

Abstract
We report results of a phase II trial of combination of melphalan, lenalidomide, and dexamethasone for the treatment of immunoglobulin light chain (AL) amyloidosis. The primary objectives were tolerability and hematologic response rate; secondary objectives were organ responses and survival. Treatment protocol consisted of melphalan 5 mg/m2/day for four days, lenalidomide 10 mg/day for 21 days and dexamethasone 20–40 mg once a week every 28 days for a total of 12 cycles. Sixteen subjects were enrolled of whom 14 completed at least 3 cycles and were evaluable for response. Grade 3/4 toxicities were experienced by 88% (n=14), the most common being myelosuppression (n=7). Dose reductions occurred in 85% (n=12 of 14) of subjects. Hematologic partial and complete responses were achieved by 43% (n=6 of 14) and 7% (n=1 of 14), respectively. The median overall survival has not been reached and median progression-free survival is 24 months. In conclusion, this combination is associated with significant myelosuppression leading to dose modifications and producing minor hematologic responses in AL amyloidosis.
Introduction
Immunoglobulin light chain amyloidosis (AL) is a plasma cell dyscrasia characterized by deposition of amyloid fibrils in various organs and tissues, derived from monoclonal light chains, leading to organ dysfunction.1–3 High-dose melphalan with autologous stem cell transplant (HDM/SCT) is an effective treatment with high complete hematologic response rates (CR) and is capable of producing durable remissions and prolonged overall survival.4–6 Only selected patients are eligible to receive HDM/SCT, and treatment-related mortality is in the range of 5–15%. More effective and widely applicable treatment modalities in AL amyloidosis are, therefore, needed.
Clinical trials of alternate treatment options have tested non-transplant melphalan-based strategies and novel therapeutics such as lenalidomide and bortezomib. Oral melphalan and dexamethasone (M-Dex) is a standard regimen for patients not eligible to receive HDM/SCT; reported complete response rates range from 13% to 33% and median overall survival ranges from 10.5 to 61.2 months.7–9 The efficacy and side effect profile of lenalidomide in multiple myeloma have prompted investigators to study its utility in AL amyloidosis. Lenalidomide and dexamethasone for AL amyloidosis have been evaluated in several phase II studies, with CR ranging from 29% to 42% by intention-to-treat analysis and a median time to response of six months.10,11 However, it should be noted that lenalidomide has unique toxicities in patients with this disease and the maximum tolerated dose is 15 mg/day, which is lower than the dose usually initially employed in the treatment of multiple myeloma.
In an effort to improve efficacy while maintaining tolerability, the opportunity for synergy is raised with melphalan and lenalidomide in combination with dexamethasone. This proof-of-concept has already been established in myeloma, and was recently studied by Moreau et al. in AL amyloidosis.12,13 Furthermore, combination regimens of alkylating agents and novel agents such as bortezomib, melphalan and dexamethasone, and, recently, lenalidomide, cyclophosphamide and dexamethasone (CRd, RdC) have also shown activity in phase II trials and are currently being compared to melphalan and dexamethasone in phase III trials.14,15
We designed a prospective phase II trial of melphalan, lenalidomide and low-dose dexamethasone (MLd) for the treatment of patients with AL amyloidosis. The primary end points were to assess the hematologic response rate, toxicity and tolerability of this regimen; secondary end points were to assess the organ response and overall survival.
