden dynamics by hydroxycarbamide in polycythaemia vera and essential thrombocythaemia patients. Br J Haematol. 2011; 152(4):413–9 [DOI] [PubMed] [Google Scholar]
- 22.Stein BL, Williams DM, Wang NY, Rogers O, Isaacs MA, Pemmaraju N, et al. Sex differences in the JAK2 V617F allele burden in chronic myeloproliferative disorders. Haematologica. 2010;95(7):1090–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polycythemia vera: the natural history of 1213 patients followed for 20 years. Gruppo Italiano Studio Policitemia Ann Intern Med. 1995;123(9):656–64 [DOI] [PubMed] [Google Scholar]
- 24.Jensen MK, de Nully Brown P, Nielsen OJ, Hasselbalch HC. Incidence, clinical features and outcome of essential thrombocythaemia in a well defined geographical area. Eur J Haematol. 2000;65(2):132–9 [DOI] [PubMed] [Google Scholar]