Design and Methods
Eligibility criteria
Eligible patients were 18 years or older with biopsy-proven amyloidosis alongside evidence of a plasma cell dyscrasia evidenced by: a) monoclonal gammopathy by serum electrophoresis, immunofixation, free light chain assay or by urine immunofixation; and/or b) plasmacytosis in bone marrow of clonal origin. Those with a history of familial amyloidosis, evidence of secondary amyloidosis or concurrent overt multiple myeloma (> 30% plasma cells in bone marrow biopsy or lytic bone lesions) were excluded. Other inclusion criteria included a platelet count over 100 x 109/L, absolute neutrophil count over 1.5 x 109/L, AST/ALT less of 1.5 mg/dL than twice the upper limit of normal, total bilirubin and performance status according to Southwest Oncology Group (SWOG) of 2 or below. Patients with end-stage renal failure on dialysis or evidence of invasive malignancy in the last five years were excluded. Previous treatment for AL amyloidosis was permitted only if it was discontinued four weeks prior to enrollment and excluded patients who received cumulative doses of oral melphalan over 200 mg or received more than one course of high-dose melphalan and stem cell transplantation. Pregnant and nursing women were excluded. All women of childbearing age were required to practice abstinence or use dual-method contraception and undergo routine pregnancy testing based on regularity of menstruation. Men were also required to use contraception. All patients were counseled every four weeks about pregnancy precautions and risks of fetal exposure. This prospective, phase II, single arm, open label study was approved by the Institutional Review Board of the Boston University Medical Center in accordance with federal regulations and the Declaration of Helsinki. (http://clinicaltrials.gov/ct2/show/NCT00679367).
Treatment schedule
The treatment schedule consisted of melphalan 5 mg/m2/day for four days, lenalidomide 10 mg/day for 21 days, and dexamethasone 40 mg once weekly for a 28-day cycle, for 12 cycles or until disease progression or development of unacceptable toxicity. All patients received aspirin 325 mg/day to decrease the risk of venous thromboembolism associated with lenalidomide, and also a proton pump inhibitor to minimize gastritis associated with steroid use. All dose reductions for non-hematologic toxicities were prompted by The National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v3.0 grade 3 or 4 toxicities and performed in a stepwise fashion, with lenalidomide and dexamethasone reduced by 50% each time and melphalan by 25% each time. For any hematologic toxicity, lenalidomide and melphalan were reduced similarly by 50% and 25%, respectively, in an alternating fashion starting with lenalidomide. Patients unable to tolerate the minimum dose of any treatment agent had that agent discontinued. Routine antibiotic, antiviral or antifungal prophylaxis was not mandatory and was left to the discretion of the treating physician. All patients meeting eligibility criteria who had signed consent and completed at least 3 cycles of treatment were evaluated for response.
Response and toxicity assessment
The response criteria for hematologic and clinical/organ response used were standards defined by the consensus opinion from the Xth International Symposium on Amyloid and Amyloidosis.16 A hematologic complete response (CR) was defined as an absence of monoclonal protein in serum and urine by immunofixation electrophoresis, a normal bone marrow biopsy with less than 5% plasma cells without clonal dominance of κ or λ isotope, and a normal immunoglobulin serum free light chain ratio. A hematologic partial response (PR) was defined as a 50% or more improvement in quantifiable measures of plasma cell dyscrasia (i.e. marrow plasmacytosis, monoclonal (M) spike, urine free light chain excretion and serum free light chain ratio).
Hematologic responses were measured at three, six, nine months and at completion of 12 cycles of protocol-directed treatment. Organ responses were measured at the same time points.
Safety analysis
Safety analysis was performed on a weekly basis during protocol directed treatment and within 30 days of protocol completion or termination. Toxicity and adverse event data were recorded and graded using CTCAE v3.0. These data were monitored by the Data Safety Monitoring Board (DSMB) which is a multidisciplinary group consisting of the principal investigator and members of the Amyloid Treatment and Research Program at Boston University to ensure toxicities did not warrant termination of the study. This clinical trial was stopped prior to completion of the accrual goal of 35 patients due to toxicities and limited efficacy.
Results and Discussion
Sixteen subjects with AL amyloidosis were enrolled from May 2008 to June 2011. Median age was 70 years (range 57–84) and there were 11 women. There were 10 (63%) patients with λ clonal plasma cell dyscrasia. The median number of organs involved was 2 (range 1–5). There were 11 subjects (69%) with cardiac, 9 (44%) with renal and 4 (25%) with hepatic involvement. Of the subjects with cardiac involvement, 9 (81%) had cardiac bio-marker stage II or III defined by either a troponin I over 0.1 ng/mL or brain natriuretic peptide (BNP) level over 100 pg/mL or both. Of subjects with renal involvement, median 24-h urine protein excretion was 4056 mg (range 856–9511). Median time from initial diagnosis to enrollment in the clinical trial was five months (range 1–50). Five subjects (31%) had received prior therapies: 2 HDM/SCT, 2 bortezomib + dexamethasone and 1 lenalidomide + dexamethasone prior to enrollment in this study (Table 1).
Table 1.
Patients’ characteristics.
Treatment duration, dose modifications and hematologic outcomes for each patient are summarized in Figure 1. Of the 16 patients enrolled, 14 completed at least 3 cycles and were evaluable for response. Two patients who could not complete 3 cycles died within three months of enrollment. The median number of cycles received was 6 (range 1–12). Four patients (25%) completed 12 cycles of treatment as planned. Dose reductions occurred in 85% (n=12 of 14) subjects. Initial dose reductions in dexamethasone, lenalidomide, and melphalan occurred at a median of 2, 3 and 6 cycles respectively. Seventy-nine percent of patients (n=11 of 14) required a dose reduction of lenalidomide, 50% (n=7 of 14) required a dose reduction of both lenalidomide and dexamethasone and 43% (n=8 of 14) required a reduction in melphalan. The average patient underwent 2.5 dose reductions (range 0–5) over the treatment duration. The most common reason for lenalidomide dose reduction was myelosuppression.
Figure 1.
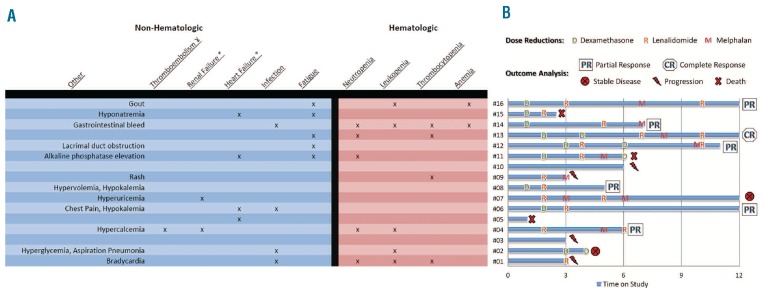
Toxicities, dose reductions and outcomes analysis by subject number. Chart A: Starts on right side with toxicities on x-axis and subject number on y-axis. #Subject; ψ: venous; *: acute. Chart B: Starts on bottom left corner with subject number on x-axis and duration of protocol-directed treatment on y-axis. Dose reductions are superimposed on individual time bars with outcome listed at the end.
Adverse events for each patient are illustrated in Table 2. A total of 4 patients were withdrawn from study: one was unable to comply with oral drug regimen due to G-tube placement, one due to grade 3 neutropenia, and 2 refused treatment by choice. All other subjects were continued on treatment to study completion, evidence of disease progression or death. Five subjects died: 3 within 100 days of treatment and 2 during follow up. Grade 3 and 4 toxicities were experienced by 88% (n=14 of 16) of subjects. Fifty-seven percent (n=8 of 14) of these subjects also had concomitant grade 3 or 4 hematologic toxicity with myelosuppression (n=7 of 8), thrombocytopenia (n=3 of 8) and/or anemia (n=2 of 8). Four patients developed infection during treatment (1 pneumonia and 3 sepsis), 3 (75%) of whom had associated grade 3 or 4 myelosuppression, and all of whom responded successfully to parenteral antibiotics. Additionally, 31% (n=5 of 16) experienced grade 3 fatigue as the most common non-hematologic side effect, 2 of 11 patients with cardiac involvement had grade 3 heart failure, and 2 patients with prior renal involvement went into grade 3 renal failure. One patient with renal failure who was compliant with prescribed aspirin thrombo-prophylaxis developed grade 3 venous thromboembolism while also receiving an erythropoietic stimulating agent.
A hematologic response was achieved by 50%, with partial responses (PR) and complete responses (CR) of 43% (n=6 of 14) and 7% (n=1 of 14), respectively. All responses were evident within three months of initiating protocol-directed treatment.
Organ response or progression was measured by comparing the final organ-specific laboratory value with the baseline value at the time of enrollment. Renal involvement was noted to be improved or stable in 8 of 9 patients with 5 patients having more than 30% reduction and one patient meeting consensus criteria with more than 50% reduction in 24-h urinary protein excretion. Progression of hepatic involvement was characterized by an over 50% increase in alkaline phosphatase in 2 of 4 patients per consensus criteria.19 Lastly, in patients with cardiac involvement, 7 of 11 were noted to have an over 30% increase in both troponin I and BNP. There was no patient with cardiac involvement with organ response based on consensus criteria. However, we and others have described a paradoxical elevation of BNP in the setting of lenalidomide use, and thus the BNP is not a reliable predictor of disease progression in this setting.20,21 Accordingly, 2 of 6 patients with both cardiac and renal involvement had an over 30% increase in troponin I and BNP despite an over 30% decrease in 24-h urine protein excretion.
The median overall survival has not been reached with a median follow up of 34 months and the median progression-free survival is 24 months (Figure 2).
Figure 2.
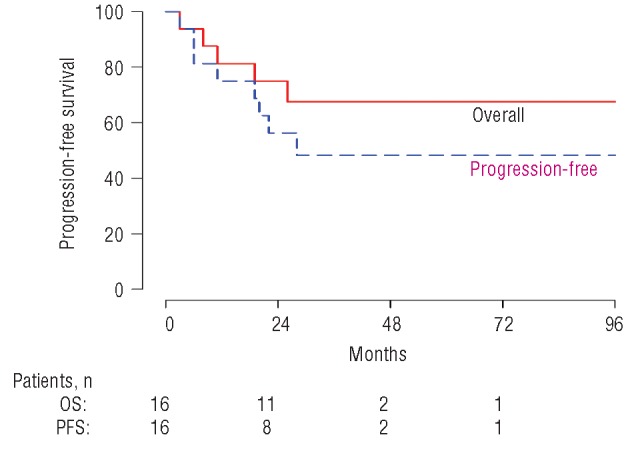
Overall and progression-free survival by Kaplan-Meier curves.
In summary, MLd led to an average of 2.5 dose reductions, frequent toxicities of myelosuppression and fatigue while providing an overall hematologic response rate of 50% in patients with AL amyloidosis.
The response rate does not appear to be superior to that seen with the use of lenalidomide plus dexamethasone alone or to melphalan plus dexamethasone alone.7,10,11 However, a higher rate of myelosuppression was seen with the combination. Side effects in this trial were greater than those seen in a previously reported study by Moreau et al. that may reflect the higher median age (70 years compared with 57 years in the French study). In that study, a higher lenalidomide dose was used, perhaps accounting for the higher response rate (75%, with a CR of 42%).13 Thus, the combination regimen may be more suitable for younger patients. Adding cyclophosphamide to lenalidomide appears to be better tolerated, and might be a more suitable combination for a wide range of patients.14,15 Of the two studies, the phase II study by Kumar et al. serves as the best comparison, as their study population had a median age of 64 years, a median of 2 organs involved, 63% of patients had cardiac involvement, and 31% were pre-treated. They achieved an overall response of 60%, with 40% very good partial response (VGPR) or greater. However, grade 3 or 4 toxicity was still 74% and dose reductions in lenalidomide were still required for most patients.
In conclusion, the combination of MLd required dose reductions to avoid high-grade myelosuppression and other toxicities and produced response rates that were no better than either melphalan or lenalidomide alone at higher doses, at least in this patient population. This clinical trial had to be stopped prior to the accrual goal by the investigators due to toxicities with limited efficacy. Addition of agents that may be synergistic with M-Dex (e.g. bortezomib) and do not cause significant myelotoxicity may provide a better tolerated 3-drug combination in AL amyloidosis.
Acknowledgments
The authors acknowledge the participation of colleagues in the Amyloid Treatment and Research Program at Boston University School of Medicine and the staff of cancer clinical trials office at Boston Medical Center. We also thank Dr. Gheorghe Doros for providing statistical analysis and survival curves. Most of all, the authors thank the patients who participated in our research program.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial and other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6): 583–96 [DOI] [PubMed] [Google Scholar]
- 2.Merlini G, Westermark P. The systemic amyloidoses: clearer understanding of the molecular mechanisms offers hope for more effective therapies. J Intern Med. 2004;255(2):159–78 [DOI] [PubMed] [Google Scholar]
- 3.Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45–59 [PubMed] [Google Scholar]
- 4.Cibeira MT, Sanchorawala V, Seldin DC, Quillen K, Berk JL, Dember LM, et al. Outcome of AL amyloidosis after high-dose melphalan and autologous stem cell transplantation: long-term results in a series of 421 patients. Blood. 2011;118(16):4346–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchorawala V, Skinner M, Quillen K, Finn KT, Doros G, Seldin DC. Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem-cell transplantation. Blood. 2007; 110(10):3561–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar S. Transplantation for amyloidosis. Curr Opin Oncol. 2007;19(2): 136–41 [DOI] [PubMed] [Google Scholar]
- 7.Palladini G, Perfetti V, Obici L, Caccialanza R, Semino A, Adami F, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103(8):2936–8 [DOI] [PubMed] [Google Scholar]
- 8.Sanchorawala V, Seldin DC, Berk JL, Sloan JM, Doros G, Skinner M. Oral cyclic melphalan and dexamethasone for patients with Al amyloidosis. Clin Lymphoma Myeloma Leuk. 2010;10(6):469–72 [DOI] [PubMed] [Google Scholar]
- 9.Lebovic D, Hoffman J, Levine BM, Hassoun H, Landau H, Goldsmith Y, et al. Predictors of survival in patients with systemic light-chain amyloidosis and cardiac involvement initially ineligible for stem cell transplantation and treated with oral melphalan and dexamethasone. Br J Haematol. 2008;143 (3):369–73 [DOI] [PubMed] [Google Scholar]
- 10.Sanchorawala V, Wright DG, Rosenzweig M, Finn KT, Fennessey S, Zeldis JB, et al. Lenalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 2 trial. Blood. 2007;109(2):492–6 [DOI] [PubMed] [Google Scholar]
- 11.Dispenzieri A, Lacy MQ, Zeldenrust SR, Hayman SR, Kumar SK, Geyer SM, et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood. 2007;109(2): 465–70 [DOI] [PubMed] [Google Scholar]
- 12.Palumbo A, Falco P, Corradini P, Falcone A, Di Raimondo F, Giuliani N, et al. Melphalan, prednisone, and lenalidomide treatment for newly diagnosed myeloma: a report from the GIMEMA--Italian Multiple Myeloma Network. J Clin Oncol. 2007;25 (28):4459–65 [DOI] [PubMed] [Google Scholar]
- 13.Moreau P, Jaccard A, Benboubker L, Royer B, Leleu X, Bridoux F, et al. Lenalidomide in combination with melphalan and dexamethasone in patients with newly diagnosed AL amyloidosis: a multicenter phase 1/2 dose-escalation study. Blood. 2010;116(23): 4777–82 [DOI] [PubMed] [Google Scholar]
- 14.Kastritis E, Terpos E, Roussou M, Gavriatopoulou M, Pamboukas C, Boletis I, et al. A phase 1/2 study of lenalidomide with low-dose oral cyclophosphamide and low-dose dexamethasone (RdC) in AL amyloidosis. Blood. 2012;119(23):5384–90 [DOI] [PubMed] [Google Scholar]
- 15.Kumar SK, Hayman SR, Buadi FK, Roy V, Lacy MQ, Gertz MA, et al. Lenalidomide, cyclophosphamide, and dexamethasone (CRd) for light-chain amyloidosis: long-term results from a phase 2 trial. Blood. 2012;119(21):4860–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol. 2005;79(4): 319–28 [DOI] [PubMed] [Google Scholar]